
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence“Disrupt and induce” intermolecular interactions to rationally design organic semiconductor crystals: from herringbone to rubrene-like pitched π-stacking†
Chengyuan
Wang
 *a,
Daisuke
Hashizume
b,
Masahiro
Nakano
a,
Takuya
Ogaki
a,
Hiroyuki
Takenaka
c,
Kohsuke
Kawabata
c and
Kazuo
Takimiya
*ac
*a,
Daisuke
Hashizume
b,
Masahiro
Nakano
a,
Takuya
Ogaki
a,
Hiroyuki
Takenaka
c,
Kohsuke
Kawabata
c and
Kazuo
Takimiya
*ac
aEmergent Molecular Function Research Team, RIKEN Center for Emergent Matter Science (CEMS), 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. E-mail: chengyuan.wang@riken.jp
bMaterials Characterization Support Team, RIKEN Center for Emergent Matter Science (CEMS), 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
cDepartment of Chemistry, Graduate School of Science, Tohoku University, 6-3 Aoba, Aramaki, Aoba-ku, Sendai, Miyagi 980-8578, Japan
First published on 7th January 2020
Abstract
The packing structures of organic semiconductors in the solid state play critical roles in determining the performances of their optoelectronic devices, such as organic field-effect transistors (OFETs). It is a formidable challenge to rationally design molecular packing in the solid state owing to the difficulty of controlling intermolecular interactions. Here we report a unique materials design strategy based on the β-methylthionation of acenedithiophenes to generally and selectively control the packing structures of materials to create organic semiconductors rivalling rubrene, a benchmark high-mobility material with a characteristic pitched π-stacking structure in the solid state. Furthermore, the effect of the β-methylthionation on the packing structure was analyzed by Hirshfeld surface analysis together with theoretical calculations based on symmetry-adapted perturbation theory (SAPT). The results clearly demonstrated that the β-methylthionation of acenedithiophenes can universally alter the intermolecular interactions by disrupting the favorable edge-to-face manner in the parent acenedithiophenes and simultaneously inducing face-to-face and end-to-face interactions in the β-methylthionated acenedithiophenes. This “disrupt and induce” strategy to manipulate intermolecular interactions can open a door to rational packing design based on the molecular structure.
Introduction
In the past few decades, both theoretical and experimental studies have proven that the packing of organic semiconductors in the solid state plays a critical role in determining the properties, in particular, the charge carrier transport properties, of various optoelectronic devices, such as organic field-effect transistors (OFETs).1–6 Organic semiconductors including the ones based on small molecules are packed through intermolecular interactions, in which different intermolecular forces, e.g., electrostatics, exchange repulsion, induction, and dispersion, act as attractive or repulsive forces in the packing process to reach a certain balance that defines the packing structure of molecules in the solid state. For this reason, it is extremely difficult to design and predict the packing structure of a given semiconducting molecule, and even a tiny molecular modification could break the existing balance of intermolecular interactions and thus result in the change of the packing in an unpredictable manner.7,8 In this respect, Anthony et al. have been highly successful in selectively changing the packing structures of linear acenes and acenedithiophenes (a family of high-performance thienoacene-based organic semiconductors in OFETs) (Fig. 1a and b) from a typical herringbone to a two-dimensional (2D) brickwork packing by attaching [(trialkyl)silyl]ethynyl groups at their peri-positions.9–11 In sharp contrast, most of the studies on the control of packing structures are still in the trial-and-error stage, and therefore, the rational design of packing structures has remained a challenging issue in the development of organic semiconductors. | ||
| Fig. 1 Molecular structures of (a) linear acenes, (b) linear acenedithiophenes, (c) rubrene, and (d) β-methylthionated acenedithiophenes. | ||
The importance of the packing structures of organic semiconductors in the solid state is attributed to the fact that the packing of molecules critically determines the electronic structures of the organic semiconducting materials.6,12 This correlation is well illustrated by rubrene (Fig. 1c), a benchmark organic semiconductor in OFETs. Rubrene packing features the characteristic “pitched π-stacking”,13 where the intermolecular electronic coupling between the HOMOs of the π-stacking molecules is very large (∼0.1 eV). As a result, the single-crystal OFETs (SC-OFETs) of rubrene show reproducible high mobilities of 15–20 cm2 V−1 s−1.14–16 Tremendous efforts have been devoted to realizing organic semiconductors with a rubrene-like pitched π-stacking in the solid state, which, however, have hardly succeeded.17–20 More generally, establishing rational materials design strategies to control the packing structure by manipulating molecular structures is highly desired. However, they remain ambitious tasks.21–23
We have recently found that methylthionation at the β-position of benzo[1,2-b:4,5-b′]dithiophene (BDT, Fig. 1b), the smallest linear acenedithiophene, can selectively alter the packing from the BDT's herringbone24 to the rubrene-like pitched π-stacking.13,25,26 Thanks to its packing structure, β-methylthionated BDT (β-MT-BDT) realizes large intermolecular electronic coupling between the HOMOs of the π-stacking molecules. Accordingly, its SC-OFETs yield a moderately high mobility of 0.38 cm2 V−1 s−1 despite having a small π-conjugated system with only 14 π-electrons. Enlightened by these results, we were curious about whether β-methylthionation is just a successful example particularly of BDT to selectively alter the packing or if it can be a general strategy to widely realize the rubrene-like pitched π-stacking. If the latter is the case, the general questions then arise: what is the origin of this selectivity and generality, and furthermore, with the strategy, is it possible to create materials rivalling rubrene? Inspired by these questions, we designed a series of β-methylthionated (β-MT)-acenedithiophenes with a gradually extended molecular π-backbone from BDT to naphtho[2,3-b:6,7-b′]dithiophene (NDT) and anthra[2,3-b:6,7-b′]dithiophene (ADT) (Fig. 1d). Here we report the synthesis, packing structure, electronic structure in the solid state, and OFET performance of a series of β-MT-acenedithiophenes. We also discuss the origin of the general and selective packing control through β-methylthionation and the potential of this materials design strategy in the development of high performance organic semiconductors in OFETs.
Results
Synthesis of molecules
Scheme 1 shows the synthesis of β-methylthionated NDT (β-MT-NDT) and ADT (β-MT-ADT). In our previous study on β-MT-BDT, we developed a straightforward method to directly introduce sulphur functional groups at the β-position of BDT by utilizing disulphur dichloride (S2Cl2) as the electrophilic reagent in Larock cyclization.25,27 This method is convenient and versatile for the β-alkylthionation of the BDT core,26 but results in only moderate yields, possibly because of the formation of oligomers as byproducts. In this work, we employed methyl thiohypochlorite (methylsulfenyl chloride, MeSCl) as the electrophilic reagent instead of S2Cl2 to avoid the formation of oligomers.28,29 Starting from the functionalized naphthalene derivative (1), β-MT-NDT could be readily synthesized by the reaction of 1 with MeSCl followed by treatment with tetrabutylammonium fluoride (TBAF). In contrast, a similar reaction of functionalized anthracene (3) with MeSCl afforded 5,11-dichlorinated ADT derivative (4) instead of the precursor (5). These unexpected results could be explained as follows: MeSCl was unstable under the reaction conditions and decomposed to liberate chlorine, which reacted with in situ formed 5 at the reactive 5,11-positions to afford 4 as the major product. Fortunately, the chlorine substituents were readily removed by the treatment with lithium aluminium hydride (LAH) to give 5, which was then converted into β-MT-ADT in a similar manner to the synthesis of β-MT-NDT. β-MT-NDT and β-MT-ADT were fully characterized by means of spectroscopic analysis (see the ESI†) and single-crystal X-ray structural analysis (vide infra). Their optical and electrochemical properties were evaluated and are fully discussed in the ESI† with the aid of theoretical calculations.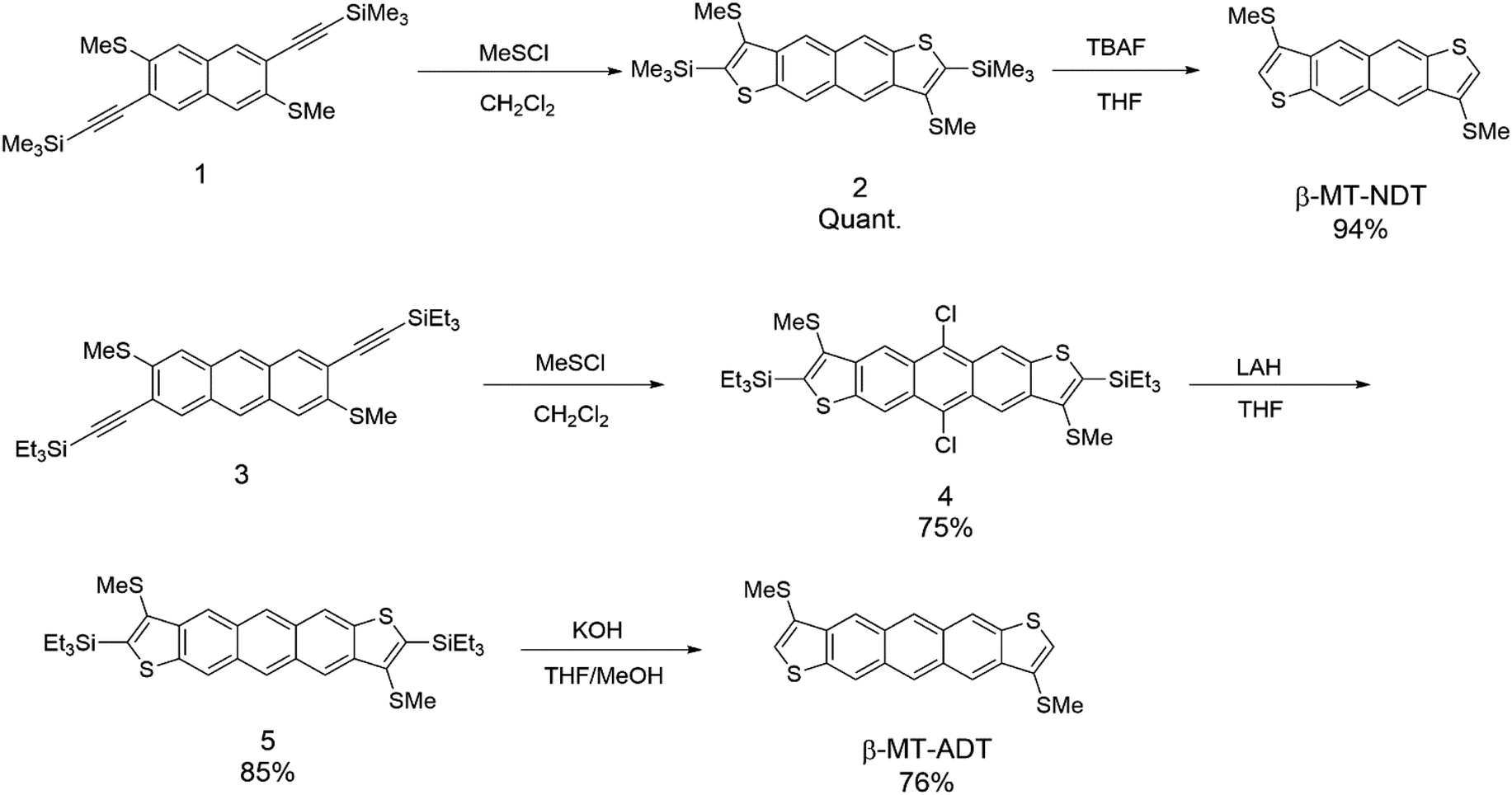 | ||
| Scheme 1 Synthetic route to the β-MT-acenedithiophenes. | ||
Packing structures in the solid state
Fig. 2 shows the packing structures of β-MT-acenedithiophenes.25 Intriguingly, with the extension of the molecular π-backbone, β-MT-NDT (with one more central benzene ring) and β-MT-ADT (two additional central benzene rings) are of isostructure with β-MT-BDT (monoclinic with P21/c space group), and all the three structures have the same pitched π-stacking (Fig. 2). This indicates that the β-methylthionation of linear acenedithiophenes is a universal strategy to tune the packing into the rubrene-like pitched π-stacking. Note that all the corresponding parent acenedithiophenes, i.e., BDT,24 NDT, and ADT,30 are crystallized into the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with the herringbone packing, similar to those of linear acenes.
space group with the herringbone packing, similar to those of linear acenes.
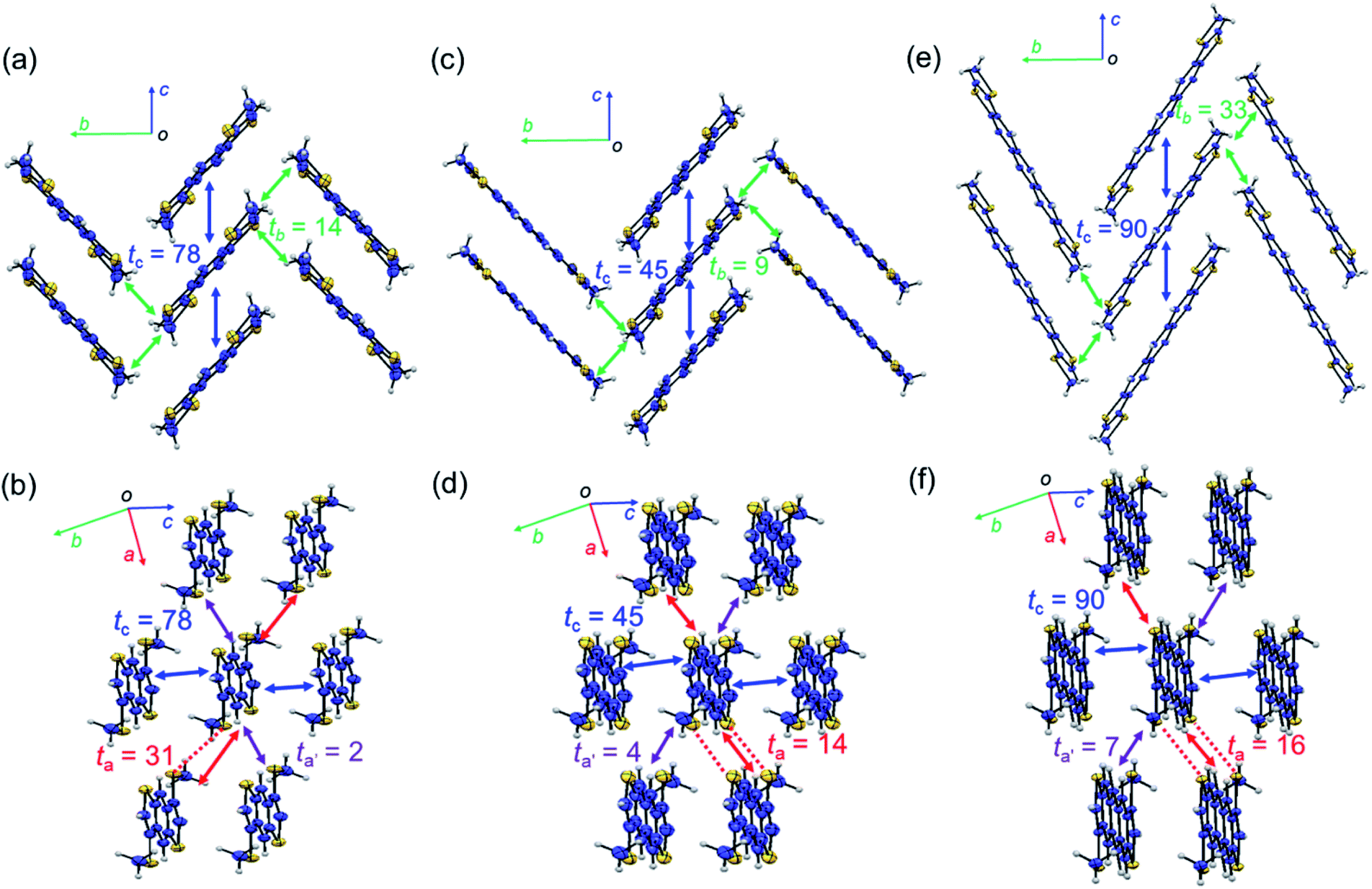 | ||
| Fig. 2 Packing structures of molecules projected along short and long molecular axes: (a and b) β-MT-BDT, (c and d) β-MT-NDT (CCDC: 1899661), and (e and f) β-MT-ADT (CCDC: 1899662). Red dashed lines represent intermolecular S–S contacts shorter than the sum of van der Waals (vdW) radii. ta, ta′, tb, and tc (meV) are calculated values of the intermolecular electronic coupling between the HOMOs of neighbouring molecules in each crystallographic axis direction. | ||
One distinct feature of the pitched π-stacking is that the displacement of neighbouring molecules occurs along the long molecular axis direction while maintaining substantial face-to-face overlap, whereas only marginal displacement is observed along the short molecular axis direction. The other feature is that the neighbouring π-stacking columns are inclined in opposite directions to each other with certain dihedral angles (68–90°) between the molecules in the adjacent stacks, which interact with each other in an end-to-face mode (Fig. 2a, c and e). As a result, the molecular network with the face-to-face and end-to-face modes in the pitched π-stacking forms a layered structure (Fig. 2b, d and f). These features are significantly different from those of the herringbone packing, where the displacement of neighbouring molecules occurs along the short molecular axis direction without face-to-face overlap, resulting in different layered structures with the face-to-edge and edge-to-edge modes.13
Intermolecular electronic coupling between the HOMOs of molecules in the solid state
One advantage of the pitched π-stacking is that large intermolecular electronic coupling between the HOMOs (or transfer integral, t) in the stacking columns likely occurs. This is well represented by the packing of β-MT-BDT, where the maximum electronic coupling is as large as 78 meV (tc) in the π-stacking direction (or the crystallographic c-axis direction) (Fig. 2a). In contrast, tb in the end-to-face direction (crystallographic b-axis direction) is only 14 meV, indicating the anisotropic nature of the electronic structure in each layer. Interestingly, moderately large electronic coupling is observed between the layers (or in the edge-to-edge direction of molecules) (ta, up to 31 meV), implying that three-dimensional (3D) electronic communication is possible (Fig. 2b).With the same packing structure and extended π-backbone, β-MT-NDT (with 18 π-electrons) is expected to have larger electronic coupling than β-MT-BDT. However, the largest electronic coupling (tc) in β-MT-NDT is only 45 meV in the face-to-face direction, which is significantly smaller than that in β-MT-BDT (Fig. 2c). The electronic couplings in the other directions are also small for β-MT-NDT. This may be because in β-MT-NDT crystals, the neighbouring dimers have disadvantageous mutual positions to achieve efficient electronic coupling, which is discussed in detail in the ESI.†
In the packing structure of β-MT-ADT with 22 π-electrons, the maximum electronic coupling in the stacking column (tc) is 90 meV (Fig. 2e), which is the highest among the three β-MT-acenedithiophenes, and is very close to the corresponding one in rubrene (96 meV) (Fig. S4†). Furthermore, the electronic coupling in the end-to-face direction (tb) in β-MT-ADT is also the largest and even larger than that of rubrene (33 meV for β-MT-ADT, 19 meV for rubrene), indicating that the anisotropic nature of β-MT-ADT in the semiconducting layer is mitigated. These features of the electronic structure of β-MT-ADT are mostly identical to those of rubrene, indicating that β-MT-ADT can be a rubrene-like semiconducting material. Especially, in the t vs. displacement plots (Fig. S5†), the actual mutual positions of neighbouring molecules along the π-stacking direction in rubrene and β-MT-ADT are both located at the extrema. Furthermore recent studies pointed out that small dynamic disorder of rubrene in the solid state is crucial to achieve high performance in SC-OFETs. The similarities of β-MT-ADT in the solid state to those of rubrene may suggest the possibility of that benefit to achieve high performance as semiconducting materials in SC-OFETs (discussed in the ESI†).31–33
SC-OFETs of β-MT-acenedithiophenes
SC-OFETs of β-MT-BDT, -NDT, and -ADT with the bottom-gate/top-contact device configuration were fabricated with single crystals grown directly on octadecyltrichlorosilane (ODTS)-modified Si/SiO2 substrates by the physical vapour transport (PVT) method.34,35 The XRD patterns of β-MT-NDT and β-MT-ADT single crystals on the substrates show similar diffraction peaks, which can be indexed as 1 0 0 and 2 0 0 diffractions by comparison with the simulated powder patterns from the single-crystal X-ray data (Fig. S8†). This indicates that both β-MT-NDT and β-MT-ADT adopt the edge-on molecular orientation on the substrates (Fig. 2d and f), consistent with the case of rubrene and β-MT-BDT (Fig. S9†).25 Such an edge-on molecular orientation is reported to be advantageous to enhance carrier mobility by reducing the coupling between molecular polarization and charge carriers.36The β-MT-BDT-based SC-OFETs were reported to show mobilities of up to 0.38 cm2 V−1 s−1 (Table 1).19 We noticed that the crystals of β-MT-BDT were very thick (∼500–800 nm), which could increase the contact resistance between the top electrodes and the transistor channel at the substrate/crystal interface, likely limiting empirical mobility values. In the case of β-MT-NDT and -ADT, on the other hand, the single crystals prepared by the PVT method were much thinner (∼100–300 nm thickness) than those of β-MT-BDT, and thus the effects of the contact resistance could be reduced.
| Compound | Mobility (cm2 V−1 s−1) | V th (V) | On/off ratio |
|---|---|---|---|
| a The charge transport directions in the SC-OFETs are along the π-stacking directions (crystallographic b-axis for rubrene and c-axis for β-MT-acenedithiophenes, Fig. S7). The mobilities were extracted from the saturation regime of the transfer curves. The average mobilities and Vths are based on more than 10 devices, and values within parentheses are the highest mobilities. | |||
| β-MT-BDT | 0.22 (0.38) | −21.5 | 105 to 106 |
| β-MT-NDT | 0.25 (0.35) | −11.8 | 105 to 106 |
| β-MT-ADT | 3.44 (4.08) | −4.5 | 104 to 106 |
| Rubrene | 3.79 (4.73) | −5.1 | 106 to 107 |
SC-OFETs based on β-MT-NDT showed mobilities of up to 0.35 cm2 V−1 s−1 (Table 1), which are similar to those of β-MT-BDT-based devices, although the intermolecular electronic couplings in β-MT-NDT are much smaller than those in β-MT-BDT, as mentioned above (see also Fig. 2). In contrast, single crystals of β-MT-ADT acted as an excellent transistor channel, showing mobilities of up to 4.08 cm2 V−1 s−1 (average 3.44 cm2 V−1 s−1, Table 1), which are almost one order of magnitude higher than those of β-MT-BDT and -NDT.
Fig. 3 shows the transfer and output curves of the SC-OFETs based on β-MT-NDT and β-MT-ADT, respectively. It has been reported that rubrene-based SC-OFETs usually show mobilities of 3–6 cm2 V−1 s−1 on Si/SiO2 substrates,37–39 and in fact, similar values (average mobility: 3.79 cm2 V−1 s−1, ∼100–300 nm thickness) were also reproduced by our group (Table 1). The reliability factor of the SC-OFETs is discussed in the ESI.†40
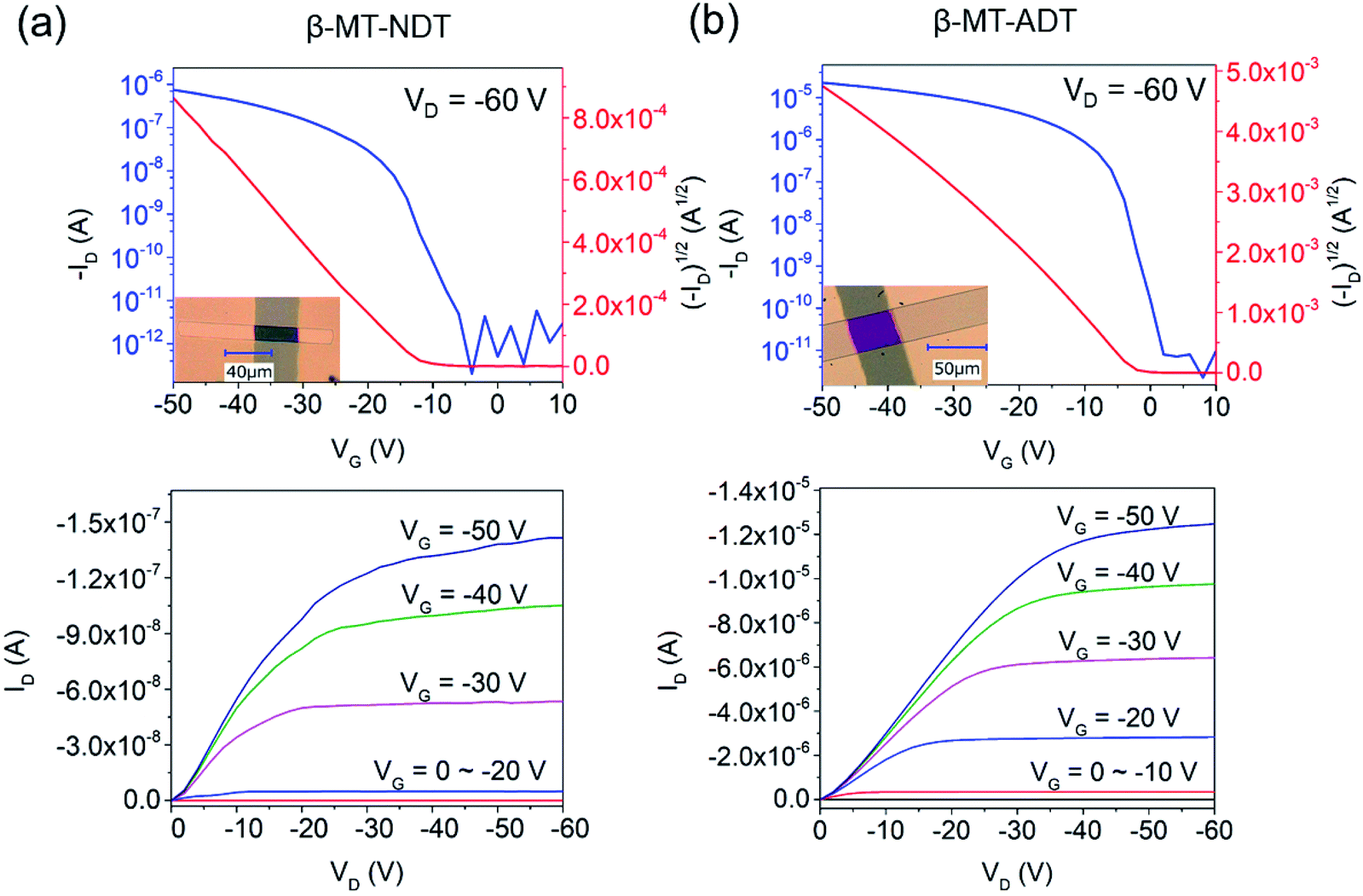 | ||
| Fig. 3 Transfer (top) and output (bottom) curves of SC-OFETs (a) β-MT-NDT and (b) β-MT-ADT. | ||
To confirm the experimental carrier motilities, theoretical mobilities of β-MT-acenedithiophenes and rubrene were calculated in the hopping regime utilizing the Marcus/Hush model based on the electronic structure in the solid state. The calculations show that β-MT-ADT can have the highest mobilities (0.65–12.38 cm2 V−1 s−1) among the β-MT-acenedithiophenes (0.014–1.62 cm2 V−1 s−1 for β-MT-BDT, 0.010–2.81 cm2 V−1 s−1 for β-MT-NDT), which are even higher than those of rubrene (0.024–7.68 cm2 V−1 s−1), suggesting its great potential in the application of OFETs (see the ESI for details†). From both the experimental (SiO2 as the dielectric layer) and theoretical investigations, it is safe to say that β-MT-ADT is a superior organic semiconducting material rivalling rubrene.
Discussion
Molecular factors that dictate packing structures
As described in the section of packing structures, it is concluded that the β-methylthio group on the linear acenedithiophenes is a reliable controller that selectively alters the packing structure from the herringbone of the parent acenedithiophenes to the pitched π-stacking of the β-MT-acenedithiophenes.The herringbone packing is known to be a dispersion-driven packing structure.41 In fact, zeroth-order symmetry-adapted perturbation theory (SAPT0) calculations with the jun-cc-pVDZ basis42,43 for the parent acenedithiophenes clearly show that the dispersion term in the edge-to-face dimer (−9.15 kcal mol−1 for BDT, −13.67 kcal mol−1 for NDT, and −18.89 kcal mol−1 for ADT) is the largest one among the decomposed intermolecular interaction energies (see Table 2). The presence of the favourable edge-to-face intermolecular interaction is further confirmed by the Hirshfeld surface mapped with de (distance from the Hirshfeld surface to the nearest nucleus outside the surface),44,45 which shows close contacts between the peri-hydrogen atoms and the π-clouds of the adjacent molecule in the edge-to-face manner (top of Fig. 4a, b and c). Furthermore, the intermolecular C–H contact accounts for up to 40–50% of the total intermolecular short contacts in the packing structure (bottom of Fig. 4a, b and c). From these analyses, we can confirm that the herringbone packing of the parent acenedithiophenes is a dispersion-driven packing structure.
| Compound | E es | E ex | E ind | E disp | E total | |
|---|---|---|---|---|---|---|
| a Obtained from zeroth-order symmetry-adapted perturbation theory (SAPT0) calculations with the jun-cc-pVDZ basis. Ees, Eex, Eind, Edisp, and Etotal are electrostatic, exchange, induction, dispersion, and total intermolecular interaction energy, respectively. Etotal = Ees + Eex + Eind + Edisp. b The rows highlight the intermolecular interaction energies in the π-stacking dimers without (w/o) methylthio groups. | ||||||
| BDT | CH–π (edge-to-face) | −2.83 | 5.48 | −0.83 | −9.15 | −7.33 |
| β-MT-BDT | π-stacking | −7.05 | 14.59 | −1.69 | −21.31 | −15.46 |
| π-stacking (w/o MT)b | −4.07 | 9.26 | −0.87 | −12.93 | −8.60 | |
| CH–π (end-to-face) | −2.48 | 5.76 | −0.98 | −7.35 | −5.06 | |
| S–H w/S–S contact (edge-to-edge) | −4.12 | 5.97 | −0.71 | −3.91 | −2.77 | |
| S–H w/o S–S contact (edge-to-edge) | −2.49 | 3.92 | −0.60 | −5.20 | −4.36 | |
| NDT | CH–π (edge-to-face) | −4.06 | 7.99 | −1.18 | −13.67 | −10.92 |
| β-MT-NDT | π-stacking | −10.58 | 21.07 | −2.28 | −30.80 | −22.59 |
| π-stacking (w/o MT)b | −6.23 | 15.66 | −1.40 | −21.93 | −13.91 | |
| CH–π (end-to-face) | −2.32 | 5.64 | −0.95 | −7.23 | −4.88 | |
| S–H w/S–S contact (edge-to-edge) | −5.36 | 8.84 | −1.12 | −8.03 | −5.68 | |
| S–H w/o S–S contact (edge-to-edge) | −1.03 | 3.49 | −0.55 | −5.66 | −3.75 | |
| ADT | CH–π (edge-to-face) | −5.63 | 11.15 | −1.66 | −18.89 | −15.03 |
| β-MT-ADT | π-stacking | −10.47 | 22.30 | −2.53 | −32.16 | −22.86 |
| π-stacking (w/o MT)b | −6.70 | 17.59 | −1.75 | −24.39 | −15.26 | |
| CH–π (end-to-face) | −2.94 | 6.62 | −1.10 | −9.63 | −7.06 | |
| S–H w/S–S contact (edge-to-edge) | −6.56 | 11.78 | −1.45 | −9.68 | −5.91 | |
| S–H w/o S–S contact (edge-to-edge) | −2.00 | 4.22 | −0.65 | −6.68 | −5.11 | |
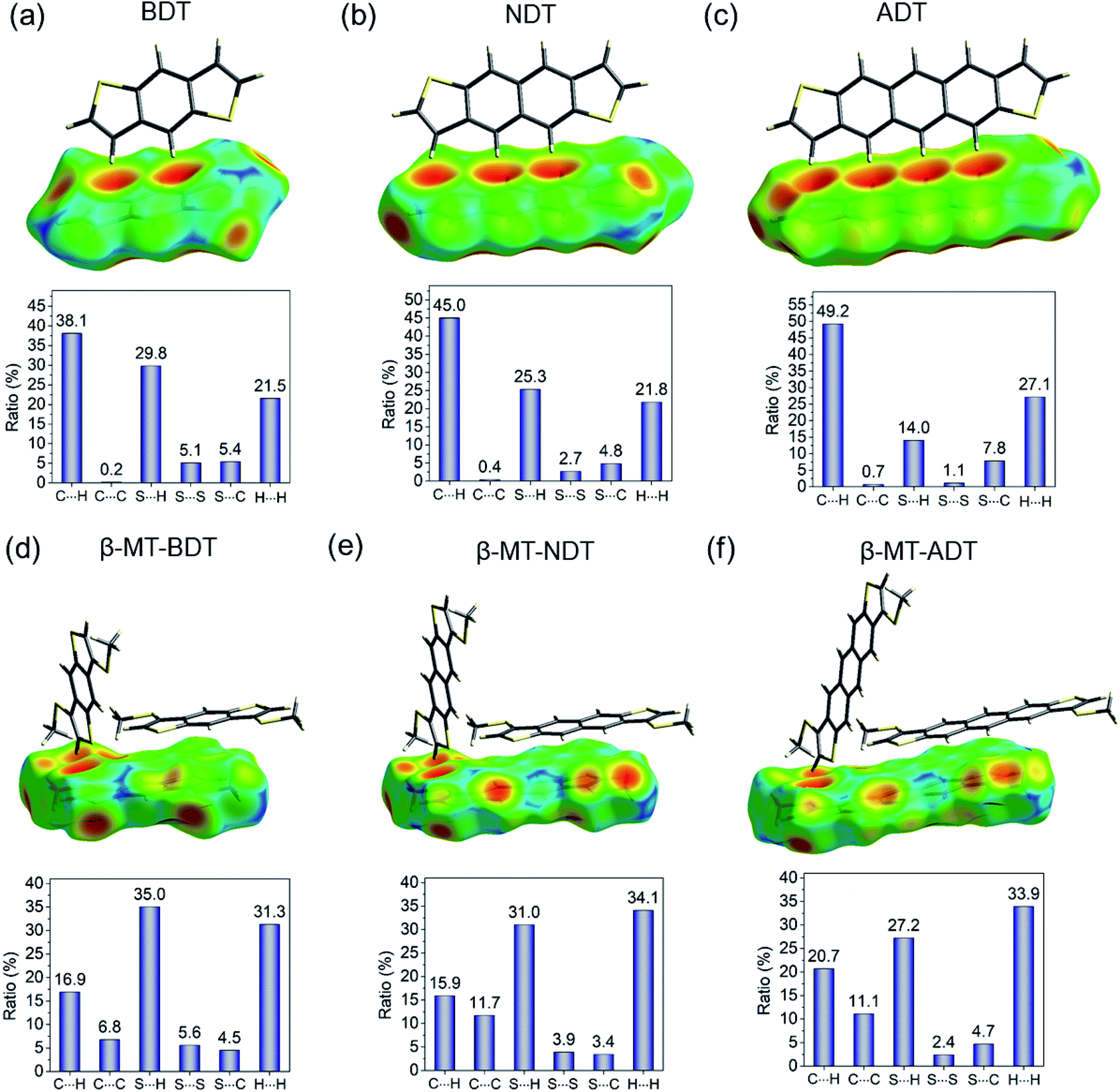 | ||
| Fig. 4 Hirshfeld surface analysis of (a) BDT, (b) NDT (CCDC: 1899660), (c) ADT, (d) β-MT-BDT, (e) β-MT-NDT, and (f) β-MT-ADT, which illustrates the distance (de) from the surface to the nucleus of external atoms in the adjacent molecules in a red-green-blue colour scheme. | ||
The β-methylthio groups on acenedithiophenes, on the other hand, can have drastic effects on the intermolecular interactions. First, the stabilizing effect induced by the edge-to-face interaction in the parent acenedithiophenes does not work owing to the steric bulkiness of the β-methylthio groups; in other words, the β-methylthio group disrupts the contacts between the peri-hydrogen atoms and the π-cloud of the adjacent molecules. As a result, instead of the edge-to-face interaction in the parent system, the face-to-face (π–π) interaction, which is largely stabilized by dispersion (−21.31 kcal mol−1 for β-MT-BDT, −30.80 kcal mol−1 for β-MT-NDT, and −32.16 kcal mol−1 for β-MT-ADT) and electrostatic interactions (−7.05 kcal mol−1 for β-MT-BDT, −10.58 kcal mol−1 for β-MT-NDT, and −10.47 kcal mol−1 for β-MT-ADT) is manifested in the packing structure of β-MT-acenedithiophenes (Table 2). The change of intermolecular interaction caused by β-methylthionation is consistent with the ratio of intermolecular contacts; the CH–π contacts decrease from 40–50% in the parent system to 15–20% of all the intermolecular contacts, whereas the C–C contacts corresponding to the face-to-face (π–π) interaction increase to around 6–12% relative to the parent system that has less than 1% contribution (Fig. 4d, e and f).
Second, the β-methylthio group induces other intermolecular interactions in a different manner; in the face-to-face dimer, the methyl moiety in the β-methylthio group is immediately above/beneath the π-face, facilitating a CH–π interaction between the π-face of the acenedithiophene core and the methyl hydrogen atom. This is clearly visualized by the Hirshfeld surface, where a large red depression directly beneath the methyl moiety shows close contact, which can be described as a “methyl-on–π” interaction. To confirm the net effect of the “methyl-on–π” interaction, we calculated the interaction energy of the face-to-face dimer without the methylthio groups. The difference of the interaction energies with and without the methylthio groups is substantially large (approximately 7–9 kcal mol−1 for the total interaction energy, including a dispersion energy of approximately 7–9 kcal mol−1 and an electrostatic energy of approximately 3–4 kcal mol−1, Table 2), indicating that the “methyl-on–π” interaction contributes to the stabilization of the face-to-face dimer. Another intermolecular interaction induced by the methylthio groups is observed in the end-to-face dimer; one of the methyl hydrogen atoms and the π-hydrogen atoms in the thiophene moiety cooperatively come into contact with the π-face of a neighbouring molecule, enabling an “end-to-face” CH–π interaction, as visualized from the Hirshfeld surface mapped with de (top of Fig. 4d, e and f). The SAPT calculations also support this attractive interaction with relatively large interaction energies (−5.06 kcal mol−1 for β-MT-BDT, −4.88 kcal mol−1 for β-MT-NDT, and −7.06 kcal mol−1 for β-MT-ADT, Table 2) contributed mainly by the dispersion term. In addition, the β-methylthio group participates in the intermolecular contacts in the edge-to-edge direction through S–H and S–S contacts (bottom of Fig. 4d, e and f) that energetically stabilize the present pitched π-stacking.
From the above analyses, it can be summarized that the β-methylthio groups play two key roles in altering the packing structure. One is to disrupt the favourable CH–π interaction along the molecular edge in the parent system and the other is to induce new intermolecular interactions in different molecular faces, i.e., methyl-on–π in the face-to-face dimer, CH–π in the end-to-face dimer, and S–S and S–H in the edge-to-edge dimers. The “disrupt and induce” effects caused by the β-methylthio groups would be a possible scenario that dictates the structural change from the herringbone in the parent system to the pitched π-stacking in β-MT-acenedithiophenes.
Conclusion
We have demonstrated that β-methylthionation of a series of acenedithiophenes can control the packing of molecules into rubrene-like pitched π-stacking. β-MT-ADT with a large π-backbone realizes large electronic couplings between neighbouring molecules, which yield high mobility comparable to that of rubrene, up to 4.1 cm2 V−1 s−1, in SC-OFETs with SiO2 as the dielectric layer. Taking these aspects, including the packing structure, the electronic structure in the solid state, and the excellent empirical and theoretical transport properties, into account, it can be said that β-MT-ADT is a material rivalling rubrene.The most important knowledge obtained from this study is that such a simple molecular modification as β-methylthionation is a powerful strategy to generally and selectively control the packing structure of molecules in the solid state. The key to the generality and selectivity in the packing design is to “disrupt and induce” intermolecular interactions along different directions of the molecular backbone. In particular, the major driving force is changed from the dispersion in the CH–π interaction along the edge-to-face direction in the parent acenedithiophenes to the dispersion together with electrostatic terms in the π–π interactions along the face-to-face direction in the β-MT-acenedithiophenes. Overall, our results strongly imply that the rational control of intermolecular interactions by the “bottom–up approach” i.e., molecular modification, enables developing new superior organic semiconductors with well-designed packing structures. We hope that the present study will open the door to the possibility of a rational design of packing structures of organic semiconductors through the manipulation of intermolecular interactions with molecular modifications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Takemichi Nakamura of the Molecular Structure Characterization Unit, RIKEN Center for Sustainable Resource Science (CSRS), for high-resolution mass spectral (HR-MS) measurements. We acknowledge the Supercomputer System in the Advanced Center for Computing and Communication (ACCC) of RIKEN and the Center for Computational Materials Science, Institute for Materials Research, Tohoku University, for the use of MASAMUNE-IMR (MAterials science Supercomputing system for Advanced MUlti-scale simulations towards NExt-generation – Institute for Materials Research) for support in theoretical calculations. This work was financially supported by JSPS KAKENHI Grant Numbers 15H02196, 17K14478 and 19H00906, the Strategic Promotion of Innovative Research and Development from JST, and the Mitsubishi Foundation.Notes and references
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- H. Huang, L. Yang, A. Facchetti and T. J. Marks, Chem. Rev., 2017, 117, 10291–10318 CrossRef CAS PubMed.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS PubMed.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 CrossRef CAS PubMed.
- H. Dong, X. Fu, J. Liu, Z. Wang and W. Hu, Adv. Mater., 2013, 25, 6158–6183 CrossRef CAS PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- Y. Tsutsui, G. Schweicher, B. Chattopadhyay, T. Sakurai, J.-B. Arlin, C. Ruzié, A. Aliev, A. Ciesielski, S. Colella, A. R. Kennedy, V. Lemaur, Y. Olivier, R. Hadji, L. Sanguinet, F. Castet, S. Osella, D. Dudenko, D. Beljonne, J. Cornil, P. Samorì, S. Seki and Y. H. Geerts, Adv. Mater., 2016, 28, 7106–7114 CrossRef CAS PubMed.
- C. Sutton, C. Risko and J.-L. Brédas, Chem. Mater., 2016, 28, 3–16 CrossRef CAS.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- J. E. Anthony, Angew. Chem., Int. Ed., 2007, 47, 452–483 CrossRef PubMed.
- M. Klues and G. Witte, CrystEngComm, 2018, 20, 63–74 RSC.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
- M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 126, 4318–4328 CrossRef CAS PubMed.
- V. C. Sundar, J. Zaumseil, V. Podzorov, E. Menard, R. L. Willett, T. Someya, M. E. Gershenson and J. A. Rogers, Science, 2004, 303, 1644–1646 CrossRef CAS PubMed.
- V. Podzorov, E. Menard, A. Borissov, V. Kiryukhin, J. A. Rogers and M. E. Gershenson, Phys. Rev. Lett., 2004, 93, 086602 CrossRef CAS PubMed.
- X. Ren, M. J. Bruzek, D. A. Hanifi, A. Schulzetenberg, Y. Wu, C.-H. Kim, Z. Zhang, J. E. Johns, A. Salleo, S. Fratini, A. Troisi, C. J. Douglas and C. D. Frisbie, Adv. Electron. Mater., 2017, 3, 1700018 CrossRef.
- K. A. McGarry, W. Xie, C. Sutton, C. Risko, Y. Wu, V. G. Young, J.-L. Brédas, C. D. Frisbie and C. J. Douglas, Chem. Mater., 2013, 25, 2254–2263 CrossRef CAS.
- M. Mamada, H. Katagiri, T. Sakanoue and S. Tokito, Cryst. Growth Des., 2015, 15, 442–448 CrossRef CAS.
- Y. Sakamoto and T. Suzuki, J. Org. Chem., 2017, 82, 8111–8116 CrossRef CAS PubMed.
- G. Xie, S. Hahn, F. Rominger, J. Freudenberg and U. H. F. Bunz, Chem. Commun., 2018, 54, 7593–7596 RSC.
- Z. Chen, P. Müller and T. M. Swager, Org. Lett., 2006, 8, 273–276 CrossRef CAS PubMed.
- H. Moon, R. Zeis, E.-J. Borkent, C. Besnard, A. J. Lovinger, T. Siegrist, C. Kloc and Z. Bao, J. Am. Chem. Soc., 2004, 126, 15322–15323 CrossRef CAS PubMed.
- T. Kimoto, K. Tanaka, M. Kawahata, K. Yamaguchi, S. Otsubo, Y. Sakai, Y. Ono, A. Ohno and K. Kobayashi, J. Org. Chem., 2011, 76, 5018–5025 CrossRef CAS PubMed.
- K. Takimiya, Y. Konda, H. Ebata, N. Niihara and T. Otsubo, J. Org. Chem., 2005, 70, 10569–10571 CrossRef CAS PubMed.
- C. Wang, H. Nakamura, H. Sugino and K. Takimiya, Chem. Commun., 2017, 53, 9594–9597 RSC.
- C. Wang, H. Nakamura, H. Sugino and K. Takimiya, J. Mater. Chem. C, 2018, 6, 3604–3612 RSC.
- D. Yue and R. C. Larock, J. Org. Chem., 2002, 67, 1905–1909 CrossRef CAS PubMed.
- J. K. Son and R. W. Woodard, J. Am. Chem. Soc., 1989, 111, 1363–1367 CrossRef CAS.
- H. Takenaka, T. Ogaki, C. Wang, K. Kawabata and K. Takimiya, Chem. Mater., 2019, 31, 6696–6705 CrossRef CAS.
- M. Mamada, H. Katagiri, M. Mizukami, K. Honda, T. Minamiki, R. Teraoka, T. Uemura and S. Tokito, ACS Appl. Mater. Interfaces, 2013, 5, 9670–9677 CrossRef CAS PubMed.
- S. Fratini, D. Mayou and S. Ciuchi, Adv. Funct. Mater., 2016, 26, 2292–2315 CrossRef CAS.
- S. Fratini, S. Ciuchi, D. Mayou, G. T. de Laissardière and A. Troisi, Nat. Mater., 2017, 16, 998 CrossRef CAS PubMed.
- S. Illig, A. S. Eggeman, A. Troisi, L. Jiang, C. Warwick, M. Nikolka, G. Schweicher, S. G. Yeates, Y. H. Geerts, J. E. Anthony and H. Sirringhaus, Nat. Commun., 2016, 7, 10736 CrossRef CAS PubMed.
- R. A. Laudise, C. Kloc, P. G. Simpkins and T. Siegrist, J. Cryst. Growth, 1998, 187, 449–454 CrossRef CAS.
- C. Kloc, P. G. Simpkins, T. Siegrist and R. A. Laudise, J. Cryst. Growth, 1997, 182, 416–427 CrossRef CAS.
- N. A. Minder, S. Ono, Z. Chen, A. Facchetti and A. F. Morpurgo, Adv. Mater., 2011, 24, 503–508 CrossRef PubMed.
- H. T. Yi, Y. Chen, K. Czelen and V. Podzorov, Adv. Mater., 2011, 23, 5807–5811 CrossRef CAS PubMed.
- A. F. Stassen, R. W. I. de Boer, N. N. Iosad and A. F. Morpurgo, Appl. Phys. Lett., 2004, 85, 3899–3901 CrossRef CAS.
- I. N. Hulea, S. Fratini, H. Xie, C. L. Mulder, N. N. Iossad, G. Rastelli, S. Ciuchi and A. F. Morpurgo, Nat. Mater., 2006, 5, 982 CrossRef CAS PubMed.
- H. H. Choi, K. Cho, C. D. Frisbie, H. Sirringhaus and V. Podzorov, Nat. Mater., 2017, 17, 2 CrossRef PubMed.
- G. Gryn’ova and C. Corminboeuf, J. Phys. Chem. Lett., 2016, 7, 5198–5204 CrossRef PubMed.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- R. M. Parrish, L. A. Burns, D. G. A. Smith, A. C. Simmonett, A. E. DePrince, E. G. Hohenstein, U. Bozkaya, A. Y. Sokolov, R. Di Remigio, R. M. Richard, J. F. Gonthier, A. M. James, H. R. McAlexander, A. Kumar, M. Saitow, X. Wang, B. P. Pritchard, P. Verma, H. F. Schaefer, K. Patkowski, R. A. King, E. F. Valeev, F. A. Evangelista, J. M. Turney, T. D. Crawford and C. D. Sherrill, J. Chem. Theory Comput., 2017, 13, 3185–3197 CrossRef CAS PubMed.
- J. J. M. M. J. Turner, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017, http://hirshfeldsurface.net Search PubMed.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627–668 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1899660, 1899661, 1899662. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05902d |
| This journal is © The Royal Society of Chemistry 2020 |