
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn umpolung approach to the hydroboration of pyridines: a novel and efficient synthesis of N-H 1,4-dihydropyridines†
Huan
Yang
,
Li
Zhang
,
Fei-Yu
Zhou
and
Lei
Jiao
 *
*
Center of Basic Molecular Science (CBMS), Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: Leijiao@mail.tsinghua.edu.cn
First published on 3rd December 2019
Abstract
The first inverse hydroboration of pyridine with a diboron(4) compound and a proton source has been realized under simple basic and catalyst-free conditions. This process consists of a formal boryl anion addition to pyridine, which produces an N-boryl pyridyl anion complex, and the subsequent protonation of the anion complex. This process enables a simple and efficient method for the synthesis of multi-substituted N-H 1,4-dihydropyridine (1,4-DHP) derivatives that are difficult to prepare using established methods. Furthermore, this method allows for facile preparation of 4-deuterated 1,4-DHPs from an easily accessible deuterium ion source. This inverse hydroboration reaction represents a new mode for pyridine functionalization.
Introduction
Dihydropyridine (DHP) represents an important class of heterocyclic skeletons which prevalently exist in biologically active agents, pharmaceutically important molecules, and synthetically useful organic reductants, such as nicotinamide adenine dinucleotide (NADH) and Hantzsch ester (HEH).1 Owing to their potential medicinal properties and broad application as reducing agents, 1,4-DHP derivatives have attracted great interest in terms of their synthesis.2 Reduction of pre-activated pyridines using strong inorganic reductants (Scheme 1a)3 and the Hantzsch cyclocondensation (Scheme 1b)4 are two conventional synthetic methods of 1,4-DHPs. In recent years, an array of catalytic reduction reactions of pyridines employing milder reducing reagents such as boranes and silanes have been developed (Scheme 1c).5,6 In these reactions, pyridine coordinates to a Lewis acid center under catalytic conditions, and then the hydride addition occurs at the C4 (or C2) position of the in situ activated pyridine.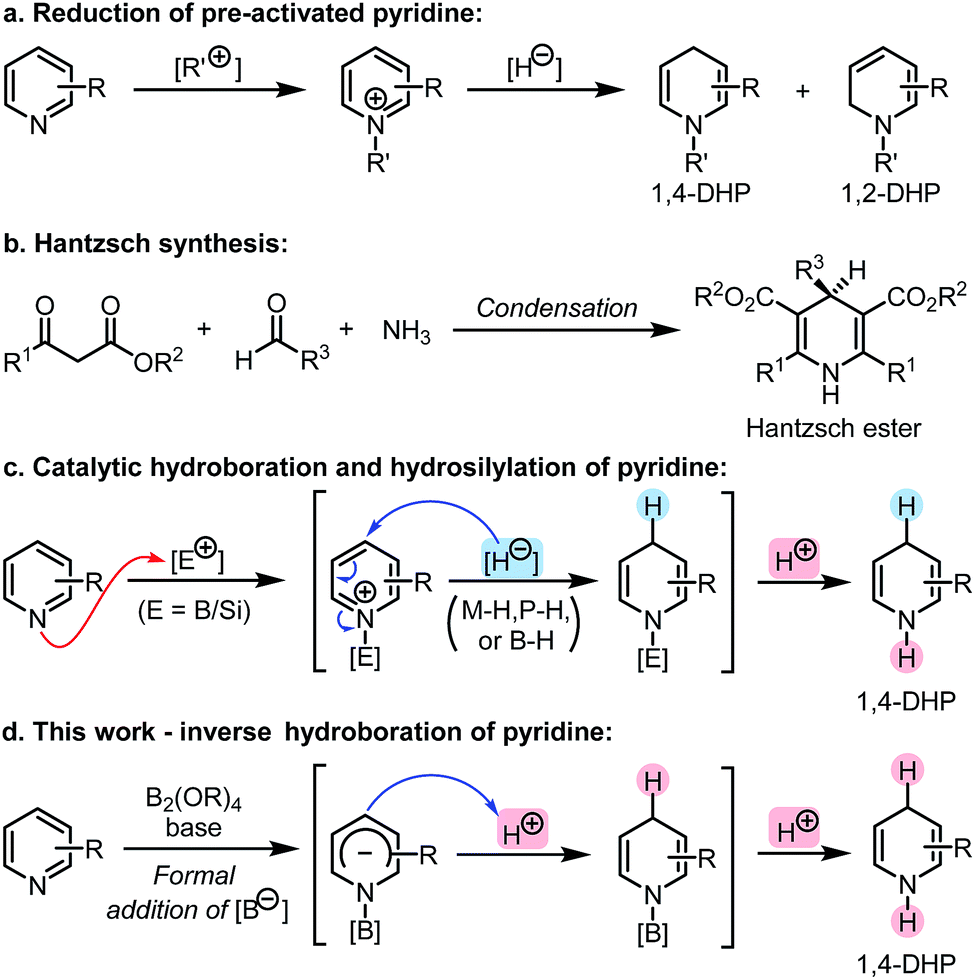 | ||
| Scheme 1 Methods for the synthesis of 1,4-dihydropyridines (1,4-DHPs). | ||
These hydroboration and hydrosilylation protocols provided ideal and flexible approaches to DHPs from easily available pyridine derivatives utilizing either metal- or organocatalysis. Despite these achievements, alternative methodologies that could produce N-H DHPs in a highly efficient and regioselective manner are still in high demand. Opposite to the Lewis acid activation–hydride addition mode involved in these protocols, we envisioned that if the hydroboration of pyridine could be inverse in its polarity, a novel approach to DHPs might be realized. This could not only bring about a conceptually new hydroboration process, but also expand the scope of pyridine reduction. Herein, we report such an umpolung approach to pyridine hydroboration, in which a formal addition of a boryl anion to pyridine occurred first, and the formed Meisenheimer complex was then protonated to form the 1,4-DHP derivative (Scheme 1d). This anion formation–protonation sequence enables an efficient approach to a variety of N-H 1,4-DHPs, which are not easily accessible by conventional methods.
Results and discussion
Reaction design
Our idea for realizing the inverse pyridine hydroboration originated from our previous discovery that 4-phenylpyridine (1) efficiently catalyses the borylation of aryl halides with bis(pinacolato)diboron (B2pin2),7a as well as the related mechanistic studies.7b It was found that the reaction of pyridine 1, B2pin2, and a methoxide anion led to the formation of a trans-2H,2′H-[2,2′-bipyridine]-1,1′-diide borate complex 2 (Scheme 2). DFT calculation suggested that the reaction between pyridine 1, diboron, and methoxide first led to the formation of an N-boryl pyridyl Meisenheimer anion (Int-B) through heterolytic cleavage of the B–B bond in the bis-coordinated diboron(4) (Int-A), which was then trapped by another molecule of 1 to form complex 2. Although Int-B was not observed experimentally, we envisioned to attempt trapping of this proposed anionic intermediate by protonation, which might produce product 3 and thus realize an intriguing umpolung of the conventional pyridine hydroboration process. Further hydrolysis of 3 might afford N-H 1,4-DHP product 4 (Scheme 2). If realized, such an inverse pyridine hydroboration process would represent a novel mode of pyridine reduction, and bring about new opportunities for the synthesis of 1,4-DHP derivatives.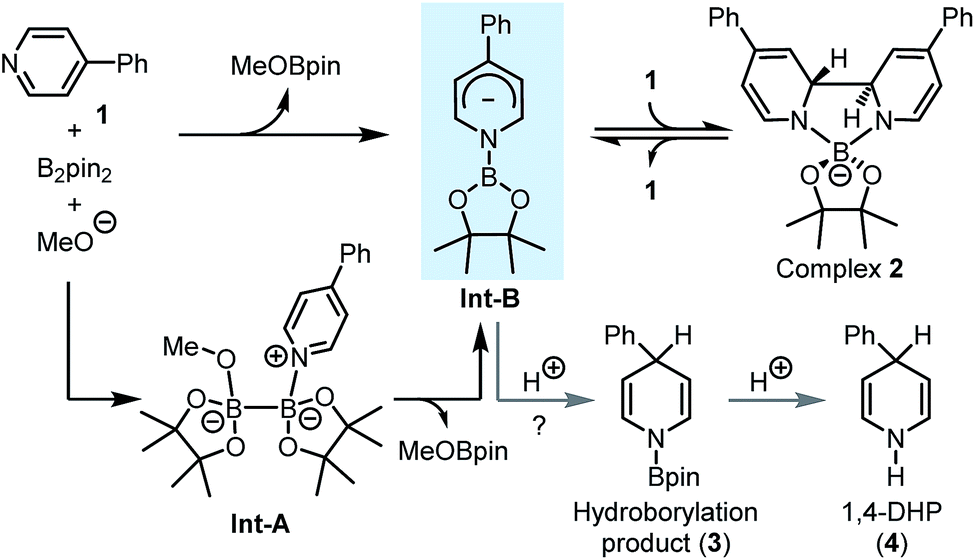 | ||
| Scheme 2 Design of the inverse hydroboration of pyridine based on our previous mechanistic study. | ||
Proof-of-concept and mechanistic understanding
We set out to test this hypothesis by employing methanol as a proton source. The reaction between pyridine 1, MeOK, and B2pin2 was carried out in the presence of 18-crown-6 and varied amounts of methanol in tetrahydrofuran (THF). 1H NMR analysis of the crude reaction mixture showed that when 1.1 equivalent of MeOH was employed, a set of peaks related to dearomatized pyridine emerged (Fig. 1a). Their coupling pattern fitted well with a 4-hydro-4-phenylpyridyl motif, in which the protons in the R–CH(CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)2N moiety showed up at 4.25 ppm (broadened triplet, 1H), 3.89 ppm (multiplet, 2H), and 6.37 ppm (doublet, 2H), respectively. Interestingly, with 1.3 equiv. of MeOH, another set of peaks of dearomatized pyridine appeared at 4.21 ppm (broadened triplet, 1H), 4.12 ppm (multiplet, 2H), and 6.15 ppm (dd, 2H), respectively (Fig. 1b), which became the only dearomatized species when excess MeOH was used (Fig. 1c). The coupling pattern of these peaks was in agreement with that of the desired 1,4-DHP molecule, and gas chromatography-mass spectrometry (GC-MS) analysis of the reaction mixture also detected the peak of the 1,4-DHP product (m/z = 157 for M˙+).
CH)2N moiety showed up at 4.25 ppm (broadened triplet, 1H), 3.89 ppm (multiplet, 2H), and 6.37 ppm (doublet, 2H), respectively. Interestingly, with 1.3 equiv. of MeOH, another set of peaks of dearomatized pyridine appeared at 4.21 ppm (broadened triplet, 1H), 4.12 ppm (multiplet, 2H), and 6.15 ppm (dd, 2H), respectively (Fig. 1b), which became the only dearomatized species when excess MeOH was used (Fig. 1c). The coupling pattern of these peaks was in agreement with that of the desired 1,4-DHP molecule, and gas chromatography-mass spectrometry (GC-MS) analysis of the reaction mixture also detected the peak of the 1,4-DHP product (m/z = 157 for M˙+).
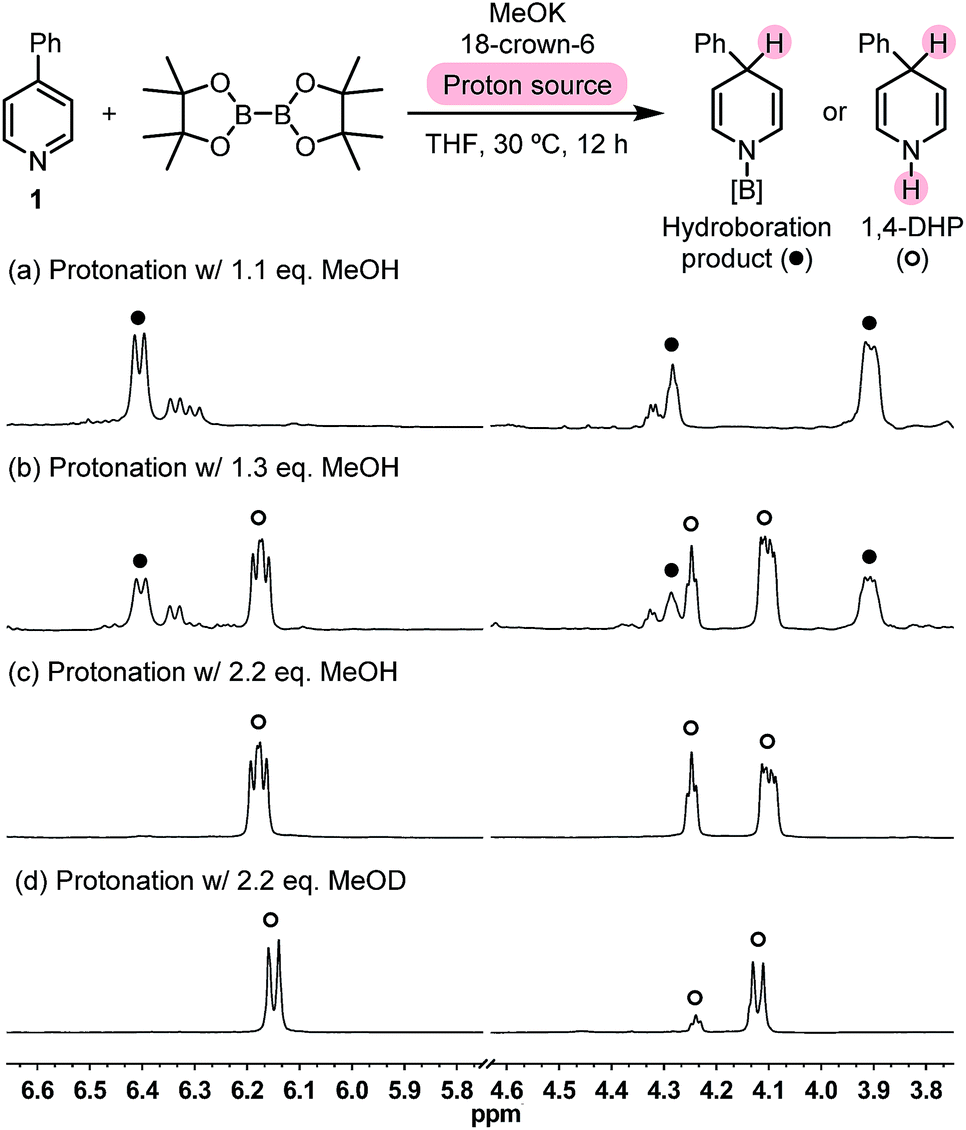 | ||
| Fig. 1 Partial crude 1H NMR spectra of the 4-PhPy/B2pin2/MeOK reaction system protonated with different amounts of methanol. | ||
When excess deuterated methanol (MeOD) was employed, the methine peak at 4.21 ppm decreased due to deuteration, and the peaks at 4.12 and 6.15 ppm became a doublet because deuterium replaces the coupling protons on both NH and methane positions (Fig. 1d). This observation, together with the GC-MS analysis of the product [m/z = 159 for (M-d2)˙+], supported the assignment of the 1,4-DHP structure. These experimental findings served as direct evidence for the formation of the N-boryl pyridyl Meisenheimer anion (Int-B) in the diboron(4)/pyridine/base system, and the regioselective protonation at its C4 position brought about a novel approach to 1,4-DHP derivatives. Because the present reaction is distinct from the established hydroboration protocols in terms of the reaction mechanism, it could be regarded as an inverse hydroboration process.
In order to gain further understanding regarding the formation of the key intermediate Int-B and the regioselective protonation at its C4-position, we performed DFT calculations. In our previous study, the B–B bond cleavage transition state (TS1·K) starting from the bis-coordinated diboron(4)8 has been located.7b In the present study, we analysed the change of NBO charge distribution during the B–B bond breaking event (Int-A·K → TS1·K, Scheme 3). It was found that during this process, negative charge transferred from the MeOBpin part (left part, in blue) to the N-borylpyridine part (right part, in red), and the NBO charge on C4 of the pyridine ring evolved from −0.001 to −0.213, implying a heterolytic cleavage of the B–B bond with a pair of electrons going to the forming N-boryl pyridyl Meisenheimer anion Int-B. The whole process could be viewed as a formal addition of a boryl anion to pyridine, which served as the key step of the inverse pyridine hydroboration. We noted that in the phosphine-catalysed β-selective hydroboration reaction with B2pin2 developed by Gulyás and Fernández, a mechanism involving a similar formal boryl anion addition towards conjugated electron-deficient substrates was proposed.9 The regioselectivity of the subsequent protonation step was also analysed using the DFT calculated HOMO and the corresponding orbital composition of Int-B (Scheme 3). It was found that C4 of the pyridine ring had the largest contribution to the HOMO of anion Int-B, which was supported by both the plot of HOMO and the calculated molecular orbital composition using the NAO method. This could account for the observed regioselectivity in the protonation step, which exhibited preference to the formation of 1,4-DHP over 1,2-DHP.
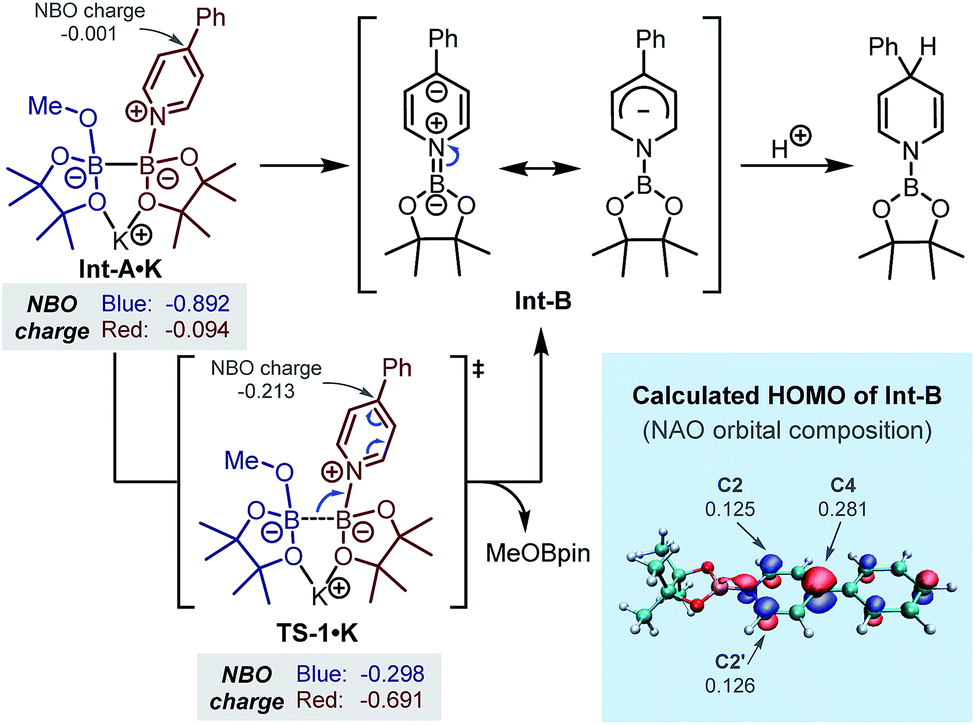 | ||
| Scheme 3 Mechanism of the formation of Meisenheimer complex Int-B and its highest occupied molecular orbital (HOMO) from DFT calculation. | ||
Optimization of reaction conditions and substrate scope
With this proof-of-concept in hand, we set out to search for suitable reaction conditions for practical synthesis of 1,4-DHP (Table 1). First, diboron(4) compounds with different steric hindrances were attempted, and it was found that the less hin-dered diboron(4) exhibits better reactivity (entries 1–3), with bis(ethylene glycolato)diboron (B2eg2) leading to the best NMR yield of 4-phenyl-1,4-dihydropyridine (4). A brief screen of bases indicated that MeONa, Cs2CO3, and K2CO3 were all compatible, and among them Cs2CO3 was the optimal one (entries 4–6). Acetonitrile was also proved to be a suitable solvent, although the yield of 4 was lower than that in THF solvent (entry 7).|
|
||||
|---|---|---|---|---|
| Entry | Diboron(4) | Base | Solvent | Conversionb |
| a Reaction conditions: 4-PhPy (0.36 mmol, 1 equiv.), diboron(4) (1.1 equiv.), base (1.1 equiv.), MeOH (11 equiv.), solvent (2 mL), sealed tube, 30 °C for 4.5 h. b Determined by 1H NMR analysis. B2pin2 = bis(pinacolato)diboron; B2neo2 = bis(neopentyl glycolato)diboron; B2eg2 = bis(ethylene glycolato)diboron. | ||||
| 1 | B2pin2 | MeOK | THF | 17% |
| 2 | B2neo2 | MeOK | THF | 63% |
| 3 | B2eg2 | MeOK | THF | 96% |
| 4 | B2eg2 | MeONa | THF | 78% |
| 5 | B2eg2 | Cs2CO3 | THF | 91% |
| 6 | B2eg2 | Cs2CO3 | THF | 73% |
| 7 | B2eg2 | Cs2CO3 | MeCN | 71% |
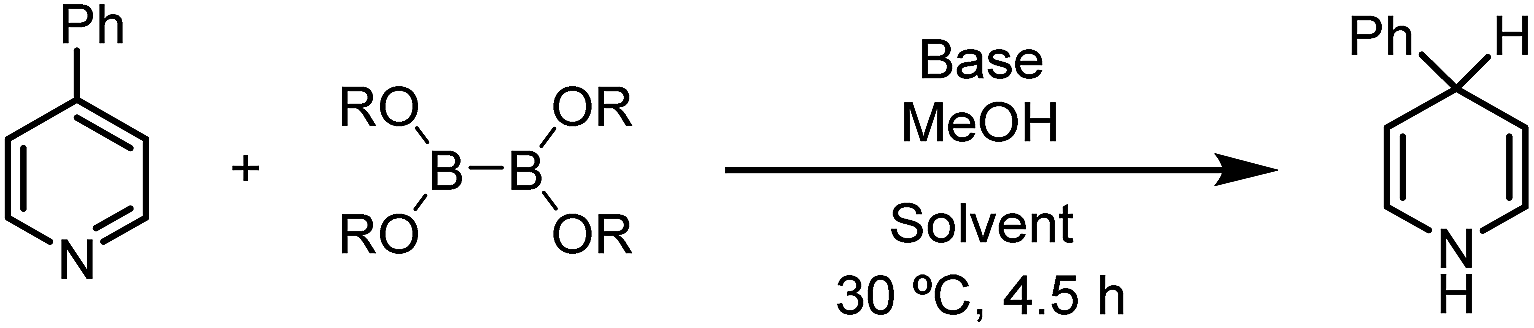
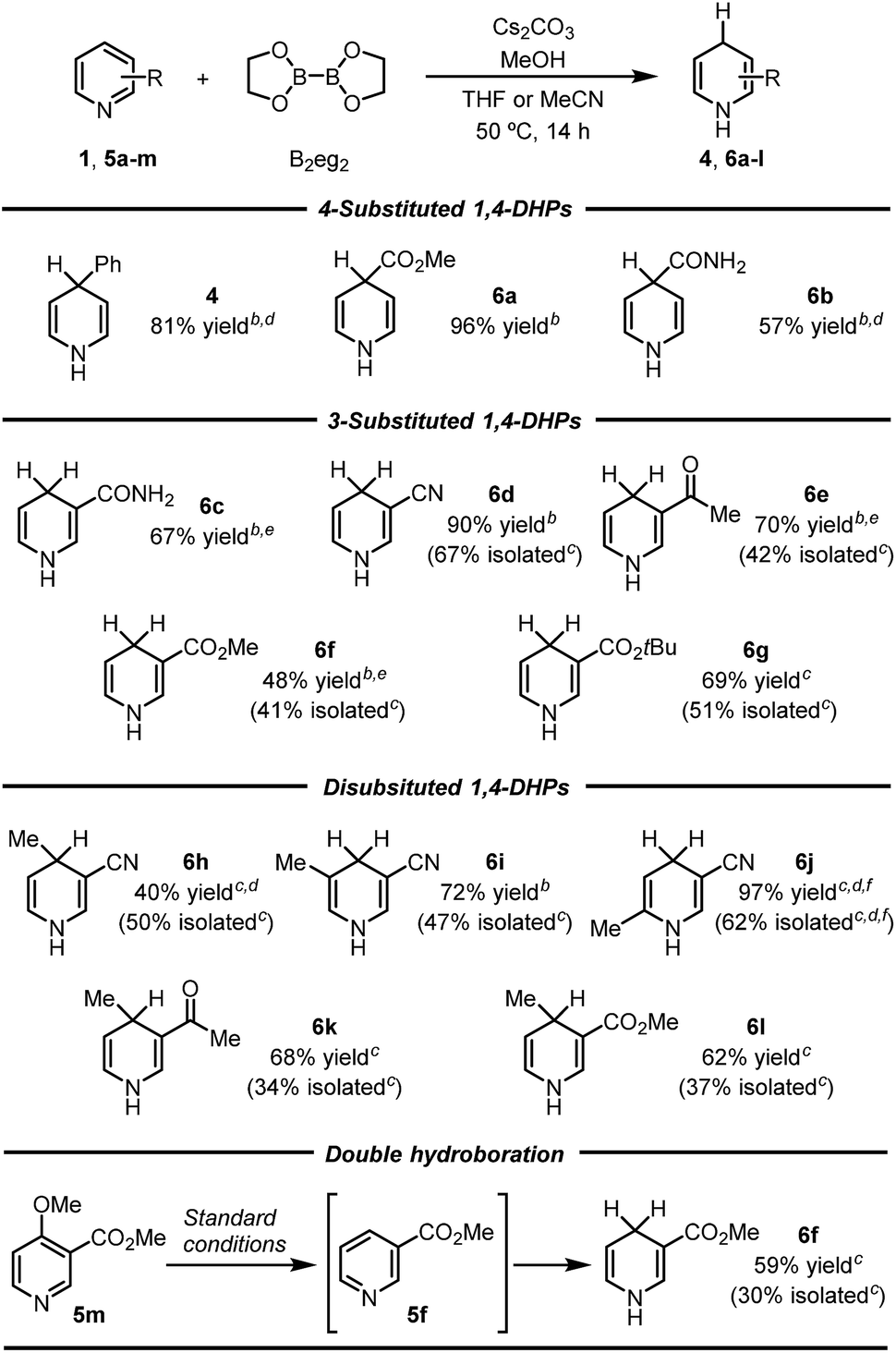
Subsequently, the scope of this reaction was tested by employing various pyridine substrates. In general, this reaction exhibits a broad substrate scope with respect to the pyridine substrate, affording 1,4-DHP derivatives with different substitution patterns in moderate to good yields (Table 2). Although their structures were far from complex, many of these N-H 1,4-DHPs were synthesized for the first time, emphasizing the synthetic value of the present protocol. Generally, the yields of these rather air-labile products were quantified by 1H NMR analyses of the reaction mixtures, and pure products could be obtained by flash column chromatography on basic alumina under an inert atmosphere. However, 4-substituted-1,4-DHPs were difficult to purify because they underwent rapid oxidation back to pyridine substrates (4, 6a, and 6b). Electron-withdrawing substituents, such as amide, ester, and cyano groups, were proved beneficial to the hydroboration process, affording the 1,4-DHP products in good yields.10 Of special note, this inverse hydroboration protocol tolerated the active hydrogen in the pyridine substrates (e.g., 5b and 5c), which complements the traditional hydroboration methods. Multi-substituted pyridines, such as 2,5-, 3,4-, and 3,5-disubstituted ones (5h-l), were all compatible substrates. Interestingly, for a nicotinate substrate bearing a 4-methoxy substitution (5m), hydroboration occurred twice to afford 1,4-DHP product 6f when 2.2 equivalents of B2eg2 and base were employed, due to the elimination of the methoxy group. In all cases, the hydroboration occurred selectively at the 1,4-position, and no 1,2-DHP product was obtained.
| a Reaction conditions: pyridine substrate (1 equiv.), B2eg2 (1.1 equiv.), Cs2CO3 (1.1 equiv.), MeOH (11 equiv.), sealed tube, 50 °C for 14 h. Yields of the 1,4-DHP products were determined by 1H NMR analysis of the reaction mixture using DMSO as the internal standard (isolated yields are shown in parentheses). b THF as the solvent. c MeCN as the solvent. d MeONa was used instead of Cs2CO3. e MeOK and 18-crown-6 (1.1 equiv. each) were used instead of Cs2CO3. f The reaction was conducted at 65 °C. |
|---|
|
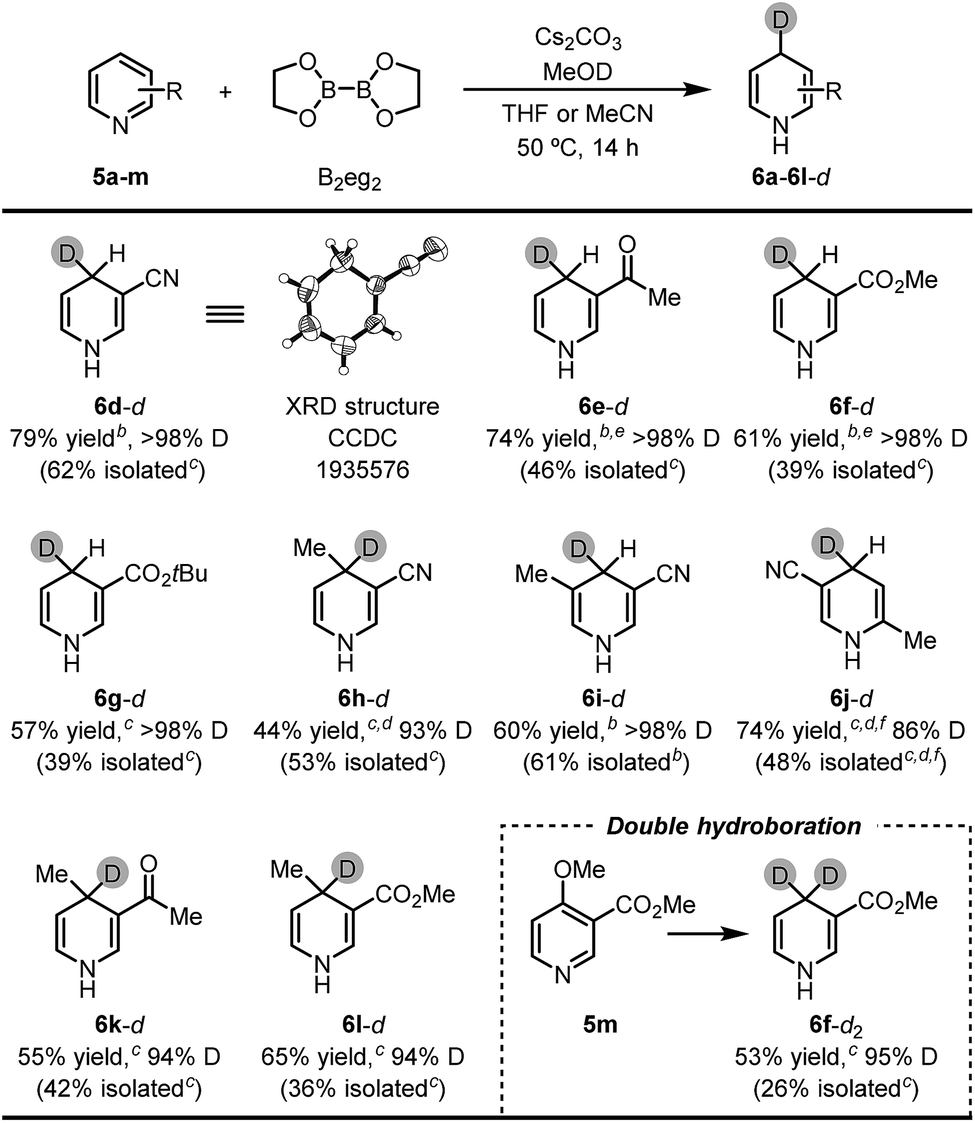
The successful umpolung of the pyridine hydroboration allowed for facile access to a series of 4-deuterated 1,4-DHPs. 1,4-DHPs are prevalently used as hydride and/or electron donors in organic chemistry, and 4-deuterium substituted 1,4-DHPs are always employed as ideal mechanistic probes and organic deuteride transfer reagents. However, following the established synthetic methods (Scheme 1a–c), less accessible deuteride reagents (e.g., NaBD4 and DBpin) and deuterated aldehydes should be employed to prepare 4-deuterated 1,4-DHPs, while the present inverse hydroboration protocol only requires a readily available D+ source for the same task. The result shown in Fig. 1d has already demonstrated this, although the deutero ratio was still not satisfactory with only 2.2 equiv. of MeOD. To our delight, by employing 22 equiv. of MeOD, 1,4-DHPs with high deuterium incorporation at the 4-position were obtained (Table 3).
| a Reaction conditions: pyridine substrate (1 equiv.), B2eg2 (1.1 equiv.), Cs2CO3 (1.1 equiv.), MeOD (22 equiv.), sealed tube, 50 °C for 14 h. Yields of the 1,4-DHP products were determined by 1H NMR analysis of the reaction mixture using DMSO as the internal standard (isolated yields are shown in parentheses). b THF as the solvent. c MeCN as the solvent. d MeONa was used instead of Cs2CO3. e MeOK and 18-crown-6 (1.1 equiv. both) were used instead of Cs2CO3. f The reaction was conducted at 65 °C. |
|---|
|
It was found that most pyridine substrates described in Table 2 could be transformed to the desired 4-deuterated 1,4-DHPs in moderate to good yields with excellent deuterium incorporation (up to >98%) at the C4 position. Furthermore, the structure of the 1,4-DHP product was unambiguously confirmed using the XRD structure of 3-cyano-4-deuterated-1,4-dihydropyridine (6d-d). Interestingly, double inverse hydroboration of pyridine 5m has been successfully utilized to prepare 4,4-dideuterated 1,4-DHP 6f-d2. These results demonstrate that our method can provide a straightforward synthetic approach to a series of 4-deuterated 1,4-DHP derivatives, where easily available and inexpensive MeOD was used as the deuterium source.
Further experiments were also performed to reveal the limitations of the present protocol (Fig. 2). We found that halogenated pyridines 5n and 5o, 3-trifluoromethylpyridine (5p), 2-substituted pyridines 5q and 5r, tetrasubstituted pyridine 5s, and other pyridine-embedded heterocycles, such as quinoline (5t), isoquinoline (5u) and acridine (5v), were not compatible with this inverse hydroboration protocol. They either exhibited a low reactivity, or produced a complex mixture of products under the reaction conditions.
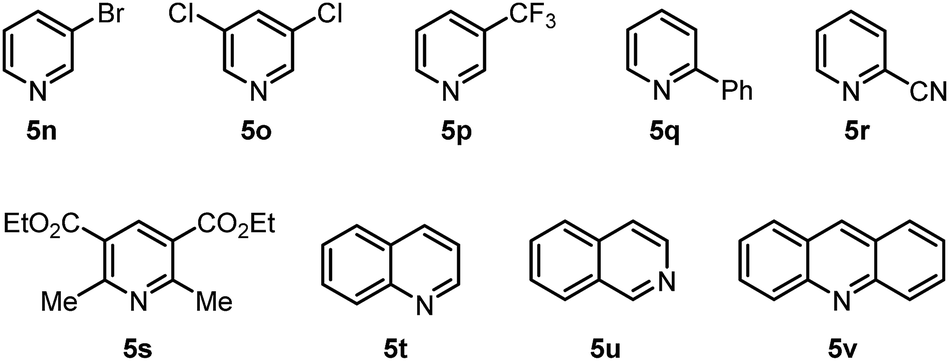 | ||
| Fig. 2 Unsuccessful substrates for the inverse hydroboration. | ||
Potential use of the 1,4-DHP product
1,4-DHPs are typical organic hydride donors in hydride transfer reactions.1g In particular, Hantzsch ester (HEH) is the most prevalently used hydride donor in asymmetric hydride transfer reactions. Since many of the newly synthesized 1,4-DHPs are structurally unprecedented in the literature, we are curious about their performance in asymmetric hydride transfer reactions as hydride donors. In this line, we explored the enantioselective hydride reduction of α,β-unsaturated aldehyde 7 in the presence of organocatalyst 8 (Scheme 4). The model reaction employing HEH 11 as the hydride donor afforded chiral aldehyde 9 in 49% yield and 83% ee under literature conditions.11 A slight decrease in both yield and ee was observed when 1,4-DHP 6f was employed in place of HEH 11. Gratifyingly, when deuterium-labeled 1,4-DHP 6f-d2 was used, deuteride transfer occurred with comparable efficiency to hydride transfer (50% yield, 80% ee), affording deuterium-enriched product 9 (90% D). These results showcased the potential use of the 1,4-DHP products of the present method, which also indicated that in asymmetric hydride transfer reactions, the densely substituted DHP core of HEH is not a key factor in stereocontrol.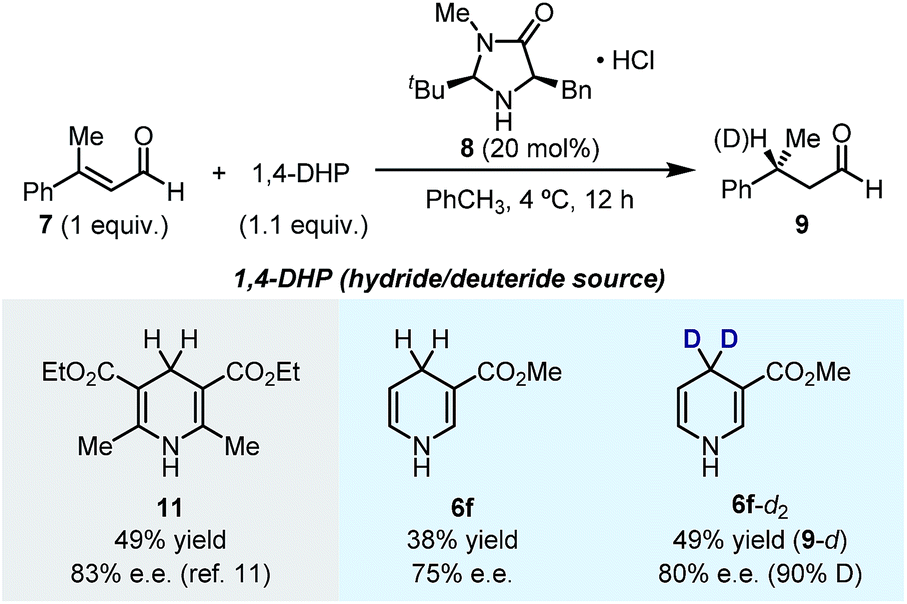 | ||
| Scheme 4 The use of the produced 1,4-DHPs in asymmetric hydride/deuteride transfer reaction. | ||
Conclusions
In conclusion, the umpolung of pyridine hydroboration has been realized for the first time by utilizing the diboron(4)/pyridine/base system in a transition-metal-free manner. The key steps include a base-promoted formal addition of a boryl anion to pyridine, and the subsequent protonation of the produced N-boryl pyridyl anion complex. This inverse hydroboration process enables a simple and efficient method for the synthesis of 1,4-dihydropyridine derivatives from pyridines, and features broad substrate scope and good functional group compatibility. A great advantage of this protocol is that 4-deuterated 1,4-DHPs could be easily prepared using easily available MeOD as the deuterium source. The produced 1,4-DHP derivatives are complementary to previously known ones, and can act as useful organic hydride/deuteride donors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge the financial support from the National Key Research and Development Plan (grant no. 2017YFA0505200) and the National Natural Science Foundation of China (grant no. 21772110 and 21822304). The technology platform of the CBMS and the Tsinghua Xuetang Talents Program are acknowledged for providing instrumentation and computational resources.Notes and references
- For selected reviews, see: (a) U. Eisner and J. Kuthan, Chem. Rev., 1972, 72, 1 CrossRef CAS; (b) D. M. Stout and A. I. Meyers, Chem. Rev., 1982, 82, 223 CrossRef CAS; (c) C. Safak and R. Simsek, Mini-Rev. Med. Chem., 2006, 6, 747 CrossRef CAS PubMed; (d) N. Pollak, C. Dölle and M. Ziegler, Biochem. J., 2007, 402, 205 CrossRef CAS PubMed; (e) N. Edraki, A. R. Mehdipour, M. Khoshneviszadeh and R. Miri, Drug Discovery Today, 2009, 14, 1058 CrossRef CAS PubMed; (f) M. Rueping, J. Dufour and F. R. Schoepke, Green Chem., 2011, 13, 1084 RSC; (g) C. Zheng and S. L. You, Chem. Soc. Rev., 2012, 41, 2498 RSC.
- For selected reviews, see: (a) R. Lavilla, J. Chem. Soc., Perkin Trans. 1, 2002, 1141 RSC; (b) S. G. Ouellet, A. M. Walji and D. W. C. MacMillan, Acc. Chem. Res., 2007, 40, 1327 CrossRef CAS PubMed; (c) A. Silva, E. Silva and P. Varandas, Synthesis, 2013, 45, 3053 CrossRef.
- (a) E. E. Knaus and K. Redda, Can. J. Chem., 1977, 55, 1788 CrossRef CAS; (b) D. L. Comins and A. H. Abdullah, J. Org. Chem., 1984, 49, 3392 CrossRef CAS; (c) R. J. Sundberg, G. Hamilton and C. Trindle, J. Org. Chem., 1986, 51, 3672 CrossRef CAS; (d) P. S. Wagenknecht, J. M. Penney and R. T. Hembre, Organometallics, 2003, 22, 1180 CrossRef CAS; (e) J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642 CrossRef CAS PubMed; (f) Y. Maenaka, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 367 CrossRef CAS PubMed; (g) L. Q. Lu, Y. Li, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 8382 CrossRef CAS PubMed.
- (a) J.-P. Wan and Y. Liu, RSC Adv., 2012, 2, 9763 RSC; (b) M. M. Khan, Saigal, S. Khan, S. Shareef and M. Danish, ChemistrySelect, 2018, 3, 6830 CrossRef CAS.
- For metal-catalysed hydroboration/hydrosilylation of pyridines, see: (a) L. Hao, J. F. Harrod, A.-M. Lebuis, Y. Mu, R. Shu, E. Samuel and H.-G. Woo, Angew. Chem., Int. Ed., 1998, 37, 3126 CrossRef CAS; (b) R. Shu, J. F. Harrod, H.-G. Woo and E. Samuel, Can. J. Chem., 2001, 79, 1075 CrossRef; (c) D. V. Gutsulyak, A. van der Est and G. I. Nikonov, Angew. Chem., Int. Ed., 2011, 50, 1384 CrossRef CAS PubMed; (d) M. Arrowsmith, M. S. Hill, T. Hadlington, G. Kociok-Kohn and C. Weetman, Organometallics, 2011, 30, 5556 CrossRef CAS; (e) C. D. F. Königs, H. F. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 10076 CrossRef PubMed; (f) A. S. Dudnik, V. L. Weidner, A. Motta, M. Delferro and T. J. Marks, Nat. Chem., 2014, 6, 1100 CrossRef CAS PubMed; (g) J. Intemann, M. Lutz and S. Harder, Organometallics, 2014, 33, 5722 CrossRef CAS; (h) J. Intemann, H. Bauer, J. Pahl, L. Maron and S. Harder, Chem.–Eur. J., 2015, 21, 11452 CrossRef CAS PubMed; (i) J. Jeong, S. Park and S. Chang, Chem. Sci., 2016, 7, 5362 RSC; (j) A. Kaithal, B. Chatterjee and C. Gunanathan, Org. Lett., 2016, 18, 3402 CrossRef CAS PubMed; (k) J. L. Lortie, T. Dudding, B. M. Gabidullin and G. I. Nikonov, ACS Catal., 2017, 7, 8454 CrossRef CAS; (l) S. Park and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 7720 CrossRef CAS PubMed; (m) F. Zhang, H. Song, X. Zhuang, C.-H. Tung and W. Wang, J. Am. Chem. Soc., 2017, 139, 17775 CrossRef CAS PubMed; (n) P. Ji, X. Feng, S. S. Veroneau, Y. Song and W. Lin, J. Am. Chem. Soc., 2017, 139, 15600 CrossRef CAS PubMed; (o) H. Liu, M. Khononov and M. S. Eisen, ACS Catal., 2018, 8, 3673 CrossRef CAS; (p) S. R. Tamang, A. Singh, D. K. Unruh and M. Findlater, ACS Catal., 2018, 8, 6186 CrossRef CAS; (q) M. W. Gribble Jr, S. Guo and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 5057 CrossRef PubMed; (r) J. Liu, J.-Y. Chen, M. Jia, B. Ming, J. Jia, R.-Z. Liao, C.-H. Tung and W. Wang, ACS Catal., 2019, 9, 3849 CrossRef CAS.
- For organocatalytic hydroboration/hydrosilylation of pyridines, see: (a) X. Fan, J. Zheng, Z. H. Li and H. Wang, J. Am. Chem. Soc., 2015, 137, 4916 CrossRef CAS PubMed; (b) N. Gandhamsetty, S. Park and S. Chang, J. Am. Chem. Soc., 2015, 137, 15176 CrossRef CAS PubMed; (c) Z. Y. Liu, Z. H. Wen and X. C. Wang, Angew. Chem., Int. Ed., 2017, 56, 5817 CrossRef CAS PubMed; (d) E. N. Keyzer, S. S. Kang, S. Hanf and D. S. Wright, Chem. Commun., 2017, 53, 9434 RSC; (e) J. E. Radcliffe, J. J. Dunsford, J. Cid, V. Fasano and M. J. Ingleson, Organometallics, 2017, 36, 4952 CrossRef CAS PubMed; (f) B. Rao, C. C. Chong and R. Kinjo, J. Am. Chem. Soc., 2018, 140, 652 CrossRef CAS PubMed; (g) T. Hynes, E. N. Welsh, R. McDonald, M. J. Ferguson and A. W. H. Speed, Organometallics, 2018, 37, 841 CrossRef CAS.
- (a) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607 CrossRef CAS PubMed; (b) L. Zhang and L. Jiao, Chem. Sci., 2018, 9, 2711 RSC; (c) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2019, 141, 9124 CrossRef PubMed.
- Bis-coordinated diboron(4) compounds have been characterized in previous studies, see: (a) S. Pietsch, U. Paul, I. A. Cade, M. J. Ingleson, U. Radius and T. B. Marder, Chem. - Eur. J., 2015, 21, 9018 CrossRef CAS PubMed; (b) I. A. Cade, W. Y. Chau, I. Vitorica-Yrezabal and M. J. Ingleson, Dalton Trans., 2015, 44, 7506 RSC ; For a review on diboron(4) compounds, see:; (c) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091 CrossRef CAS PubMed.
- A. Bonet, H. Gulyás and E. Fernández, Angew. Chem., Int. Ed., 2010, 49, 5130 CrossRef CAS PubMed.
- It was found that the pyridine substrate without an electron-withdrawing substituent could also undergo the hydroboration reaction, although affording a lower yield of the 1,4-DHP product. See the ESI† for details.
- For selected examples, see: (a) J. W. Yang, M. T. Hechavarria Fonseca, N. Vignola and B. List, Angew. Chem., Int. Ed., 2004, 44, 108 CrossRef PubMed; (b) S. G. Ouellet, J. B. Tuttle and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 32 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional information, and characterization data. CCDC 1935576. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05627k |
| This journal is © The Royal Society of Chemistry 2020 |