
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimetic nanochannels for the discrimination of sialylated glycans via a tug-of-war between glycan binding and polymer shrinkage†‡
Minmin
Li
ab,
Yuting
Xiong
ab,
Dongdong
Wang
a,
Yunhai
Liu
b,
Bing
Na
b,
Haijuan
Qin
c,
Jinxuan
Liu
 d,
Xinmiao
Liang
*a and
Guangyan
Qing
*a
d,
Xinmiao
Liang
*a and
Guangyan
Qing
*a
aKey Laboratory of Separation Science for Analytical Chemistry, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. E-mail: qinggy@dicp.ac.cn; liangxm@dicp.ac.cn
bJiangxi Province Key Laboratory of Polymer Micro/Nano Manufacturing and Devices, East China University of Technology, 418 Guanglan Avenue, Nanchang 330013, China
cResearch Centre of Modern Analytical Technology, Tianjin University of Science and Technology, Tianjin 300457, China
dInstitute of Artificial Photosynthesis, State Key Laboratory of Fine Chemicals, Dalian University of Technology, Dalian 116024, China
First published on 3rd December 2019
Abstract
Sialylated glycans that are attached to cell surface mediate diverse cellular processes such as immune responses, pathogen binding, and cancer progression. Precise determination of sialylated glycans, particularly their linkage isomers that can trigger distinct biological events and are indicative of different cancer types, remains a challenge, due to their complicated composition and limited structural differences. Here, we present a biomimetic nanochannels system integrated with the responsive polymer polyethyleneimine-g-glucopyranoside (Glc-PEI) to solve this problem. By using a dramatic “OFF–ON” change in ion flux, the nanochannels system achieves specific recognition for N-acetylneuraminic acid (Neu5Ac, the predominant form of sialic acid) from various monosaccharides and sialic acid species. Importantly, different “OFF–ON” ratios of the conical nanochannels system allows the precise and sensitive discrimination of sialylated glycan linkage isomers, α2–3 and α2–6 linkage (the corresponding ion conductance increase ratios are 96.2% and 264%, respectively). Analyses revealed an unusual tug-of-war mechanism between polymer-glycan binding and polymer shrinkage. The low binding affinity of Glc-PEI for the α2–6-linked glycan caused considerable shrinkage of Glc-PEI layer, but the high affinity for the α2–3-linked glycan resulted in only a slight shrinkage. This competition mechanism provides a simple and versatile materials design principle for recognition or sensing systems that involve negatively charged target biomolecules. Furthermore, this work broadens the application of nanochannel systems in bioanalysis and biosensing, and opens a new route to glycan analysis that could help to uncover the mysterious and wonderful glycoworld.
Introduction
Mammalian cell surfaces are covered with a dense layer of glycans, that are attached to various glycoconjugates, constituting essential mediators of diverse cellular processes.1 As a key component, sialic acid is typically linked to glycan non-reducing termini with an α2–3 or α2–6 linkage.2 This outmost position and ubiquitous distribution enable sialylated glycans, serving as critical binding sites for lectins or pathogens, to be involved in numerous cellular processes.3 For example, the avian influenza H7N9 virus specifically recognizes sialylated glycans with an α2–3 linkage (α2–3-linked glycans). However, in 2013, a reassortant avian-origin H7N9 acquired the ability to bind α2–6-linked glycans that are dominant in human respiratory epithelial cells and infect humans (Scheme 1A).4 In addition, aberrant protein sialylation has been closely correlated with various cancers.5 For instance, prostate cancer features an elevated expression of α2–3-linked glycans,5c,d whereas breast cancer exhibits overexpression of α2–6-linked glycans (Scheme 1B).5e,f Additionally, HIV-1 disguises itself with a glycan-shield to escape the recognition by the host immune system.6 Virion-incorporated, host-derived α2–3-linked glycans mediate the capture of mature dendritic cells to HIV-1, constituting a key early step in HIV-1 infection.7 Therefore, precisely recognizing sialylated glycans, discriminating different glycan linkage types, and determining the glycan fine structure can facilitate both the understanding of disease progression and the improvement of early diagnoses and targeted treatments.8 However, due to complicated composition, subtle structural differences, and ultralow abundance of these glycans,2,9a current glycan analysis and determination methods rely excessively on multistage mass spectrum (MS),9a,b ultra-high field nuclear magnetic resonance spectroscopy (NMR),9c or various informatics approaches,9d which usually require significant expertise in complex operation and data analysis, complicated glycans derivatization, and high sample amounts. Thus, developing highly sensitive, easily accessible, and label-free glycan analysis methods and devices represents a major challenge, albeit one with potential rewards.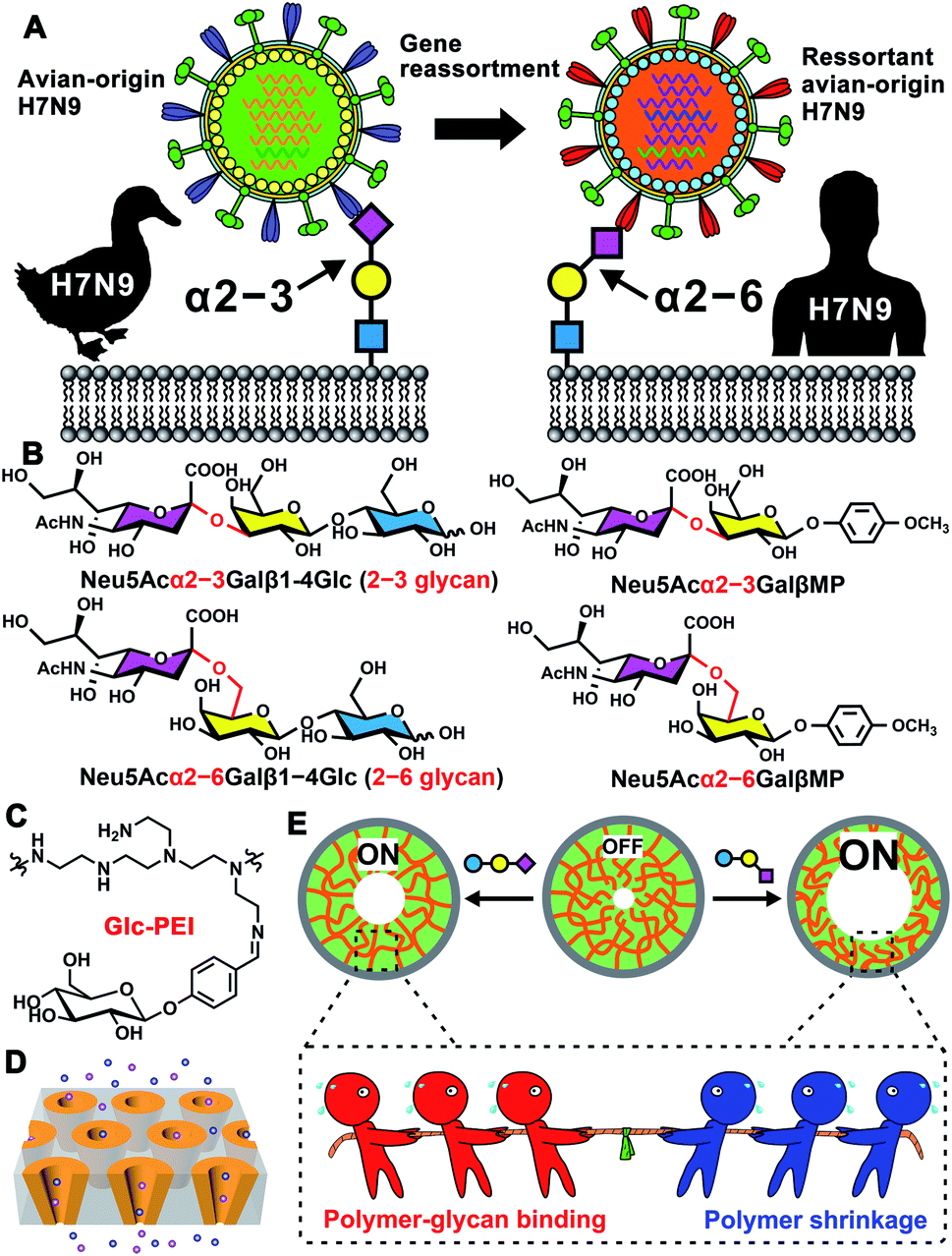 | ||
| Scheme 1 Schematics illustration of the biomimetic nanochannels for glycan recognition. (A) H7N9 virus binds to avian α2–3-linked sialylated glycan receptor, while the reassortant H7N9 virus binds to human α2–6-linked glycan receptor. (B) Chemical structures of two sialylated glycan models (2–3 glycan and 2–6 glycan), and two sialylated disaccharides with same linkage types. (C) Chemical structure of Glc-PEI. (D) Schematic illustration of the Glc-PEI-modified nanochannels. (E) Glycan-induced nanochannel “OFF–ON” changes, and the tug-of-war cartoon to illustrate the competition between polymer-glycan binding and the polymer shrinkage. | ||
Biomimetic ion nanochannels offer an ideal solution to the challenge.10 Over the past decade, various exquisite functional nanochannel systems (resulting from integration of the functional elements onto the inner wall of nanochannel) have been developed for detection, sensing, and molecular transport applications.11 For example, Rant's group reported that a solid nanochannel modified with nitrilotriacetic acid receptors can be used to detect His-tagged proteins and discriminate subclasses of rodent IgG antibodies.11d Xia's group used DNA nanostructures as the functional element to prepare the nanochannel-based sensor and detected the target DNA with a detection limit of 10 fM in complex matrices.11e Jiang's group constructed a series of biomimetic nanochannels integrated with peptides or DNA aptamers as selective molecule transport platforms, and explored the prospect of drug delivery with a feedback function.11f,g However, few reports exist for the detection of biologically significant glycan molecules using this functional nanochannels platform. The gap in complex glycan-sensing devices has persisted despite several excellent research papers describing glucose recognition based on a pillar[6]arene- and an enzyme-functionalized nanochannel,12a,b and several other similarly creative research papers involving the analyses of hyaluronan12c and heparin12dvia a solid nanopore approach. Compared with monosaccharides, disaccharides, and polysaccharides with periodic repeated units, the recognition of complex glycans is substantially more challenging, owing to their complicated monosaccharide compositions, branched structures and microheterogeneity,2,9a which poses higher demands in the design of the functional elements of nanochannels. To solve this challenge, we propose a simple but versatile strategy that combines a responsive polymer with artificial nanochannels, and report that the precise recognition and discrimination of glycan linkage isomers can be achieved with this biomimetic nanochannels system.
As shown in Scheme 1C, the polymer polyethyleneimine-g-glucopyranoside (Glc-PEI) was prepared by grafting the glucopyranoside unit onto the polyethyleneimine chain, which was further integrated into the nanochannels to produce a functional nanochannels system (Scheme 1D). This system achieved recognition of monosaccharides, sialic acids, and sialylated glycans through “OFF–ON” changes in ion flux through the nanochannel (Scheme 1E). Notably, different “OFF–ON” ratios enable precise discrimination of glycan linkage isomers. Binding affinity studies of Glc-PEI with glycans have indicated that the competition between Glc-PEI-glycan binding and Glc-PEI shrinkage leads to varying shrinkage levels of Glc-PEI, finally causing increases in nanopore size and ion conductance. This system exhibits high selectivity, high sensitivity, and label-free features, indicating its potential for applications of functional nanochannel platforms in glycomics and the development of glycan-related devices.
Results and discussion
Cylindrical nanochannels for glycan recognition
The polymer Glc-PEI comprises two parts. Polycationic PEI functions as the flexible main chain, with a conformation transition feature.13 Inspired by emerging trends in carbohydrate–carbohydrate interactions,14 the glucose unit (Glc) from glucopyranoside was chosen as the recognition element for the glycans, which could participate in multiple hydrogen bonding interactions via its four hydroxyl groups.15 A cylindrical porous anodic alumina (PAA) membrane was chosen as the nanochannels substrate, due to its regular and parallel porous structure, which aids investigations of recognition effects and controlled transport of molecules and ions.16 After being constructed by grafting Glc units onto PEI chains through aldimine condensation (the grafted ratio of Glc was approximately 9%, Scheme S1, Fig. S1‡), the resulting Glc-PEI was anchored onto the inner walls of the PAA nanochannels through a two-step coupling reaction (Scheme S2, Fig. S2 and S3‡). The Glc-PEI-modified PAA membrane was then mounted in a home-made electrochemical device (Fig. 1A, inset), and the transmembrane ionic current was recorded 10 min after the addition of a NaCl electrolyte containing the target analyte.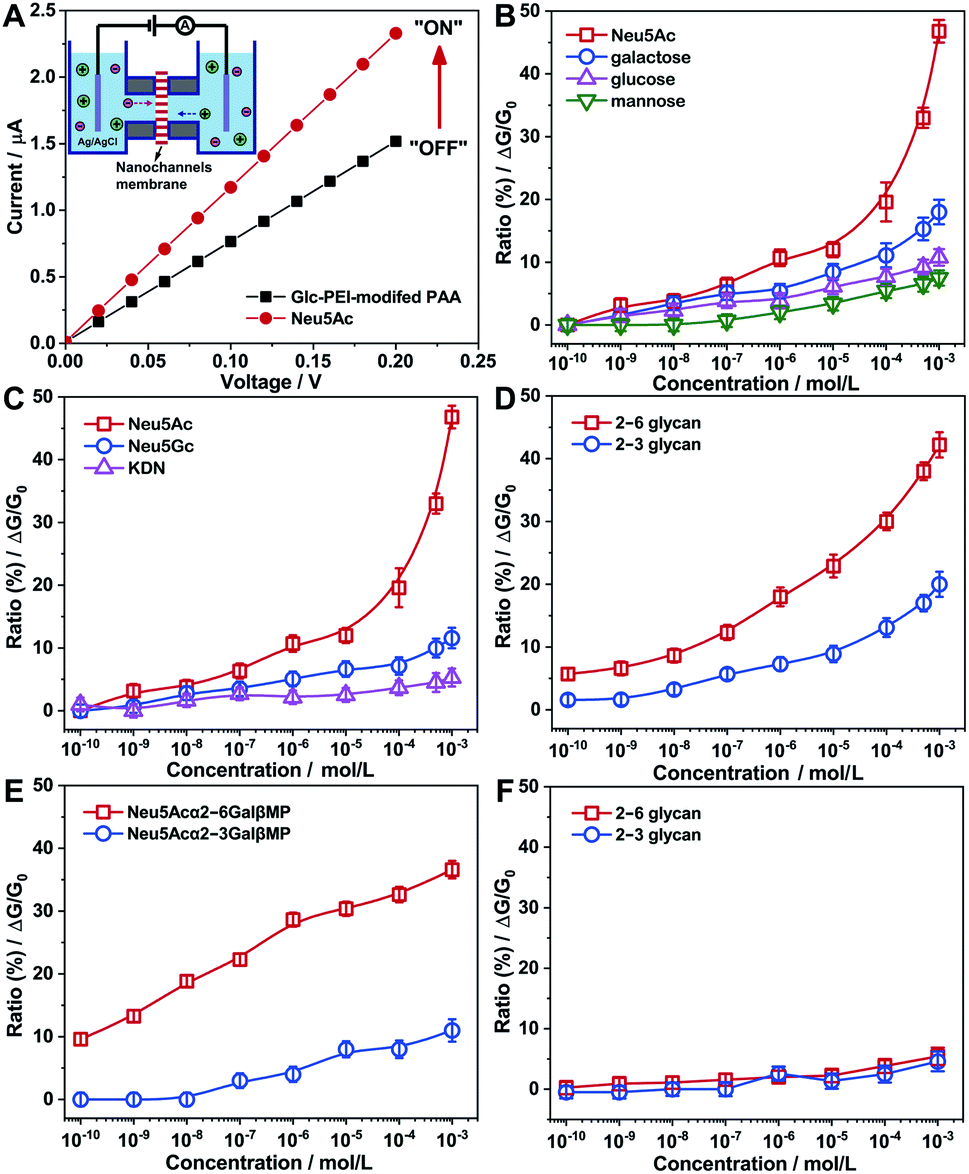 | ||
| Fig. 1 Transmembrane ionic current measurements based on the PAA nanochannels. (A) I–V curves of Glc-PEI-modified PAA in 0.01 M NaCl solution before and after addition of Neu5Ac (1 mM). The inset represents the electrochemical device. (B–E) Concentration-dependent transmembrane ion conductance increase ratios (ΔG/G0) of Glc-PEI-modified PAA upon treatment by various monosaccharides (B), sialic acids (C), sialylated glycan linkage isomers (D), sialylated disaccharide linkage isomers (E). (F) Control experiment based on the PEI-modified PAA membrane. | ||
The addition of Neu5Ac caused an increase of the ionic current (at +0.2 V) from an initial 1.58 μA to 2.35 μA (Fig. 1A), that is, the ion conductance (G) increased from 7.9 μS (“OFF” state) to 11.75 μS (“ON” state). The corresponding ion conductance increase ratio (defined as [G − G0]/G0, i.e., ΔG/G0) was approximately 48% (the concentration was 1 mM, and similarly hereinafter). Concentration-dependent measurements demonstrate that the increase ratios of ion conductance with the addition of Neu5Ac are more pronounced than with the addition of galactose, mannose, and glucose (Fig. 1B). These different conductance increases demonstrate a distinct recognition effect. Moreover, the ion conductance measurement results from three sialic acids, namely Neu5Ac, N-glycolylneuraminic acid (Neu5Gc), and 2-keto-3-deoxyonic acid (KDN), also revealed a significant discrimination effect. The conductance increase ratio of Neu5Ac (47%) was higher than those of Neu5Gc (11.6%) and KDN (5.3%) (Fig. 1C). These results reveal that the Glc-PEI-modified PAA has a strong specific recognition ability for Neu5Ac, which cannot be attributed to the acidity of Neu5Ac.
Next, we turned our attention to Neu5Ac-capped glycans. Neu5Acα2–3Galβ1–4Glc (abbreviated as 2–3 glycan) and Neu5Acα2–6Galβ1–4Glc (abbreviated as 2–6 glycan), a pair of model glycan linkage isomers with similar composition to the glycan fragments found in H7N9 receptor (Scheme 1B), was chosen to perform the transmembrane ionic current measurement. The ion conductance change of Glc-PEI-modified PAA was concentration dependent for both 2–3 glycan and 2–6 glycan (Fig. 1D). Notably, the effect of increasing the concentration of 2–6 glycan on the increase in ion conductance was greater than that of 2–3 glycan. The maximum increase ratios of 2–6 glycan and 2–3 glycan were 42.5% and 20%, respectively. This implies that Glc-PEI-modified PAA exhibited a strong recognition preference for the sialylated trisaccharide with α2–6 linkage. This preference for α2–6 linkage was further confirmed in the evaluations of two sialylated disaccharide linkage isomers, Neu5Acα2–6GalβMP and Neu5Acα2–3GalβMP (two kinds of methoxyphenyl pyranoside disaccharides, MP denotes the 4-methoxyphenyl, the chemical structures are shown in Scheme 1B). A series of ion conductance increase ratios observed for the 2–6-linked disaccharide were considerably higher than those observed for the 2–3-linked disaccharide (Fig. 1E). In addition, control experiments to explore the glycan-recognition capacity of PEI-modified PAA nanochannels in the absence of the grafted Glc unit was performed. The treatment of either 2–6 glycan or 2–3 glycan produced small, nearly indistinguishable changes in ion conductance (Fig. 1F). This indicates that the Glc unit of the Glc-PEI polymer is necessary to achieve efficient glycan recognition.
Asymmetric conical nanochannels for glycan recognition
The polyethylene terephthalate (PET) conical nanoporous membrane features a significant ionic current rectification effect that can facilitate the study of recognition sensitivity.17 Thus, the PET membrane was chosen as substrate to prepare functional nanochannels system. The PET conical nanoporous membranes was prepared using an asymmetric ion track-etching technique based on the home-made electrochemical device (Fig. S4‡). The diameters of the large opening (base side) and the small opening (tip side) of the nanochannels were determined to be approximately 323 nm and 31 nm, respectively, by scanning electron microscope (Fig. S5‡). Then, the Glc-PEI polymer was anchored to the PET conical nanochannel inner wall through a one-step coupling reaction (Scheme S3, Fig. S6 and S7‡). The resulting Glc-PEI-modified PET membrane was used to conduct the same transmembrane ionic current measurement. The results show that, addition of 2–6 glycan caused an obvious change in ionic current (Fig. 2A). Taking the current value at −2 V for example, the current change shows a concentration-dependent increase trend. The ionic rectification effect (|I−2V|/|I+2V|) of Glc-PEI-modified PET also becomes evident as the concentration of 2–6 glycan increases (Fig. S8‡). More specifically, for the 2–6 glycan solution with a concentration of 0.1 nM (1 × 10−8 M), the recorded current value was 3.49 μA at −2 V. The current increase ratio (conductance increase ratio) was approximately 102%, representing a considerably higher recognition sensitivity than that observed for the Glc-PEI-modified PAA nanochannels, where the maximum ratio was only approximately 42.5% and the corresponding concentration was as high as 1 mM (Fig. 1D). Comparative analyses revealed that Glc-PEI-modified PET achieved a significant discrimination effect for 2–6 glycan and 2–3 glycan, and the ion conductance increase ratios (at −2 V, concentration = 10 nM) were approximately 264%, and 96.2%, respectively (Fig. 2B). Under the same conditions, the ion conductance increase ratio that was observed for Neu5Ac reached approximately 367% (see inset, Fig. 2B). These results indicated that the Glc-PEI-modified PET conical nanochannels system could significantly elevate the recognition sensitivity, and amplify the discrimination effect, presumably owing to the ionic rectification property of the conical nanochannel.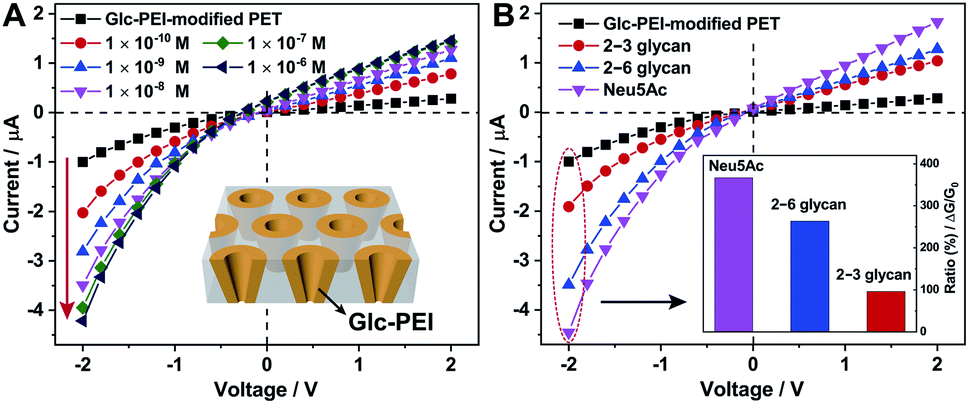 | ||
| Fig. 2 Transmembrane ionic current measurements based on the PET conical nanochannels. (A) I–V curves of Glc-PEI-modified PET in 0.1 M NaCl solution before and after addition of 2–6 glycan solutions with different concentration. (B) I–V curves of Glc-PEI-modified PET before and after addition of 2–3 glycan, 2–6 glycan, and Neu5Ac solution (1 × 10−8 M). The inset is the corresponding current change ratios at −2 V. | ||
Given these results, we performed a Laser Scanning Confocal Microscopy (LSCM) experiment to investigate the response of the Glc-PEI-modified PET nanochannel when it encountered different glycan molecules. First, fluorescein-labelled sialylated glycans were obtained by reductive amination of the glycan reducing end with fluoresceinamine under the agency of sodium cyanoborohydride (Fig. 3A and S9, Scheme S4‡). Three-dimensional (3D) reconstructed LSCM images obtained by a layer-by-layer scanning way are shown Fig. 3B and C. Many green cylinders are apparent on the image of Glc-PEI-modified PET nanochannels after treatment with the fluorescein-labelled 2–3 glycan and subsequent rinsing with water. As the Glc-PEI-modified PET membrane itself does not fluoresce, the green fluorescent signals must be the result of the fluorescein-labelled 2–3 glycan adsorption. A 3D reconstructed image of a single, bright green signal shows a conical shape (right panel, Fig. 3B). This implies that the Glc-PEI, that were anchored to the inner wall of conical nanochannels, had adsorbed a large amount of the fluorescein-labelled 2–3 glycan molecules. These disperse fluorescence signals reflect the conical nanochannels of the PET membrane. However, for the Glc-PEI-modified PET membrane that was treated with the fluorescein-labelled 2–6 glycan, there were fewer green spots observed (Fig. 3C), and they were less intense in strength compared with that observed with the fluorescein-labelled 2–3 glycan treatment. A similar difference was observed in different areas of the membrane surface (Fig. S10‡). In addition, the intrinsic weak fluorescence signal obtained from the fluorescein-labelled 2–6 glycan obscured the distinctive cone shapes from view (Fig. 3C). Thus, in the 3D reconstructed image of single fluorescent feature (right panel, Fig. 3C), the shape of a partial cone is only faintly discernible. The results indicate that the Glc-PEI has stronger adsorbing capability towards α2–3-linked glycan compared with α2–6-linked isomer.
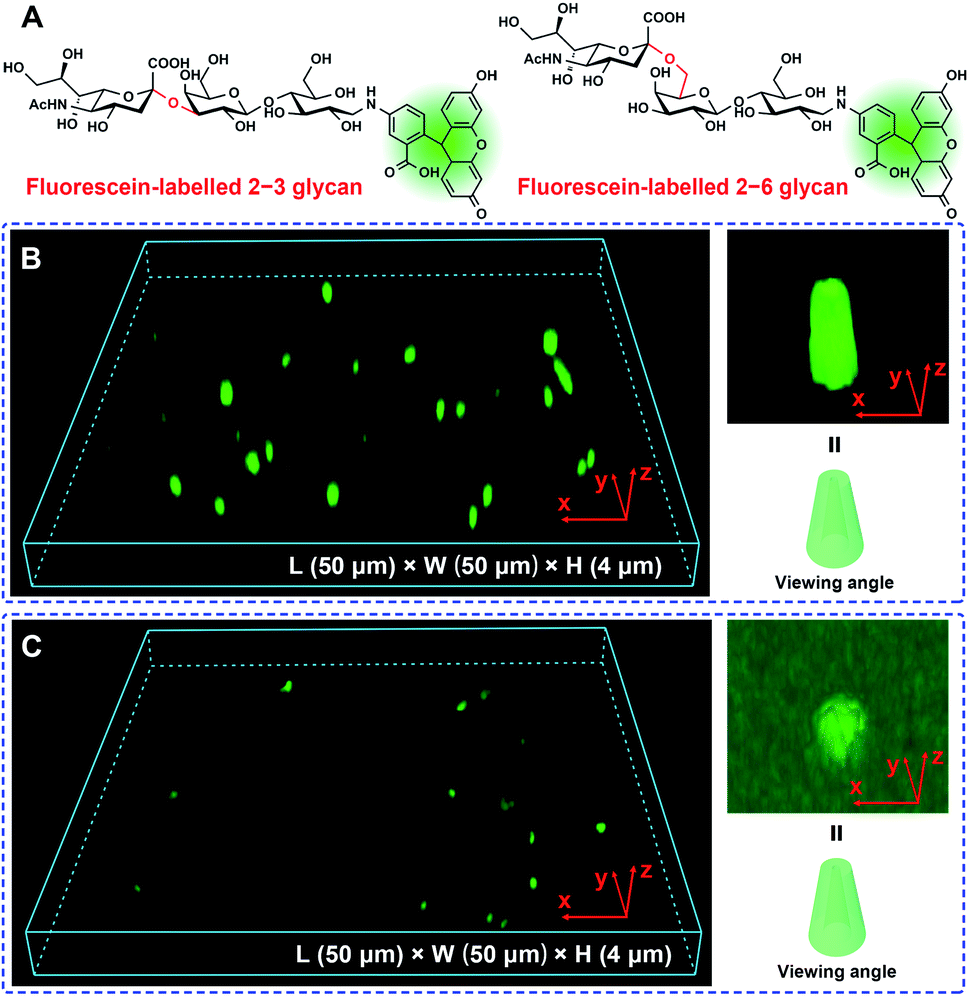 | ||
| Fig. 3 LSCM observation of the Glc-PEI-modified PET conical nanochannels. (A) Chemical structures of the fluorescein-labelled 2–3 glycan and 2–6 glycan. (B and C) 3D reconstructed LSCM images of the Glc-PEI-modified PET membrane and a single conical nanochannels upon interaction with the fluorescein-labelled 2–3 glycan (B), and the fluorescein-labelled 2–6 glycan (C). The drawn conical nanochannel model shown in the bottom right is used to indicate the viewing angle of the 3D reconstructed LSCM image. | ||
Investigation of conformational changes of the Glc-PEI polymer
LSCM observation provided direct evidence for the different adsorption behaviours of 2–3 glycan and 2–6 glycan onto the asymmetric nanochannels grafted with the polymer; however, this did not explain the change of the transmembrane ion conductance. Generally, changes in transmembrane ion conductance depend on a change in the effective aperture of nanochannel.18 Thus, here we used Atomic Force Microscopy (AFM) to study the surface morphology change of the nanochannels based on the PAA substrate. The AFM image of the Glc-PEI-modified PAA nanochannels is shown in Fig. 4A. After treatment with the 2–6 glycan solution for 10 min, the average aperture changed from approximately 37 to 55 nm (Fig. 4B), implying that the Glc-PEI layer had shrunk, following adsorption of the 2–6 glycan molecules. This offers direct evidence that Glc-PEI shrinkage was responsible for the “OFF–ON” change of the nanochannel, and the corresponding increase in ion flux, i.e., ion conductance.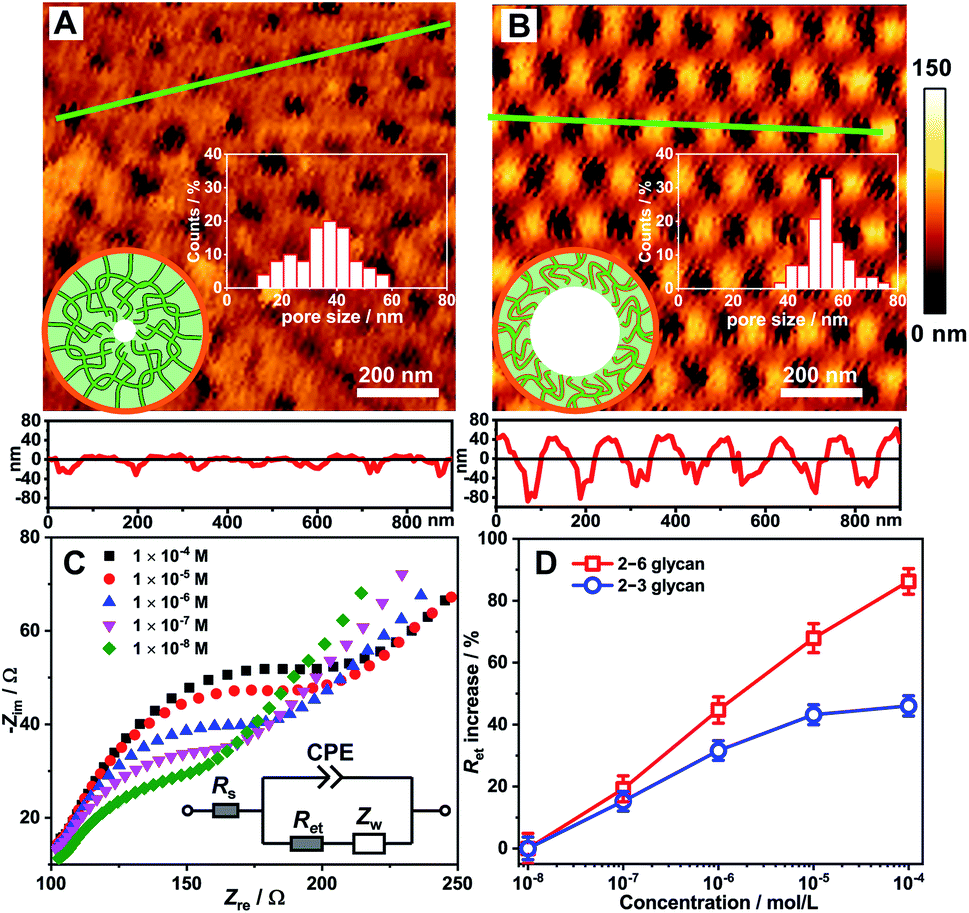 | ||
| Fig. 4 Investigation of conformational changes of the Glc-PEI polymer. AFM images of the Glc-PEI-modified PAA membrane (A) before and (B) after being immersed in the 2–6 glycan solution (1 mM) for 10 min. The low panels are the corresponding section profiles along the green lines. The inserts represent the distribution of pore size and schematic of a Glc-PEI-modified PAA nanochannel. (C) EIS measurements of the Glc-PEI-modified gold electrode in 0.1 M KCl solution containing 5 mM Fe(CN)63−/4−, after treatment with 2–6 glycan solution with different concentration for 10 min. The inset represents the equivalent circuit for fitting the impedance spectrum to offer the electron transfer resistance. Rs: solution resistance; Ret: electron transfer resistance; Zw: Warburg impedance; CPE: constant phase element. (D) Concentration-dependent Ret increase. | ||
This distinct shrinkage behaviour was confirmed by Electrochemical Impedance Spectroscopy (EIS) measurements using a gold electrode decorated with the Glc-PEI film (see Section 5 in ESI‡). The EIS spectrum of Glc-PEI-modified electrode was recorded in an electrolyte solution after immersion in the target solution for 10 min. The EIS spectra from the treatment with 2–6 glycan are shown in Fig. 4C. The semicircle diameters correspond to electron transfer resistance (Ret) through the Glc-PEI layer. This indicates that Ret increases as the concentration of 2–6 glycan increases. This means that the Glc-PEI layer, after the treatment of 2–6 glycan, changed from an initial swollen state to a compact shrunken state, blocking the electron transfer process through the polymer layer and causing the increase of Ret.19 Notably, under the same conditions, the Ret increase ratios due to the treatment of 2–6 glycan are higher than those of 2–3 glycan (Fig. 4D). This implies Glc-PEI shrinkage upon the adsorption of 2–6 glycan was much more substantial than that of 2–3 glycan. The different shrinkage levels are fully consistent with the corresponding ion conductance changes of the Glc-PEI-modified nanochannels (Fig. 1D).
Investigations of the binding affinities of Glc-PEI with glycans
Generally, in a recognition or sensing system, a stronger binding affinity of the material to the target of interest results in a more dramatic recognition effect; examples of such an effect include more obvious colour changes, or larger current changes.20 However, in this study, a unique phenomenon was observed for glycan recognition based on the Glc-PEI-modified nanochannels system. Specifically, the high binding affinity between the Glc-PEI and the target caused a slight shrinkage of the Glc-PEI layer and a small increase in ion conductance. On the other hand, low binding affinity caused an obvious shrinkage and a much larger increase in ion conductance. This unusual scenario was initially observed in affinity measurements using an Isothermal Titration Calorimetry (ITC) method. With the titration of Neu5Ac in the Glc-PEI solution, strong exothermic data were recorded, and the optimal fitting curve yielded a binding constant (Ka) value of approximately 3.52 × 103 M−1 (Fig. 5A). Under the same conditions, Glc-PEI with either 2–6 glycan or 2–3 glycan exhibited much stronger, and gradually increasing affinity levels, giving Ka values of approximately 1.52 × 104 M−1 and 5.33 × 104 M−1, respectively (red columns, Fig. 5B). The affinity results of Glc-PEI with 2–6 glycan and 2–3 glycan are in agreement with the strength of the fluorescent signals observed in the LSCM experiments (Fig. 3B and C).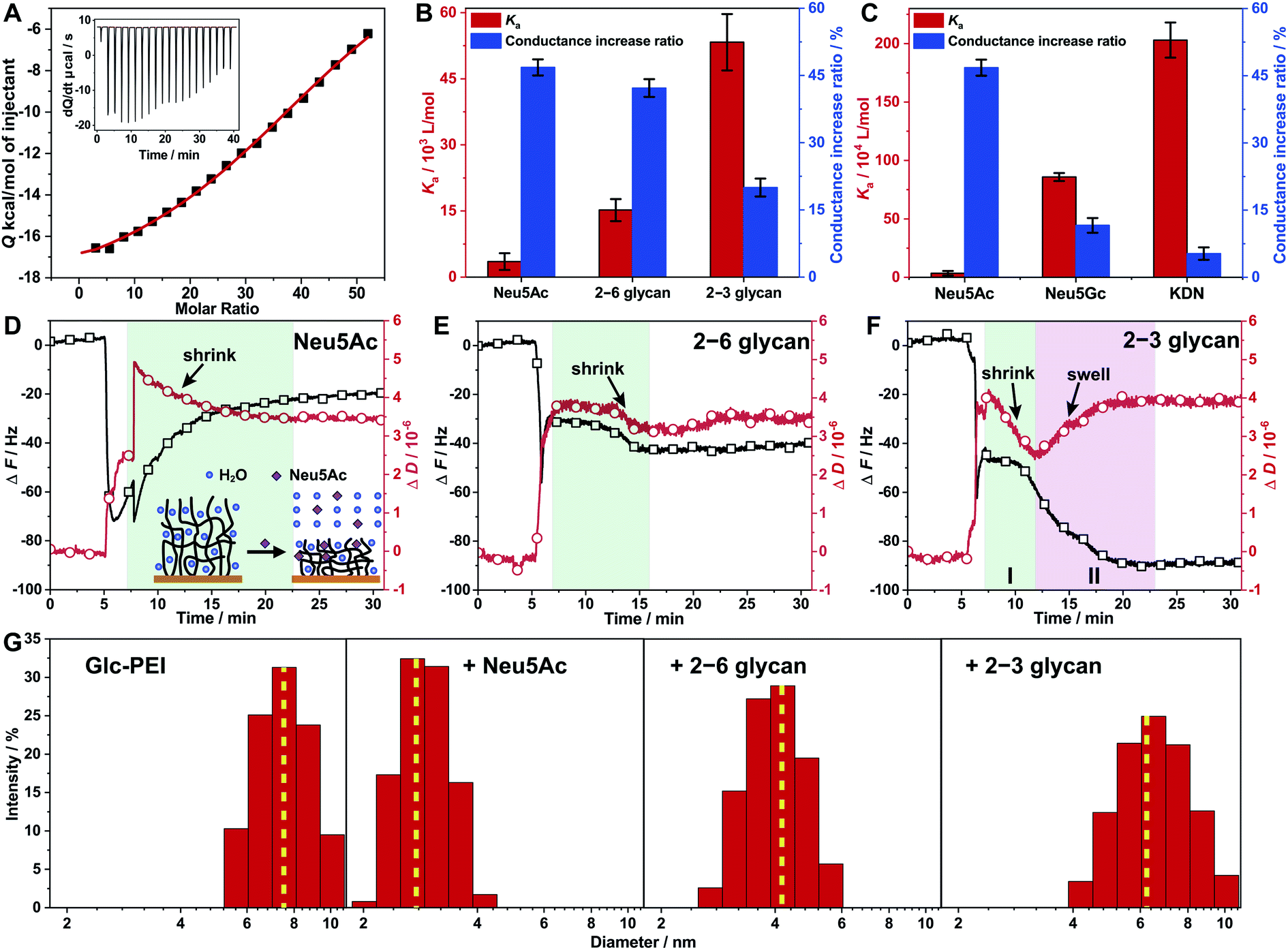 | ||
| Fig. 5 Investigations of the binding affinity of the Glc-PEI polymer with glycans and conformational changes of Glc-PEI. (A) ITC data recorded for Glc-PEI titrated with Neu5Ac. (B and C) Comparison between Ka values of Glc-PEI with different targets and corresponding ion conductance increase of the Glc-PEI-modified PAA nanochannels, where the conductance increase ratios are derived from Fig. 1D and C. Note, these two charts are double Y axis chart, the colour of the column corresponds to the colour of the Y axis. (D–F) Frequency shift (ΔF) and energy dissipation shift (ΔD) of the Glc-PEI film modified on QCM-D sensor upon injection of Neu5Ac, 2–6 glycan, and 2–3 glycan solution; all data were from the third overtone (n = 3). The initial sharp shifts or oscillations for both curves observed resulted from the switching of solution with different density and viscosity, which were unaccounted in analysis.23 (D) After initial sharp shifts and oscillations, the frequency curve is shown to gradually increase and level off, the dissipation curve is shown to gradually decrease and level off in 15 min. This indicates the Glc-PEI film, upon adsorbing Neu5Ac, gradually extruded water and was shrunk considerably to a more compact conformation. (E) After initial sharp shifts, the frequency shift is shown to decrease weakly and level off immediately, whereas the dissipation shift is shown to decrease abnormally and level off in 8 min, implying that both 2–6 glycan adsorption and water extrusion occurred simultaneously. However, the dehydration and the shrinkage were short-lived, finally resulting in a moderate shrinkage of the Glc-PEI film. (F) Both the frequency shift and dissipation shift decreased in stage I, implying that 2–3 glycan adsorption occurred along with dehydration and shrinkage of the Glc-PEI film. Subsequently, the dissipation shift is shown to turn upward and level off, the frequency shift is shown to decrease and level off in stage II, indicating the Glc-PEI film gradually swelled instead. (G) Hydrodynamic diameter distribution determined by DLS of the Glc-PEI polymer before and after addition of Neu5Ac, 2–6 glycan, and 2–3 glycan. | ||
Surprisingly, when the corresponding ion conductance increase ratio obtained from ionic current measurements based on Glc-PEI-modified PAA nanochannels was plotted side by side with the Ka value in the chart, a negative correlation between binding affinity (Ka value) and conductance increase was observed (Fig. 5B). In other words, the higher the affinity of Glc-PEI with the analyte is, the smaller the increase in conductance increase of Glc-PEI-modified PAA nanochannels upon the treatment with the analyte is, and vice versa. More specifically, the highest binding affinity (5.33 × 104 M−1) of Glc-PEI with 2–3 glycan corresponds to the smallest conduction increase (20%). The lowest affinity (3.52 × 103 M−1) of Glc-PEI with Neu5Ac is associated with the largest conductance increase (47%). Not coincidentally, a similar negative correlation was also observed for the comparison of Neu5Ac, Neu5Gc, and KDN (Fig. 5C). Given that the shrinkage of the Glc-PEI polymer is responsible for the increase of ion conductance of the Glc-PEI-modified nanochannel (which has been proven by AFM and EIS experiments), these results indicate the existence of an unusual negative correlation between the binding affinity of Glc-PEI with the analyte and the shrinkage level of Glc-PEI caused by the binding to the analyte, which warrants further investigation.
Correlation of the binding affinity and conformational change
The correlation between the binding affinity and conformation change of the Glc-PEI polymer was investigated using Quartz Crystal Microbalance with Dissipation monitoring (QCM-D). First, the QCM-D sensor was modified with Glc-PEI via EDC coupling to the exposed carboxylic acid groups of a gold-immobilized mercaptoacetic acid, yielding a Glc-PEI film on the sensor surface (see Section 7 in ESI‡). The different adsorption behaviours of the Glc-PEI film towards different analytes are shown in Fig. 5D–F. The overall frequency shifts (ΔF) for Neu5Ac, 2–6 glycan, and 2–3 glycan were approximatively 20, 40, and 85 Hz, respectively. ΔF is dependent on the mass absorbed on polymer films, and thus can serve as a direct reporter for binding affinity of the analyte with the polymer.21 Accordingly, the affinity difference implied in those ΔF values is in good agreement with the Ka values obtained from ITC assays (red columns, Fig. 5B).On the other hand, for energy dissipation shift (ΔD), a downward curve generally signifies the shrinkage and densification of the polymer film, whereas an upward curve signifies the swelling of the polymer film.22 In this study, ΔD curves and the implied conformational change information of the Glc-PEI polymer for the entire dynamic adsorption process are detailed in Fig. 5D–F. In short, under the same conditions, the adsorption of Neu5Ac caused a significant shrinkage of the Glc-PEI film, yielding a ΔD value of approximately 3.4 × 10−6 (Fig. 5D). However, the adsorption of 2–6 glycan resulted in only moderate shrinkage with a ΔD value of approximately 3.5 × 10−6 (Fig. 5E). In contrast, the adsorption of 2–3 glycan produced a short-lived shrinking process and subsequent swelling process, finally giving a ΔD value of approximately 4.0 × 10−6 (Fig. 5F). The comparison of the dynamic processes, as well as ΔD values, indicated that Neu5Ac caused the highest shrinkage level of Glc-PEI, 2–6 glycan led to a moderate shrinkage, and 2–3 glycan caused the lowest shrinkage level. The different levels of Glc-PEI shrinkage were determined to be highly consistent with the observed changes in the corresponding ion conductance increase (blue columns, Fig. 5B). Furthermore, the polymer particle size determined by Dynamic Light Scattering (DLS) in solution confirmed these different shrinking behaviours, where the average diameter of the Glc-PEI changed from approximatively 7 nm initially to about 3, 4.1, and 6.2 nm after the addition of Neu5Ac, 2–6 glycan, and 2–3 glycan, respectively (Fig. 5G). Thus, the results of QCM-D experiments from two aspects of frequency shift and energy dissipation shift confirmed the difference in binding affinity and ion conductance increase, respectively. In other words, the results offer direct evidence that the negative correlation exists between Glc-PEI-analyte binding affinity and Glc-PEI shrinkage.
Discussion of the underlying tug-of-war competition mechanism
Quantum chemistry calculations were performed to reveal the binding modes between Glc-PEI and glycan isomers, and the corresponding binding difference. First, a Glc-PEI fragment with two Glc units was designed as a model for ease of calculation. Prior to calculation, glycans were placed in the middle of two Glc arms of the model. The result shows that the 2–3 glycan molecule was trapped by the two Glc arms via nine sets of H-bonds (Fig. 6A). This multivalent H-bonding model implies a strong binding affinity of Glc-PEI with 2–3 glycan. Meanwhile, the multiple H-bonds could significantly increase polymer network rigidity. On the other hand, 2–6 glycan showed a semi-inclusive binding involving only one Glc arm via four sets of H-bonds, with the aid of an amine from the PEI chain (Fig. 6B). The other Glc arm hung freely on the outside. The affinity resulting from this semi-inclusive binding mode was relatively low, and the Glc-PEI polymer still retained partial flexibility. Furthermore, the binding difference with a preference for the α2–3 linkage was confirmed by the calculated binding modes of the Glc-PEI fragment with Neu5Acα2–3GalβMP and Neu5Acα2–6GalβMP (Fig. S11‡).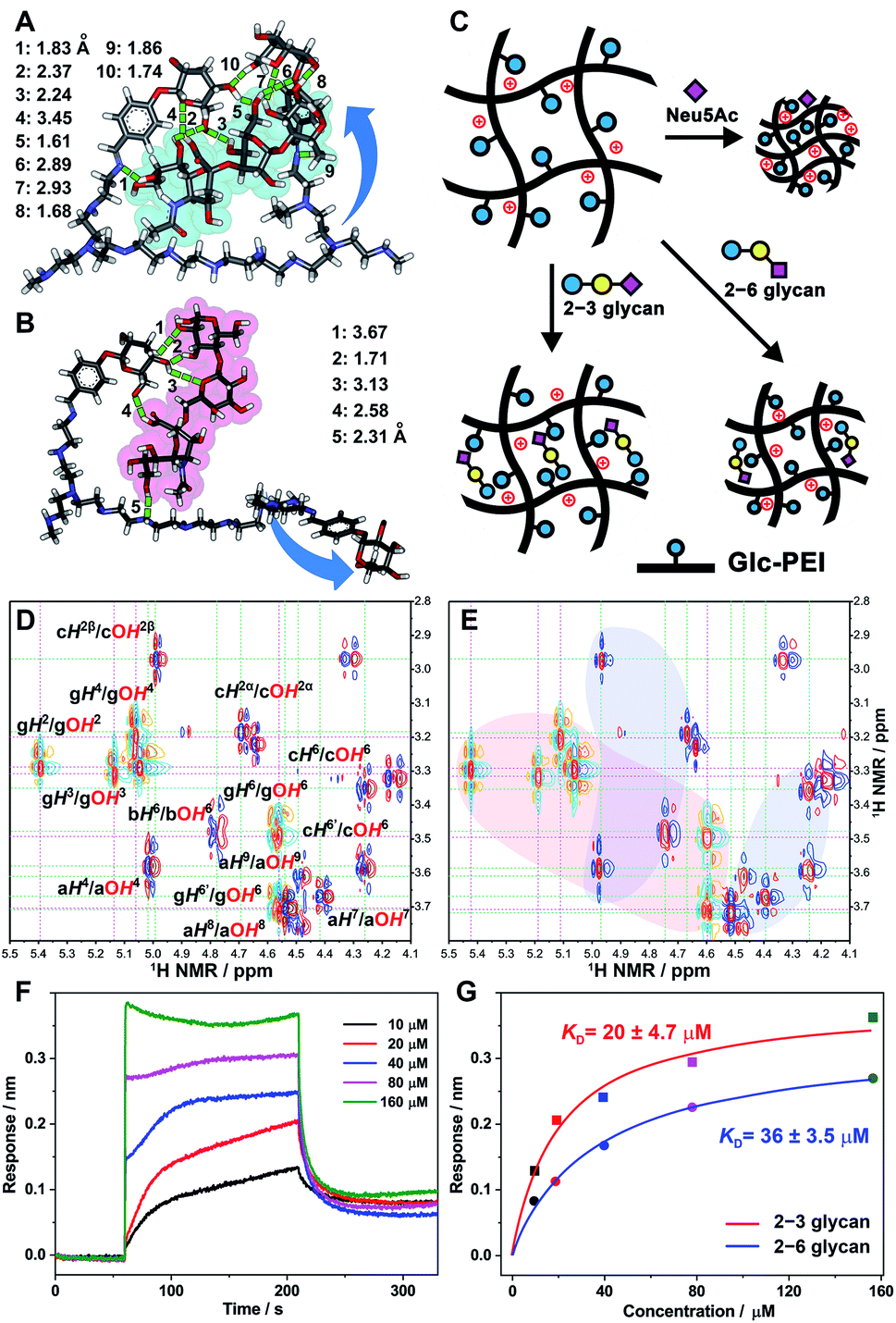 | ||
Fig. 6 Discussion of the underlying tug-of-war competition mechanism. (A and B) Optimized binding models of the Glc-PEI fragment with 2–3 glycan (A) and 2–6 glycan (B) from quantum chemistry calculations (Gaussian, density functional theory (DFT), B3LYP, at the 3-21G level); hydrogen bonds are indicated with green dashed lines of different lengths. (C) Possible conformation models of Glc-PEI upon binding to target molecules. (D) Stack of the partial 1H–1H COSY NMR spectrum of the glucopyranoside (orange-cyan contours) and 2–3 glycan (red-blue contours). (E) Partial 1H–1H COSY NMR spectrum of mixture of the glucopyranoside and 2–3 glycan at a molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in DMSO-d6. “g” denotes the glucopyranoside unit, “a”, “b”, and “c” denote Neu5Ac, Gal, and Glc unit of 2–3 glycan, respectively. The light-red area means that, for OH protons of glucopyranoside, shifts were observed. The light-blue areas mean that, for OH protons of 2–3 glycan, shifts were observed. (F) Sensorgrams of the Glc-PEI-modified senor binding to 2–3 glycan from BLI measurements. (G) Response curves of the steady-state fitting to provide dissociation constant (KD) of Glc-PEI with 2–3 glycan and 2–6 glycan, respectively. :1 in DMSO-d6. “g” denotes the glucopyranoside unit, “a”, “b”, and “c” denote Neu5Ac, Gal, and Glc unit of 2–3 glycan, respectively. The light-red area means that, for OH protons of glucopyranoside, shifts were observed. The light-blue areas mean that, for OH protons of 2–3 glycan, shifts were observed. (F) Sensorgrams of the Glc-PEI-modified senor binding to 2–3 glycan from BLI measurements. (G) Response curves of the steady-state fitting to provide dissociation constant (KD) of Glc-PEI with 2–3 glycan and 2–6 glycan, respectively. | ||
Based on the preceding results, possible conformational change models to explain the distinct recognition effects of the Glc-PEI polymer for Neu5Ac and two glycans were proposed (Fig. 6C). Initially, Glc-PEI exhibited a loose conformation due to the electrostatic repulsions among protonated amines. In the presence of Neu5Ac, the binding of Glc-PEI to Neu5Ac caused the introduction of negatively charged Neu5Ac into the inside of the Glc-PEI network, causing a considerable shrinkage of the Glc-PEI network induced by electrostatic attraction. For the 2–3 glycan molecule, its strong affinity with Glc-PEI and the multisite binding mode significantly increased the rigidity of the Glc-PEI network. Thus, most of the shrinkage potential of the Glc-PEI network that was induced by the introduction of the counter-charge from the 2–3 glycan was counteracted, finally resulting in a slight shrinkage. For the 2–6 glycan molecule, the semi-inclusive mode and the relatively weak binding enabled retention of the partial shrinking potential of the Glc-PEI film; thus, a moderate shrinkage was exhibited. The relationship between polymer-target binding, and the shrinkage of polymer itself, exhibited a tug-of-war competition mechanism (Scheme 1E).
The above calculation regarding the difference in binding affinity were further verified by NMR spectroscopy and Bio-layer Interferometry (BLI) tests. When glucopyranoside (Glc component of Glc-PEI) was mixed with 2–3 glycan, obvious shifts of several OH protons of glucopyranoside was observed (Fig. 6D, light-red area in Fig. 6E and S12‡). For example, OH4 and OH6 protons shifted downfield from 5.06 and 4.56 ppm to 5.11 and 4.60 ppm, respectively. At the same time, evident shifts of partial OH protons of 2–3 glycan were also observed (light-blue areas in Fig. 6E). In contrast, although similar OH protons' shifts were observed, the corresponding shift ranges were small, and the number of OH moieties of 2–6 glycan involved in the binding was relatively small (Fig. S13 and S14‡). These results proved the binding preference of the grafted Glc units for 2–3 glycan, and confirmed the recognition role of Glc unit at the molecular level. Additionally, BLI tests of the Glc-PEI film with 2–3 glycan revealed an equilibrium dissociation constant (KD) of 20 ± 4.7 μM, which was evidently smaller than the KD value (36 ± 3.5 μM) of the Glc-PEI film with 2–6 glycans (Fig. 6F, G, and S15‡). Because the KD is the inverse of Ka, this result demonstrates the binding preference of Glc-PEI for 2–3 glycan at the polymer level.
Conclusions
This work presented a biomimetic nanochannels system integrated with a biomolecule-responsive polymer, which achieved precise recognition and discrimination of sialylated glycan linkage isomers based on dramatic “OFF–ON” ion flux changes. Notably, the “OFF–ON” change range was attributed to the result of tug-of-war competition between polymer-glycan binding and the shrinkage of polymer itself. The findings of this study demonstrated that a functional nanochannel platform constitutes an excellent tool for glycan recognition and analysis. The high selectivity, high sensitivity, and label-free features of this nanochannels system render it attractive as a basis for the development of devices that can improve the detection of glycan-related disease biomarkers. This simple yet effective design rationale for functional nanochannel offers a general strategy for the analysis or detection of other biologically significant molecules. In addition, the underlying tug-of-war competition mechanism also provides inspiration for material design of those recognition or sensing systems that involve the recognition of negatively charged target biomolecules. For example, those system can be designed by combining diverse polycationic polymers and receptor molecules. In particular, this work highlights the great potential of a single nanochannel or nanopore as a powerful complement for glycomics techniques for glycans structure determination at the single-molecule level.12c,dConflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51473131, 51533007, 21775116 and 21922411), DICP Innovation Funding (DICP-RC201801). G. Qing acknowledges Wuhan Morning Light Plan of Youth Science and Technology. The authors are thankful for Energy Biotechnology Platform-DICP for their technical support.Notes and references
- (a) J. Alper, Science, 2003, 301, 159–160 CrossRef CAS PubMed; (b) S. Weinbaum, J. M. Tarbell and E. R. Damiano, Annu. Rev. Biomed. Eng., 2007, 9, 121–167 CrossRef CAS.
- T. Hayashi, G. Kehr, K. Bergander and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 3814–3818 CrossRef CAS.
- (a) A. Varki, Nature, 2007, 446, 1023–1029 CrossRef CAS; (b) S. Ghosh, Glycoconjugate J., 2015, 32, 79–85 CrossRef CAS; (c) Y. Chen, H. Liu, Y. Xiong and H. Ju, Angew. Chem., Int. Ed., 2018, 57, 785–789 CrossRef CAS.
- (a) Y. Shi, W. Zhang, F. Wang, J. Qi, Y. Wu, H. Song, F. Gao, Y. Bi, Y. Zhang and Z. Fan, et al. , Science, 2013, 342, 243–247 CrossRef CAS; (b) J. Zhou, D. Wang, R. Gao, B. Zhao, J. Song, X. Qi, Y. Zhang, Y. Shi, L. Yang and W. Zhu, et al. , Nature, 2013, 499, 500–503 CrossRef CAS.
- (a) C. Büll, M. A. Stoel, M. H. den Brok and G. J. Adema, Cancer Res., 2014, 74, 3199–3204 CrossRef; (b) A. Magalhães, H. O. Duarte and C. A. Reis, Cancer Cell, 2017, 31, 733–735 CrossRef; (c) S. Gilgunn, P. J. Conroy, R. Saldova, P. M. Rudd and R. J. O'Kennedy, Nat. Rev. Urol., 2013, 10, 99–107 CrossRef CAS PubMed; (d) R. Saldova, Y. Fan, J. M. Fitzpatrick, R. William, G. Watson and P. M. Rudd, Glycobiology, 2011, 21, 195–205 CrossRef CAS; (e) W. R. Alley and M. V. Novotny, J. Proteome Res., 2010, 9, 3062–3072 CrossRef CAS; (f) S. Wang, X. Chen, A. Wei, X. Yu, B. Niang and J. Zhang, Tumor Biol., 2015, 36, 885–892 CrossRef CAS.
- J. R. Mascola and D. C. Montefiori, Nat. Med., 2003, 9, 393–394 CrossRef CAS.
- W. B. Puryear, X. Yu, N. P. Ramirez, B. M. Reinhard and S. Gummuluru, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7475–7480 CrossRef CAS.
- (a) D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2005, 4, 477–488 CrossRef CAS; (b) X. P. He, Y. L. Zeng, X. Y. Tang, N. Li, D. M. Zhou, G. R. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 13995–13999 CrossRef CAS PubMed; (c) R. Xing, S. Wang, Z. Bie, H. He and Z. Liu, Nat. Protoc., 2017, 12, 964–987 CrossRef CAS PubMed; (d) L. Yang, X. Du, Y. Peng, Y. Cai, L. Wei, Y. Zhang and H. Lu, Anal. Chem., 2019, 91, 1486–1493 CrossRef CAS PubMed.
- (a) P. Both, A. P. Green, C. J. Gray, R. Šardzík, J. Voglmeir, C. Fontana, M. Austeri, M. Rejzek, D. Richardson, R. A. Field, G. Widmalm, S. L. Flitsch and C. E. Eyers, Nat. Chem., 2014, 6, 65–74 CrossRef CAS PubMed; (b) C. Pett, W. Nasir, C. Sihlbom, B.-M. Olsson, V. Caixeta, M. Schorlemer, R. P. Zahedi, G. Larson, J. Nilsson and U. Westerlind, Angew. Chem., Int. Ed., 2018, 57, 9320–9324 CrossRef CAS; (c) M. Lundborg and G. Widmalm, Anal. Chem., 2011, 83, 1514–1517 CrossRef CAS; (d) M.-Q. Liu, W.-F. Zeng, P. Fang, W.-Q. Cao, C. Liu, G.-Q. Yan, Y. Zhang, C. Peng, J.-Q. Wu and X.-J. Zhang, et al. , Nat. Commun., 2017, 8, 438 CrossRef.
- (a) M. Tagliazucchi and I. Szleifer, Mater. Today, 2015, 18, 131–142 CrossRef CAS; (b) A. de la Escosura-Muñiz and A. Merkoçi, Trends Anal. Chem., 2016, 79, 134–150 CrossRef; (c) G. Pérez-Mitta, A. G. Albesa, C. Trautmann, M. E. Toimil-Molares and O. Azzaroni, Chem. Sci., 2017, 8, 890–913 RSC.
- (a) H. Zhang, Y. Tian and L. Jiang, Nano Today, 2016, 11, 61–81 CrossRef CAS; (b) M. Ali, I. Ahmed, P. Ramirez, S. Nasir, C. M. Niemeyer, S. Mafe and W. Ensinger, Small, 2016, 12, 2014–2021 CrossRef CAS; (c) Q. Ma, Z. Si, Y. Li, D. Wang, X. Wu, P. Gao and F. Xia, Trends Anal. Chem., 2019, 115, 174–186 CrossRef CAS; (d) R. Wei, V. Gatterdam, R. Wieneke, R. Tampé and U. Rant, Nat. Nanotechnol., 2012, 7, 257–263 CrossRef CAS; (e) N. Liu, Y. Jiang, Y. Zhou, F. Xia, W. Guo and L. Jiang, Angew. Chem., Int. Ed., 2013, 52, 2007–2011 CrossRef CAS; (f) K. Xiao, K. Wu, L. Chen, X.-Y. Kong, Y. Zhang, L. Wen and L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 151–155 CrossRef CAS PubMed; (g) P. Li, G. Xie, P. Liu, X.-Y. Kong, Y. Song, L. Wen and L. Jiang, J. Am. Chem. Soc., 2018, 140, 16048–16052 CrossRef CAS; (h) G. Nie, Y. Sun, F. Zhang, M. Song, D. Tian, L. Jiang and H. Li, Chem. Sci., 2015, 6, 5859–5865 RSC; (i) Q. Li, Y.-L. Ying, S.-C. Liu, Y. Lin and Y.-T. Long, ACS Sens., 2019, 4, 1185–1189 CrossRef CAS.
- (a) Y. Sun, F. Zhang, J. Quan, F. Zhu, W. Hong, J. Ma, H. Pang, Y. Sun, D. Tian and H. Li, Nat. Commun., 2018, 9, 2617 CrossRef; (b) G. Hou, H. Zhang, G. Xie, K. Xiao, L. Wen, S. Li, Y. Tian and L. Jiang, J. Mater. Chem. A, 2014, 2, 19131–19135 RSC; (c) F. Rivas, O. K. Zahid, H. L. Reesink, B. T. Peal, A. J. Nixon, P. L. DeAngelis, A. Skardal, E. Rahbar and A. R. Hall, Nat. Commun., 2018, 9, 1037 CrossRef; (d) B. Karawdeniya, J. W. Nichols, R. B. Chevalier, J. R. Dwyer and Y. M. N. D. Y. Bandara, Nat. Commun., 2018, 9, 3278 CrossRef.
- S. Suriyanarayanan, H.-H. Lee, B. Liedberg, T. Aastrup and I. A. Nicholls, J. Colloid Interface Sci., 2013, 396, 307–315 CrossRef CAS.
- I. Bucior and M. M. Burger, Curr. Opin. Struct. Biol., 2004, 14, 631–637 CrossRef CAS.
- X. Li, Y. Xiong, G. Qing, G. Jiang, X. Li, T. Sun and X. Liang, ACS Appl. Mater. Interfaces, 2016, 8, 13294–13302 CrossRef CAS.
- (a) G. Rajeev, B. Prieto Simon, L. F. Marsal and N. H. Voelcker, Adv. Healthcare Mater., 2018, 7, 1700904 CrossRef; (b) S. B. Lee, D. T. Mitchell, L. Trofin, T. K. Nevanen, H. Söderlund and C. R. Martin, Science, 2002, 296, 2198–2200 CrossRef CAS.
- (a) M. Ali, B. Yameen, R. Neumann, W. Ensinger, W. Knoll and O. Azzaroni, J. Am. Chem. Soc., 2008, 130, 16351–16357 CrossRef CAS; (b) W.-J. Lan, M. A. Edwards, L. Luo, R. T. Perera, X. Wu, C. R. Martin and H. S. White, Acc. Chem. Res., 2016, 49, 2605–2613 CrossRef CAS.
- (a) Z. Long, S. Zhan, P. Gao, Y. Wang, X. Lou and F. Xia, Anal. Chem., 2018, 90, 577–588 CrossRef CAS; (b) M. Wang, H. Meng, D. Wang, Y. Yin, P. Stroeve, Y. Zhang, Z. Sheng, B. Chen, K. Zhan and X. Hou, Adv. Mater., 2019, 31, 1805130 CrossRef.
- M. Motornov, R. Sheparovych, E. Katz and S. Minko, ACS Nano, 2008, 2, 41–52 CrossRef CAS.
- (a) F. Zhang, D. Wang, H. Qin, L. Feng, X. Liang and G. Qing, ACS Appl. Mater. Interfaces, 2019, 11, 13114–13122 CrossRef CAS; (b) H. Okuyama, Y. Oshiba, H. Ohashi and T. Yamaguchi, Small, 2018, 14, 1702267 CrossRef PubMed; (c) H. Peng, X.-F. Li, H. Zhang and X. C. Le, Nat. Commun., 2016, 8, 14378 CrossRef PubMed.
- N.-J. Cho, C. W. Frank, B. Kasemo and F. Höök, Nat. Protoc., 2010, 5, 1096–1106 CrossRef CAS.
- (a) G. Zhang and C. Wu, Macromol. Rapid Commun., 2009, 30, 328–335 CrossRef CAS; (b) J.-G. Wu, S.-C. Wei, Y. Chen, J.-H. Chen and S.-C. Luo, Langmuir, 2018, 34, 943–951 CrossRef CAS.
- (a) E. Guzmán, F. Ortega, N. Baghdadli, C. Cazeneuve, G. S. Luengo and R. G. Rubio, ACS Appl. Mater. Interfaces, 2011, 3, 3181–3188 CrossRef PubMed; (b) Y. Hou, G. Liu, W. Wu and G. Zhang, Phys. Chem. Chem. Phys., 2011, 13, 2880–2886 RSC.
Footnotes |
| † Dedicated to the 70th anniversary of Dalian Institute of Chemical Physics, CAS. |
| ‡ Electronic supplementary information (ESI) available: Materials and instruments, preparation and characterization of functional nanochannels, other experimental details, and supplementary schemes and figures. See DOI: 10.1039/c9sc05319k |
| This journal is © The Royal Society of Chemistry 2020 |