
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient trinuclear Ru(II)–Re(I) supramolecular photocatalysts for CO2 reduction based on a new tris-chelating bridging ligand built around a central aromatic ring†
Ambra M.
Cancelliere
a,
Fausto
Puntoriero
 a,
Scolastica
Serroni
a,
Sebastiano
Campagna
*a,
Yusuke
Tamaki
b,
Daiki
Saito
b and
Osamu
Ishitani
*b
a,
Scolastica
Serroni
a,
Sebastiano
Campagna
*a,
Yusuke
Tamaki
b,
Daiki
Saito
b and
Osamu
Ishitani
*b
aDipartimento di Scienze Chimiche, Biologiche, Farmaceutiche ed Ambientali, Università di Messina, Centro di Ricerca Interuniversitario per la Conversione Chimica dell'energia Solare (SOLAR-CHEM, sezione di Messina), Viale Ferdinando Stagno D'Alcontres, 31, Messina, 98166, Italy. E-mail: campagna@unime.it
bDepartment of Chemistry, School of Science, Tokyo Institute of Technology, 2-12-1-NE-1, O-okayama, Meguro-ku, Tokyo, 152-8550, Japan. E-mail: ishitani@chem.titech.ac.jp
First published on 14th December 2019
Abstract
We have designed and synthesized a new tris-chelating polypyridine ligand (bpy3Ph) suitable to be used as a bridging ligand (BL) for constructing various supramolecular photocatalysts. This BL is a phenylene ring with three ethylene chains at 1, 3, and 5 positions, of which the other terminals are connected to 2,2′-bipyridine moieties. The ligand bpy3Ph has been used to prepare, according to a multi-step synthetic protocol, trinuclear supramolecular photocatalysts containing different metal subunits. In particular, the compounds Ru2Re and RuRe2 have been prepared, containing different ratios of components based on Ru(dmb)32+-type and Re(dmb)(CO)3Cl-type units (dmb = 4,4′-dimethyl-2,2′-bipyridine), which can play the roles of photosensitizers and catalyst units for photocatalytic CO2 reduction, respectively. The trinuclear model Ru3 and mononuclear and dinuclear Ru and Ru2 precursor metal complexes, containing free chelating sites, have also been synthesized using the same bridging ligand. The absorption spectra, redox behaviour and photophysical properties of the new species indicate that there is no strong electronic interaction among the Ru and Re units. The trinuclear complexes Ru2Re and RuRe2 could photocatalyze CO2 reduction to CO with high selectivity (up to 97%), high efficiency (ΦCOs of 28% and 25%, respectively: BIH as a reductant), and high durability (TONCOs of 5232 and 6038, respectively: BIH as a reductant) which are the largest TONs for CO2 reduction using supramolecular photocatalysts in homogeneous solutions. The absence of negligible accumulation of the mono-reduced form of the photosensitizer indicates fast electron transfer to the catalyst unit(s) through the relatively large bridging ligand and is proposed to contribute to the outstanding photocatalytic properties of the new species, including their durability. The relevant photocatalytic behaviour of the new systems indicates new avenues for the design of extended bridging ligands capable of efficiently and functionally integrating photosensitizers and catalysts towards the preparation of new, larger supramolecular photocatalysts for selective CO2 reduction.
Introduction
Increase of atmospheric CO2 concentration arising from the mass consumption of “fossil resources” as an energy source causes serious problems, i.e., global warming and shortage of carbon and energy resources.1,2 This situation, in connection with the increasing global energy consumption, makes it essential for our society to find new, sustainable ways to obtain a huge amount of energy without the increase of CO2 emission. Sunlight is a promising candidate as a renewable energy source.3,4 Conversion of CO2 into energy-rich chemicals, such as CO and HCOOH, by using solar light as the energy source possibly can provide a solution to both shortage of carbon and energy resources and global warming.5For this goal, photocatalytic reduction of CO2 using metal complexes which can act as a redox photosensitizer (PS) and a catalyst (CAT) has been extensively investigated.6,7 The PS promotes photochemical electron transfer from an electron donor to the CAT. A suitable PS, which induces an efficient photocatalytic reaction via the reductive quenching process of its excited state, should exhibit strong absorption in the visible region for efficiently utilizing solar light, and its excited state should have a relatively long lifetime, strong oxidation power in the excited state, and high stability of the one-electron reduced species.8 Ru(II) polypyridine complexes fulfil all the above mentioned requirements and are widely employed in photocatalytic CO2 reduction. The CAT for CO2 reduction accepts and accumulates multiple electrons from the PS and should show high selectivity of CO2 reduction against competitive H2 evolution. Various Re(I) diimine carbonyl complexes have been reported as such CATs.9
In this research area, multinuclear metal complexes, the so-called “supramolecular photocatalysts”, have been widely studied in the recent years.10 The supramolecular photocatalysts contain both units, i.e. PS and CAT units, in one supramolecular photocatalyst, connected to one another by a bridging ligand (BL).11 Rapid electron transfer from the photochemically reduced PS unit – which can be produced upon electron transfer from a sacrificial reductant or from an electrode in a photoelectrochemical cell12 – to the CAT unit can proceed across the BL:13 this can lead to higher performances of the supramolecular photocatalysts and their higher durability compared to a mixed system of the corresponding mononuclear metal complexes. The photocatalysis behaviour of the supramolecular photocatalysts strongly depends on the properties of the BL: for example, most of the efficient supramolecular photocatalysts reported in the literature for CO2 reduction are made of a Ru(II) PS and Re(I) CAT containing a non-conjugated bridging ligand in which the metal chelating fragments (usually polypyridine ligands) are connected by an alkyl chain,14 typically an ethylene group. In fact, the introduction of a conjugated chain as a substituent of the diimine ligand of the Re(I) CAT unit induces shift of its reduction potential to more positive values, which causes lower catalysis activity of the Re unit. This limitation in BL design inhibits diversification of structures of supramolecular photocatalysts and imposes severe restrictions to the development of supramolecular photocatalytic systems, thereby causing a paradigmatic problem for the development of the whole research field. On the other side, because of the shortness of ethylene chains, back electron transfer between the PS and CAT can occur, thereby limiting the efficiency of the overall process: extended, large bridging ligands would therefore be desirable.
We now propose a new BL (bpy3Ph) suitable for constructing various supramolecular photocatalysts. This BL is a phenylene ring with three ethylene chains at 1, 3, and 5 positions, of which the other terminals are connected with 2,2′-bipyridine moieties (Chart 1). In the BL, all of the 2,2′-bipyridine moieties are equivalent, and various trinuclear metal complexes with different ratios of different metal centers can be selectively synthesized by using relatively simple step-by-step synthetic methods as described below. The distances among the metal-complex units connected by this new BL are much longer compared to those of the reported supramolecular photocatalysts in which each metal-complex unit is connected by ethylene chain(s).15–17 This and the rigidity of the phenyl ring should reduce back electron transfer via direct collision of the photosensitizer unit with the reduced catalyst unit. A non-conjugated chain such as the ethylene chain between the PS and CAT units is necessary to maintain the reduction power of the CAT.15 However, since the three ethylene chains bearing the metal center units are covalently linked to one another through one aromatic ring of which carbons at the 1, 3, and 5 positions have strong electronic interactions, trough-bond forward electron transfer could be promoted by the large electronic interactions between the metal-based units.13b We can anticipate that this is indeed what occurs. Syntheses, photophysical and electrochemical properties, and photocatalytic abilities of trinuclear Ru(II)–Re(I) complexes based on this new BL, in which the ratio of the Re(I) and Ru(II) centers is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 or 2:1 (Chart 1), are reported.
:2 or 2:1 (Chart 1), are reported.
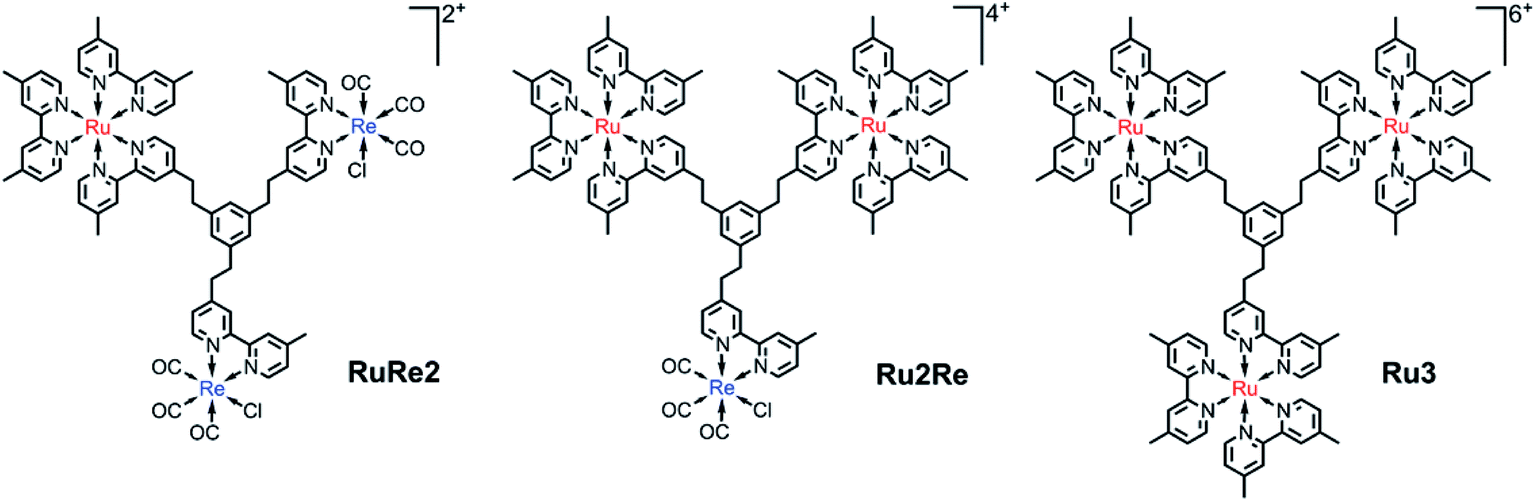 | ||
| Chart 1 Structures and abbreviations of the synthesized trinuclear complexes used as PS–CAT supramolecular photocatalysts (RuRe2 and Ru2Re), together with Ru3, used as a model. The counter anions are PF6−. | ||
Results and discussion
Synthesis
The synthesis of the novel bridging ligand bpy3Ph was accomplished in one step using 4,4′-dimethyl-2,2′-bipyridine (dmb) and 1,3,5-tris(bromomethyl)benzene. One of the methyl positions of dmb was lithiated using commercially available lithiumdiisopropylamide (LDA) slowly added to the dmb solution at −33 °C. After that, a THF solution of 1,3,5-tris(bromomethyl)benzene was added dropwise and the resulting mixture was left stirring overnight at room temperature.18A chromatography column on silica of the crude resulted in the isolation of the product bpy3Ph. The isolated yield was 57%. The characterization was performed by NMR spectroscopy (Fig. S1†). The signal of the aliphatic protons of 1,3,5-tris(bromomethyl)benzene (δ = 4.45 ppm) was absent in the product spectrum, but it presents a singlet signal at δ = 2.82 ppm for all the twelve protons of the ethylene chain.The ligand bpy3Ph has been employed for the preparation of three different multinuclear complexes by using step-by-step coordination of Ru(II) and/or Re(I) precursor complexes (Scheme 1). By using different molar ratios between the ligand and the cis-[Ru(dmb)2Cl2·2H2O] metal complex, the Ru, Ru2 and Ru3 species have been prepared. In all the cases, the reaction procedure gave a mixture of complexes, subsequently separated by ion-exchange chromatography. The successive reactions of the complexes, Ru and Ru2, containing respectively two and one free dmb-type moieties, with Re(CO)5Cl gave the desired RuRe2 and Ru2Re complexes (Scheme 1). All of yields of the trinuclear complexes were reasonably high as shown in Scheme 1.
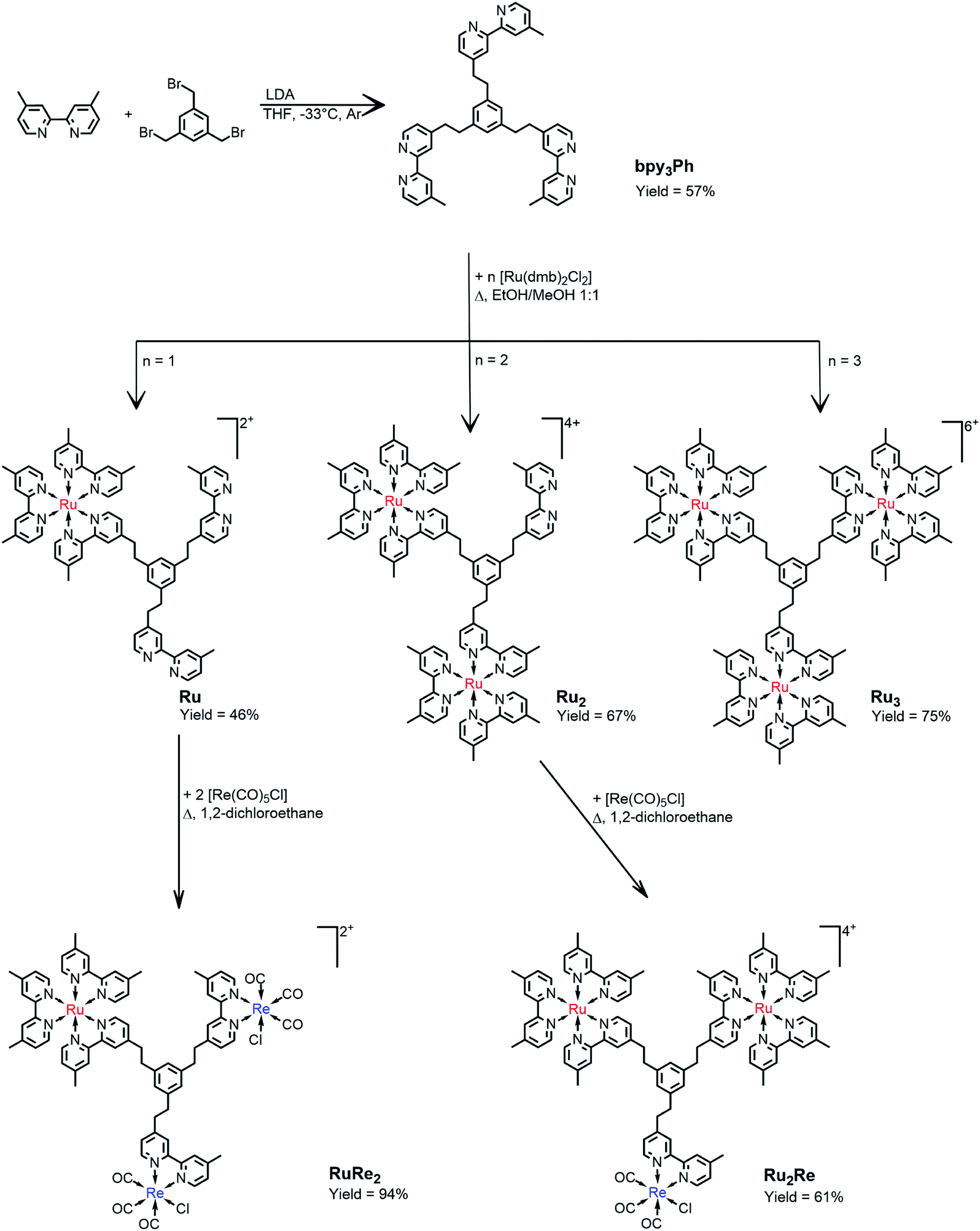 | ||
| Scheme 1 Schematization of the synthetic routes. | ||
Absorption spectra and photophysical properties
UV-vis absorption spectra of the complexes are shown in Fig. 1. The band at λmax = 460 nm was observed in all the complexes and is attributed to the spin-allowed metal-to-ligand charge transfer (MLCT) band of the Ru(II) centre(s). The shapes of the absorption spectra of all the new compounds are very similar to one another and the absorption coefficient is dependent on the number of Ru(II) units (see Table 1). The stronger absorption band at λmax = ∼285 nm can be assigned mainly to the π–π* transition of the diimine moieties. The 1MLCT band of the Re(I) subunit(s) should contribute to the absorption in the range λmax = 350–390 nm.19,20 It is noteworthy that the absorption spectrum of each multinuclear species closely overlaps the calculated weighted sum of the absorption spectra of the relevant individual monomer components (see Fig. 1), indicating that there is no strong electronic interaction among metal centres in the multicomponent compounds.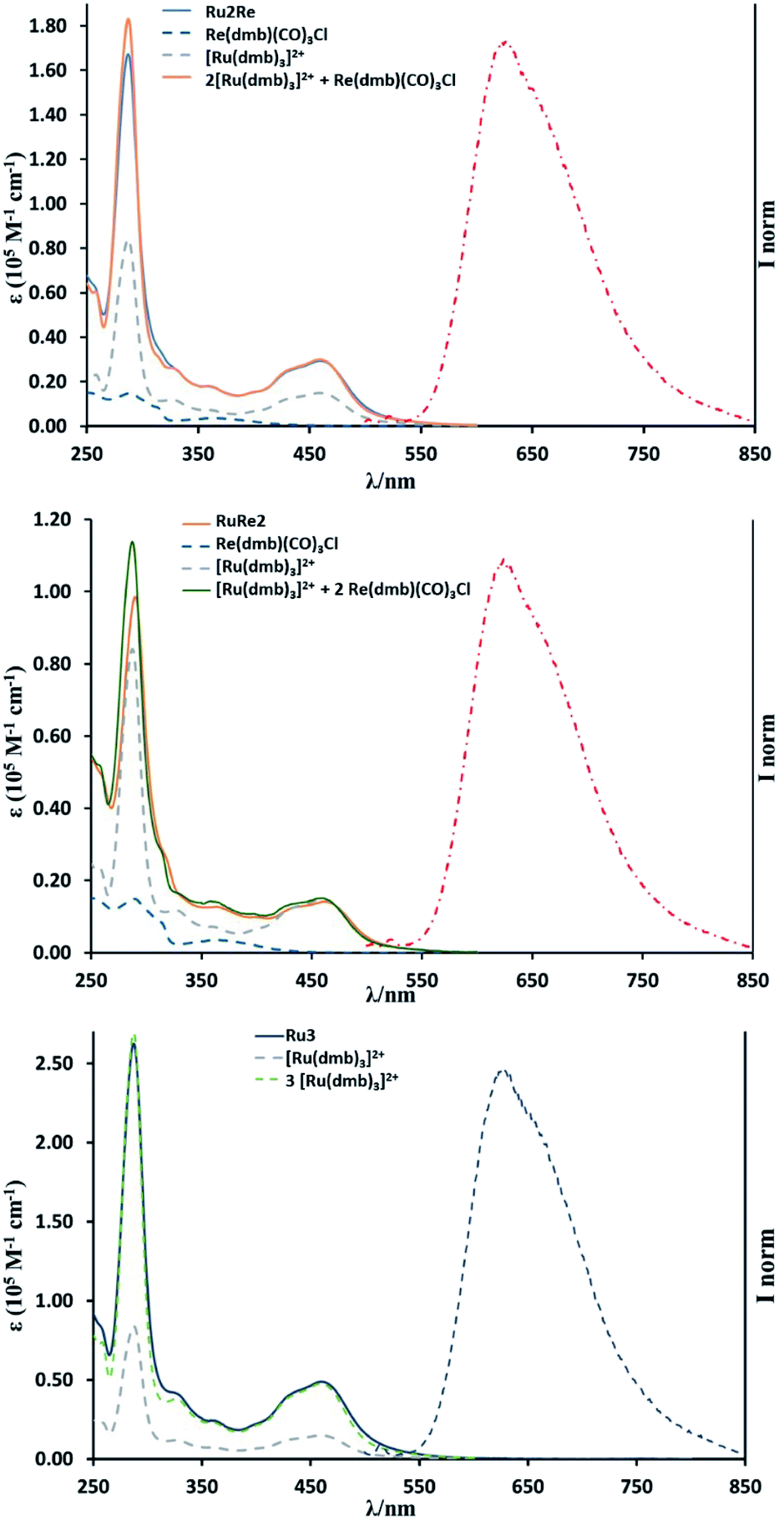 | ||
| Fig. 1 UV-vis absorption spectra of RuRe2 (top), Ru2Re (middle) and Ru3 (bottom) in MeCN at room temperature. The absorption spectra of the model species Ru(dmb)32+ and Re(dmb)(CO)3Cl are also shown for comparison purposes, along with the calculated weighted sum spectra of the isolated components (see text for details). Emission spectra of RuRe2 (top), Ru2Re (middle), and Ru3 (bottom) in MeCN are also shown on the right side of each panel. Excitation wavelength is 450 nm. | ||
| Absorption | Luminescence | |||
|---|---|---|---|---|
| λ max /nm (ε460/M−1 cm−1) | λ max/nm | τ /ns | Φ em | |
| a All data are measured in MeCN at 293 K. b Only the low energy maximum is reported. c Data within parenthesis refer to air-equilibrated solution. d From ref. 21. e From ref. 22. f From ref. 23. g From ref. 24. h In MeTHF. | ||||
| Ru | 460 (14760) |
624 | 878 (120) | 0.085 (0.016) |
| Ru2 | 460 (31180) |
625 | 851 (115) | 0.086 (0.016) |
| Ru3 | 460 (44500) |
625 | 847 (117) | 0.087 (0.016) |
| RuRe2 | 460 (14500) |
624 | 867 (119) | 0.085 (0.016) |
| Ru2Re | 460 (28550) |
625 | 852 (117) | 0.086 (0.016) |
| [Ru(dmb)3]2+d | 458 (16300) |
622 | 875 | 0.089e |
| Re(dmb)CO3Cl | 364 (3630)f | 600g | 49g | 0.0057h |
Fig. 1 also shows the emission spectra of RuRe2, Ru2Re, and Ru3 excited at λex = 450 nm: all the trinuclear complexes exhibit an emission band at around 625 nm, similar to that of [Ru(dmb)3]2+, which is attributed to the 3MLCT excited state of the Ru(II) unit(s). Table 1 summarizes photophysical data of all the complexes. The emission spectra, lifetimes and quantum yields of RuRe2 and Ru2Re, independent of the presence of Re(I) units in their structure, are very similar to those of Ru3, Ru2, and Ru. The shapes of the emission spectra of the multinuclear compounds are independent of the excitation wavelength, at least in the range 350–530 nm. These results clearly indicate that there is no strong electronic interaction between the excited Ru unit and the others, and intramolecular electron transfer from the excited Ru unit to the Re unit as well as self-quenching of the excited Ru units do not occur in the trinuclear species. Excitation spectra of Ru2Re and RuRe2 overlap with their respective absorption spectra, suggesting that energy transfer from the 3MLCT state of the Re(I) unit(s) (responsible for the 600 nm emission of the model species Re(dmb)(CO)3Cl reported in Table 1) to the 3MLCT state of the low-lying Ru(II) unit(s) largely takes place in RuRe2 and Ru2Re when the Re unit is excited. However, due to the small absorption of the Re(I) unit in comparison to the absorption of the Ru(II) units (see data in Table 1) and the less intense intrinsic emission of the Re units, we cannot state if such an energy transfer is effectively quantitative.
Redox properties
The redox behaviour of all the metal complexes synthesized is summarized in Table 2, together with data of model species. In the cyclic voltammograms (CVs) and differential pulse voltammograms (DPVs) of the complexes (Fig. S2–S5†), there are different ligand-based reduction and metal-based oxidation processes. Comparing the redox data of the new complexes with those of [Ru(dmb)3]2+ and [Re(dmb)(CO)3Cl] reported in the literature,23–25 it is possible to safely assign the various processes to specific subunits. On oxidation, the first reversible and second irreversible waves of RuRe2 and Ru2Re are attributable to the Ru(II) and Re(I) units, respectively. In all the other complexes, only a single oxidation process takes place until +1.60 V vs. SCE, with an E1/2 within 1.14–1.17 V, which can be attributed to the oxidation of the Ru(II) centre(s). Interestingly, the number of exchanged electrons, measured by comparison of the area of DPVs, corresponds to the number of Ru(II) centres in the structures, indicating that negligible interaction takes place between the metal centres in the multinuclear compounds from an electrochemical viewpoint, a common behaviour for multiruthenium species built around a multichelating ligand with reduced conjugation.26| E 1/2, V vs. SCEa | |||||
|---|---|---|---|---|---|
| E Ox2 | E Ox1 | E Red1 | E Red2 | E Red3 | |
| a Electrochemical properties measured at room temperature in MeCN containing 0.1 M TBAH. All values are obtained using the redox couple ferrocene/ferrocenium (395 mV vs. SCE in acetonitrile) as the internal reference. The numbers within parentheses refer to the number of exchanged electrons. Irr indicates an irreversible process: in this case, the E values reported in the table refer to peak potentials in pulse voltammetry experiments. b From ref. 27. c From ref. 24. | |||||
| Ru | +1.15[1] | −1.44[1] | −1.63[1] | −1.85[1] | |
| Ru2 | +1.15[2] | −1.43[2] | −1.61[2] | −1.89[2] | |
| Ru3 | +1.17[3] | −1.45[3] | −1.60[3] | −1.92[3] | |
| RuRe2 | +1.36 irr | +1.14[1] | −1.41[3] | −1.67[1] | −1.88[1] |
| Ru2Re | +1.38 irr | +1.14[2] | −1.42[3] | −1.58[2] | −1.85[2] |
| [Ru(dmb)3]2+b | +1.10[1] | −1.45[1] | |||
| [Re(dmb)(CO)3Cl]c | +1.36 irr | −1.43 [1] | |||
On reduction, a series of reversible processes takes place for all the complexes, at potentials that are typical of successive reductions of the various dmb ligands coordinated to a single metal centre. For example, in Ru, the three successive reduction processes are assigned to successive one-electron reduction of the three polypyridine ligands coordinated to the Ru(II) centre, including the one belonging to the bridging ligand. For Ru2 and Ru3, all the reduction processes involve two and three electrons, respectively: in both cases, the first process is assigned to simultaneous one-electron reduction of polypyridine ligands coordinated to different metal centres (two and three metal centres in Ru2 and Ru3, respectively); the second and third reduction processes are assigned to simultaneous one-electron reduction of the not yet reduced coordinated dmb-type ligands. In the mixed-metal Ru2Re and RuRe2 complexes, the assignment of the first reduction process – involving three simultaneous one-electron processes – follows that for Ru3, whereas the number of exchanged electrons for the second and third reduction processes is reduced compared to the number of electrons involved in the first reduction process, because the Re(I) centre(s) has a single dmb-type ligand. It is noteworthy that the reduction potential of the Re unit and the first reduction potential of the Ru unit(s) are very similar to each other in Ru2Re and RuRe2. The reduction patterns of the new compounds, like their oxidation behaviour, confirm that each metal-based subunit is electrochemically independent of the presence of the others in the multicomponent arrays.
Photocatalysis for CO2 reduction
Photocatalytic reactions for the reduction of CO2 were performed using both Ru2Re and RuRe2 as supramolecular photocatalysts. In order to achieve the same light-absorbing abilities between two systems, thereby normalizing the light absorption and allowing a direct comparison of the properties, the concentrations of the PS units were adjusted to 50 μM in the photocatalytic reactions, i.e. [Ru2Re] = 25 μM and [RuRe2] = 50 μM were selected. In a typical run, a 3 mL CO2-saturated mixed solution of N,N-dimethylacetamide–triethanolamine (DMA–TEOA; 5:1 v/v) containing RuRe2 (50 μM) and 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH; 0.1 M) as a sacrificial electron donor was irradiated using a LED light source (530 nm, 4 mW), giving CO almost selectively (Fig. 2b). After 20 h of irradiation, the turnover number for CO production (TONCO) reached 1850 based on RuRe2 used. The selectivity of CO formation was ΓCO = 92%. Since CO is a two-electron reduced compound of CO2 and BIH has been reported to function as a two-electron donor,28 CO formation of TONCO = 1850 means the consumption of 92.5 mM (=0.05 × 1850) BIH, which is 92.5% of BIH added originally (100 mM). This should be one of the reasons for slower CO formation with longer irradiation. Therefore, to observe the potential durability of RuRe2, the photocatalytic reaction was conducted using a 1/5 concentration of RuRe2 (10 μM) with longer irradiation time (Fig. S6†). After 60 h of irradiation, TONCO reached 5232. After irradiation for 60 h, further irradiation did not produce additional CO in both cases using Ru2Re and RuRe2 even though an enough amount of BIH still remained in the reaction solution. This is one of the largest turnover numbers of CO2 reduction using photocatalysts in homogeneous solutions. It is noteworthy that the mixed systems of separated PSs and CATs, which reported tens of thousands of TONs only based on the catalyst, contain hundreds-to-thousand-times concentration of the PS compared with the CAT (in the present case, obviously the PS–CAT ratio is 1:2 for RuRe2 and 2:1 in Ru2Re). In such mixed systems, TONs based on the PS concentration used are only tens or less than that, which is a much smaller TON compared to the present case for the supramolecular photocatalyst RuRe2. The quantum yield for CO production was determined as ΦCO = 25% using 480 nm light (light intensity: 1.0 × 10−8 einstein s−1). Fig. 3 shows absorption spectral changes during irradiation; it can be noted that only negligible changes occur.
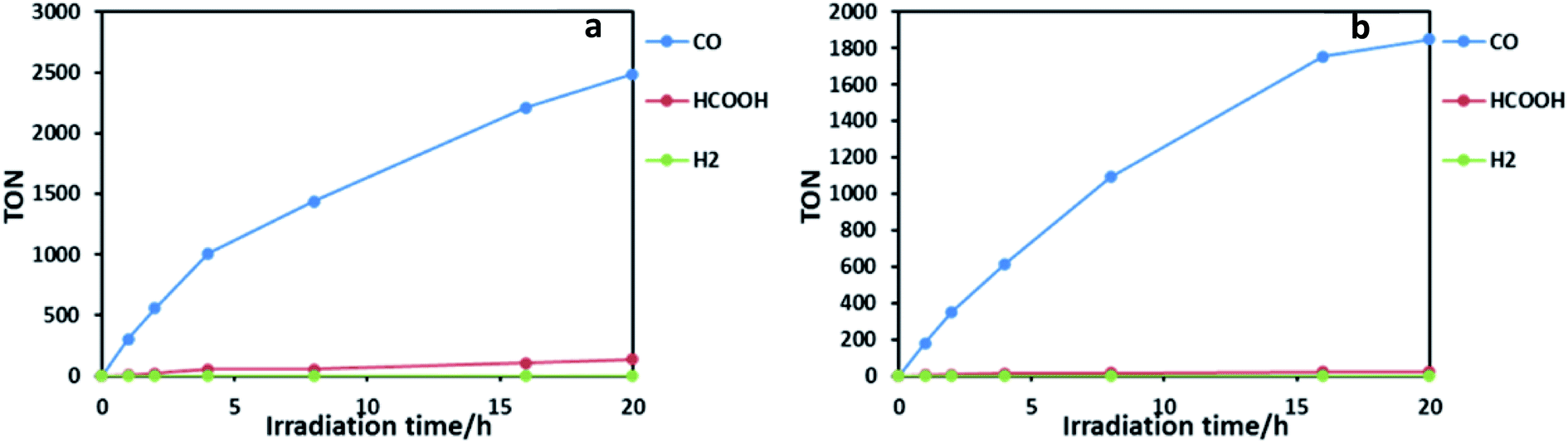 | ||
| Fig. 2 Photocatalytic formation of CO (blue line), formic acid (red line) and H2 (green line) as a function of irradiation time using (a) Ru2Re and (b) RuRe2:CO2-saturated DMA–TEOA (5:1 v/v, 3 mL) solutions containing Ru2Re (25 μM) or RuRe2 (50 μM) and BIH (0.1 M) irradiated at λex = 490–615 nm. | ||
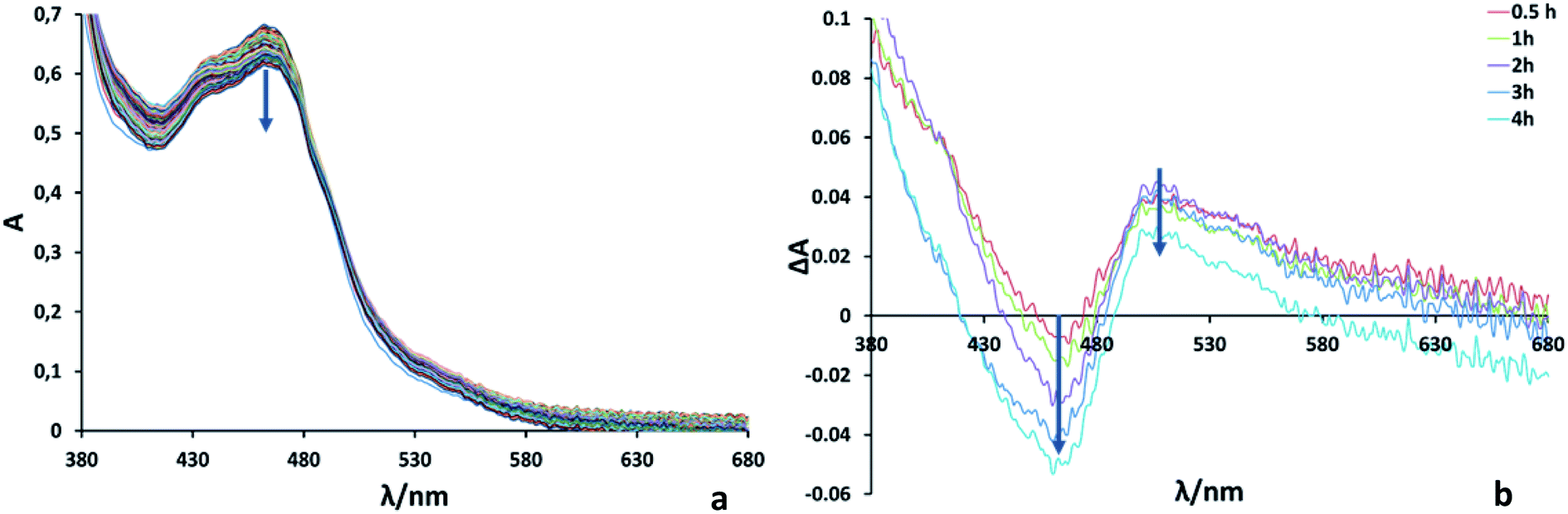 | ||
| Fig. 3 (a) UV-vis absorption spectra during irradiation of a DMA–TEOA (5:1 v/v) solution containing RuRe2 (50 μM) and BIH (0.1 M) at λex = 480 nm for 240 min at 1 min intervals. (b) UV-vis differential absorption spectra between before and after irradiation. | ||
In the system using [Ru(dmb)3]2+-type PSs and BIH as a sacrificial electron donor, it has been reported that the initial step of the photocatalytic reaction is photo-induced electron transfer from BIH to the excited PS giving its one-electron reduced species (OERS) which exhibits the characteristic shape of the spectrum at λmax ∼500–540 nm.10 Since, in Fig. 3, the accumulation of the OERS species was not significantly observed during the photocatalytic reaction, intramolecular electron transfer from OERS of the PS unit to the CAT units should be fast and not the rate-limiting step.
Ru2Re also displayed superior photocatalytic activities: TONCO = 2486, ΓCO = 90%, and ΦCO = 28% in the case of [Ru2Re] = 25 μM (Fig. 2a), and TONCO = 6038 and ΓCO = 81% in the case of [Ru2Re] = 5 μM. During photocatalysis, no significant UV-vis absorption spectral change was also observed. For comparison, we also checked photocatalysis of the reported binuclear complex (RuRe) of which one Ru(II) photosensitizer and one Re(I) catalyst unit are connected with an ethylene chain without the phenyl group under the same reaction conditions: TONCO = 3657 in the case of [RuRe] = 10 μM (Fig. S8†). It is noteworthy that TONCO of Ru2Re based on the Re unit was over 6000, which is much higher compared to that in the case of RuRe even though both supramolecular photocatalysts have the same number of Re units. The research studies related to this difference of durability among the supramolecular photocatalysts are in progress in our laboratory.
Table 3 summarizes the photocatalytic abilities of Ru2Re and RuRe2 by using BIH, which is a two-electron donor, or 1-benzyl-1,4-dihydronicotinamide (BNAH), which is a one-electron donor.28 In the cases using BNAH as a sacrificial electron donor instead of BIH, photocatalytic CO2 reduction also proceeded with relatively high durability and efficiency even though these values are less than those using BIH. The reason for the difference in photocatalytic activities between using BNAH and BIH has already been reported as follows: (i) because of stronger reduction power of BIH (Eox1/2(BIH/BIH˙+) = 0.33 V vs. SCE)29 than BNAH (E°ox(BNAH/BNAH˙+) = 0.57 V),30 quenching efficiencies of the excited Ru unit using BNAH (ηq = 67% and 63% in the case using RuRe2 and Ru2Re, respectively) were lower than that using BIH (ηq = 99% in both cases); (ii) BIH is a two-electron donor while BNAH is a one-electron donor (eqn (1) and (2)); (iii) The one-electron-oxidized, deprotonated, and dimerizing compounds of BNAH, i.e., 4,4′- and 4,6′-BNA2s, efficiently and reductively quench the excited Ru unit. However, they do not work as reductants owing to high stability of their one-electron-oxidized product (BNA2+˙) and rapid back-electron transfer from the reduced Ru unit to BNA2+˙ (eqn (3)). Therefore, accumulation of BNA2 should lower the yield of reduction of the excited Ru unit using BNAH, as competitive quenchers for excited Ru quenching. In the cases using BIH, on the other hand, the oxidized compound of BIH (BI+) does not affect the photocatalytic reactions. Note also that it is known that proton release is much faster for oxidized BIH than for oxidized BNAH,28 and this can also contribute to the best performance of BIH as a sacrificial donor.
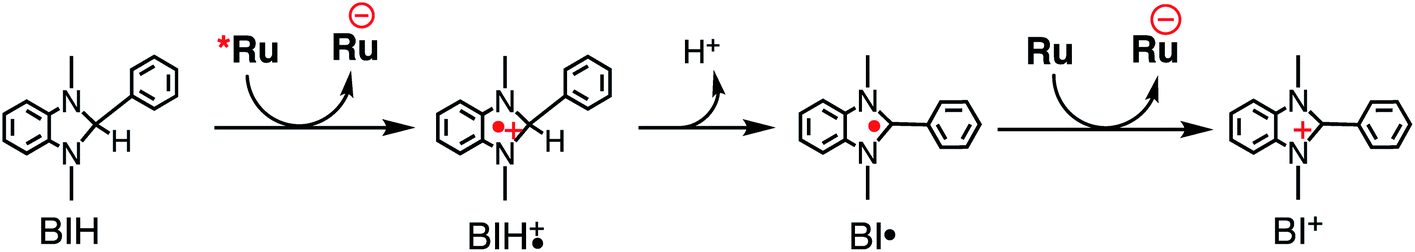 | (1) |
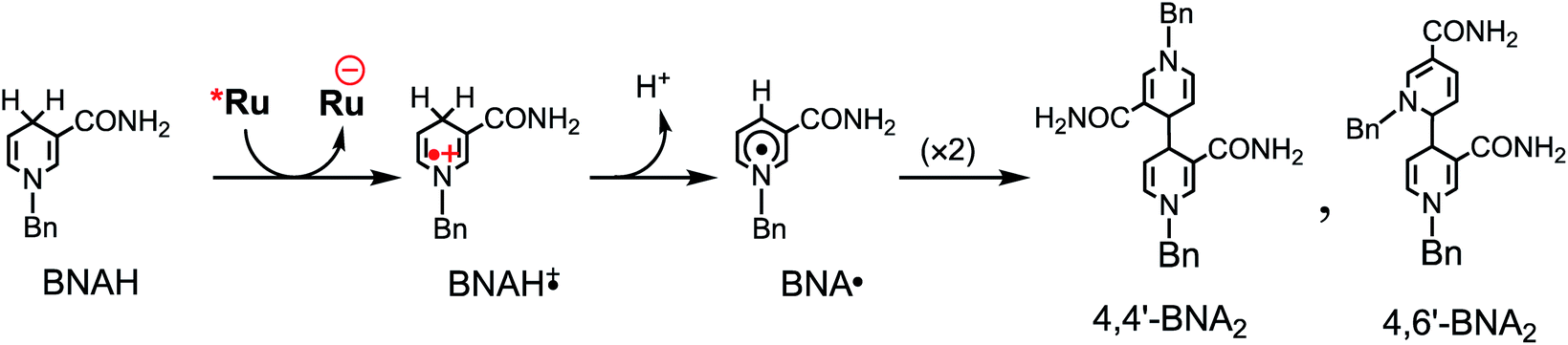 | (2) |
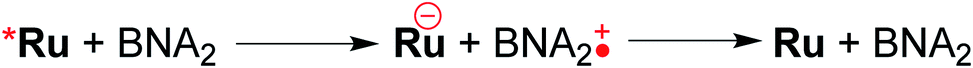 | (3) |
| Productb/μmol (TON) | Φ CO /% | Γ CO , , % | k q , 107 M−1 s−1 | η q % | ||||
|---|---|---|---|---|---|---|---|---|
| CO | HCOOH | H2 | ||||||
|
a CO2-saturated DMA–TEOA (5:1 v/v) solution containing Ru2Re (25 μM) or RuRe2 (50 μM) and a sacrificial electron donor (0.1 M) was irradiated.
b 3 mL solution was irradiated for 20 h using a LED (530 nm, 4 mW) as a light source. TONs are calculated based on the photocatalyst used.
c 4 mL solution was irradiated at λex = 480 nm (light intensity: 1 × 10−8 einstein per s).
d The selectivity for CO production.
e Quenching rate constants for emissions from the photosensitizer unit by a sacrificial electron donor obtained from linear Stern–Volmer plots and their lifetimes.
f Quenching fractions for emissions from the photosensitizer unit by a sacrificial electron donor (0.1 M) calculated as 0.1kqτem/(1 + 0.1kqτem).
g [Ru2Re] = 5 μM, 60 h irradiation.
h [RuRe2] = 10 μM, 60 h irradiation.
i [RuRe] = 10 μM, 60 h irradiation.
|
||||||||
| BIH | Ru2Re | 186.5 (2486 ± 12) | 20.2 (269) | 0.01 | 28 ± 0.6 | 90 | 83 ± 0.8 | 99 |
| Ru2Re | 90.6 (6038 ± 18) | 21.7 (1447) | 0.02 | — | 81 | 83 ± 0.8 | 99 | |
| RuRe2 | 277.4 (1850 ± 10) | 3.3 (22) | ∼0 | 25 ± 0.5 | 99 | 80 ± 2.6 | 99 | |
| RuRe2 | 157.0 (5232 ± 14) | 5.3 (177) | ∼0 | — | 97 | 80 ± 2.6 | 99 | |
| RuRe | 110.0 (3657 ± 29) | 1.1 (36) | 0.03 | 30 ± 1.1 | 99 | 120 ± 0.5 | 99 | |
| BNAH | Ru2Re | 17.5 (216 ± 5) | 13.0 (77) | 0.8 | 6.9 ± 0.9 | 56 | 2.0 ± 0.3 | 63 |
| RuRe2 | 33.7 (225 ± 6) | 8.6 (57) | 0.2 | 8.3 ± 1.2 | 79 | 2.2 ± 0.5 | 67 | |
Conclusions
A series of trinuclear metal complexes have been synthesized, based on a new tris-chelating bridging ligand, in which three bipyridine-decorated ethylene chains are connected to one another by a central phenyl ring. The trinuclear metal complexes so prepared contain Ru(dmb)3-type chromophores and Re(dmb)(CO)3Cl-type catalysts for CO2 reduction in different ratios, by made-to-order synthesis, therefore giving rise to supramolecular photocatalysts for CO2 photoreduction. The results indicate that:- The various metal subunits of the supramolecular catalysts maintain their own light absorption and redox properties in the supermolecular assemblies and electronic interactions between the various metal centres are negligible;
- Fast electron transfer between reduced chromophores and the catalyst unit(s) occurs, in spite of the relatively large metal–metal separation due to the bridging ligand structure;
- Quite efficient visible light-induced catalytic CO formation occurs in mixed-metal Ru(II)–Re(I) supramolecular photocatalysts, with outstanding turnover numbers (over 6000 in one case), high selectivity and durability: such high TONs are among the highest values for photocatalytic CO2 reduction in homogeneous solutions. This TONCO is almost twice as large as the previous most durable photocatalyst, the binuclear complex consisting of the Ru(II) photosensitizer and Re(I) catalyst units, which are similar units but are connected with an ethylene chain without the phenyl group.
All together, these achievements represent a breakthrough in the design of novel supramolecular photocatalysts, allowing us to overcome the size limit of bridging ligands represented by the ethylene bridges most commonly used in efficient PS–CAT supramolecular photocatalysts reported up to now. Actually, the present results indicate that new bridging ligands in which aromatic moieties are judiciously incorporated within the bridging ligand structure can allow us to obtain fast long-range electron transfer, suitable for the photocatalytic process, without affecting the behaviour of the catalytic subunit(s). This can open new avenues for the design of new, more efficient and stable supramolecular photocatalysts for selective CO2 photoreduction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Ministero degli Affari Esteri e della Cooperazione Internazionale, Direzione Generale per la Promozione del Sistema Paese, is gratefully acknowledged for an Italy-Japan Collaborative Grant (project title: “A supramolecular approach to artificial photosynthesis”). The Tokyo Tech group thanks JSPS KAKENHI Grant Number JP17H06440 in Scientific Research on Innovative Areas ‘Innovations for Light-Energy Conversion (I4LEC)’ and JST CREST Grant Number JPMJCR13L1 in ‘Molecular Technology’ for their financial support. AMC thanks the Regione Sicilia for a PhD grant.References
- T. R. Karl and K. E. Treberth, Science, 2003, 302, 1719–1723 CrossRef CAS PubMed.
- H. Akimoto, Science, 2003, 302, 1716–1719 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- S. Berardi, S. Drouet, L. Francàs, C. Gimbrt-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- S. Das and W. M. A. Wan Daud, RSC Adv., 2014, 40, 20856–20893 RSC.
- A. Ito and T. J. Meyer, Phys. Chem. Chem. Phys., 2012, 14, 13731–13745 RSC.
- (a) J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC; (b) J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS; (c) K. Koike, H. Hori, M. Ishizuka, J. R. Westwell, K. Takeuchi, T. Ibusuki, K. Enjouji, H. Konno, K. Sakamoto and O. Ishitani, Organometallics, 1997, 16, 5724–5729 CrossRef CAS; (d) H. Hori, F. P. A. Johnson, K. Koike, O. Ishitani and T. Ibusuki, J. Photochem. Photobiol., A, 1996, 96, 171–174 CrossRef CAS.
- Y. Tamaki and O. Ishitani, ACS Catal., 2017, 7, 3394–3409 CrossRef CAS and references therein.
- V. Balzani and F. Scandola, Supramolecular Photochemistry, Horwood, Chichester (UK), 1991 Search PubMed.
- A. Juris, V. Balzani, F. Barigalletti, S. Campagna, P. Belser and A. von Zelewski, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- (a) K. Koike, D. C. Grills, Y. Tamaki, E. Fujita, K. Okubo, Y. Yamazaki, M. Saigo, T. Mukuta, K. Onda and O. Ishitani, Chem. Sci., 2018, 9, 2961–2974 RSC; (b) Y. Yamazaki, K. Ohkubo, D. Saito, T. Yatsu, Y. Tamaki, S. Tanaka, K. Koike, K. Onda and O. Ishitani, Inorg. Chem., 2019, 58, 11480–11492 CrossRef CAS PubMed.
- K. Ohkubo, Y. Yamazaki, T. Nakashima, Y. Tamaki, K. Koike and O. Ishitani, J. Catal., 2016, 343, 278–289 CrossRef CAS.
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326–2336 CrossRef CAS PubMed.
- Y. Kuramochi and O. Ishitani, Inorg. Chem., 2016, 55, 5702–5709 CrossRef CAS PubMed.
- S. Meister, R. O. Reithmeier, A. Ogrodnik and B. Rieger, ChemCatChem, 2015, 7, 3562–3569 CrossRef CAS.
- R. Dorta, R. Dorta, L. J. W. Shimon and D. Mistein, Inorg. Chem., 2004, 43, 7180–7186 CrossRef CAS PubMed.
- (a) A. Kirgan, B. P. Sullivan and D. P. Rillema, Photochemistry and Photophysics of Coordination Compounds: Rhenium, in Photochemistry and Photophysics of Coordination Compounds II, ed. V. Balzani and S. Campagna, Springer, Berlin, 2007, vol. 281, pp. 45–100 Search PubMed; (b) A. J. Lees, Chem. Rev., 1987, 87, 711–743 CrossRef CAS.
- (a) J. Rohacovaa and O. Ishitani, Dalton Trans., 2017, 46, 8899–8919 RSC; (b) A. Juris, S. Campagna, I. Bidd, J. M. Lehn and R. Ziessel, Inorg. Chem., 1988, 27(22), 4007–4011 CrossRef CAS.
- D. S. Tyson and F. N. Castellano, J. Phys. Chem. A, 1999, 103(50), 10955–10960 CrossRef CAS.
- Y. Tamaki, K. Watanabe, K. Koike, H. Inoue, T. Morimoto and O. Ishitani, Faraday Discuss., 2012, 155, 115–127 RSC.
- L. A. Worl, R. Duesing, P. Chen, L. Della Ciana and T. J. Meyer, J. Chem. Soc., Dalton Trans., 1991, 849–858 RSC.
- M. Furue, M. Naiki, Y. Kanematsu, T. Kushida and M. Kamachi, Coord. Chem. Rev., 1991, 111, 221–226 CrossRef CAS.
- K. Koike, S. Naito, S. Sato, Y. Tamaki and O. Ishitani, J. Photochem. Photobiol., A, 2009, 207, 109–114 CrossRef CAS.
- (a) E. La Mazza, F. Puntoriero, F. Nastasi, B. Laramée-Milette, G. S. Hanan and S. Campagna, Dalton Trans., 2016, 45, 19238–19241 RSC; (b) B. Laramée-Milette, F. Nastasi, F. Puntoriero, S. Campagna and G. S. Hanan, Chem.–Eur. J., 2017, 23, 16497–16504 CrossRef PubMed; (c) F. Puntoriero, S. Serroni, F. Nastasi and S. Campagna, in Electrochemistry of Functional Supramolecular Systems, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2010, pp. 121–143 Search PubMed.
- Y. Kawanishi, N. Kitamura, Y. Kim and S. Tazuke, Sci. Pap. Inst. Phys. Chem. Res. (Jpn.), 1984, 78, 212–219 CAS.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- E. Hasegawa, S. Takizawa, T. Seida, A. Yamaguchi, N. Yamaguchi, H. Chiba, T. Takahashi, H. Ikeda and K. Akiyama, Tetrahedron, 2006, 62, 6581–6588 CrossRef CAS.
- S. Fukuzumi, S. Koumitsu, K. Hironaka and T. Tanaka, J. Am. Chem. Soc., 1987, 109, 305–316 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04532e |
| This journal is © The Royal Society of Chemistry 2020 |