
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel photoredox-active group for the generation of fluorinated radicals from difluorostyrenes†
Mikhail O.
Zubkov
ab,
Mikhail D.
Kosobokov
a,
Vitalij V.
Levin
 a,
Vladimir A.
Kokorekin
ac,
Alexander A.
Korlyukov
de,
Jinbo
Hu
f and
Alexander D.
Dilman
*a
a,
Vladimir A.
Kokorekin
ac,
Alexander A.
Korlyukov
de,
Jinbo
Hu
f and
Alexander D.
Dilman
*a
aN. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, 47 Leninsky prosp., 119991 Moscow, Russia. E-mail: adil25@mail.ru
bHigher Chemical College, D. Mendeleev University of Chemical Technology of Russia, 9 Miusskaya sq., 125047 Moscow, Russia
cI. M. Sechenov First Moscow State Medical University, 8-2 Trubetskaya st., 119991 Moscow, Russia
dA. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 28 Vavilova st., 119991 Moscow, Russia
ePirogov Russian National Research Medical University, 1 Ostrovitianov st., 117997 Moscow, Russia
fKey Laboratory of Organofluorine Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Ling-Ling Road, 200032 Shanghai, China
First published on 26th November 2019
Abstract
A 4-tetrafluoropyridinylthio group was suggested as a new photoredox-active moiety. The group can be directly installed on difluorostyrenes in a single step by the thiolene click reaction. It proceeds upon visible light catalysis with 9-phenylacridine providing various difluorinated sulfides as radical precursors. Single electron reduction of the C–S bond with the formation of fluoroalkyl radicals is enabled by the electron-poor azine ring. The intermediate difluorinated sulfides were involved in a series of photoredox reactions with silyl enol ethers, alkenes, nitrones and an alkenyl trifluoroborate.
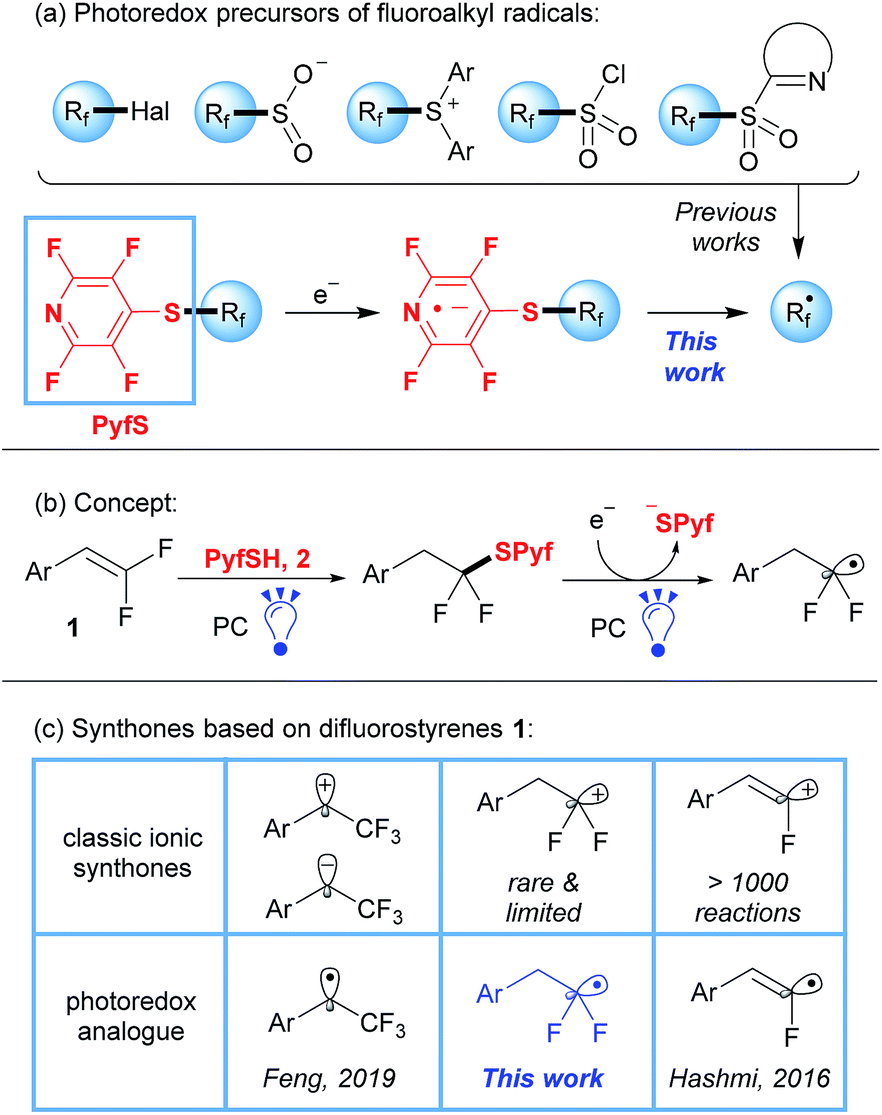
Organofluorine compounds have gained increasing attention due to their utility in medicinal chemistry and agrochemistry in the last few decades.1 Among various methods of fluorine incorporation, major attention in recent years has been devoted to radical pathways of fluoroalkylation by visible light photoredox catalysis.2,3 This approach has attracted much attention because of the exceptionally mild reaction conditions and functional group tolerance. Known reagents, which are suitable for efficient radical fluoroalkylation such as halides, sulfonyl chlorides, sulfones, sulfinates, and Umemoto and Togni reagents (Scheme 1a),3 suffer from limited structural diversity and complicated preparation. Indeed, besides a significant amount of CF3 and CF2H derivatives, most of the other Rf radical precursors require multistep preparation under harsh conditions.3c,4 As a result, operationally simple methods are still needed.
 | ||
| Scheme 1 Generation of fluorinated radicals. | ||
Herein we report a simple and diversity-oriented strategy based on the direct introduction of a photoredox-active group into the fluorinated substrate. Our approach involves the addition of tetrafluoropyridine-4-thiol (2, PyfSH) to readily accessible difluorostyrenes followed by photocatalytic reduction with fluoroalkyl radical formation (Scheme 1b). As is well known, the thiol-ene reaction is an excellent instrument for difluorostyrene functionalization leading to sulfides.5 However, C–S bond reduction in common sulfides is challenging owing to their unfavorable redox potential compared to their S-oxygenated counterparts.6,7 To overcome this obstacle, we propose to use an electron-withdrawing fluorinated pyridine moiety, which would be susceptible to SET reduction (Scheme 1a).
Thus, we report the application of sulfides as a new class of readily available, bench-stable and easy-to-handle reagents for radical fluoroalkylation under visible-light photoredox catalysis. It is worth mentioning that our concept allows the synthesis of precursors of various Rf radicals in a single step from easily accessible compounds, which is often hard in practice for other photoredox-active groups.3c,4 Thiol 2 can be easily prepared from commercially available pentafluoropyridine8 and the initial difluorostyrenes come from aldehydes by the Wittig-type reaction.9
Styrenes 1 serve as a basis for a number of ionic synthones9b,10,11 and recent developments in visible light photoredox catalysis allowed us to replace most of them with complementary radical synthones (Scheme 1c).12,13 Herein, we disclose the last “blind spot” in the map of radical analogues of ionic difluorostyrene synthones. It should be noted that due to facile elimination of the fluorine atom in polar reactions, only a few examples of difluoroalkyl cation synthons have been described previously.11
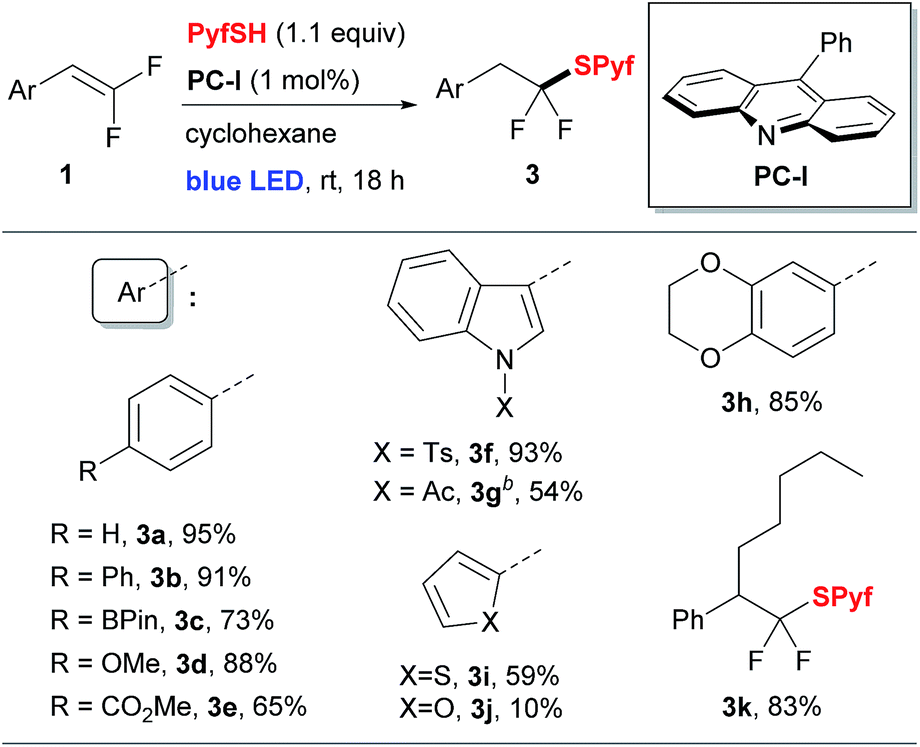
To perform the addition of thiol 2 to styrenes 1, we applied a protocol involving the activation of thiols based on proton coupled electron transfer recently developed by our group.5,14 Thus, screening of reaction conditions allowed us to identify the optimal system: 9-phenylacridine (PC-I) as the photocatalyst under blue light irradiation (see the ESI for details†). The reaction is performed in cyclohexane and requires a virtually stoichiometric amount (1.1 equiv.) of the thiol 2. A series of styrenes 1 were reacted with thiols 2 leading to difluorinated sulfides 3 (Table 1). Aromatic and heteroaromatic substrates provided sulfides 3 in good to excellent yields. The only exception was the furan substituted product 3j, which had a low yield due to instability of 1j.15 The product 3k derived from α-pentyl-substituted difluorostyrene was also obtained. In contrast to the reactions with styrenes, only traces of sulfides were obtained with aliphatic gem-difluoroalkenes (see the ESI for details†). It should also be pointed out that all reactions shown in Table 1 were performed on a 5 mmol scale.
A suggested mechanism for thiol–alkene addition is shown in Scheme 2. Thus, upon interaction of colorless compounds 9-phenylacridine (PC-I) and PyfSH, a red colored salt A is instantly formed (proton transfer, PT). The structure of this salt was studied by X-ray analysis indicating a π–π stacking-type structure, in which positively charged acridinium cations and negatively charged thiolate anions are arranged in parallel planes. The reaction is believed to proceed via light induced electron transfer (ET) thereby representing the proton-coupled electron transfer (PCET) manifold.16 The generated S-centered radical attacks the double bond of styrene 1, and the resulting benzyl radical abstracts the hydrogen atom either from the N–H acridinium radical or from the starting PyfSH.
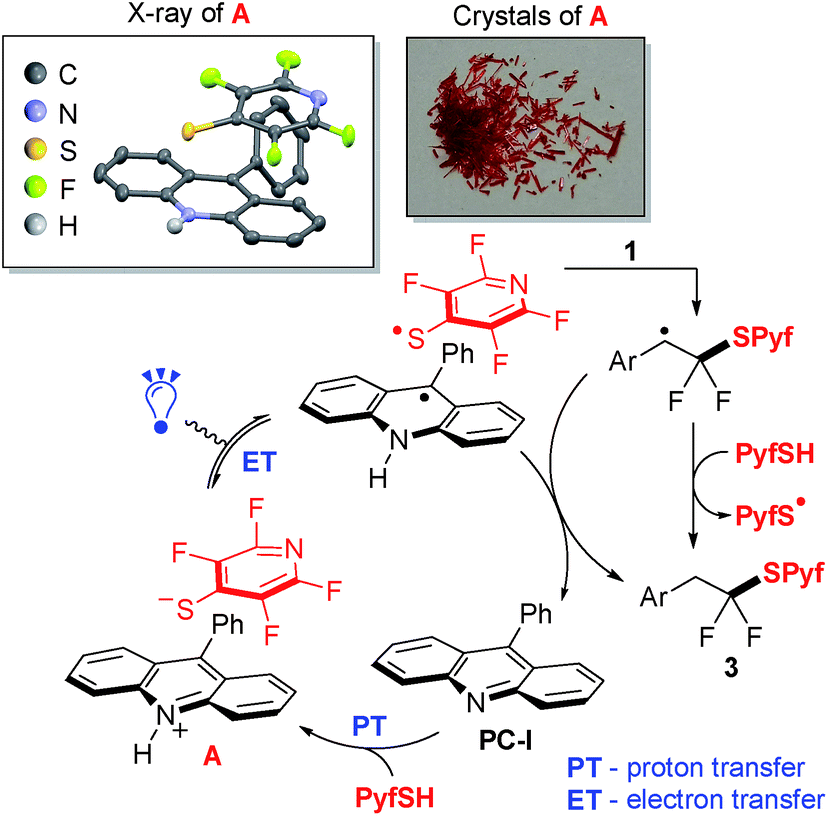 | ||
| Scheme 2 Plausible mechanism of thiol 2 addition to gem-difluorostyrenes 1. | ||
Measurement of the reduction potential of 3a by cyclic voltammetry provided a value of −1.36 V (vs. SCE), which supports the single electron reduction of compounds 3 by means of light activated photocatalysts. After the reduction of sulfide 3a, peaks corresponding to the oxidation of the thiolate anion are observed in the reverse scan (see the ESI for details†).
Using sulfide 3a as a model substrate, we evaluated its reactions with silyl enol ethers17,18 (see the ESI† for optimization details). The reactions were performed in the presence of 20 mol% triphenylphosphine, which, as we noted previously, exerts a beneficial effect on some photoredox reactions.17b,19 Two sets of optimal conditions, both operating using blue LED irradiation, were identified. In the first system, an organic photocatalyst, 12-phenyl-12H-benzo[b]phenothiazine20 (PC-II, 5 mol%), and zinc acetate (0.6 equiv.) as a scavenger of the thiolate byproduct were used (method A).
Method A provided good results for the difluoroalkylation of electron-donor-substituted aromatic silyl enolates. Thus, products 5aa, 5ef, 5ad, and 5ai were obtained with excellent yield. Unfortunately, the approach showed poor results for some other substrates. For instance, it is incompatible with aryl halide moieties due to concomitant carbon–halogen bond reduction induced by the phenothiazine catalyst (PC-II).20b For EWG-containing silyl enolates, we observed low yields along with radical polymerization by-products. To overcome these drawbacks, the second system involving an iridium based catalyst [Ir(ppy)2(dtbbpy)]PF6 (PC-III, 0.5 mol%) in combination with 50 mol% tetrabutylammonium iodide (method B) was suggested. The iodide ion is believed to induce reductive quenching of the photoexcited Ir(III) catalyst generating Ir(II) species behaving as a reductant of the substrate. Indeed, Stern–Volmer studies demonstrated that the iodide anion serves as a good fluorescence quencher of the iridium photocatalyst (see the ESI†). Under optimized conditions, a series of sulfides 3 were coupled with silyl enol ethers 4 (Table 2).
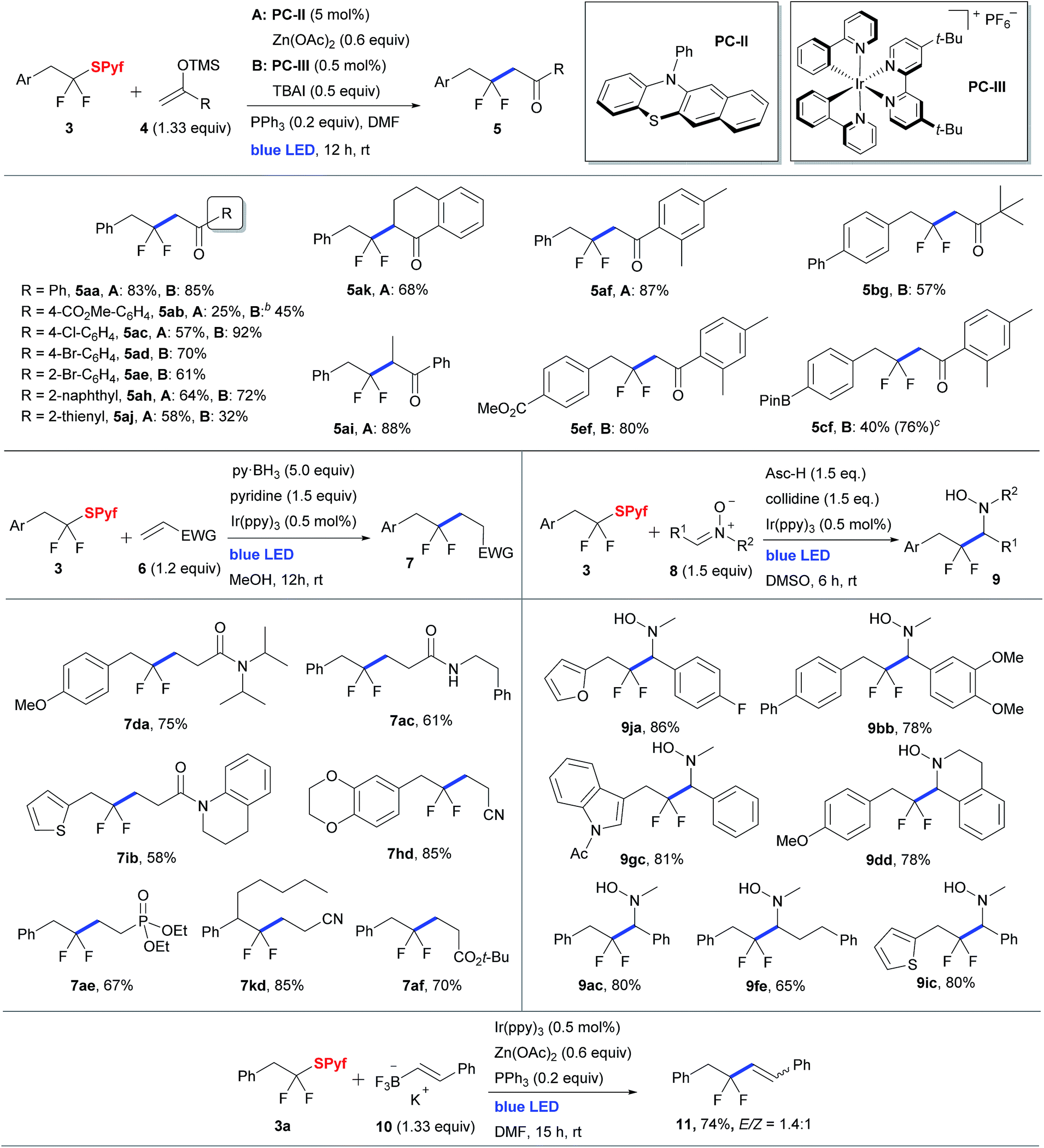
Generally, the procedure involving the iridium catalyst provided higher yields of products 5. In the case of 4-chloro-substituted silyl enol ether, the yield of the product 5ac was only 57% with phenothiazine, likely due to the formation of radical oligomerization by-products. However, switching to the iridium/iodide system gave an increased yield of 92%. Presumably, the ability of the latter system to cope with oligomerization is associated with the capture of the intermediate silyloxy-substituted radical by iodine followed by the formation of the carbonyl group.
Besides silyl enol ethers, other classes of compounds, which could be expected to trap fluorinated radicals, were evaluated (Table 2). To perform hydroperfluoroalkylation of alkenes bearing an electron withdrawing group, borane reagents were evaluated as sources of the hydrogen atom.21 In this regard, by using excess of pyridine–borane complex in combination with fac-Ir(ppy)3 as the photocatalyst, sulfides 3 were successfully combined with acrylamides, acrylonitrile, tert-butyl acrylate and vinyl phosphonate. Nitrones are known to be good traps for radicals, and reductive addition of fluorinated halides to nitrones has recently been developed.22 Sulfides 3 proved to be competent partners for coupling with nitrones (Table 2). Ascorbic acid in the presence of collidine was employed as the stoichiometric reducing agent using fac-Ir(ppy)3 as the catalyst leading to gem-difluorinated hydroxylamines 9 in good yields.
Even nitrones derived from enolizable aldehydes afforded the expected addition product 9fe. We also demonstrated that sulfide 3a can alkylate styryltrifluoroborate 10 in the presence of an iridium photocatalyst under visible light, affording the product 11 as a mixture of cis and trans isomers (Table 2, bottom).
Control experiments confirmed that the reaction does not proceed without a photocatalyst or light. Moreover, in the presence of TEMPO, the formation of the product was totally suppressed (Scheme 3). Finally, the gem-difluorinated radical was trapped by a nitrone spin trap, and the nitroxyl radical was detected by EPR spectroscopy.
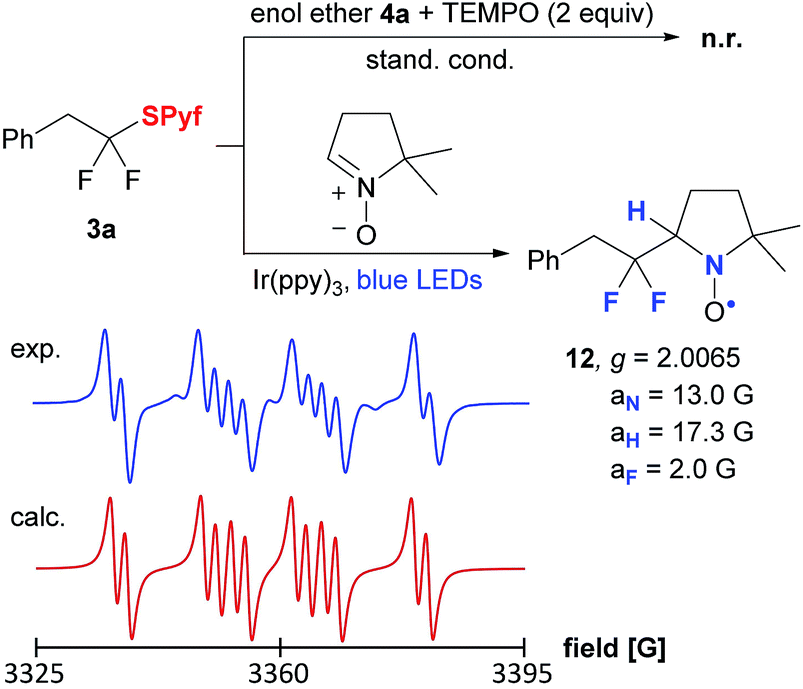 | ||
| Scheme 3 Mechanistic experiments. | ||
Conclusions
In summary, a new 4-tetrafluoropyridinylthio group suitable for mild activation of difluorostyrenes is described. The utility of this group is enabled by a combination of two unique features: first, the pyridinethiol readily gives fluoroalkyl sulfides by the thiolene reaction, and second, the sulfides have favorable potential for radical generation under photoredox catalysis conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Russian Foundation for Basic Research (project 18-53-53009). X-ray studies of compounds 3a and A were carried out with financial support from the Ministry of Science and Higher Education of the Russian Federation using the equipment of the Center for Molecular Composition Studies of INEOS RAS.Notes and references
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (c) J. Wang, M. Sanchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (d) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- (a) X. Pan, H. Xia and J. Wu, Org. Chem. Front., 2016, 3, 1163–1185 RSC; (b) T. Koike and M. Akita, Top. Catal., 2014, 57, 967–974 CrossRef CAS; (c) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (d) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- (a) C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed; (b) D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed; (c) J. Rong, L. Deng, P. Tan, C. Ni, Y. Gu and J. Hu, Angew. Chem., Int. Ed., 2016, 55, 2743–2747 CrossRef CAS PubMed; (d) J. Fang, Z.-K. Wang, S.-W. Wu, W.-G. Shen, G.-Z. Ao and F. Liu, Chem. Commun., 2017, 53, 7638–7641 RSC; (e) D. B. Vogt, C. P. Seath, H. Wang and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 13203–13211 CrossRef CAS PubMed; (f) C. Le, T. Q. Chen, T. Liang, P. Zhang and D. W. C. Macmillan, Science, 2018, 360, 1010–1014 CrossRef CAS PubMed; (g) A. E. Allen and D. W. C. Macmillan, J. Am. Chem. Soc., 2010, 132, 4986–4987 CrossRef CAS PubMed.
- (a) R. Gianatassio, S. Kawamura, C. L. Eprile, K. Foo, J. Ge, A. C. Burns, M. R. Collins and P. S. Baran, Angew. Chem., Int. Ed., 2014, 53, 9851–9855 CrossRef CAS; (b) V. Matoušek, J. Václavík, P. Hájek, J. Charpentier, Z. E. Blastik, E. Pietrasiak, A. Budinská, A. Togni and P. Beier, Chem.–Eur. J., 2016, 22, 417–424 CrossRef PubMed.
- (a) V. V. Levin and A. D. Dilman, J. Org. Chem., 2019, 84, 8337–8343 CrossRef CAS PubMed; (b) S. S. Ashirbaev, V. V. Levin, M. I. Struchkova and A. D. Dilman, Fluorine Notes, 2017, 115, 1–2 CrossRef CAS.
- (a) For reduction of the C–S bond of fluorinated sulfides with elemental magnesium, see: G. K. S. Prakash, J. Hu and G. A. Olah, J. Org. Chem., 2003, 68, 4457–4463 CrossRef CAS; (b) For reduction of sulfides with samarium diiodide, see: Z. Abdallah, G. Doisneau and J.-M. Beau, Angew. Chem., Int. Ed., 2003, 42, 5209–5212 CrossRef CAS PubMed.
- The C–S bond of dithiocarbamates can be cleaved under visible-light irradiation; however, synthesis of these compounds from difluorostyrenes via thiol-ene reaction would be problematic, see: (a) S. Cuadros, M. A. Horwitz, B. Schweitzer-Chaput and P. Melchiorre, Chem. Sci., 2019, 10, 5484–5488 RSC; (b) D. Mazzarella, G. Magagnano, B. Schweitzer-Chaput and P. Melchiorre, ACS Catal., 2019, 9, 5876–5880 CrossRef CAS; (c) B. Schweitzer-Chaput, M. A. Horwitz, E. de Pedro Beato and P. Melchiorre, Nat. Chem., 2019, 11, 129–135 CrossRef CAS PubMed.
- R. E. Banks, R. N. Haszeldine, D. R. Karsa, F. E. Rickett and I. M. Young, J. Chem. Soc. C, 1969, 1660–1662 RSC.
- (a) J. Zheng, J. Cai, J.-H. Lin, Y. Guo and J.-C. Xiao, Chem. Commun., 2013, 49, 7513–7515 RSC; (b) X. Zhang and S. Cao, Tetrahedron Lett., 2017, 58, 375–392 CrossRef CAS.
- (a) L. Yang, W.-X. Fan, E. Lin, D.-H. Tan, Q. Li and H. Wang, Chem. Commun., 2018, 54, 5907–5910 RSC; (b) P. Tian, C.-Q. Wang, S.-H. Cai, S. Song, L. Ye, C. Feng and T.-P. Loh, J. Am. Chem. Soc., 2016, 138, 15869–15872 CrossRef CAS PubMed.
- (a) T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390–402 CrossRef CAS PubMed; (b) D. L. Orsi, B. J. Easley, A. M. Lick and R. A. Altman, Org. Lett., 2017, 19, 1570–1573 CrossRef CAS PubMed; (c) J. Li, C. Xu, N. Wei and M. Wang, J. Org. Chem., 2017, 82, 11348–11357 CrossRef CAS; (d) D. L. Orsi, M. R. Yadav and R. A. Altman, Tetrahedron, 2019, 75, 4325–4336 CrossRef CAS.
- H. Liu, L. Ge, D.-X. Wang, N. Chen and C. Feng, Angew. Chem., Int. Ed., 2019, 58, 3918–3922 CrossRef CAS PubMed.
- J. Xie, J.-T. Yu, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 9416–9421 CrossRef CAS PubMed.
- For earlier report on radical addition of thiols to difluorostyrenes, see: M. Suda, Tetrahedron Lett., 1981, 22, 2395–2396 CrossRef CAS.
- (a) C. S. Thomoson, H. Martinez and W. R. Dolbier, J. Fluorine Chem., 2013, 150, 53–59 CrossRef CAS; (b) S. A. Fuqua, W. G. Duncan and R. M. Silverstein, J. Org. Chem., 1965, 30, 1027–1029 CrossRef CAS.
- (a) J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed; (b) T. T. Eisenhart and J. L. Dempsey, J. Am. Chem. Soc., 2014, 136, 12221–12224 CrossRef CAS PubMed.
- (a) P. V. Pham, D. A. Nagib and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2011, 50, 6119–6122 CrossRef CAS PubMed; (b) G. N. Chernov, V. V. Levin, V. A. Kokorekin, M. I. Struchkova and A. D. Dilman, Adv. Synth. Catal., 2017, 359, 3063–3067 CrossRef CAS.
- For this methodology, we also evaluated pentafluorothio-phenol (C6F5SH). However, the photoredox reaction of the corresponding gem-difluorinated sulfide (BnCF2SC6F5) with silyl enol ether 4a was inefficient. Besides being slow, another pathway corresponding to the conversion of the aromatic C–F bond into C–H bond was observed along with the desired process (see ESI† for details). Such a photoredox reduction of the pentafluorophenylthio-group has been described, see: S. M. Senaweera, A. Singh and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 3002–3005 CrossRef CAS PubMed.
- L. I. Panferova, V. O. Smirnov, V. V. Levin, V. A. Kokorekin, M. I. Struchkova and A. D. Dilman, J. Org. Chem., 2017, 82, 745–753 CrossRef CAS.
- (a) S. Dadashi-Silab, X. Pan and K. Matyjaszewski, Chem.–Eur. J., 2017, 23, 5972–5977 CrossRef CAS PubMed; (b) D. Liu, M.-J. Jiao, Z.-T. Feng, X.-Z. Wang, G.-Q. Xu and P.-F. Xu, Org. Lett., 2018, 20, 5700–5704 CrossRef CAS.
- (a) V. I. Supranovich, V. V. Levin, M. I. Struchkova, A. A. Korlyukov and A. D. Dilman, Org. Lett., 2017, 19, 3215–3218 CrossRef CAS PubMed; (b) V. I. Supranovich, V. V. Levin, M. I. Struchkova, J. Hu and A. D. Dilman, Beilstein J. Org. Chem., 2018, 14, 1637–1641 CrossRef CAS PubMed; (c) T. Kawamoto and I. Ryu, Org. Biomol. Chem., 2014, 12, 9733–9742 RSC.
- (a) V. I. Supranovich, V. V. Levin, M. I. Struchkova and A. D. Dilman, Org. Lett., 2018, 20, 840–843 CrossRef CAS PubMed; (b) I. A. Dmitriev, V. I. Supranovich, V. V. Levin, M. I. Struchkova and A. D. Dilman, Adv. Synth. Catal., 2018, 360, 3788–3792 CrossRef CAS; (c) I. A. Dmitriev, V. I. Supranovich, V. V. Levin and A. D. Dilman, Eur. J. Org. Chem., 2019, 2019, 4119–4122 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1947115 and 1947116. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04643g |
| This journal is © The Royal Society of Chemistry 2020 |