
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A robust photoluminescence screening assay identifies uracil-DNA glycosylase inhibitors against prostate cancer†
Guodong
Li‡
a,
Stuart Adam
Henry‡
b,
Hao
Liu‡
c,
Tian-Shu
Kang‡
a,
Sang-Cuo
Nao‡
a,
Yichao
Zhao
d,
Chun
Wu
c,
Jianwen
Jin
d,
Jia-Tong
Zhang
a,
Chung-Hang
Leung
 *a,
Philip
Wai Hong Chan
*bd and
Dik-Lung
Ma
*c
*a,
Philip
Wai Hong Chan
*bd and
Dik-Lung
Ma
*c
aState Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macau. E-mail: duncanleung@um.edu.mo
bDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK
cDepartment of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong. E-mail: edmondma@hkbu.edu.hk
dSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: phil.chan@monash.edu
First published on 10th January 2020
Abstract
Many cancers have developed resistance to 5-FU, due to removal by the enzyme uracil-DNA glycosylase (UDG), a type of base excision repair enzyme (BER) that can excise uracil and 5-fluorouracil (5-FU) from DNA. However, the development of UDG inhibitor screening methods, especially for the rapid and efficient screening of natural product/natural product-like compounds, is still limited so far. We developed herein a robust time-resolved photoluminescence method for screening UDG inhibitors, which could significantly improve sensitivity over the screening method based on the conventional steady-state spectroscopy, reducing the substantial fluorescence background interference. As a proof-of-concept, two potential UDG inhibitors were identified from a database of natural products and approved drugs. Co-treatment of these two compounds with 5-FU showed synergistic cytotoxicity, providing the basis for treating drug-resistant cancers. Overall, this method provides an avenue for the rapid screening of small molecule regulators of other BER enzyme activities that can avoid false negatives arising from the background fluorescence.
Introduction
Prostate cancer is a malignancy that can remain latent for extended periods of time, resulting in a high disease burden.1,2 Prostate cancer ranks second among cancers in incidence among men, with 1 million new incidences of cancer reported every year.3 Radiotherapy, which involves using high frequency waves to destroy tumor cells,4 is commonly employed for prostate cancer treatment.5 However, radiotherapy exhibits certain adverse effects, including loss of appetite, vomiting, nausea, hair loss and sore skin.6 5-Fluorouracil (5-FU), first patented in 1956 and entered into medical use in 1962,7 is effective against different cancers including prostate cancer through its ability to target thymidylate synthase, which leads to the incorporation of uracil and 5-FU into the genome.8,9 However, many cancers have also developed resistance to 5-FU, due to its removal from genomic DNA by the enzyme uracil-DNA glycosylase (UDG),10–12 a type of base excision repair enzyme (BER) that can excise uracil and 5-FU from DNA13 The depletion of UDG sensitizes tumor cells to 5-FU.14 The uracil-DNA glycosylase inhibitor (UDGI), produced in of Bacillus subtilis bacteriophage PBS1, is a ∼9.5 kDa protein that is used in the literature as a model inhibitor of UDG.15 Other inhibitors of UDG have been reported, such as SSP0047, p56, and uracil aldehyde small molecules,13,16–20 however none of these have undergone further in-depth disease application research. The discovery of new inhibitors of UDG and the development of methods for their identification could offer the potential for synergistic therapeutic strategies with 5-FU against cancer, including prostate cancer.The combination of radioactive labeling with gel electrophoresis is deemed to be the “gold standard” for assaying DNA-modulating enzyme activity and for the identification of their modulators.21 Other reported methods for identifying UDG inhibitors include fragment-substrate tethering, bioinformatics, radioisotopic labeling, chemical cross-linking and affinity chromatography techniques.13,16–20 However, these methods tend to be time-consuming, unwieldy and/or may necessitate stringent safety measures to control radiographic exposure.22 Therefore, new in vitro strategies for the rapid and efficient screening of UDG inhibitors are still desired. In comparison, steady-state fluorescence spectroscopy23,24 has attracted interest as a tool to detect DNA repair enzyme activity, as optical strategies are more convenient and simpler.25–27 However, the use of oligonucleotides labeled with organic dyes is limited by the high cost of synthesis of labeled DNA.28,29 Moreover, the nanosecond lifetime of organic dyes is generally too short to allow their fluorescence to be separated from the high background fluorescence of samples and can result in false negatives, which greatly limits their drug screening applications.30 Consequently, the problem of background fluorescence is a major concern for the screening of small molecule inhibitors.31 Time-resolved emission spectroscopy (TRES) is a technique that measures the emission at discrete times during the fluorescence decay process,32 which provides an potential route to overcome short-lived fluorescence signals. Recently, TRES has been used to detect a variety of analytes, such as mercury ions, aluminum ions, and mRNA.33–35 However, reports describing the detection of enzymes with DNA-modulating activities by TRES are still limited.
In this study, we developed a robust UDG inhibitor screening method through combining a G-quadruplex-specific long-lived luminescent iridium(III) probe with a DNA-switching strategy and TRES. In this method, the G-quadruplex-forming sequence (ON1, 5′-G3TAG3A3T2CT2A2GTGCG3T2G3-3′) is initially hybridized with a partially complementary, uracil-containing DNA strand (ON2, 5′-CGCACU2A2GA2U2TC-3′) to form a duplex substrate (Fig. 1). Uracil, an undesired component of DNA that is produced from the hydrolysis of cytosine, is excised by UDG to form abasic sites.36–38 In our system, the presence of UDG is expected to create four abasic sites on ON2, which will greatly weaken the interaction between ON1 and ON2 due to the loss of four A–U complementary base pairs. This allows liberation of ON1, which is then able to fold into a G-quadruplex conformation that is subsequently recognized by the G-quadruplex-selective iridium(III) complex with an enhanced luminescence response. However, if an UDG inhibitor is present, the release of ON1 will be prevented and thus the emission of the iridium(III) complex will remain low. Our assay also exploits the long-lived phosphorescence emission and large Stoke shifts of the triplet-state energy levels of iridium(III) complexes display,39 allowing their emission to be discriminated even in a highly fluorescent background.40,41
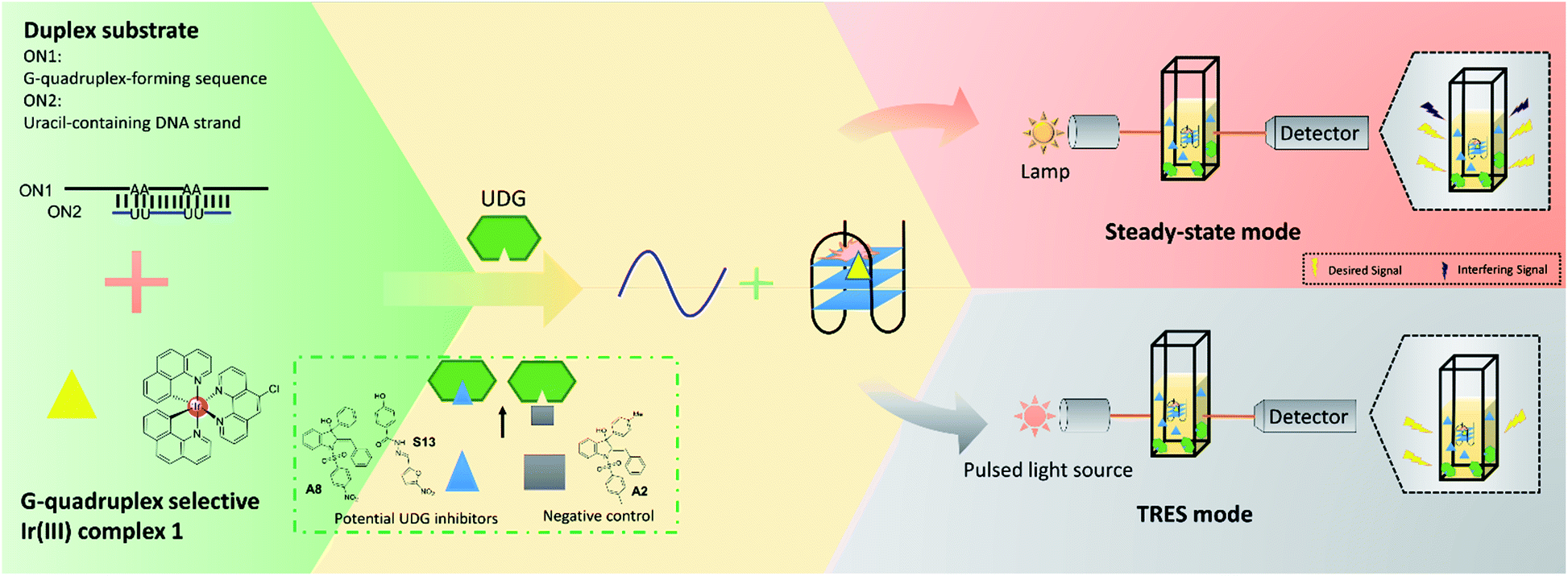 | ||
| Fig. 1 Schematic diagram of the novel UDG inhibitor screening method. In the assay mechanism, the G-quadruplex-forming motif (ON1, 5′-G3TAG3A3T2CT2A2GTGCG3T2G3-3′) is hybridized initially with a partly complementary, uracil-containing DNA sequence (ON2, 5′-CGCACU2A2GA2U2TC-3′) to form a double-stranded DNA substrate. The addition of UDG excises uracil bases from ON1–ON2, forming abasic sites on ON2 which releases ON1. ON1 converts into a G-quadruplex motif that is then bound by the iridium(III) complex 1 with enhanced emission. In the presence of a UDG inhibitor, less ON1 would be liberated, resulting in a weaker luminescence intensity from probe 1. However, this decrease might become completely swamped by the background fluorescence, resulting in a false negative result (upper panel). By using TRES, the short-lived fluorescence of inhibitors would be eliminated, allowing the reducing in luminescence intensity of probe 1 to be clearly detected (lower panel). | ||
As a proof-of-concept, we used this screening method to identify potential UDG inhibitors from a privileged library of 400 natural product-like or FDA/EMA-approved compounds (Fig. 2). These compounds span an extensive array of potential bioactive chemical scaffolds that may be involved in regulating the DNA damage response or possess antitumor activity, including saturated and unsaturated heterocycles, β-amino acids, sulfonamides, carboxylic acids, among other classes. These compounds may generate a strong background fluorescence when screening in steady-state mode, which would interfere with the screening of UDG inhibitors. Therefore, we developed a TRES method to screen our library, which eventually led to the identification of the natural product-like indole derivative (A8) and an FDA-approved antibiotic drug nifuroxazide (S13), currently used against colitis and diarrhea, as inhibitors of UDG. In this study, we used our developed TRES screening method to identify a natural product-like indole derivative (A8) and an FDA-approved antibiotic drug nifuroxazide (S13), currently used against colitis and diarrhoea, as inhibitors of UDG. Both compounds also showed synergistic cytotoxicity with 5-FU, providing a basis for treating drug-resistant cancers, such as advanced prostate cancer. A schematic of the screening method for UDG inhibitors is presented in Fig. 1. We anticipate that this study could encourage the development of future TRES platforms for screening inhibitors of DNA repair enzymes, especially for molecules that are naturally fluorescent, enabling the rapid discovery of bioactive compounds from scaffolds that were previously neglected because of their high false positive rate.
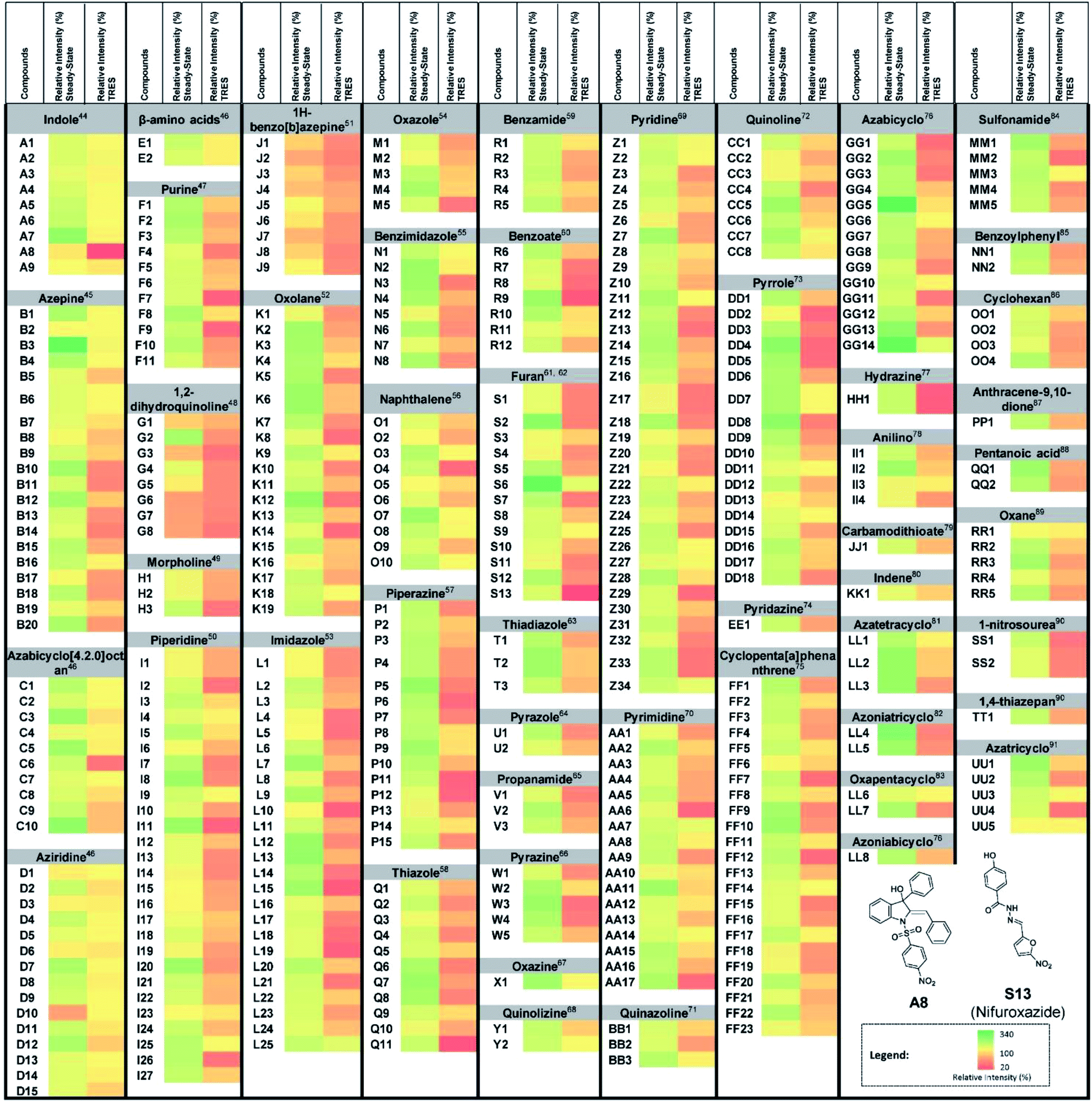 | ||
| Fig. 2 UDG inhibitor screening from a privileged library. Screening results for 400 natural product-like or FDA/EMA-approved compounds (10 μM) on UDG activity by the steady-state emission and TRES methods, with 100 U mL−1 UDGI as positive control. The duplex substrate was incubated with the indicated concentrations of UDG and 10 μM natural product-like compounds. The mixture was heated to 37 °C for 30 min to allow the base cleavage reaction to take place. The mixture was cooled down and was subsequently diluted using Tris buffer to a final volume of 500 μL. 0.5 μM of complex 1 was added to the mixture. A8 and S13 were identified as potential UDG inhibitors. | ||
Results
Development of TRES methodology for screening UDG inhibitors
To confirm the formation of the ON1 G-quadruplex structure, circular dichroism (CD) spectroscopy was performed. CD spectroscopy of the ON1 sequence showed a strong positive peak at 287 nm with a shoulder at 266 nm and a weaker negative peak at 245 nm, consistent with a hybrid-type G-quadruplex (Fig. S1†).42,43 We further evaluated the selectivity of 1 for the G-quadruplex DNA sequence. We found that 1 displayed significantly enhanced luminescence in the presence of G-quadruplex DNA (ON1 and c-kit87), while only slight luminescence changes were observed in the presence of single-stranded DNA or double-stranded DNA (Fig. S2†). These results demonstrate that complex 1 can selectively recognize the ON1 G-quadruplex leading to an enhanced luminescence response, and is consistent with previous reports.38In steady-state mode, almost all of the test samples showed higher emission signals than the vehicle control (Fig. 2), indicating that the steady-state mode is highly susceptible false negatives arising from fluorescence interference. However, we determined that complex 1 had a luminescence lifetime of 931 ns, which was over nine times as long as the 400 natural product-like or FDA/EMA-approved compounds, which had lifetimes less than 100 ns. Therefore, we anticipated that the fluorescence of the samples could be avoided by screening the library under TRES conditions. As expected, the obvious false signals in steady-state mode could be effectively eliminated by using TRES mode (Fig. 2), demonstrating that the TRES-based screening method is a viable technique to screen libraries with a larger quantity and variety of scaffolds. Overall, these data suggest the TRES-based screening method that we developed in this work could effectively eliminate the problem of background fluorescence, allowing false negatives to be avoided.
In the TRES screen, two potent UDG inhibitors, the natural product-like indole compound A8 and the FDA/EMA-approved drug nifuroxazide (S13) were identified. A8 showed the highest inhibition of UDG activity (80.7% at 10 μM), compared to 78.7% for S13 and 65.6% for the positive control compound UDGI.92 Complex 1 benefited from a larger Stokes shifts and a longer emission lifetime than A8 (Fig. S3A–D†). Hence, in TRES mode, the long-lived luminescence lifetime of complex 1 can be easily separated from the short-lived fluorescence of compound A8 (Fig. S3E and F†). Importantly, although compounds A8 and S13 showed promising UDG inhibitory activity in TRES mode, they displayed no apparent activity under steady-state conditions, and thus would be registered as an apparent false negative arising from fluorescence interference. Under TRES conditions, the ability of A8 to inhibit UDG activity could be clearly observed, as revealed by a marked decrease in the emission levels of probe 1 (Fig. 3A and B). A8 showed an IC50 of 1.9 μM at inhibiting UDG activity, while S13 showed an IC50 of ca. 4.0 μM at inhibiting UDG activity (Fig. 3C and D).
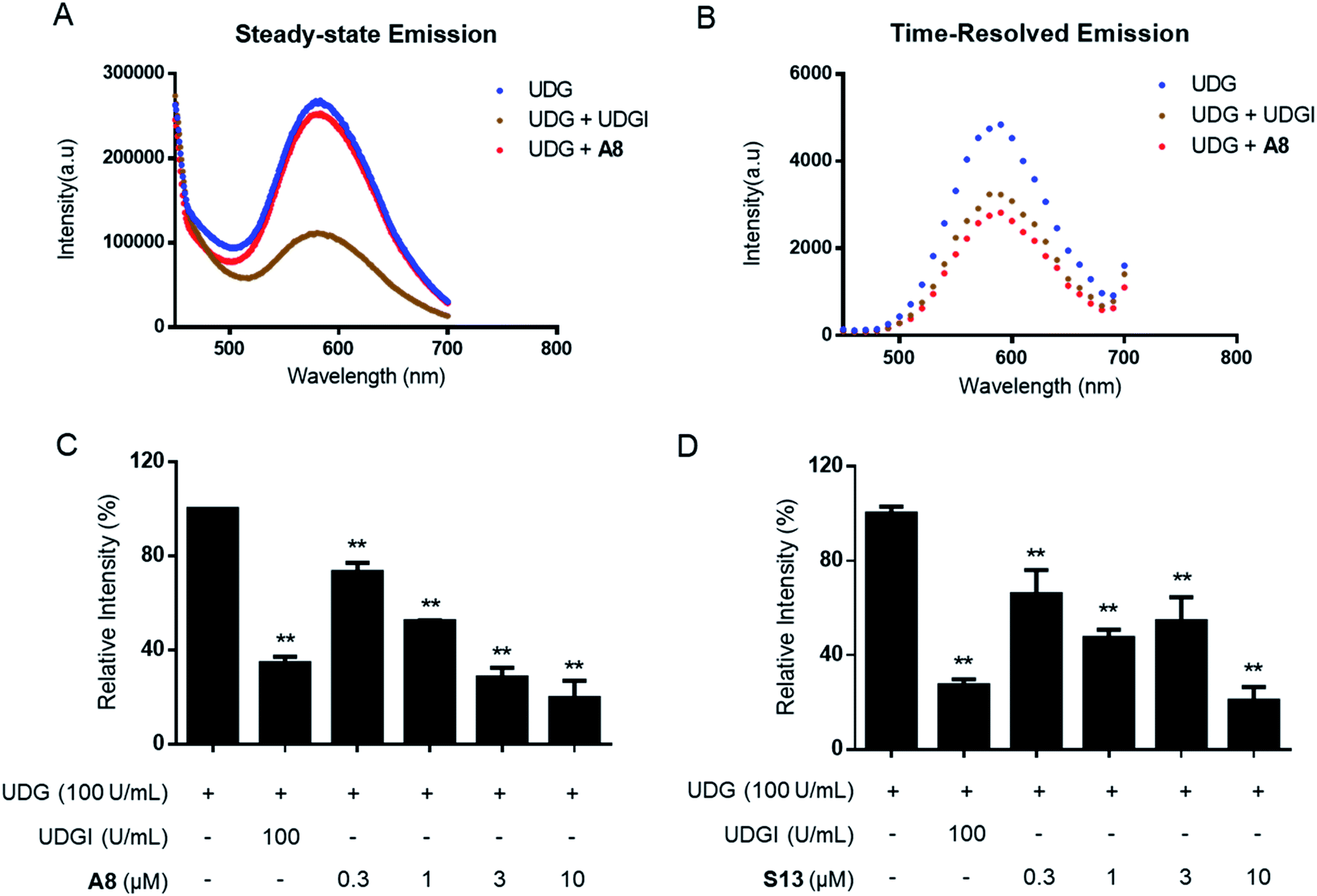 | ||
| Fig. 3 TRES-based screening method could effectively avoid false negatives. (A and B) G-quadruplex/1 method in steady-state emission mode or TRES mode in the presence of 100 U mL−1 UDG, 100 U mL−1 UDG + 100 U mL−1 UDGI, or 100 U mL−1 UDG + 10 μM A8. Luminescence emission intensity was recorded at 450–700 nm with excitation at 355 nm. (C) Dose effect of A8 on UDG activity as measured by TRES. (D) Dose effect of S13 on UDG activity as measured by TRES. Error bars represent the standard deviations of the results from three independent experiments. P values were calculated using a two-sided t-test. *P < 0.05, **P < 0.01 vs. UDG group, respectively. | ||
Verification of UDG activity inhibition by UDG inhibitors by DNA polyacrylamide gel electrophoresis
Non-denaturing polyacrylamide gel electrophoresis (PAGE) was performed to verify the ability of compound A8 and S13 to inhibit UDG (Fig. 4A and S5A†). Isolated ON1 and ON2 oligonucleotides migrate at different speeds on the agarose gel (Lanes 1 and 2), and both move faster than the ON1–ON2 duplex (Lane 3). In the presence of UDG from cell lysates, the uracil bases on ON2 will be excised to liberate ON1 and cleaved ON2 (Lane 4), however, this process could be blocked by the presence of both A8 (Lane 5) or positive control UDGI (Lane 6). A2, a negative control compound, was unable to prevent the release of ON1 (Lane 7). These results verify that the decrease in the emission of the G-quadruplex/1 system by A8 is likely due to the ability of the compound to inhibit UDG activity rather than via other mechanisms, such as emission quenching. In order to distinguish the cleaved DNA band more clearly, we further designed the sequence ON3 containing only a single uracil (5′-CGCACTUA2GA2T3C-3′). PAGE analysis showed that ON1–ON3 duplex (Lane 3) could be cleaved by UDG with or without A2 (Lane 4 and Lane 7), as revealed by the appearance of the cleaved ON3 band, but that this was blocked by the presence of A8 (Lane 5) or UDGI (Lane 6) (Fig. S4†). Compared with the band cleaved by UDG with or without A2 (Lane 4 and Lane 7), S13 could also reduce the cleaved ON3 band (Lane 5), although it slightly less effective than the positive control UDGI (500 U mL−1) or A8 (Fig. S5A†).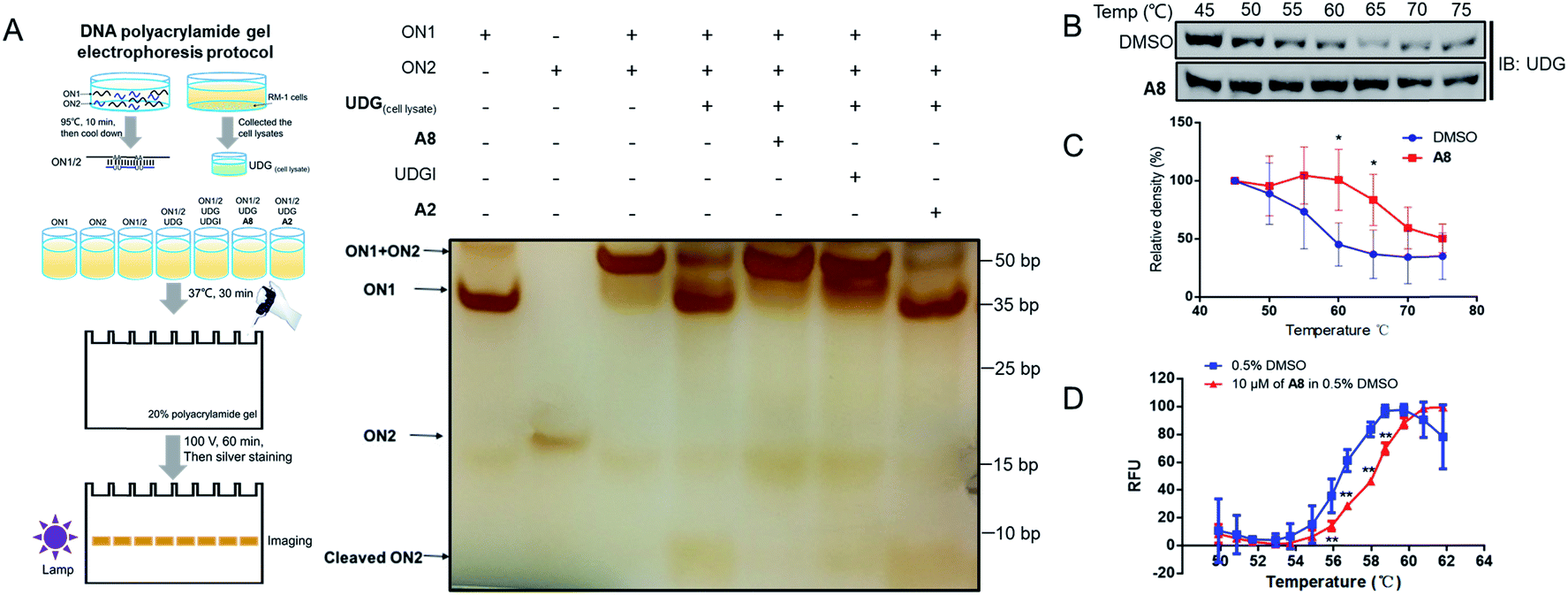 | ||
| Fig. 4 A8 could engage UDG and inhibit its activity. (A) PAGE analysis of DNA assay reaction products in the absence or presence of complexes. RM-1 cell lysates were collected and products treated with UDG in presence of A8, UDGI (positive control), or A2 (negative control), followed by resolution on 20% polyacrylamide gel to separate the cleaved products from the substrate. The separated products were visualized by using ChemiDoc™ MR Imaging System (Bio-Rad), following sliver staining. (B) Stabilization of UDG by A8in cellulo. RM-1 cell lysates were treated with 3 μM of A8 or DMSO at room temperature for 30 min and then heated at different temperature ranging from 45 °C to 75 °C for 5 min. The supernatants of protein samples were collected and detected by western blotting using UDG antibody. (C) Densitometry analysis of CETSA for the level of remaining soluble protein of UDG at different incubation temperatures for treatment and DMSO-treated control samples. Error bars represent the standard deviations of the results obtained from four independent experiments. (D) Shown are plots of the fluorescence changes of UDG (1000 U mL−1) as the temperature was increased in the presence or absence of compound A8 (10 μM). Error bars represent the standard deviations (SD) of the results from three independent experiments. P values were calculated using a two-sided t-test. *P < 0.05, **P < 0.01 vs. DMSO group. | ||
In a cellular thermal shift assay (CETSA), obvious shifts in melting curve of UDG in RM-1 advanced prostate cancer cell lysates were observed in the presence of compound A8 and S13, indicating that both of them directly engaged and stabilized UDG (Fig. 4B, C and S5B, C†). These were corroborated using a fluorescence-based protein thermal shift assay (FTSA), which revealed by a marked shift of the melting curve (ca. 2.0 °C for A8 and ca. 1.2 °C for S13) (Fig. 4D and S5D†).
Combination studies of UDG inhibitors and 5-FU against prostate cancer
Since UDG depletion is known to sensitize cancer cells to chemotherapy,93 the ability of the identified UDG inhibitor A8 to synergize with the anticancer drug, 5-FU, was next investigated in prostate cancer cells. Using the Chou-Talalay method, significant synergism was shown between A8 and 5-FU at inhibiting RM-1 cell proliferation (Fig. 5A). In wells with significant synergy for which there were equipotent proportions of A8 and 5-FU in the mixture, a 2.4-fold lower A8 concentration (IC50 = 3.56 ± 0.35 μM) and 6-fold lower 5-FU concentration (IC50 = 13.09 ± 0.46 μM) were required together to achieve 50% growth inhibition, versus treatment with each single agent alone (IC50 = 8.37 ± 2.55 μM for A8 and IC50 = 77.16 ± 21.84 μM for 5-FU, respectively) (Fig. 5B–D, and Table 1). An apparent synergism was also shown between nifuroxazide and 5-FU with 2.7-fold lower S13 concentration (IC50 = 8.39 ± 0.86 μM) and 3.6-fold lower 5-FU concentration (IC50 = 26.33 ± 2.67 μM) to reach 50% growth inhibition, respectively (Fig. S6†). For UDGI, 4.8- and 2.5-fold lower concentrations were needed in combination to reach 50% growth inhibition (IC50 = 25.23 ± 2.90 U mL−1 for UDGI and IC50 = 24.93 ± 3.71 μM for 5-FU) relative to treatment with the single agent for UDGI (IC50 = 121.84 ± 31.76 U mL−1) and 5-FU (IC50 = 61.80 ± 5.04 μM), respectively (Fig. S7†). Collectively, these results indicate that both of A8 and S13 could synergize with 5-FU in lowering the proliferation of prostate tumour cells in cellulo.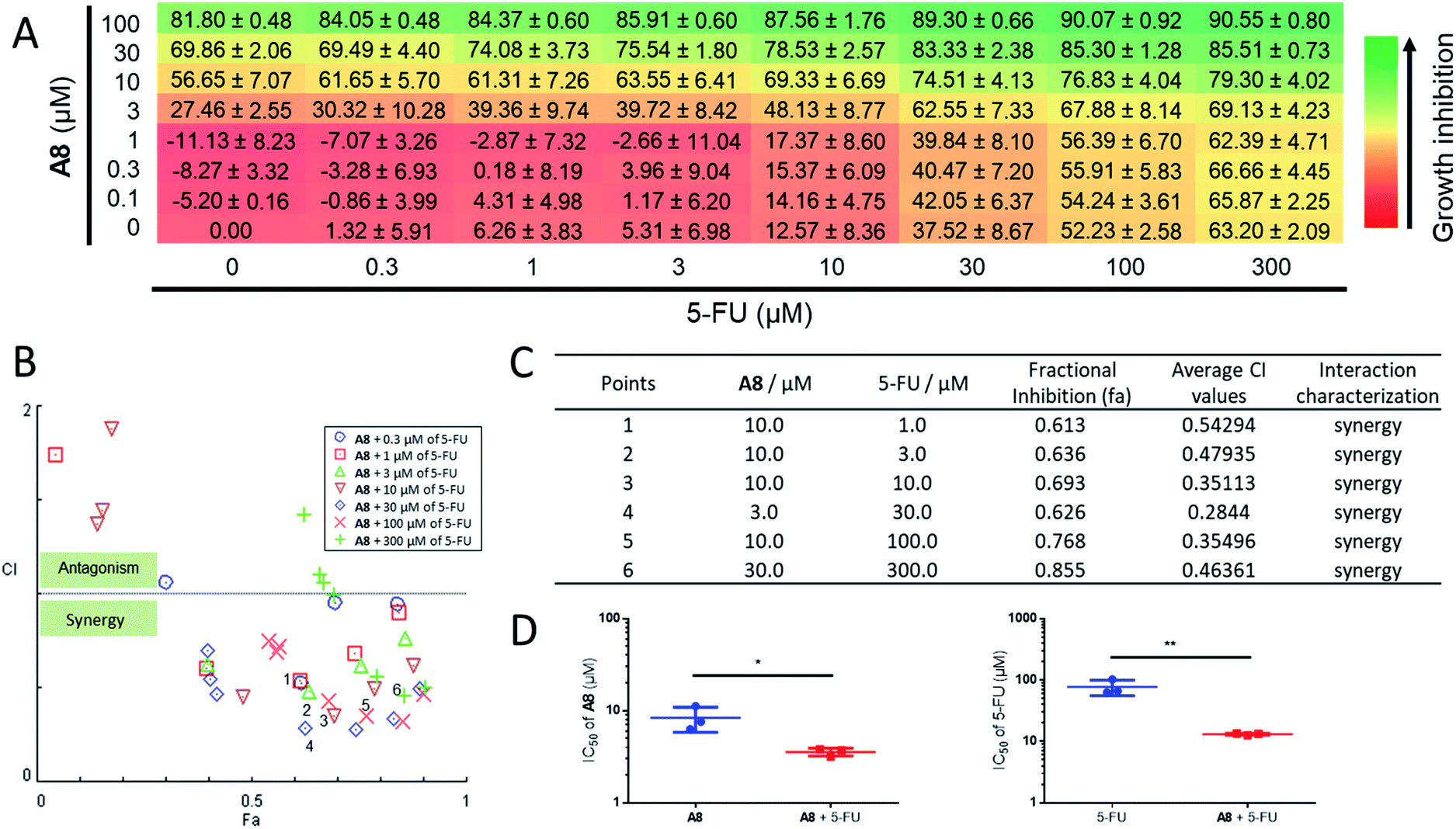 | ||
Fig. 5 Combination assay for A8 and 5-FU. RM-1 cells were treated with combinations of A8 and 5-FU for 48 h and growth inhibition was determined using the MTT assay. (A) Checkerboard data showing viability of RM-1 cells with varying A8 (0–100 μM) and 5-FU (0–300 μM) concentrations as a percentage of untreated cells. Data are expressed as means ± SD (n = 3). (B and C) Combination effect analysis for A8 and 5-FU. Combination index (CI, measure of drug synergy) was determined using the Chou-Talalay method. CI values of <1 indicate drug synergy. (D) The concentration needed to reach 50% inhibition (IC50) of cell proliferation is indicated. The left histograms indicate the IC50 of A8 as a single agent and in combination with 5-FU in the RM-1 cell line. The right histograms indicate the IC50 of 5-FU as a single agent and in combination with A8. The IC50 values for the combination of the two compounds were determined using rays with an effective fraction ∼0.5, corresponding to compounds that are in equipotent proportion (A8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5-FU, 1:3) in the mixture. Error bars represent the standard deviations of the results obtained from three independent experiments. P values were calculated using a two-sided t-test. *P < 0.05, **P < 0.01 UDGI or 5-FU vs. UDG + 5-FU group, respectively. :5-FU, 1:3) in the mixture. Error bars represent the standard deviations of the results obtained from three independent experiments. P values were calculated using a two-sided t-test. *P < 0.05, **P < 0.01 UDGI or 5-FU vs. UDG + 5-FU group, respectively. | ||
Interaction between the UDG inhibitor and the binding pocket of UDG
The above studies demonstrated that A8 and S13 could inhibit the UDG activity and enhance 5-FU-induced growth inhibition. To further understand the interaction of small molecule inhibitors with UDG, molecular modelling was performed with A8, as the more potent of the two compounds. The model of UDG was constructed from the X-ray crystal structure (PDB:3FCI) of UDG in complex with the reported UDG inhibitor, compound P (Fig. S8A†),16 using the molecular conversion procedure implemented in the ICM-pro 3.6-1d program (Molsoft). Compound A8 was predicted to bind snugly in the pocket of UDG (Fig. S8B†), forming hydrogen bonding interactions to Asp145 and Ser247 via its sulfonamide and nitro groups respectively. The binding pose of A8 overlapped significantly with the binding pose of compound P with UDG (Fig. S8C†), with the 4-nitrophenyl ring of A8 occupying roughly the same location as the terminal benzoic acid group of compound P. Notably, both compounds form hydrogen bonding interactions with Ser247, although compound P also makes additional interactions to Gln144, Phe158, Asn204 and Tyr248 that are not replicated in A8. The interaction with Ser247 may be a key determinant for UDG inhibitory activity, as A2, which has a similar structure to A8 but lacks the nitro group that A8 uses to hydrogen bond to Ser247, showed no activity in the UDG inhibitor assay.
Inhibition of UDG activity promotes 5-FU-induced DNA damage and cell cycle arrest in advanced prostate cancer cells
UDG expression is coordinated with DNA replication.94,95 Two well-recognized molecular indicators of DNA damage, phosphorylation of histone H2AX (γH2AX), and cleavage of poly(ADP-ribose) polymerase (cleaved-PARP), were then analyzed. Immunoblotting results showed a significant increase (ca. 2.5 fold) in the co-treated group compared with only the 5-FU treated group (Fig. S9A†), and an increasing accumulation of Ser139-phosphorylated γ-H2AX and cleaved-PARP in the combination group over 0, 4, 8, 12, 16, and 20 h of treatment (Fig. S9B†), suggesting that A8 could enhance the ability of 5-FU to induce DNA damage. This finding was further confirmed by immunofluorescence analysis, which showed increased expression of γH2AX level in the A8/5-FU combination group (Fig. S9C†). To visualize DNA damage more clearly, a comet assay was carried out, which showed more long-tail migrated DNA, indicative of DNA damage, in the A8/5-FU combination group compared with the single-treatment groups and the control group (Fig. S9D†).5-FU is well-known to affect the cell cycle.96,97 To further verify the synergistic effect of A8 and 5-FU, and also to evaluate the selectivity of A8 interacting with UDG in cells, cell cycle analysis combined with siRNA knockdown experiments were performed. As shown Fig. S9E,† the percentages of G0/G1 and S-phase cells were relatively unaffected in the presence of A8 and 5-FU in UDG knockdown RM-1 cells, while co-treatment of A8 and 5-FU resulted in a significant decrease of G0/G1 phase cells and an increase in the percentage of S-phase cells. This suggests that the cell cycle modulation by 5-FU is enhanced by A8 and also requires the presence of UDG in RM-1 cells.
Discussion
Many biochemical small inhibitor screening assays have been developed based on reactions to produce conventional fluorophores, such as commercial steady-state fluorescent inhibitor screening assay kits.98 Moreover, conventional steady-state fluorometric detection is also central to many other screening applications, including, but not limited to, enzyme-linked immunosorbent assay (ELISA), the AlphaScreen assay, flow cytometry and immunohistochemistry (IHC). These inhibitor screening methods provide a very effective technique for high-throughput screening (HTS) of proteins or kinase inhibitors. However, they also share several limitations. Firstly, high background signals are often raised by the simultaneous excitation and emission processes. Secondly, many commercially available dyes suffer from small Stokes shifts, which can result in poor signal-to-noise (S:N) ratios, as well as self-quenching due to overlap between their absorption and emission spectra.99 Thirdly, autofluorescent substances of biological matrices (cell or tissue samples) often act as a source of background signal in assays,100 because it is very hard to eliminate their presence completely prior to measurement. Finally, the fluorescent nature of certain classes of small-molecule compounds can generate false negatives or positives in a fluorescence-based HTS strategy.101 In this study, we employed a privileged library of 400 natural product-like or approved dug compounds covering over 50 types of potential bioactive chemical scaffolds, including saturated and unsaturated heterocycles, β-amino acids, sulfonamides, and carboxylic acids. Significantly, most of these compounds were known to exhibit higher background fluorescence in steady-state mode which makes them unsuitable for most photoluminescence screening methods. Thus, we anticipated that TRES could be an effective strategy to eliminate or even avoid the problem of background fluorescence for the screening of small molecule inhibitors.
Different from standard steady-state fluorometric methods, TRES detection provides an effective strategy to overcome fluorescence interference by recording the luminescence decay profile with high temporal resolution after a pulsed excitation.102 TRES relies on the use of specific luminescent probes, such as iridium complexes, that have long-lived emission which allows detection of the emitted light to take place long after excitation has occurred. Specifically, the long luminescence lifetime (typically microseconds) of iridium probes are usually several orders of magnitude greater than nonspecific background fluorescence (typically nanoseconds), which enables the emission to be read at a time well after any background fluorescence has decayed. Iridium probes also have large Stokes shifts, which can also help to increase the S:N ratio.103 Finally, the high luminescence intensity and stable emission signal of iridium probes also significantly improves assay sensitivity, robustness and dynamic range.
UDG is emerging as a very interesting pharmacologic target for therapeutic intervention due to its important function in regulating various physiological activities, including DNA replication within viruses, and the generation of DNA strand breaks during chemotherapy.13 Many cancers have developed resistance to 5-FU, due to the ability of UDG to continuously repair 5-FU induced DNA damage.104,105 Therefore, the combination of 5-FU with a UDG inhibitor could be a potential strategy to overcome 5-FU resistance and generate effective anticancer effect, including against prostate cancer. Several potent small molecule UDG inhibitors with IC50 values between 1.1 to 315 μM have been identified by screening linked libraries using 6-formyl uracil as the substrate fragment.13,16,17 However, none of these have undergone further in-depth disease application research, such as the treatment of drug-resistant cancers, including in combination therapy with clinical drugs. In addition, besides the general strategy of substrate fragment tethering, effective methods for the discovery of novel UDG inhibitors are still lacking. In this work, we have demonstrated for the first time the use of TRES to overcome the background fluorescence of samples in order to identify UDG inhibitors from a privileged library of natural product/natural product-like compounds. The method exploits the long phosphorescence lifetime (931 ns) of iridium probe 1, which is much longer than the fluorescence lifetimes of library. Using a delay time of 500 ns in TRES mode, the emission of the complex 1 could be clearly distinguished and the problem of false negatives in steady-state emission mode could be avoided.38 In particular, we note that the promising UDG inhibitors A8 and S13 identified in this work would have been recorded as a non-active under steady-state mode, whereas both of them showed clear UDG inhibition via TRES. One drawback of this strategy is that it is relatively time-consuming compared with traditional steady-state fluorescent screening method. Moreover, while it can overcome interference from short-lived background fluorescence, it is still susceptible to interference from inhibitors with longer fluorescence lifetimes.
The indole derivative A8 showed 81% inhibition of UDG activity at 10 μM, compared to 65% for UDGI and 79% for S13, and displayed an IC50 value of 1.9 μM against UDG activity in a dose–response experiment. More importantly, we demonstrated that inhibition of UDG activity by A8 and S13 significantly promotes the anti-tumor effect of 5-FU in advanced prostate cancer cells. Non-denaturating PAGE analysis showed that both of them compromised the ability to cleave ON2 and liberate ON1, consistent with the proposed mechanism of the assay. Molecular docking analysis further elaborated that the high affinity of lead compound A8 binding to binding pocket of UDG by forming two H-bondings and hydrophobic effect with the binding pocket of UDG.
Biological experiments showed the potential of indole derivative A8 and furan derivative S13 to synergize with 5-FU for prostate cancer therapy. In the checkerboard assay, A8 and S13 could synergize with 5-FU in lowering the proliferation of prostate tumour cells in cellulo. Moreover, comparison of IC50 fold-changes suggested that A8 might be better than UDGI at synergizing with 5-FU. However, it should be noted that the in vitro potency of these compounds against UDG kinase can be different from their in cellulo activities, due to variations in cell absorption, metabolism, or other factors. The combination of A8 and 5-FU increased DNA damage markers, γ-H2AX and cleaved-PARP, in RM-1 cells, suggesting that A8 could enhance the ability of 5-FU to induce DNA damage. This finding was further confirmed by immunofluorescence analysis and a comet assay. Meanwhile, the cell cycle data showed that A8 could enhance the cell cycle regulation of 5-FU by selective inhibition of UDG activity in RM-1 cells.
In conclusion, we have successfully developed a TRES-based UDG activity detection method that can overcome the background fluorescence of samples. This work led to the identification of the indole derivative A8 and the FDA/EMA-approved drug S13, as only the second and third classes overall of UDG small molecule inhibitors reported to date. Moreover, A8 showed comparable potency to the report UDG small molecule inhibitor, and also showed promising synergism with 5-FU at inducing DNA damage and impairing the proliferation of prostate cancer cells. Notably, drug S13 has anti-cancer properties that have been elucidated in previous studies,106–108 and the discovery that S13 targets UDG target will facilitate drug repurposing against resistance of antineoplastic drugs. We envision that this rapid screening method could be easily adapted for the screening of other BER enzyme inhibitors for potential therapeutic applications, including previously neglected scaffolds classes that could not be screened because of their high background fluorescence leading to false negatives.
Experimental
Cells and reagents
Dulbecco's modified Eagle's medium (DMEM) and Fetal bovine serum (FBS) were obtained from Gibco BRL (Gaithersburg, MD, USA). UDG and UDG inhibitor (UDGI) were obtained from New England Biolabs Inc. All oligonucleotides were synthesized by IGE Bio Inc (Guangzhou, China), and the sequences were as follows: ON1: 5′-GGGTAGGGAAATTCTTAAGTGCGGGTTGGG-3′; ON2: 5′-CGCACUUAAGAAUUT C-3′. All the antibodies were obtained from Cell Signaling Technology. Luciferase assay reagent and passive lysis buffer were purchased from Promega Corporation (Madison, WI, USA). All the natural compounds were provided by Prof. Philip Wai Hong Chan (Monash University) in dimethyl sulfoxide (DMSO) at a stock concentration of 10 mM. Complex 1 was synthesized according to a literature method.109UDG inhibitor screening
The solution containing ON1 (100 μM) and ON2 (100 μM) sequences in a hybridization buffer (50 mM Tris, 150 mM NaCl, pH 7.0), was heated to 95 °C for 10 min and then cooled at 0.1 °C s−1 to room temperature to allow the formation of the duplex substrate (ON1–ON2). The annealed product was stored at −20 °C before use. The duplex substrate was incubated with the indicated concentrations of UDG and 10 μM natural product-like compounds. The mixture was heated to 37 °C for 30 min to allow the base cleavage reaction to take place. The mixture was cooled down and was subsequently diluted using Tris buffer (50 mM Tris, 20 mM KCl, 150 mM NH4OAc, pH 7.0) to a final volume of 500 μL. Complex 1 was added to a final concentration of 0.5 μM. Steady-state photoluminescence spectra were obtained in a HORIBA Fluorolog-3 spectrophotometer, with excitation at 355 nm. Time-resolved studies were performed using a time-correlated single photon counting (TCSPC) technique on this Fluorolog-3 spectrophotometer using a delay time of 500 ns. The use of a 500 ns delay time eliminates the short-lived fluorescence of the screened compounds, while the long-lived phosphorescence of complex 1 remains to be detected.110 The luminescence emission intensity at 450–700 nm was then monitored after excitation of the sample at 355 nm.DNA polyacrylamide gel electrophoresis
This experiment was carried as previously described with minor modification.111 The UDG-treated product was obtained according to the procedure described above. The products were resolved on 20% polyacrylamide gel to separate the cleaved products from the substrate using 1× TBE as running buffer at a constant voltage of 100 V for 120 min. The separated products were visualized by using the ChemiDoc™ MR Imaging System (Bio-Rad), following silver staining according to protocol of Fast Sliver Stain kit for Nucleic Acids (Real-Times, China).Immunofluorescence assay
The cell damage and prolonged mitosis were analyzed according to previous methods with minor modifications.112 RM-1 cells, an advanced prostate cancer cell line, were seeded in 35 mL plates at a density of (6 × 104) cells per mL overnight and treated with DMSO, A8 (3.0 μM), 5-FU (10.0 μM) and the combination treatment for 12 h. As for γH2AX, after fixation in 4% paraformaldehyde and permeabilization in 0.2% Triton X-100 in PBS for 30 minutes. Cells were incubated with fluorescein isothiocyanate (FITC)-conjugated mouse anti-γH2AX(Ser139) monoclonal antibody (1:500) (BioLegend) overnight. Cell nuclei were counterstained with 4,6-diaidino-2-phenylindole (DAPI), and then stained with DAPI for 15 min. The photos and density values of fluorescence were acquired on a Leica confocal microscope.
Combination assay
RM-1 (5000 cells per well) were seeded in 96-well plates with regular media and allowed to adhere for 24 h. The following day, the media was replaced with serum free media and cells were treated as indicated for an additional 48 h. Cell proliferation was measured using the MTT method. 20 μL 5× MTT was added in each well, and the wells were incubated at 37 °C for 4 h. The absorbance at 570 nm was measured with a plate reader. Each experiment was performed in triplicate, and the experiments are shown with standard deviation from the experiment as error bars. The error bars in reported IC50 values represent the standard deviation from three different experiments. Combination index (CI) was determined using the Chou-Talalay method.113Author contributions
G. L., T. S. K., J. T. Z., and S. C. N. carried out the in vitro and in cellulo experiments. G. L., T. S. K., H. L., and C. W. wrote the manuscript. S. A. H., Y. Z. and J. J. carried out synthesis complexes. P. W. H. C., D. L. M. and C. H. L. designed the experiments and analyzed the results.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We would like to thank Feng Chen (University of Macau) for technical assistance. This work was supported by Hong Kong Baptist University (FRG2/17-18/003), the Health and Medical Research Fund (HMRF/14150561), the National Natural Science Foundation of China (21575121 and 21775131), the Guangdong Province Natural Science Foundation (2015A030313816), the Hong Kong Baptist University Century Club Sponsorship Scheme 2018, the Interdisciplinary Research Matching Scheme (RC-IRMS/16-17/03), Interdisciplinary Research Clusters Matching Scheme (RC-IRCs/17-18/03), Collaborative Research Fund (C5026-16G), SKLEBA and HKBU Strategic Development Fund (SKLP_1718_P04), Funded by The Science and Technology Development Fund, Macau SAR (File no. 077/2016/A2), the University of Macau (MYRG2018-00187-ICMS and MYRG2019-00002-ICMS), and a Discovery Project Grant (DP160101682, Australia) from the Australian Research Council.References
- Z. Q. Tao, A. M. Shi, K. X. Wang and W. D. Zhang, Rev. Eur. Sci. Med. Pharmacol., 2015, 19, 805–812 Search PubMed
.
- H. I. Scher, K. Solo, J. Valant, M. B. Todd and M. Mehra, PLoS One, 2015, 10, e0139440 CrossRef PubMed
- E. T. Chang, P. Boffetta, H. O. Adami, P. Cole and J. S. Mandel, Eur. J. Epidemiol., 2014, 29, 667–723 CrossRef PubMed
- H.-S. Lee, M. Carmena, M. Liskovykh, E. Peat, J.-H. Kim, M. Oshimura, H. Masumoto, M.-P. Teulade-Fichou, Y. Pommier and W. C. Earnshaw, Cancer Res., 2018, 78, 6282–6296 CrossRef CAS PubMed
- D. E. Spratt, X. Pei, J. Yamada, M. A. Kollmeier, B. Cox and M. J. Zelefsky, Int. J. Radiat. Oncol., Biol., Phys., 2013, 85, 686–692 CrossRef PubMed
- J. A. Langendijk, P. Lambin, D. De Ruysscher, J. Widder, M. Bos and M. Verheij, Radiother. Oncol., 2013, 107, 267–273 CrossRef CAS PubMed
-
J. Fischer, C. R. Ganellin, A. Ganesan and J. Proudfoot, Analogue-based drug discovery, Wiley-VCH, 2010 Search PubMed
- S. Maruyama, M. Ando and T. Watayo, Gan to Kagaku Ryoho, 2003, 30, 1635–1638 Search PubMed
- F. Ghaemmaghami, N. Behtash, F. Yarandi, A. Moosavi, M. Modares, G. Toogeh and N. Khanafshar, J. Obstet. Gynaecol., 2003, 23, 422–425 CrossRef CAS PubMed
- D. J. Mauro, J. De Riel, R. J. Tallarida and M. A. Sirover, Mol. Pharmacol., 1993, 43, 854–857 CAS
- M. T. Nguyen, Y. Yan, Y. Fedorvo, J. Pink, D. Adams and S. Gerson, AACR, 2018, 1663 Search PubMed
- Y. Yan, X. Han, Y. Qing, A. G. Condie, S. Gorityala, S. Yang, Y. Xu, Y. Zhang and S. L. Gerson, Oncotarget, 2016, 7, 59299 Search PubMed
- Y. L. Jiang, D. J. Krosky, L. Seiple and J. T. Stivers, J. Am. Chem. Soc., 2005, 127, 17412–17420 CrossRef CAS PubMed
- A. M. Huehls, C. J. Huntoon, P. M. Joshi, C. A. Baehr, J. M. Wagner, X. Wang, M. Y. Lee and L. M. Karnitz, Mol. Pharmacol., 2016, 89, 53–62 CrossRef CAS PubMed
- N. Acharya, S. Roy and U. Varshney, J. Mol. Biol., 2002, 321, 579–590 CrossRef CAS PubMed
- S. Chung, J. B. Parker, M. Bianchet, L. M. Amzel and J. T. Stivers, Nat. Chem. Biol., 2009, 5, 407 CrossRef CAS PubMed
- D. J. Krosky, M. A. Bianchet, L. Seiple, S. Chung, L. M. Amzel and J. T. Stivers, Nucleic Acids Res., 2006, 34, 5872–5879 CrossRef CAS PubMed
- H.-C. Wang, K.-C. Hsu, J.-M. Yang, M.-L. Wu, T.-P. Ko, S.-R. Lin and A. H.-J. Wang, Nucleic Acids Res., 2013, 42, 1354–1364 CrossRef PubMed
- J. I. Banos-Sanz, L. Mojardín, J. Sanz-Aparicio, J. M. Lazaro, L. Villar, G. Serrano-Heras, B. Gonzalez and M. Salas, Nucleic Acids Res., 2013, 41, 6761–6773 CrossRef CAS PubMed
- G. Serrano-Heras, M. Salas and A. Bravo, J. Biol. Chem., 2006, 281, 7068–7074 CrossRef CAS PubMed
- N. M. Gueddouda, O. Mendoza, D. Gomez, A. Bourdoncle and J.-L. Mergny, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 1382–1388 CrossRef CAS PubMed
- K.-H. Leung, H.-Z. He, B. He, H.-J. Zhong, S. Lin, Y.-T. Wang, D.-L. Ma and C.-H. Leung, Chem. Sci., 2015, 6, 2166–2171 RSC
- M. H. Lim, D. Xu and S. J. Lippard, Nat. Chem. Biol., 2006, 2, 375 CrossRef CAS PubMed
- H. J. Lee, Y. G. Lee, J. Kang, S. H. Yang, J. H. Kim, A. B. Ghisaidoobe, H. J. Kang, S.-R. Lee, M. H. Lim and S. J. Chung, Chem. Sci., 2019, 10, 1000–1007 RSC
- J. Campbell and H. Girault, J. Electroanal. Chem. Interfacial Electrochem., 1989, 266, 465–469 CrossRef CAS
- J. Liu, C. Roussel, G. Lagger, P. Tacchini and H. H. Girault, Anal. Chem., 2005, 77, 7687–7694 CrossRef CAS PubMed
- Y. Xiao, A. A. Lubin, A. J. Heeger and K. W. Plaxco, Angew. Chem., 2005, 117, 5592–5595 CrossRef
- A. De Rache and J.-L. Mergny, Biochimie, 2015, 115, 194–202 CrossRef CAS PubMed
- S. Zou, G. Li, T. W. Rees, C. Jin, J. Huang, Y. Chen, L. Ji and H. Chao, Chem.–Eur. J., 2018, 24, 690–698 CrossRef CAS PubMed
- D.-L. Ma, H.-Z. He, K.-H. Leung, H.-J. Zhong, D. S.-H. Chan and C.-H. Leung, Chem. Soc. Rev., 2013, 42, 3427–3440 RSC
- M. Hintersteiner and M. Auer, Methods Appl. Fluoresc., 2013, 1, 017001 CrossRef PubMed
- J. H. Easter, R. P. DeToma and L. Brand, Biophys. J., 1976, 16, 571–583 CrossRef CAS PubMed
- J. Zheng, Y. Nie, Y. Hu, J. Li, Y. Li, Y. Jiang and R. Yang, Chem. Commun., 2013, 49, 6915–6917 RSC
- W. Wang, Z. Mao, M. Wang, L.-J. Liu, D. W. Kwong, C.-H. Leung and D.-L. Ma, Chem. Commun., 2016, 52, 3611–3614 RSC
- A. A. Martí, X. Li, S. Jockusch, Z. Li, B. Raveendra, S. Kalachikov, J. J. Russo, I. Morozova, S. V. Puthanveettil and J. Ju, Nucleic Acids Res., 2006, 34, 3161–3168 CrossRef PubMed
- S. S. Parikh, G. Walcher, G. D. Jones, G. Slupphaug, H. E. Krokan, G. M. Blackburn and J. A. Tainer, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5083–5088 CrossRef CAS PubMed
- T. Ono, S. Wang, C. K. Koo, L. Engstrom, S. S. David and E. T. Kool, Angew. Chem., 2012, 51, 1689–1692 CrossRef CAS PubMed
- K.-H. Leung, H.-Z. He, V. P.-Y. Ma, H.-J. Zhong, D. S.-H. Chan, J. Zhou, J.-L. Mergny, C.-H. Leung and D.-L. Ma, Chem. Commun., 2013, 49, 5630–5632 RSC
- C.-N. Ko, G. Li, C.-H. Leung and D.-L. Ma, Coord. Chem. Rev., 2019, 381, 79–103 CrossRef CAS
- Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568–1600 RSC
- X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202–224 RSC
- A. I. Karsisiotis, N. M. a. Hessari, E. Novellino, G. P. Spada, A. Randazzo and M. Webba da Silva, Angew. Chem., 2011, 50, 10645–10648 CrossRef CAS PubMed
- J. Mitra, M. A. Makurath, T. T. Ngo, A. Troitskaia, Y. R. Chemla and T. Ha, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8350–8359 CrossRef CAS PubMed
- D. Susanti, F. Koh, J. A. Kusuma, P. Kothandaraman and P. W. H. Chan, J. Org. Chem., 2012, 77, 7166–7175 CrossRef CAS PubMed
- W. Rao, M. J. Koh, P. Kothandaraman and P. W. H. Chan, J. Am. Chem. Soc., 2012, 134, 10811–10814 CrossRef CAS PubMed
- T. M. U. Ton, C. Tejo, D. L. Y. Tiong and P. W. H. Chan, J. Am. Chem. Soc., 2012, 134, 7344–7350 CrossRef CAS PubMed
- A. D. Zelenetz, J. C. Barrientos, J. R. Brown, B. Coiffier, J. Delgado, M. Egyed, P. Ghia, Á. Illés, W. Jurczak and P. Marlton, Lancet Oncol., 2017, 18, 297–311 CrossRef CAS PubMed
- W. Rao, P. Kothandaraman, C. B. Koh and P. W. H. Chan, Adv. Synth. Catal., 2010, 352, 2521–2530 CrossRef CAS
- R. Bentli, H. Parlakpinar, A. Polat, E. Samdanci, M. E. Sarihan and M. Sagir, Arch. Med. Res., 2013, 44, 521–528 CrossRef CAS PubMed
- P. Wang, J. Cai, J. Chen and M. Ji, Eur. J. Med. Chem., 2015, 93, 1–8 CrossRef CAS PubMed
- E. M. L. Sze, M. J. Koh, Y. M. Tjia, W. Rao and P. W. H. Chan, Tetrahedron, 2013, 69, 5558–5565 CrossRef CAS
- S. Alas, B. Bonavida and C. Emmanouilides, Anticancer Res., 2000, 20, 2961–2966 CAS
- M. Iradyan, N. Iradyan, G. Stepanyan, F. Arsenyan and B. Garibdzhanyan, Pharm. Chem. J., 2010, 44, 175–182 CrossRef CAS
- K. K. Valluri, T. R. Allaka, I. K. Viswanath and P. Nagaraju, Asian J. Res. Chem., 2018, 11, 617–627 CrossRef
- T. W. Dennie and J. M. Kolesar, Clin. Ther., 2009, 31, 2290–2311 CrossRef CAS PubMed
- M. H. Alkhatib, D. K. Zahim and W. S. Backer, Int. J. Drug Deliv. Technol., 2017, 7, 63–70 Search PubMed
-
P. L. Toogood and N. D. Ide, Innovative Drug Synthesis, 2016, pp. 167–195 Search PubMed
- S. Takano, T. Kubota, H. Nishibori, H. Hasegawa, Y. Ishii, N. Nitori, H. Ochiai, K. Okabayashi, Y. Kitagawa and M. Watanabe, Anticancer Res., 2008, 28, 3593–3599 CAS
- S.-H. Kim, H. Yoo, J. H. Chang, C.-Y. Kim, D. S. Chung, S. H. Kim, S.-H. Park, Y. S. Lee and S. H. Yang, J. Korean Med. Sci., 2018, 33, e167 CrossRef PubMed
- K. Shirakawa, L. Wang, N. Man, J. Maksimoska, A. W. Sorum, H. W. Lim, I. S. Lee, T. Shimazu, J. C. Newman and S. Schröder, eLife, 2016, 5, e11156 CrossRef PubMed
- E. A. Nelson, S. R. Walker, A. Kepich, L. B. Gashin, T. Hideshima, H. Ikeda, D. Chauhan, K. C. Anderson and D. A. Frank, Blood, 2008, 112, 5095–5102 CrossRef CAS PubMed
- C. Bailly, Drug Discovery Today, 2019, 24, 1930–1936 CrossRef CAS PubMed
- S. Karakuş, F. Tok, S. Türk, E. Salva, G. Tatar, T. Taskın-Tok and B. Kocyigit-Kaymakcıoglu, Phosphorus, Sulfur Silicon Relat. Elem., 2018, 193, 528–534 CrossRef
- S. T. Lim, Y. W. Jeon, H. Gwak, S. Y. Kim and Y. J. Suh, Mol. Med. Rep., 2018, 17, 5581–5588 CAS
- L. Choucha Snouber, A. Bunescu, M. Naudot, C. Legallais, C. Brochot, M. E. Dumas, B. Elena-Herrmann and E. Leclerc, Toxicol. Sci., 2012, 132, 8–20 CrossRef PubMed
- D. Chen, M. Frezza, S. Schmitt, J. Kanwar and Q. P. Dou, Curr. Cancer Drug Targets, 2011, 11, 239–253 CrossRef CAS PubMed
- S. Ananthula, P. Parajuli, F. A. Behery, A. Y. Alayoubi, K. A. El Sayed, S. Nazzal and P. W. Sylvester, Anticancer Res., 2014, 34, 2715–2726 CAS
- V. Srinivasulu, P. Schilf, S. Ibrahim, M. A. Khanfar, S. M. Sieburth, H. Omar, A. Sebastian, R. A. AlQawasmeh, M. J. O'Connor and T. H. Al-Tel, Nat. Commun., 2018, 9, 4989 CrossRef PubMed
- K. Connolly, D. Brungs, E. Szeto and R. Epstein, Curr. Oncol. Rep., 2014, 21, e151 CrossRef CAS PubMed
- O. Fathalla, I. Zeid, M. Haiba, A. Soliman, S. I. Abd-Elmoez and W. El-Serwy, World J. Chem., 2009, 4, 127–132 CAS
- G. Marzaro, A. Guiotto and A. Chilin, Expert Opin. Ther. Pat., 2012, 22, 223–252 CrossRef CAS PubMed
-
J. Courcambeck, P. Halfon, F. Bassissi, B. Sonia, G. Nicolas, A. Beret, S. Petit, C. Camus and J. P. Nallet, US Pat., No. 10,179,770, 2019
- D. Bandyopadhyay, S. Mukherjee, J. C. Granados, J. D. Short and B. K. Banik, Eur. J. Med. Chem., 2012, 50, 209–215 CrossRef CAS PubMed
- A. K. El-Ansary, A. M. Kamal and M. A. Al-Ghorafi, Med. Chem. Res., 2013, 22, 2589–2601 CrossRef CAS
- A. H. Banday, B. P. Mir, I. H. Lone, K. Suri and H. S. Kumar, Steroids, 2010, 75, 805–809 CrossRef CAS PubMed
- R. Kayarmar, G. Nagaraja, M. Bhat, P. Naik, K. Rajesh, S. Shetty and T. Arulmoli, Med. Chem. Res., 2014, 23, 2964–2975 CrossRef CAS
- P. Çıkla, D. Özsavcı, Ö. Bingöl-Özakpınar, A. Şener, Ö. Çevik, S. Özbaş-Turan, J. Akbuğa, F. Şahin and Ş. G. Küçükgüzel, Arch. Pharm., 2013, 346, 367–379 CrossRef PubMed
- A. Kamal, B. A. Kumar, P. Suresh, N. Shankaraiah and M. S. Kumar, Bioorg. Med. Chem. Lett, 2011, 21, 350–353 CrossRef CAS PubMed
- S. E. Sadat-Ebrahimi, L. Karim, S. Moghimi, A. Yahya-Meymandi, M. Mahdavi, M. Vosooghi, A. Foroumadi and A. Shafiee, J. Sulfur Chem., 2017, 38, 43–51 CrossRef CAS
- C. A. Herdman, T. E. Strecker, R. P. Tanpure, Z. Chen, A. Winters, J. Gerberich, L. Liu, E. Hamel, R. P. Mason and D. J. Chaplin, MedChemComm, 2016, 7, 2418–2427 RSC
- A. Kryshchyshyn, D. Atamanyuk, D. Kaminskyy, P. Grellier and R. Lesyk, Biopolymers and Cell, 2017, 33, 183–205 Search PubMed
- V. M. Golubovskaya, S. Figel, B. T. Ho, C. P. Johnson, M. Yemma, G. Huang, M. Zheng, C. Nyberg, A. Magis and D. A. Ostrov, Carcinogenesis, 2012, 33, 1004–1013 CrossRef CAS PubMed
- R. W. Huigens III, K. C. Morrison, R. W. Hicklin, T. A. Flood Jr, M. F. Richter and P. J. Hergenrother, Nat. Chem., 2013, 5, 195 CrossRef PubMed
- T. Han, M. Goralski, N. Gaskill, E. Capota, J. Kim, T. C. Ting, Y. Xie, N. S. Williams and D. Nijhawan, Science, 2017, 356, eaal3755 CrossRef PubMed
- A. M. Forbes, H. Lin, G. G. Meadows and G. P. Meier, Int. J. Oncol., 2014, 45, 831–842 CrossRef CAS PubMed
- P. Liu, Y. Lu, X. Gao, R. Liu, D. Zhang-Negrerie, Y. Shi, Y. Wang, S. Wang and Q. Gao, Chem. Commun., 2013, 49, 2421–2423 RSC
- A. E. Shchekotikhin, V. A. Glazunova, L. G. Dezhenkova, Y. N. Luzikov, Y. B. Sinkevich, L. V. Kovalenko, V. N. Buyanov, J. Balzarini, F.-C. Huang and J.-J. Lin, Bioorg. Med. Chem., 2009, 17, 1861–1869 CrossRef CAS PubMed
- F. Xu, Z.-Z. Yang and S.-J. Zhang, Phosphorus, Sulfur Silicon Relat. Elem., 2013, 188, 1312–1319 CrossRef CAS
- M. V. Zbancioc, G. N Zbancioc, C. Tanase, A. Miron, C. Ursu and I. I Mangalagiu, Lett. Drug Des. Discovery, 2010, 7, 644–649 CrossRef
- A.-K. Larsson, A. Shokeer and B. Mannervik, Arch. Biochem. Biophys., 2010, 497, 28–34 CrossRef CAS PubMed
- B. Grodner, J. Łukaszkiewicz, B. Kuran and M. Krawiecka, J. Sep. Sci., 2014, 37, 3564–3570 CrossRef CAS PubMed
- M. Chen, W. Li, C. Ma, K. Wu, H. He and K. Wang, Microchim. Acta, 2019, 186, 110 CrossRef PubMed
- Y. Yan, Y. Qing, J. J. Pink and S. L. Gerson, Mol. Cancer Res., 2018, 16, 212–221 CrossRef CAS
- L. Weeks, G. Zentner, P. Scacheri and S. Gerson, Cell Death Dis., 2014, 5, e1045 CrossRef CAS PubMed
- E. Parlanti, G. Locatelli, G. Maga and E. Dogliotti, Nucleic Acids Res., 2007, 35, 1569–1577 CrossRef CAS PubMed
- M.-H. Li, D. Ito, M. Sanada, T. Odani, M. Hatori, M. Iwase and M. Nagumo, Oral Oncol., 2004, 40, 63–70 CrossRef CAS PubMed
- L. Seiple, P. Jaruga, M. Dizdaroglu and J. T. Stivers, Nucleic Acids Res., 2006, 34, 140–151 CrossRef CAS PubMed
-
A. Simeonov and M. I. Davis, in Assay Guidance Manual [Internet], Eli Lilly & Company and the National Center for Advancing Translational Sciences, 2018 Search PubMed
- T.-B. Ren, W. Xu, W. Zhang, X.-X. Zhang, Z.-Y. Wang, Z. Xiang, L. Yuan and X.-B. Zhang, J. Am. Chem. Soc., 2018, 140, 7716–7722 CrossRef CAS PubMed
- B. del Rosal and A. Benayas, Small Methods, 2018, 2, 1800075 CrossRef
- J. Comley, Drug Discov. World, 2003, 4, 91–98 Search PubMed
- L. Wei, W. Yan and D. Ho, Sensors, 2017, 17, 2800 CrossRef PubMed
- Q. Zhang, X. Lu, H. Wang, X. Tian, A. Wang, H. Zhou, J. Wu and Y. Tian, Chem. Commun., 2018, 54, 3771–3774 RSC
- S. Kummar, M. S. Copur, M. Rose, S. Wadler, J. Stephenson, M. O'Rourke, W. Brenckman, R. Tilton, S.-H. Liu and Z. Jiang, Clin. Colorectal Cancer, 2011, 10, 85–96 CrossRef CAS PubMed
- C. M. Sánchez, J. M. P. Martín, J.-S. Jin, A. Dávalos, W. Zhang, G. de la Peña, J. Martínez-Botas, S. Rodríguez-Acebes, Y. Suárez and M. J. Hazen, Biochim. Biophys. Acta, 2015, 1851, 1240–1253 CrossRef PubMed
- Y. Zhu, T. Ye, X. Yu, Q. Lei, F. Yang, Y. Xia, X. Song, L. Liu, H. Deng and T. Gao, Sci. Rep., 2016, 6, 20253 CrossRef CAS PubMed
- F. Yang, M. Hu, Q. Lei, Y. Xia, Y. Zhu, X. Song, Y. Li, H. Jie, C. Liu and Y. Xiong, Cell Death Dis., 2015, 6, e1701 CrossRef CAS
- T.-H. Ye, F.-F. Yang, Y.-X. Zhu, Y.-L. Li, Q. Lei, X.-J. Song, Y. Xia, Y. Xiong, L.-D. Zhang and N.-Y. Wang, Cell Death Dis., 2018, 8, e2534 CrossRef PubMed
-
S. Amrane, M.-a. Andreola, G. Pratviel and J.-L. Mergny, US Pat., Application No. 15/772,355, 2018
- W. Wang, K. Vellaisamy, G. Li, C. Wu, C.-N. Ko, C.-H. Leung and D.-L. Ma, Anal. Chem., 2017, 89, 11679–11684 CrossRef CAS PubMed
- J. M. Stewart, H. K. Subramanian and E. Franco, Nucleic Acids Res., 2017, 45, 5449–5457 CrossRef CAS PubMed
- J. Yan, Y. Xie, Q. Zhang, L. Gan, F. Wang, H. Li, J. Si and H. Zhang, J. Cell. Physiol., 2019, 234, 13014–13020 CrossRef CAS PubMed
- T.-C. Chou, Cancer Res., 2010, 70, 440–446 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05623h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |