
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetabolic inhibitors of bacterial glycan biosynthesis†
Daniel A.
Williams‡
a,
Kabita
Pradhan‡
b,
Ankita
Paul
b,
Ilana R.
Olin
a,
Owen T.
Tuck
a,
Karen D.
Moulton
a,
Suvarn S.
Kulkarni
 *b and
Danielle H.
Dube
*a
*b and
Danielle H.
Dube
*a
aDepartment of Chemistry & Biochemistry, Bowdoin College, 6600 College Station, Brunswick, ME 04011, USA. E-mail: ddube@bowdoin.edu
bDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, India. E-mail: suvarn@chem.iitb.ac.in
First published on 8th January 2020
Abstract
The bacterial cell wall is a quintessential drug target due to its critical role in colonization of the host, pathogen survival, and immune evasion. The dense cell wall glycocalyx contains distinctive monosaccharides that are absent from human cells, and proper assembly of monosaccharides into higher-order glycans is critical for bacterial fitness and pathogenesis. However, the systematic study and inhibition of bacterial glycosylation enzymes remains challenging. Bacteria produce glycans containing rare deoxy amino sugars refractory to traditional glycan analysis, complicating the study of bacterial glycans and the creation of glycosylation inhibitors. To ease the study of bacterial glycan function in the absence of detailed structural or enzyme information, we crafted metabolic inhibitors based on rare bacterial monosaccharide scaffolds. Metabolic inhibitors were assessed for their ability to interfere with glycan biosynthesis and fitness in pathogenic and symbiotic bacterial species. Three metabolic inhibitors led to dramatic structural and functional defects in Helicobacter pylori. Strikingly, these inhibitors acted in a bacteria-selective manner. These metabolic inhibitors will provide a platform for systematic study of bacterial glycosylation enzymes not currently possible with existing tools. Moreover, their selectivity will provide a pathway for the development of novel, narrow-spectrum antibiotics to treat infectious disease. Our inhibition approach is general and will expedite the identification of bacterial glycan biosynthesis inhibitors in a range of systems, expanding the glycochemistry toolkit.
Introduction
The main therapies targeting bacterial glycans have had remarkable success in the clinic. Indeed, our antibiotic arsenal contains small molecule inhibitors of enzymes involved in bacterial glycoconjugate biosynthesis.3 Among the most prominent of these are the blockbusters penicillin,4 vancomycin,5 and bacitracin,6 all of which interfere with peptidoglycan biosynthesis. Although these antibiotics have saved countless lives, the emergence of World Health Organization-listed antibiotic resistant “priority pathogens”, including Helicobacter pylori and Campylobacter jejuni, has prompted the search for alternative treatments.7 A preponderance of biochemical and genetics evidence suggests that glycoprotein biosynthesis enzymes within priority pathogens are compelling potential targets of therapeutic intervention.1,3Bacterial glycoproteins, and the glycosyltransferases that synthesize them, have emerged as intriguing targets because they are produced only by select bacteria, they contain distinctive structures that are markedly different from their eukaryotic counterparts, and they often play important roles in colonization and pathogenesis.1–3 Indeed, an analysis of bacterial glycan structures reveals the presence of rare, exclusively bacterial deoxy amino monosaccharides that are absent from eukaryotes.8 For example, pseudaminic acid,9 bacillosamine,10 2,4-diacetamido-2,4,6-trideoxygalactose (DATDG),11N-acetylfucosamine (FucNAc),12 and legionaminic acid9 are a sampling of the unusual building blocks used by medically significant priority pathogens (Fig. 1A). Pathogenic bacteria that cannot synthesize glycoproteins have attenuated virulence. For example, Pseudomonas aeruginosa and Haemophilus influenzae glycosylation mutants have reduced adhesion to host cells,13,14Staphylococcus aureus and H. influenzae glycosylation mutants are defective at forming biofilms,15 and C. jejuni, H. pylori, and P. aeruginosa mutants with interruptions in flagellin glycosylation are immotile and cannot colonize the host.9,16,17 Small molecules that inhibit glycosylation enzymes and recapitulate the effects of genetics experiments have the potential to expand our antibiotic arsenal with agents that are minimally disruptive to beneficial microflora. Pathogen-specific glycan interference could circumvent problems caused by broad-spectrum agents, which alter the gut microbiome and precipitate deleterious health consequences.18–20
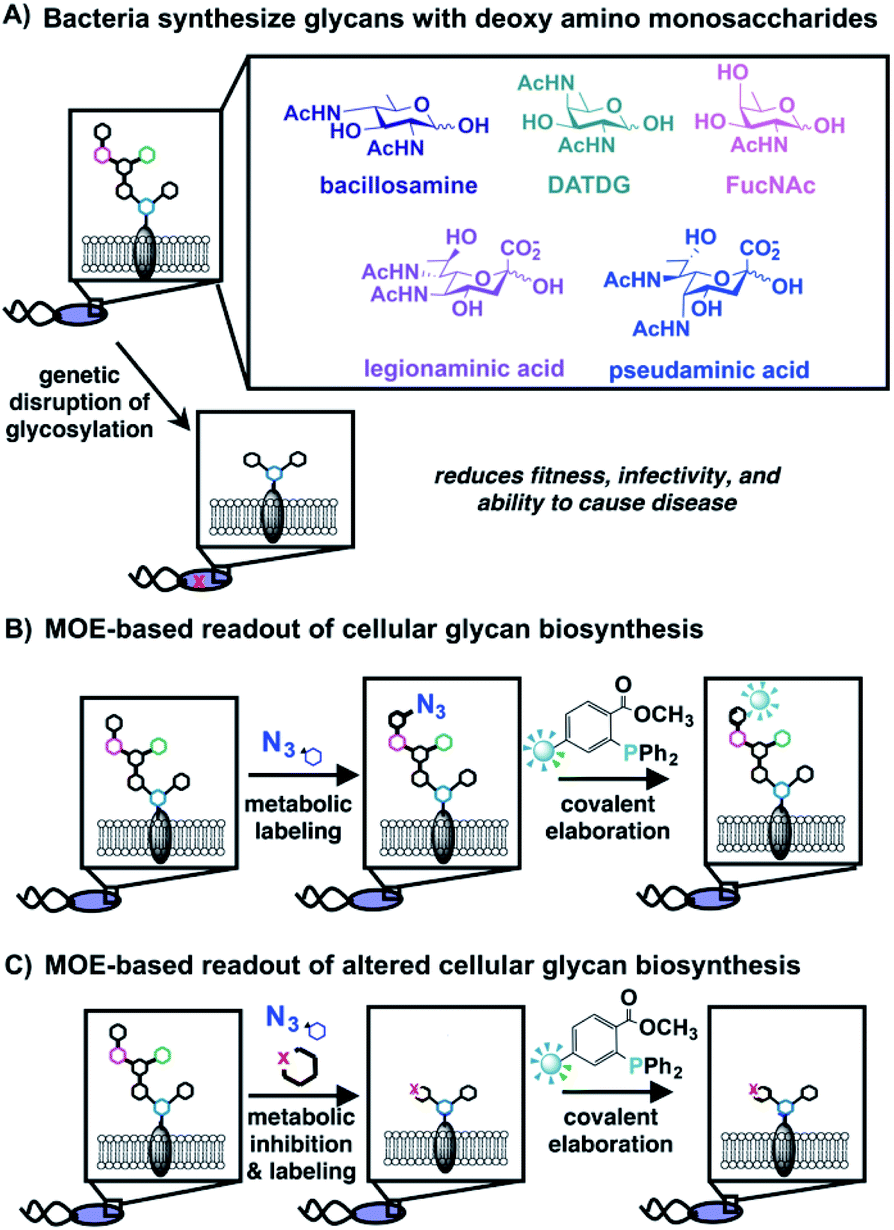 | ||
| Fig. 1 Bacteria synthesize complex glycans bearing distinctive monosaccharides that can be detected via metabolic oligosaccharide engineering (MOE); disrupted glycan biosynthesis yields a diminished MOE-based signal. (A) Bacteria coat their cells with glycans comprised of rare, deoxy amino monosaccharides that are crucial for fitness and absent from human cells. (B) Schematic overview of MOE, which facilitates detection and visualization of cellular glycan biosynthesis. Cells are first metabolically labeled with an unnatural azide-containing sugar, then azide-labeled glycans are covalently elaborated via Staudinger ligation with a phosphine-based probe to yield detectable glycans on cells or in cell lysates. (C) MOE was used as a cell-based readout of interrupted glycan biosynthesis. The effect of metabolic inhibitors on glycan biosynthesis was detected by diminished metabolic incorporation of unnatural, azide-containing sugars into cellular structures. | ||
Numerous strategies including transition state analogue design, structure-based design, fragment-based and library screening approaches have successfully yielded inhibitors of eukaryotic glycan biosynthesis enzymes.21 Inspired by these precedents, Walker, Kahne, and coworkers used structure-based design to produce a novel inhibitor of peptidoglycan glycosyltransferase that displayed potent antibacterial activity,22 Imperiali and coworkers employed fragment-based and library screening approaches to develop nM inhibitors of UDP-bacillosamine biosynthesis,23 while Logan and coworkers invoked a library-based screen of pseudaminic acid biosynthesis enzymes to discover two potent inhibitors of flagellin glycosylation.24 While these strategies are directly transferable to enzymes involved in bacterial glycan synthesis, they typically require in-depth knowledge of the targets, including the structures of substrates and products as well as robust in vitro assays to monitor enzyme-catalyzed product formation. A lack of precise glycomics information has impeded the development of suitable inhibitors of glycoprotein biosynthesis in many priority pathogens.
A method that sheds light on bacterial glycoprotein biosynthesis in intact cells could circumvent the need for full pathway and glycan characterization to screen inhibitors. Metabolic oligosaccharide engineering (MOE), which was pioneered by Bertozzi, Reutter, and coworkers, is well suited to this task as it tracks metabolic incorporation of unnatural monosaccharides into cellular glycans via endogenous carbohydrate biosynthetic pathways.25–28 In essence, azide-containing sugars are taken up by cells, then metabolically incorporated into cellular glycans. Azide-containing glycans are subsequently tagged with bioorthogonal reaction partners (e.g. alkynes, phosphines) to enable detection, identification, and visualization in live cells and animals (Fig. 1B).25,26,29 This approach has been expanded to bacterial systems3,30,31 and led to, for example, the discovery of a general protein glycosylation system in Helicobacter pylori using the azidosugar peracetylated N-azidoacetylglucosamine (Ac4GlcNAz) as a metabolic substrate.32,33
In addition to yielding a readout of cellular glycan biosynthesis, MOE provides information about which monosaccharides are metabolically processed by particular bacterial species. Azide-containing analogues of the rare bacterial sugars bacillosamine, 2,4-diacetamido-2,4,6-trideoxygalactose (DATDG), N-acetylfucosamine (FucNAc),34 altrose,35 trehalose,36,37 pseudaminic acid,38N-acetylmuramic acid (MurNAc),39,40 and 3-deoxy-D-manno-octulosonic acid (Kdo)41 have been developed and deployed in metabolic labeling experiments. Experiments have revealed differential incorporation of rare bacterial monosaccharides into glycans on bacterial cells, presumably due to species-specific differences in carbohydrate biosynthetic pathways. These observations provide the basis for the development of inhibitors based on rare bacterial monosaccharide scaffolds.
Here, we report a strategy for the production of bacterial glycan biosynthesis inhibitors in the absence of detailed pathway or glycan information. We designed and synthesized two classes of metabolic inhibitors based on rare bacterial monosaccharide scaffolds, then used a cellular MOE-based assay to assess the ability of these substrate-based analogue inhibitors to terminate or divert glycan biosynthesis (Fig. 1C). The “hits” were then screened for their ability to induce fitness defects in a variety of bacterial pathogens. Importantly, features involved in the ability of bacteria to initiate and maintain infections within a host were scored. These assays yielded metabolic inhibitors that interfere with glycan biosynthesis in the gastric pathogen H. pylori and precipitate functional defects in growth, motility, and biofilm formation. Intriguingly, these metabolic inhibitors have differential effects on glycan biosynthesis in other bacteria explored in these studies. The selectivity of these inhibitors has the potential to set the stage for the development of novel, narrow-spectrum antibiotics to treat infectious disease. This approach is a general one that promises to enable systematic study of bacterial glycosylation enzymes and expedite the identification of bacterial glycan biosynthesis inhibitors in a range of systems, further expanding the glycochemistry toolkit.
Results and discussion
Design of substrate-based metabolic inhibitors
Motivated by the successful development of small molecule inhibitors of glycosyltransferases in mammalian systems,42–45 two classes of metabolic inhibitors were explored. The first class of inhibitors was inspired by Esko's seminal work with metabolic substrate decoys such as benzyl-α-GalNAc (Bn-GalNAc).45–48 Metabolic substrate decoys are small molecule analogues of endogenous glycoproteins that are recognized as acceptor substrates by glycosyltransferases, ultimately diverting glycan synthesis onto decoy substrates and leading to the synthesis of truncated glycans on proteins. Successful decoys in mammalian systems led us to design benzyl glycosides of Bac, DATDG, and FucNAc as substrate decoys of bacterial glycosyltransferases (Fig. 2A; compounds 1, 2 and 3). We focused on derivatives of the rare bacterial monosaccharides Bac, DATDG, and FucNAc due to expedient syntheses of these scaffolds and their utilization in priority pathogens.34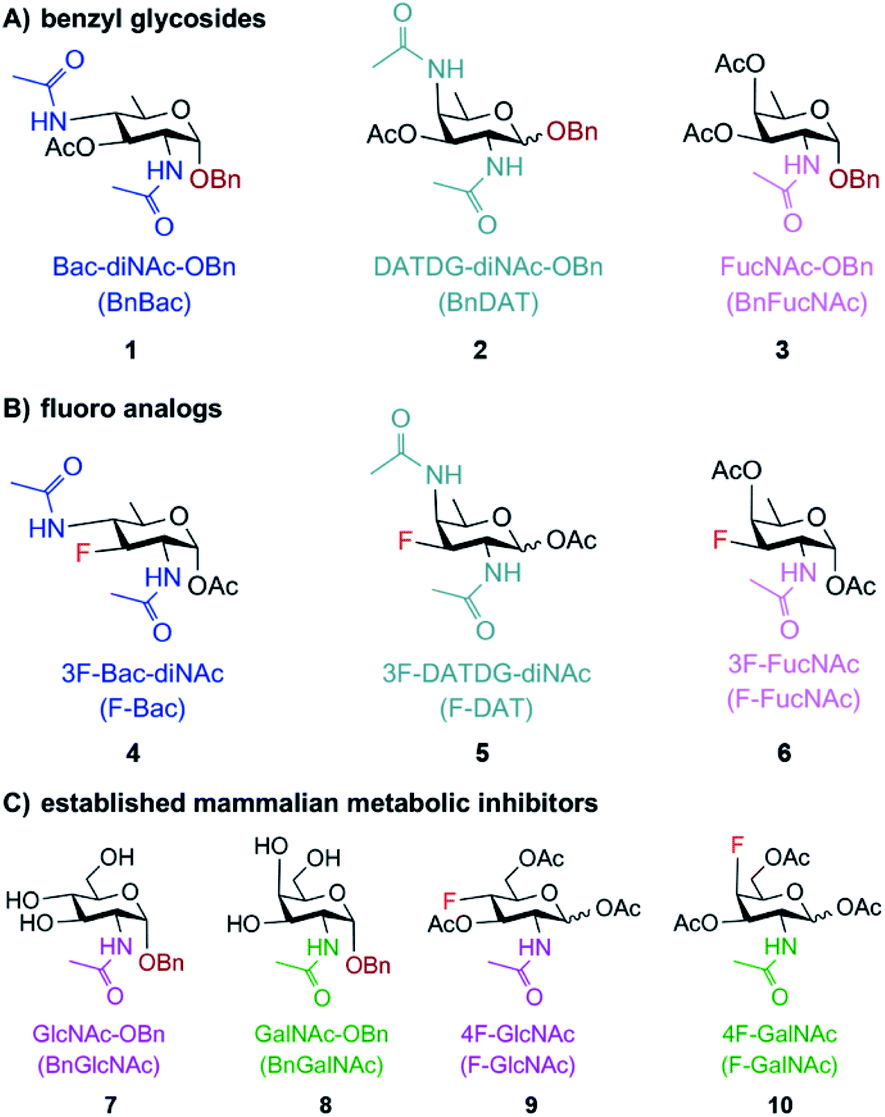 | ||
| Fig. 2 Potential metabolic inhibitors based on rare bacterial monosaccharide scaffolds (A and B) or common monosaccharide scaffolds (C) used in this study. Substrate-based metabolic inhibitors were designed based on the rare bacterial monosaccharide scaffolds bacillosamine, DATDG, and FucNAc. (A) Benzyl glycosides 1, 2 and 3 were created to compete with and divert glycan biosynthesis onto decoy substrates. (B) Fluoro analogues 4, 5 and 6 were designed to interrupt glycan biosynthesis. (C) Established mammalian glycosylation inhibitors 7–10, based on the common monosaccharide scaffolds GlcNAc and GalNAc, were also explored as potential inhibitors of bacterial glycosylation. | ||
The second class of inhibitors was inspired by classic and modern examples of chain-terminating substrates, including Sanger's pioneering use of di-deoxy chain terminators of DNA synthesis and more recent extensions of this concept with deoxy- and fluoro-sugars as terminators of glycan biosynthesis.43,44,49–51 Based on these successful precedents, we designed 3-fluoro analogues of Bac, DATDG, and FucNAc to act as global metabolic inhibitors of bacterial glycoprotein biosynthesis (Fig. 2B; compounds 4, 5, and 6). Fluoro-sugars are expected to be converted into the corresponding nucleotide-activated monosaccharide within cells, then impede glycan elaboration.
According to the original design, the presence of these fluoro sugars in glycans should terminate subsequent elaboration by glycosyltransferases as they lack sites (C3-OHs) for additional extension. Recent work in mammalian systems with fluoro analogues of sialic acid, fucose, GlcNAc and GalNAc has demonstrated that these compounds indeed act as global metabolic inhibitors, though by a different mechanism than originally intended.44,52,53 In mammalian systems, fluoro sugars are activated as nucleotide sugars in situ but not effectively utilized by glycosyltransferases, leading to a buildup of the activated unnatural nucleotide sugars in cells and metabolic feedback inhibition that prevents de novo synthesis of natural activated substrates.44,53 Regardless of the precise molecular mechanism, fluoro sugars based on rare bacterial monosaccharide scaffolds are likely to serve as global metabolic inhibitors of bacterial glycan biosynthesis.
Following these rational design criteria, benzyl glycoside and 3-F analogues of Bac, DATDG, and FucNAc were our target metabolic inhibitors of bacterial glycosylation (Fig. 2). As a key design element of putative inhibitors, the hydrophilic hydroxyl groups were temporarily masked with hydrophobic acetyl groups47 to maximize efficiency of uptake by bacteria.32 We previously validated this transient protection strategy in H. pylori by demonstrating via mass spectrometry that H. pylori treated with Ac4GlcNAz metabolically incorporated the free sugar GlcNAz into glycoproteins,33 in line with reports that this bacteria has endogenous esterase activity.54,55 In addition to these novel compounds, we included commercially available substrate decoys and chain terminators based on the common monosaccharide scaffolds N-acetylglucosamine (GlcNAc) and N-acetylgalactosamine (GalNAc) (Fig. 2C; compounds 7–10) in our studies due to their ability to elicit glycosylation defects in mammalian systems and the ubiquity of these monosaccharides in bacterial glycans.
Synthesis of bacterial monosaccharide analogue inhibitors
We synthesized a panel of substrate decoy OBn analogues 1–3 by adaptation of our published protocols for synthesis of Bac, DATDG, and FucNAc, but with slight improvements (using inexpensive KNO2 in place of expensive TBANO2).56–58 The synthesis of benzyl glycosides involved one-pot nucleophilic displacements of 2,4-bis-trifluoromethanesulfonates (triflates, OTf) to access the corresponding thioglycoside donors, followed by glycosylation with benzyl alcohol under standard conditions. Synthesis of chain terminator 3-fluoro analogues 4–6 demanded a novel approach. Fluorination of a sugar hydroxyl involves inversion at the chosen carbon. Therefore one has to gain access to its epimeric precursor, in our case, the C3 epimer 6-deoxy-D-allosamine thioglycoside derivative. Accordingly, the synthesis of 3-F analogues involved regioselective and orthogonal protection of D-rhamnosyl triol and sequential nucleophilic displacements of the C2-OTf and C3-OTf by azide and nitrite anions as nucleophiles, respectively, to access the key D-allo-3-OH intermediate. Subsequently, inversion at the C3 position with the fluorinating agent diaminoethyl sulfur trifluoride (DAST)52,59–61 afforded the C3–F quinovose as a common synthon. Finally, inversions at C4 by various nucleophiles, followed by functional group transformations, furnished compounds 4–6.To synthesize benzyl glycoside analogues 1–3, the common precursor D-rhamnosyl-2,4-diol 11 was readily prepared starting from commercially available D-mannose in six steps.57 Synthesis of Bac-diNAc-OBn (Bn-Bac) 1 is shown in Scheme 1. Triflation of diol 11 with Tf2O and pyridine in dry CH2Cl2 gave the corresponding 2,4-bis-triflate intermediate, which upon a brief work-up was subjected to a one pot, double nucleophilic displacement reaction. The crude 2,4-bis-triflate derivative of compound 11 was transformed to the corresponding C2 azide displaced product by treatment with 0.85 equiv. of TBAN3 in CH3CN at −30 °C for 18 h. Concomitant treatment with 10 equiv. of KNO2 in DMF at room temperature displaced the remaining C4-OTf to afford compound 12 in 70% yield over three steps after a single column chromatographic purification.
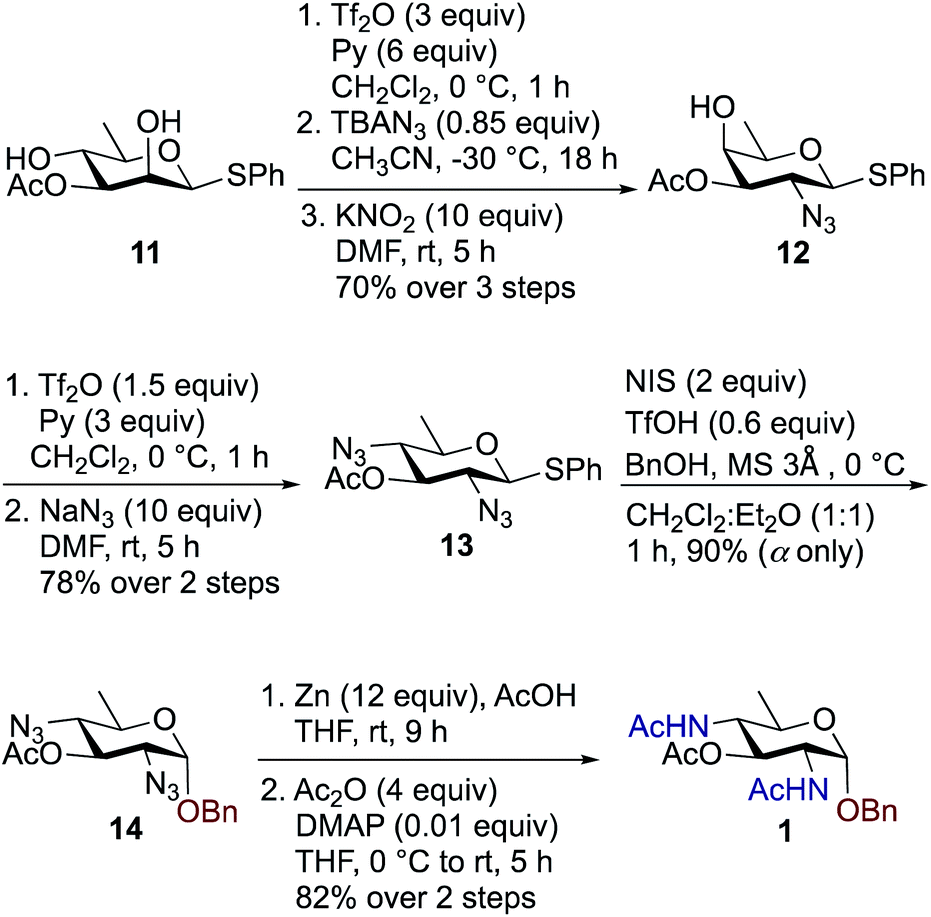 | ||
| Scheme 1 Synthesis of Bac-diNAc-OBn (Bn-Bac) 1. | ||
The free C4-hydroxyl group in compound 12 was treated with Tf2O and pyridine in dry CH2Cl2 at 0 °C followed by inversion using 10 equiv. of NaN3 in DMF at room temperature to afford 2,4-diazido-2,4,6-trideoxy-D-glucose (Bac) derivative 13 in 78% over two steps. Donor 13 was further glycosylated with 1 equiv. of benzyl alcohol (BnOH) by using NIS, TfOH as promoter at 0 °C in CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Et2O (1:1 ratio) to deliver exclusively α-linked OBn glycoside 14 in 90% yield. Azide groups of OBn glycoside 14 were reduced with Zn, AcOH in THF, which was followed by acetamide formation with Ac2O and catalytic amount of DMAP in THF to furnish benzyl-2,4-diacetamido-2,4,6-trideoxy-D-glucose 1 (Bac-diNAc-OBn) in 82% yield over two steps.
:Et2O (1:1 ratio) to deliver exclusively α-linked OBn glycoside 14 in 90% yield. Azide groups of OBn glycoside 14 were reduced with Zn, AcOH in THF, which was followed by acetamide formation with Ac2O and catalytic amount of DMAP in THF to furnish benzyl-2,4-diacetamido-2,4,6-trideoxy-D-glucose 1 (Bac-diNAc-OBn) in 82% yield over two steps.
Synthesis of the rare sugar analogue DATDG-diNAc-OBn 2 is outlined in Scheme 2. The known 2,4-diol substrate 11 was reacted with Tf2O and pyridine in dry CH2Cl2 at 0 °C to provide the corresponding 2,4-bis-triflate compound, which upon double displacement with 10 equiv. of NaN3 in DMF at room temperature afforded di-azido derivative 1557 in 88% yield over two steps. The thioglycoside donor 15 was then glycosylated with 1 equiv. of BnOH by using NIS, TfOH as promoter at 0 °C in CH2Cl2:Et2O (1:1 ratio) to obtain benzyl glycoside 16 in 93% yield as a 1:1 mixture of α:β isomers. Subsequent reduction of azide groups in 16 with Zn, AcOH in THF, followed by treatment with Ac2O and catalytic DMAP in THF, furnished benzyl-2,4-diacetamido-2,4,6-trideoxy-D-galactose 2 (DATDG-diNAc-OBn) in 86% yield over two steps.
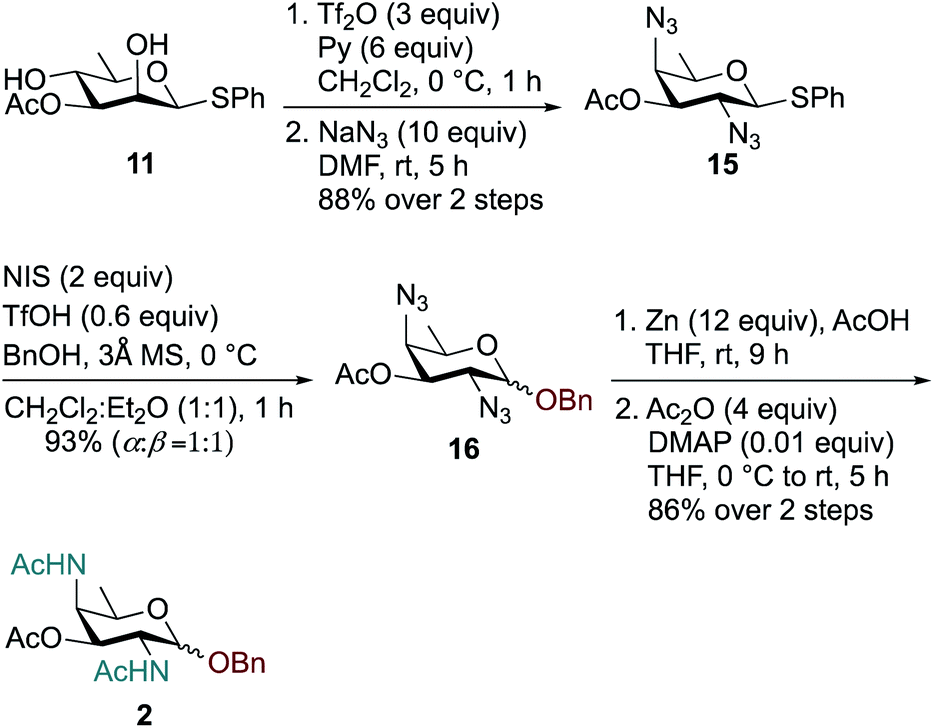 | ||
| Scheme 2 Synthesis of DATDG-diNAc-OBn (Bn-DAT) 2. | ||
To synthesize FucNAc-OBn (Bn-FucNAc) 3, we started from compound 12 (Scheme 3). The free hydroxyl group in compound 12 was acetylated with AcCl and pyridine in CH2Cl2 to give 3,4-diacetylated compound 17 in 92% yield. Donor 17 was then activated with NIS, TfOH in CH2Cl2:Et2O (1:1 ratio) and coupled with BnOH at 0 °C to provide exclusively α-glycosylated compound 18 in 87% yield. Subsequently, the azide was converted to acetamide using Zn, AcOH in THF followed by treatment with Ac2O, and catalytic amount of DMAP in THF to obtain benzyl-2-acetamido-2,6-dideoxy-D-galactose (FucNAc-OBn) 3 over two steps in 82% yield.
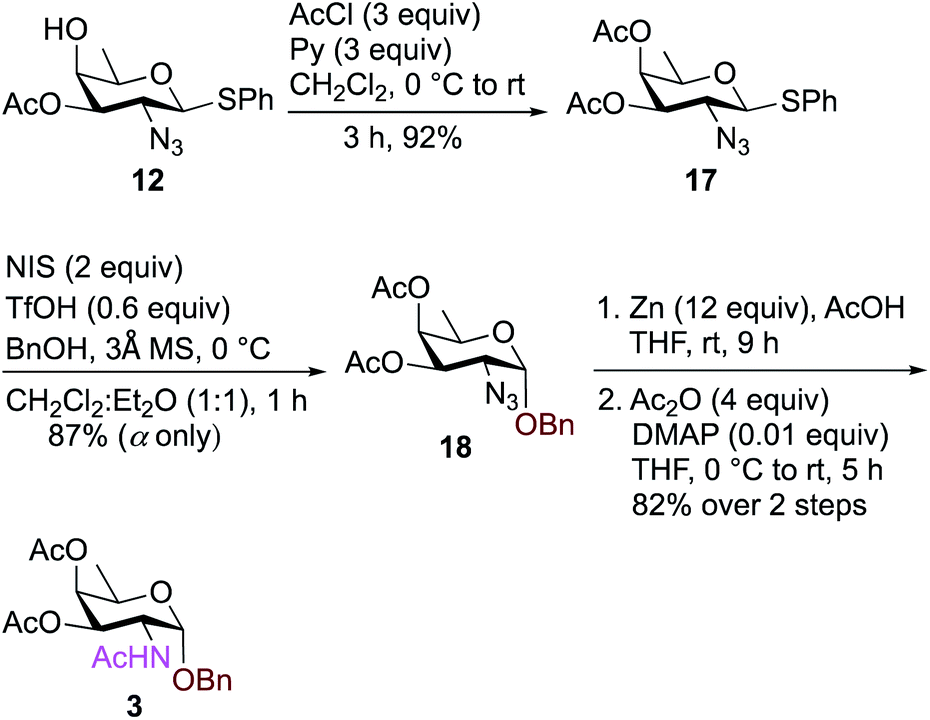 | ||
| Scheme 3 Synthesis of FucNAc-OBn (Bn-FucNAc) 3. | ||
The synthesis of fluoro analogues 4–6 involved two stages – (1) the synthesis of common precursor 3-fluoro-quinovosamine derivative 26, and (2) its C4 functionalization with inversion using azide or nitrite anions, followed by functional group inter conversions. The known D-rhamnose triol 1957 was deemed to be the appropriate starting material for the synthesis of the common 3-fluoro building block 26 (Scheme 4). First, the C2 and C3 hydroxyl groups in 19 were simultaneously capped by acetonide protecting group. Next, the remaining C4-hydroxyl group was naphthylmethylated with NapBr and NaH to obtain compound 20 in 82% yield over two steps. Acetonide deprotection upon acid hydrolysis followed by a highly regioselective acetylation at O3 using 5 mol% Me2SnCl2 afforded compound 21 in very good yield over two steps. The C2–OH was converted to the corresponding O-triflate, which was subsequently displaced by azide to give compound 22 in 89% yield. Deacetylation of 22 followed by its O-triflation and concomitant treatment with 15 equiv. of KNO2 transformed 2-azido-2,6-dideoxy glucose to 2-azido-2,6-dideoxy-D-allose 24 in 53% yield over two steps. Fluoride was installed at the C3 position using 3 equiv. of DAST at −20 °C to room temperature to afford 2-azido-3-fluoro-2,3,6-trideoxy-D-glucose 25 in 71% yield. Finally, removal of Nap group using 1.8 equiv. of DDQ in CH2Cl2 and H2O fashioned the common D-quinovosamine 3-F building block 26 in 93% yield.
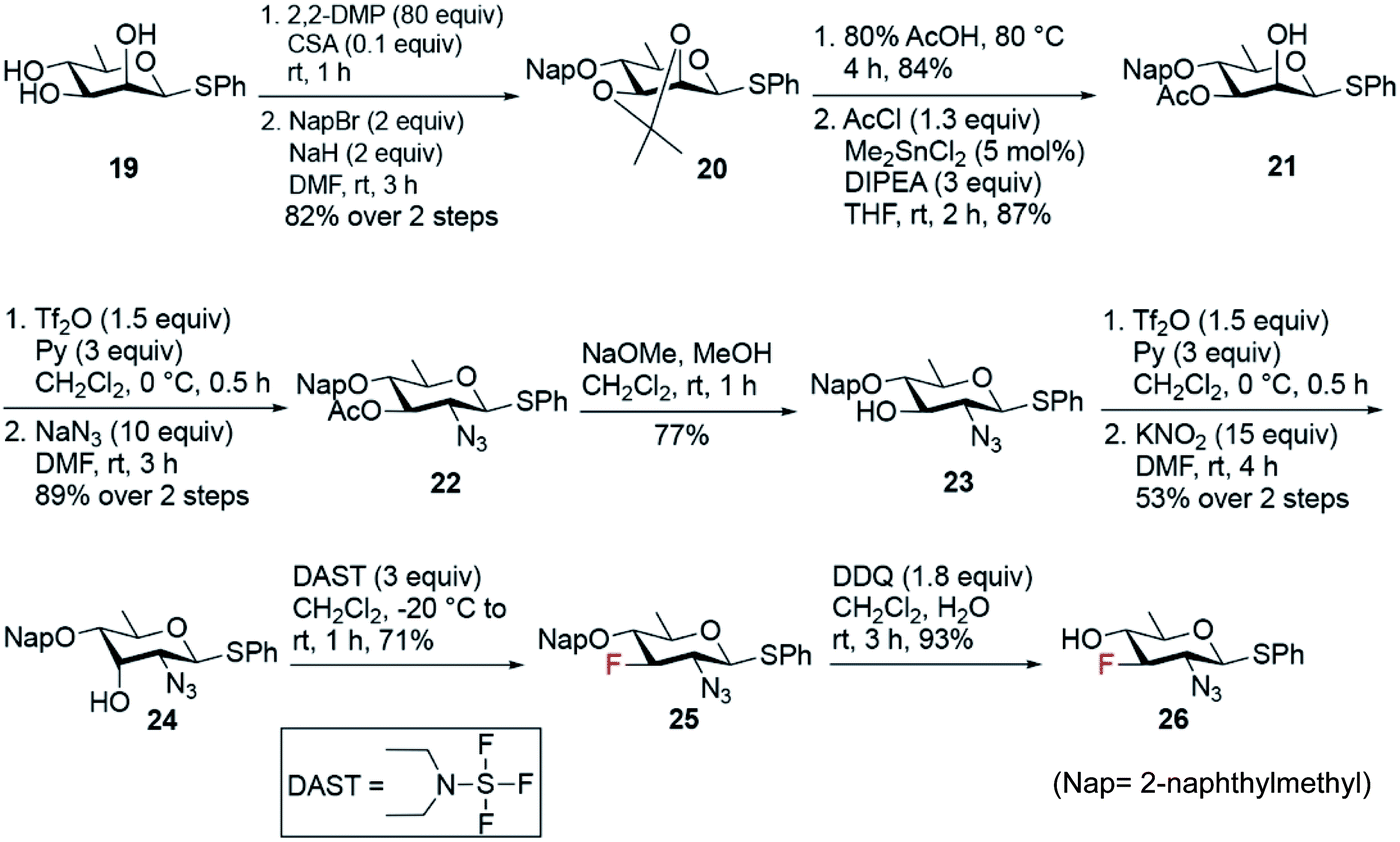 | ||
| Scheme 4 Synthesis of common 3-fluoro-D-quinovosamine 26. | ||
To access the fluoride containing rare sugar analogues 4–6, the C4 center of compound 26 was triflated and inverted using different nucleophiles (NaN3 and KNO2). The synthesis of 3F-Bac-diNAc (F-Bac) 4 is outlined in Scheme 5. The free C4–OH of compound 26 was subjected to O-triflation using triflic anhydride and pyridine, followed by inversion of O-triflate with 15 equiv. of KNO2 to afford compound 27 in 60% yield over two steps. Then, triflation of 27 and subsequent nucleophilic displacement by 10 equiv. of NaN3 furnished 3-fluoro bacillosamine derivative 28 with 80% yield. The C2 and C4 azide groups in compound 28 were reduced by Zn and AcOH to give the corresponding diamine compound, which was then treated with Ac2O and catalytic amount of DMAP in THF to afford 3-fluoro compound 29 in 75% yield over two steps. Thioglycoside 29 was hydrolyzed to hemiacetal upon treatment with NBS, THF and H2O followed by acetylation using Ac2O, Et3N and catalytic amount of DMAP in CH2Cl2 to obtain 3F-Bac-diNAc 4 in 75% yield (α only) over two steps.
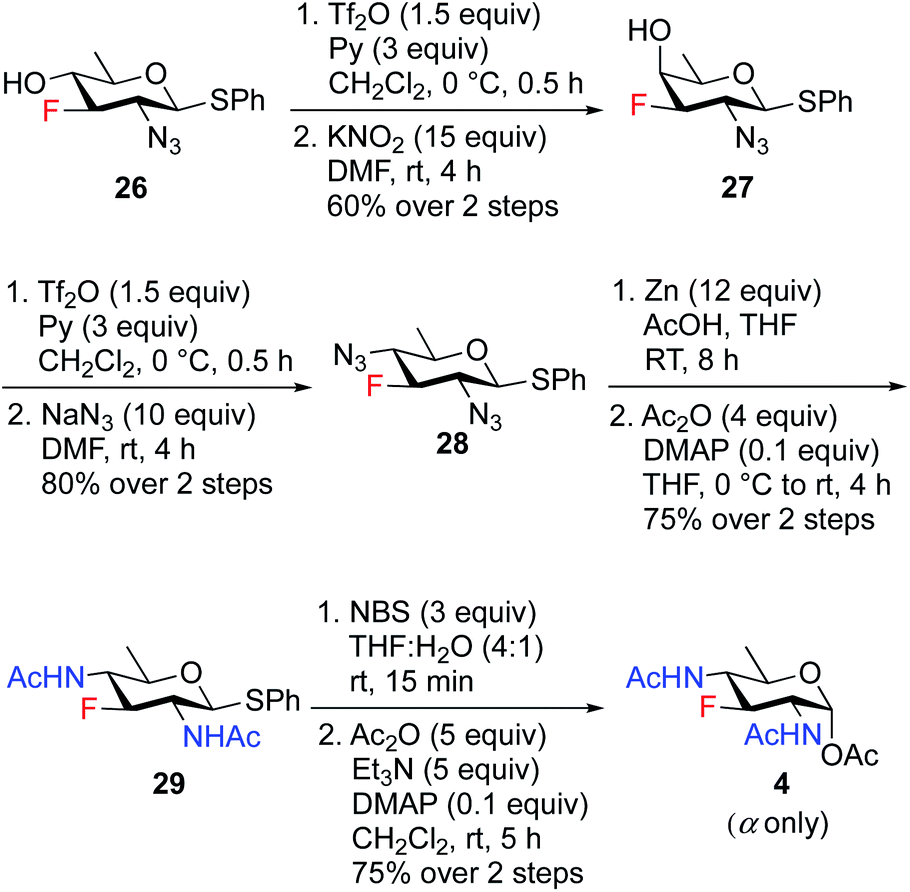 | ||
| Scheme 5 Synthesis of 3F-Bac-diNAc (F-Bac) 4. | ||
Scheme 6 outlines our straightforward synthesis of F-DAT compound 5. Starting from compound 26, the C4 hydroxyl group was subjected to O-triflation and concomitant nucleophilic displacement using NaN3, DMF to afford diazido derivative 30 in 70% yield over two steps. The C2 and C4 azide groups were then reduced to the corresponding diamine, which was then acetylated to obtain 2,4 di-NHAc derivative 31 in 72% yield. Finally, thioglycoside 31 was activated and hydrolyzed to hemiacetal followed by treatment with Ac2O, Et3N and catalytic amount of DMAP in CH2Cl2 to furnish 3F-DATDG-diNAc (F-DAT) analogue 5 in 70% yield (α:β = 5:1) over two steps.
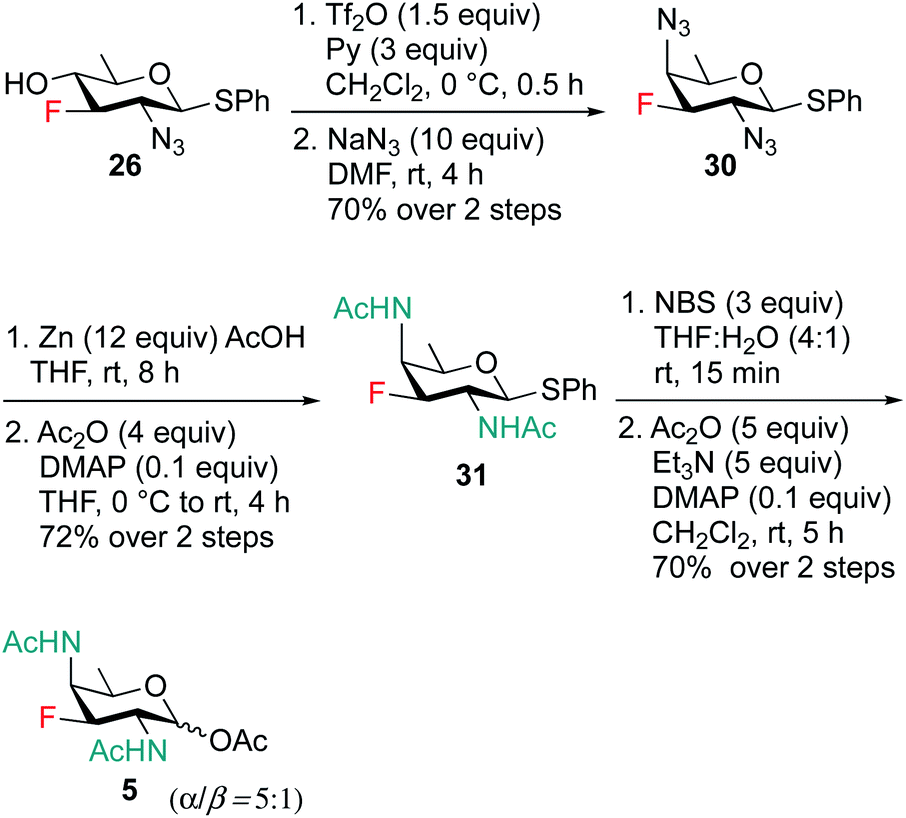 | ||
| Scheme 6 Synthesis of 3F-DATDG-diNAc (F-DAT) 5. | ||
To synthesize 3F-FucNAc 6 (Scheme 7), the C4 hydroxyl group of compound 27 was protected with AcCl and pyridine to render compound 32 in 73% yield. On treatment with Zn and AcOH, the azide group in 32 was converted to amine, then acetylated to give compound 33 in 73% yield. The anomeric thioglycoside was converted to corresponding hemiacetal which was acetylated using Ac2O, Et3N and catalytic amount of DMAP in CH2Cl2 to deliver 3F-FucNAc (F-FucNAc) 6 in 75% yield (α only) over two steps.
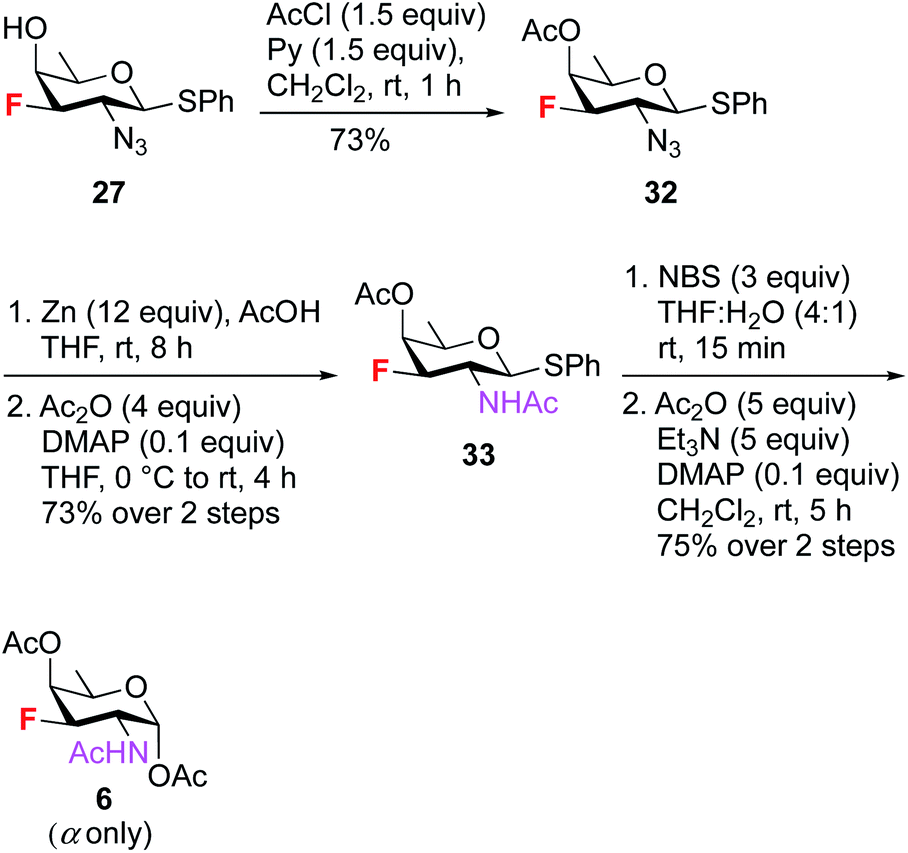 | ||
| Scheme 7 Synthesis of 3F-FucNAc (F-FucNAc) 6. | ||
Glycoprotein biosynthesis is impeded in H. pylori
To ascertain whether the newly developed 3-F and benzyl glycoside analogues of rare bacterial monosaccharides (1–6, Fig. 2) and the established mammalian metabolic inhibitors (7–10) act as metabolic inhibitors of bacterial glycosylation, we took advantage of the established utility of Ac4GlcNAz,62 an azide-containing analogue of the common monosaccharide GlcNAc, to detect the 125 glycoproteins synthesized by H. pylori's general protein glycosylation system32,33 (Fig. 1B). Though the identify of these glycosylated proteins has been revealed by mass spectrometry analyses,33 the structure of the higher-order glycan on these glycoproteins is not yet characterized. Thus, this system is a test bed for structural perturbation in the absence of full information. In essence, we assessed the ability of each of these analogues to inhibit azide-labeled glycoprotein biosynthesis (Fig. 1C) in H. pylori. Following our published methods,29 metabolic labeling experiments were performed with Ac4GlcNAz as a positive control, with the azide-free sugar peracetylated N-acetylglucosamine (Ac4GlcNAc) as a negative control, or with Ac4GlcNAz in the presence of increasing concentrations (0.5–2 mM) of putative inhibitors 1–10 as experimental samples. After four days of metabolic labeling, proteins were harvested from lysed cells and reacted via Staudinger ligation with a phosphine probe comprising a FLAG peptide63 (Phos-FLAG). As expected, analysis of positive control samples from H. pylori treated with Ac4GlcNAz yielded the full complement of azide-labeled glycoproteins detectable by western blot analysis with anti-FLAG antibody, whereas control samples from cells treated with the azide-free compound Ac4GlcNAc yielded negligible azide-dependent signal (Fig. 3). These samples benchmark the most and least signal, respectively, that we would expect to observe in any given sample. We performed an additional control in which H. pylori cells were treated with Ac4GlcNAz in the presence of increasing concentrations (0.5–2 mM) of the control sugar Ac4GlcNAc, which lacks fluoro and OBn modifications and therefore was not expected to act as a glycosylation inhibitor. This control was included due to the possibility that 2 mM of any non-azide containing sugar might be a sufficiently high concentration to outcompete the enzymes (e.g. deacetylases, carbohydrate biosynthesis enzymes, glycosyltransferases) that process the azidosugar and install it into glycoproteins. Samples treated with up to 2 mM of Ac4GlcNAc had no appreciable effect on H. pylori's ability to synthesize azide-labeled glycoproteins, as indicated by the robust azide-dependent glycoprotein profile present in these samples (Fig. 3A). This result allayed concerns that a diminishment in azide-dependent signal results from competition alone.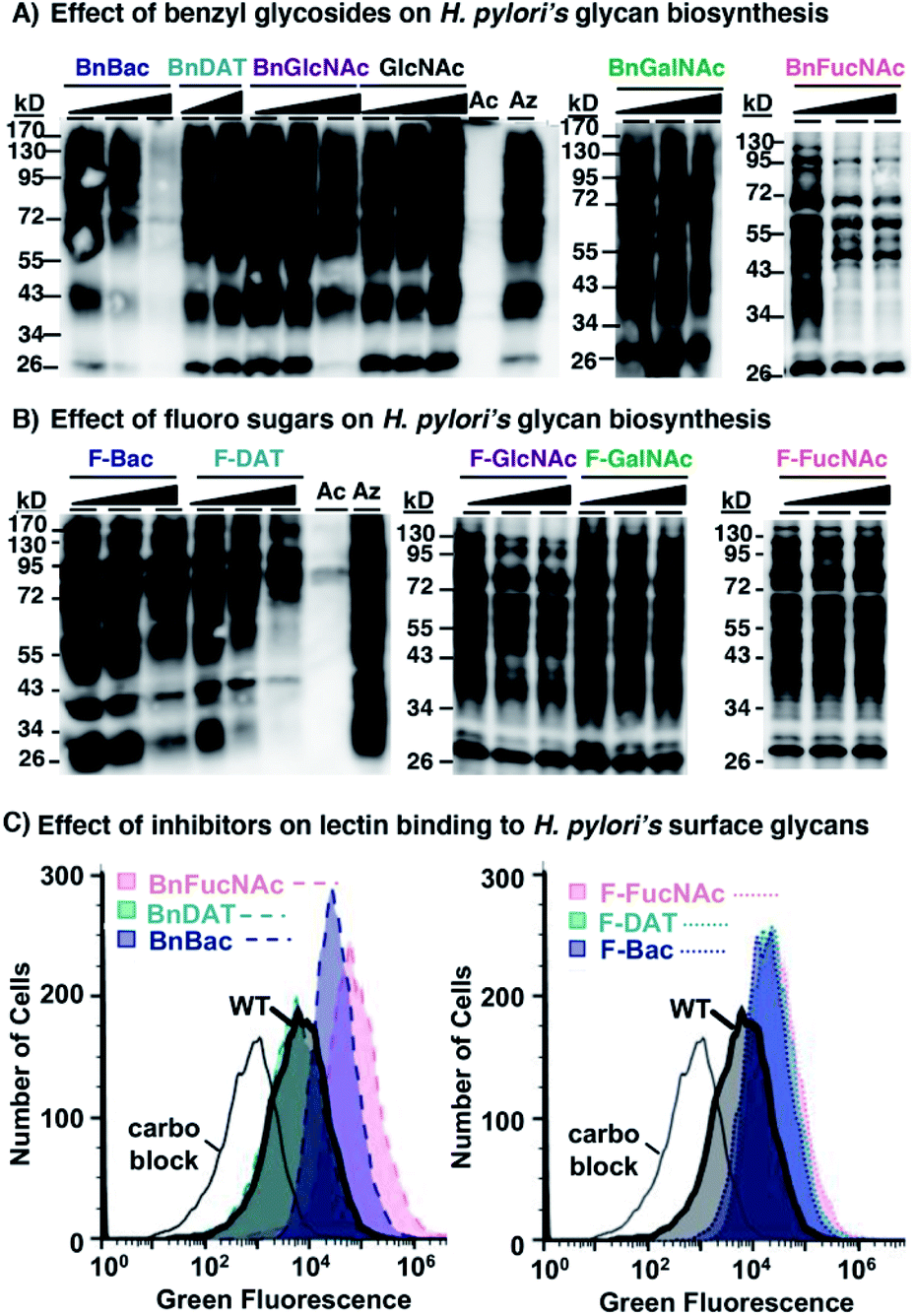 | ||
| Fig. 3 Western blot and flow cytometry analyses indicate select monosaccharide analogues act as metabolic inhibitors of H. pylori's glycan biosynthesis. (A) Benzyl glycosides (1, 2, 3, 7, 8) and (B) fluoro analogues (4, 5, 6, 9, 10) were screened for their ability to interfere with biosynthesis of the full profile of azide-labeled glycoproteins in H. pylori treated with Ac4GlcNAz. H. pylori were grown for four days in media supplemented with 0.5 mM Ac4GlcNAz (Az), Ac4GlcNAc (Ac), or with 0.5 mM Ac4GlcNAz (Az) alongside increasing concentrations (0.5, 1.0, or 2.0 mM; indicated by the increasing shaded triangle, with the shortest point representing 0.5 mM and the tallest part representing 2 mM except for the BnDAT (2) samples, which are 0.5 and 1.0 mM) of compounds 1–10 or Ac4GlcNAc (GlcNAc), then harvested by lysis. The presence of azides in cellular glycoproteins was detected by reacting lysates with 250 μM Phos-FLAG for 12 h at 37 °C and then analyzing samples via western blot with anti-FLAG antibody. (C) Benzyl glycosides (1, 2, 3) and fluoro analogues (4, 5, 6) were screened for their ability to alter lectin binding to H. pylori. H. pylori were grown for four days in media supplemented with 2.0 mM of compounds 1–6 or left as untreated wildtype (WT), then probed with ConA conjugated to Alexa Fluor 488 and analysed by flow cytometry. Alternatively, ConA was pre-treated with 400 mM mannose (carbo block) prior to probing untreated H. pylori. The data shown are representative of replicates (n ≥ 2). | ||
With the MOE-based assay validated, we turned to our experimental samples. Of the previously reported common monosaccharide analogues, BnGalNAc (8) and F-GalNAc (10) had no appreciable effect on the azide-labeled glycoprotein fingerprint detected, whereas BnGlcNAc (7) and F-GlcNAc (9) appeared to elicit only a very minor effect on detected signal intensity (Fig. 3). Therefore, previously reported metabolic inhibitors do not appear to dramatically impact protein glycosylation in H. pylori. Similarly, three rare bacterial monosaccharide analogues, BnDAT (2), F-Bac (4), and F-FucNAc (6), had little to no apparent impact on H. pylori's ability to synthesize the full profile of azide-labeled glycoproteins (Fig. 3). By contrast, the rare bacterial monosaccharide analogues BnBac (1), BnFucNAc (3), and F-DAT (5) elicited a concentration-dependent diminishment in the azide-dependent glycoprotein profile (Fig. 3). Coomassie staining of electrophoresed samples confirmed that all samples contain proteins (Fig. S1†), indicating that the lack of detectable azides in some samples was not due to low protein levels. Taken together, these results suggest that three of the novel compounds are metabolic inhibitors that impede H. pylori's ability to synthesize glycoproteins.
We next sought to confirm via an orthogonal method that H. pylori's glycosylation is disrupted upon treatment with select metabolic inhibitors. Toward this end, we assessed the binding of the carbohydrate-binding lectin concanavalin A (ConA) to untreated wildtype H. pylori versus H. pylori treated with 2 mM of compounds 1–6. Flow cytometry analysis of intact cells revealed that ConA bound to wildtype H. pylori (Fig. 3C). Pre-incubation of ConA with its known ligand mannose mitigated binding to wildtype H. pylori, consistent with the carbohydrate-dependent nature of this binding interaction (Fig. 3C). H. pylori treated with BnDAT (2) exhibited ConA binding comparable to untreated wildtype H. pylori (Fig. 3), suggesting no appreciable alteration in cell wall glycans. By contrast, treatment of H. pylori with the rare bacterial monosaccharide analogues BnBac (1), BnFucNAc (3), F-Bac (4), F-DAT (5), and F-FucNAc (6) elicited an increase in ConA binding relative to untreated H. pylori (Fig. 3C and S1†). Enhanced ConA binding is consistent with metabolic glycan inhibitors altering cell wall glycan architecture, perhaps by unmasking underlying glycan epitopes.
The combination of western blot and flow cytometry data suggest that BnBac (1), BnFucNAc (3), and F-DAT (5) alter cell wall glycan architecture by inhibiting glycoprotein biosynthesis. By contrast, F-Bac (4) and F-FucNAc (6) did not appreciably alter glycoprotein biosynthesis but did alter global cell wall glycan architecture; these data suggest that these analogs may impact biosynthesis of a different glycoconjugate in H. pylori. Collectively, these data support the hypothesis that metabolic inhibitors alter H. pylori's cell wall glycan architecture.
Metabolic inhibitors elicit functional defects in H. pylori
With compelling evidence that this panel of metabolic inhibitors interferes with glycan biosynthesis in H. pylori, we next explored whether glycosylation inhibitors impact fitness. We queried effects on critical functions that are absolutely required for H. pylori to colonize the host and sustain an infection. In particular, we scored the effect of analogues on growth by monitoring OD600, motility by quantifying halo formation on soft agar,64 and biofilm formation by measuring crystal violet staining.65Untreated wildtype H. pylori grew to stationary phase (Fig. 4A), were viable (Fig. S3†), were motile (Fig. 4B), and formed a dense biofilm that stained with crystal violet (Fig. 4C). H. pylori treated with 1 mM BnGlcNAc (7), F-Bac (4), and F-FucNAc (6) exhibited growth, motility, and biofilm formation comparable to untreated H. pylori. By contrast, H. pylori treated with BnBac (1), F-DAT (5) and BnFucNAc (3) displayed slowed growth, decreased viability, diminished motility, and impaired biofilm formation relative to untreated H. pylori (Fig. 4 and S3†). These fitness defects were also observed with an H. pylori protein glycosylation mutant containing an insertional inactivation of a putative H. pylori glycosyltransferase gene (ΔGT, HPG27_580::CmR)(Fig. 4); inactivating this gene impedes H. pylori's general protein glycosylation system (Fig. S2†) by an as-yet uncharacterized mechanism, yet has no effect on lipopolysaccharide biosynthesis.66 Small molecule-induced functional defects appeared even more striking than those induced via genetic interruption. Fitness appeared to be most impaired in H. pylori treated with BnBac (1), F-DAT (5) and BnFucNAc (3) (Fig. 3), and quantification of motility and biofilm production confirmed this qualitative finding (Fig. S3†). Profound defects in motility were sustained even up to fifteen days after allowing cells to swarm (data not shown). Intriguingly, the three compounds that impaired all fitness attributes also produced the most pronounced glycoprotein biosynthesis defects. Somewhat surprisingly, F-GlcNAc (9) and F-GalNAc (10) led to disrupted motility (Fig. 4B) despite not appreciably altering H. pylori's general glycosylation system (Fig. 3B). This effect could be due, potentially, to inhibition of flagellin glycosylation, which impacts only motility.9 Small-molecule induced functional defects in attributes that are absolutely critical for establishment and maintenance of infection within a host suggest the potential functional importance of H. pylori's protein glycosylation machinery and ensuing suitability as a drug target.
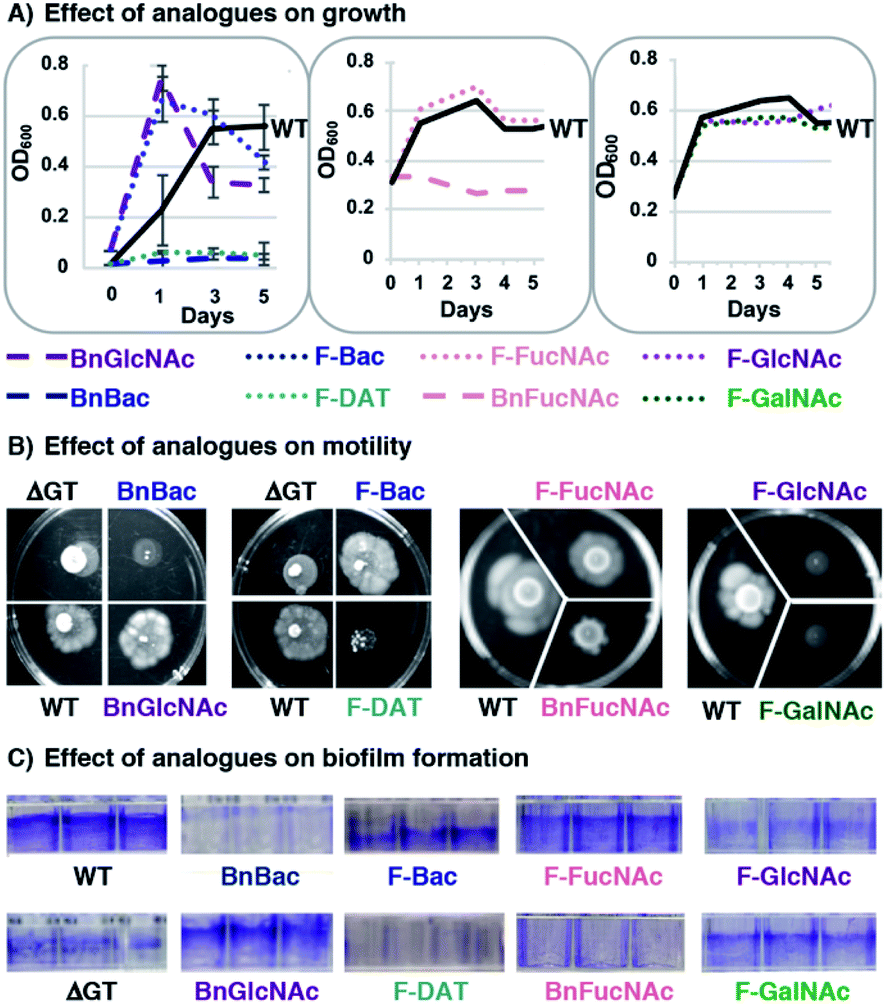 | ||
| Fig. 4 Fitness assays reveal defects in growth, motility and biofilm formation for H. pylori treated with metabolic inhibitors. H. pylori were treated with 1 mM of benzyl glycosides (1, 3, 7) or fluoro analogues (4, 5, 6, 9, 10) and scored for (A) growth by monitoring optical density at 600 nm (OD600), (B) motility on soft agar (shown at day 8), and (C) biofilm formation using a crystal violet assay. Untreated wildtype H. pylori (WT) and an H. pylori protein glycosylation mutant containing an insertional inactivation in a predicted glycosyltransferase gene (ΔGT, HPG27_580::CmR) were also scored in these assays. The data shown are representative of replicates (n ≥ 2). | ||
Metabolic inhibitors act in a bacteria-selective manner
Encouraged by the observation that metabolic inhibitors based on rare bacterial monosaccharide scaffolds interfere with H. pylori's glycan biosynthesis and induce functional defects, we next evaluated the effect of these metabolic inhibitors on an additional Gram-negative pathogen that colonizes the human gut, Campylobacter jejuni. C. jejuni is a close relative of H. pylori that was selected because it synthesizes well-characterized glycoproteins and incorporates azide-containing sugars into its glycans via MOE.34,67 Analogous to the experiments performed with H. pylori, we assessed the ability of our novel monosaccharide analogues to inhibit azide-labeled glycoprotein biosynthesis in C. jejuni. For these experiments, peracetylated N-azidoacetylgalactosamine (Ac4GalNAz),68 an azide-containing analogue of N-acetylgalactosamine, was used to metabolically label cellular glycans. Robust azide-labeled glycan biosynthesis was observed in C. jejuni treated with Ac4GalNAz, whereas control samples from cells treated with the azide-free compound Ac4GlcNAc benchmark background antibody binding that occurs in an azide-independent manner (Fig. 5A). None of the metabolic inhibitors elicited an appreciable concentration-dependent diminishment of the azide-labeled glycan profile (Fig. 5A) nor an alteration in protein expression (Fig. S4A†). Moreover, an analysis of C. jejuni's growth (Fig. S4B†), motility (Fig. 5B) and biofilm formation (Fig. 5C and S4C†) yielded comparable fitness for untreated C. jejuni versus cells treated with 2 mM BnBac (1), BnDAT (2), F-Bac (4), F-DAT (5) F-FucNAc (6), or BnGlcNAc (7). These data indicate that metabolic inhibitors act in a cell-selective manner; BnBac (1) and F-DAT (5) elicit glycosylation and phenotypic effects in H. pylori yet appears to have a negligible effect on C. jejuni. Therefore, bacteria-selective effects are possible with metabolic glycan inhibitors. This result suggests the potential for narrow-spectrum approaches to sensitize particular bacterial populations for destruction.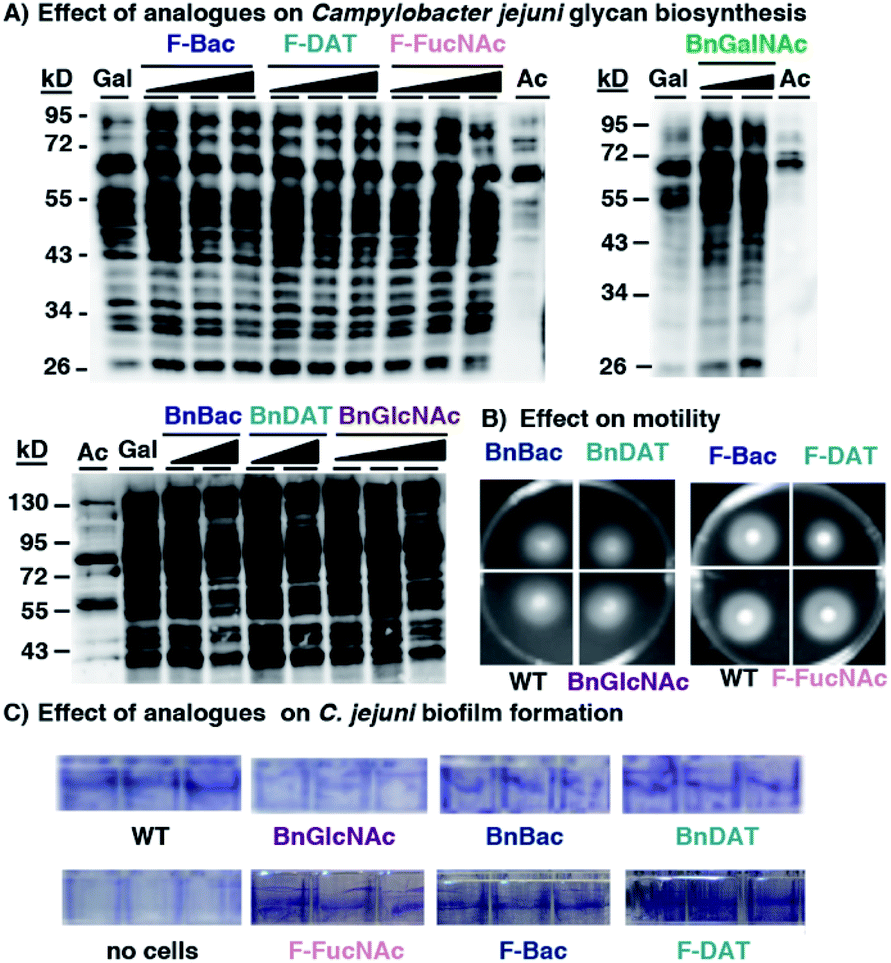 | ||
| Fig. 5 Metabolic inhibitors have a negligible effect on Campylobacter jejuni's glycan biosynthesis or fitness. C. jejuni were treated with the indicated metabolic inhibitors (1–8) and scored for (A) synthesis of azide-labeled glycoproteins upon metabolic labeling with 1 mM Ac4GalNAz (Gal), (B) motility on soft agar, and (C) biofilm formation using a crystal violet assay. (A) C. jejuni were grown for four days in media supplemented with 1 mM Ac4GalNAz (Gal), Ac4GlcNAc (Ac), or with 1 mM Ac4GalNAz alongside increasing concentrations (0.5, 1.0, or 2.0 mM; indicated by the increasing shaded triangle, with the shortest point representing 0.5 mM and the tallest part representing 2 mM except for the BnGalNAc (8) samples, which are 0.5 and 1.0 mM; BnBac (1) samples, which are 1.0 and 2.0 mM; and BnDAT (2) samples, which are 0.5 and 2.0 mM) of compounds 1–8, then harvested by lysis. The presence of azides in cellular glycoproteins was detected by reacting lysates with 250 μM Phos-FLAG for 12 h at 37 °C and then analyzing samples via western blot with anti-FLAG antibody. (B and C) C. jejuni were treated with 2 mM of the indicated metabolic inhibitors (1, 2, 4, 5, 6, 7) and scored for (B) motility on soft agar after 4 days, and (C) biofilm formation after 4 days using a crystal violet assay. Untreated wildtype C. jejuni (WT) served as a reference point in motility and biofilm formation assays. The data shown are representative of replicates (n ≥ 2). | ||
With compelling evidence of bacteria-selective effects, we next explored whether analogues impact glycan biosynthesis in the Gram-negative gut symbiont Bacteroides fragilis. This information is critical to assess the potential undesired impacts of glycan modulators on innocent bystanders. Based on the previous demonstration by Kasper and coworkers that B. fragilis robustly incorporates Ac4GalNAz into polysaccharide A,69 we turned to this azidosugar as a metabolic reporter of intact polysaccharide A biosynthesis. B. fragilis supplemented with Ac4GalNAz displayed robust azide-dependent glycan labeling.
Additional supplementation with 0.5–2 mM of select metabolic inhibitors (1–6, 8) caused no appreciable diminishment of the prominent azide-dependent signal observed ∼72–95 kDa (Fig. 6A). A subtle diminishment of the high molecular weight signal, >95 kDa, was noted in a concentration-dependent manner with several of the analogues (Fig. 6A). In all cases Coomassie staining of electrophoresed samples confirmed equivalent protein levels (Fig. S5A†), indicating that subtle changes in detectable azides in some samples was not due to low protein content. Since western blot data were difficult to interpret, we turned to a complementary approach to quantify changes in cell surface glycans.
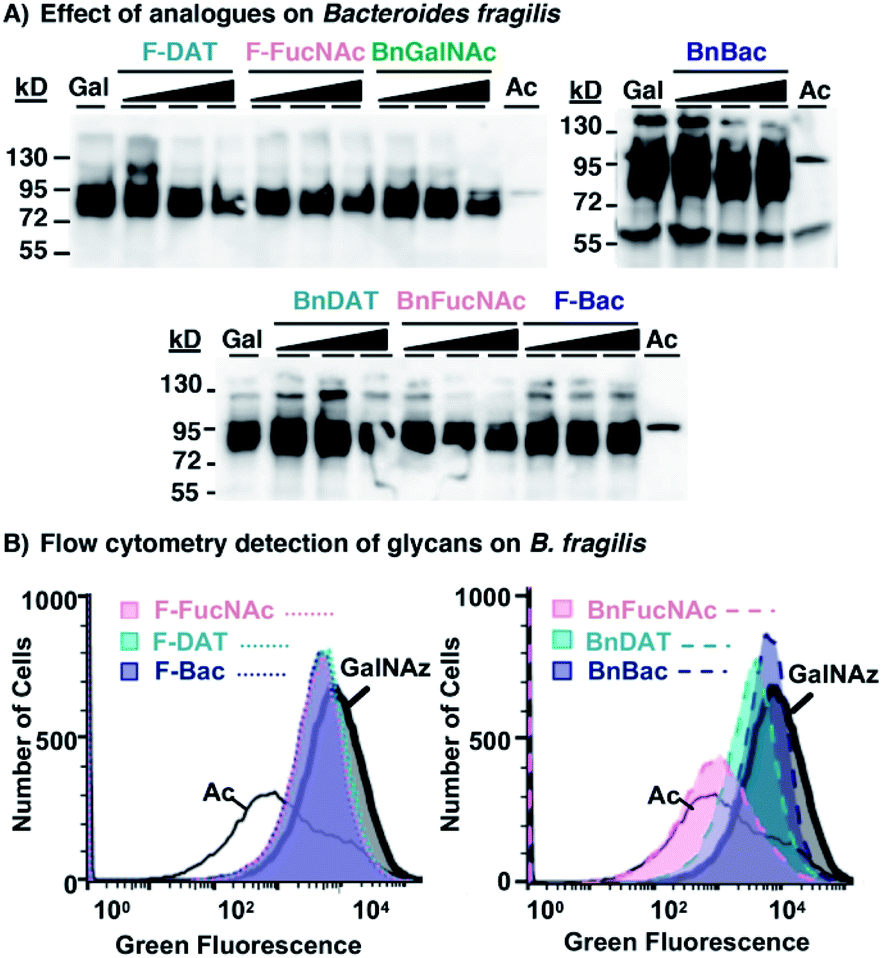 | ||
| Fig. 6 Metabolic inhibitors have a subtle effect on Bacteroides fragilis' glycan biosynthesis. B. fragilis were treated with 0.5–2 mM metabolic inhibitors (1–6, 8) and 0.1 mM Ac4GalNAz for three days, with 0.1 mM Ac4GalNAz (Gal or GalNAz) in the absence of inhibitor, or with the azide-free control sugar Ac4GlcNAc (Ac), then probed for the presence of azide-labeled glycans within lysates or on cells. (A) Lysates from treated cells were reacted with 250 μM Phos-FLAG and subsequently detected with anti-FLAG antibody via western blot analysis. (B) Treated cells were reacted with 20 μM AF488-DBCO and fluorescence intensity of each cell population was measured by flow cytometry analysis. | ||
To explore the possibility that these analogues may impact synthesis of fully-elaborated glycans, we probed cell surface azide expression using a flow cytometry-based assay.69 Cells were metabolically labeled with Ac4GalNAz or the azide-free control sugar Ac4GlcNAc, and surface accessible azides were probed on live cells using strain-promoted azide–alkyne cycloaddition with fluorescent AF488-DBCO.34B. fragilis treated with Ac4GalNAz exhibited a high fluorescence intensity, corresponding to abundant expression of azides and consistent with previous reports34,69 (Fig. 6B and S5B†). Treatment with Ac4GalNAz in tandem with 1 mM BnFucNAc (3), and to a lesser-extent BnDAT (2), led to a substantial diminishment in the fluorescence intensity that was comparable to the Ac4GlcNAc control (Fig. 6B) and occurred in a concentration-dependent manner (Fig. S5D†). In contrast, relatively subtle shifts in fluorescence intensity inconsistent with concentration-dependent inhibition were observed with the other analogues tested (Fig. 6B and S5C†). These data indicate that modifications in B. fragilis glycan biosynthesis can be induced by treatment with the metabolic inhibitors BnDAT (2) or BnFucNAc (3). Taken together, the extent of glycan disruption appears to be dependent on both the bacterial strain explored and the choice of metabolic inhibitor. Thus, tailoring of glycans in a bacteria-selective manner should be possible.
Discussion
Bacterial glycans are intriguing molecular targets as they contain rare monosaccharides that are expressed on a small number of bacterial pathogens, are absent from human cells, and are frequently linked to pathogenesis.1,3,70 Thus, expanding the repertoire of tools to experimentally modulate their biosynthesis should be a high priority. Such efforts will aid in defining the functions of these molecules and have the potential to guide the development of urgently needed antibiotics. Although systematic efforts to screen for inhibitors of glycan biosynthesis have been undertaken in mammalian systems,71–74 there is a relative paucity of bacterial glycosylation inhibitors. The progress that has been made in this area has relied on in-depth characterization of glycosyltransferases,75–78 information that is often a rate-limiting step in inhibitor development. As an alternative to direct screening of glycosyltransferases, Vocadlo,42 Paulson44 and others21,45 have demonstrated the potential of unnatural monosaccharides or glycosides bearing key structural features (e.g. O-benzyl glycosides, fluorinated analogues) that act as global metabolic inhibitors of glycosylation within mammalian cells. Inspired by these precedents, we produced a series of benzyl glycosides and fluoro-containing analogues of rare bacterial monosaccharides to metabolically inhibit bacterial glycan biosynthesis.Here we explored metabolic oligosaccharide engineering as an approach to guide the design and discovery of substrate-based metabolic inhibitors of bacterial glycosylation. Our data indicate that established mammalian glycosylation inhibitors have minimal effect on glycan biosynthesis and fitness in H. pylori, C. jejuni, and B. fragilis, yet metabolic inhibitors based on rare bacterial monosaccharide scaffolds elicit species-selective defects in glycan biosynthesis and function. For example, BnBac (1), BnFucNAc (3), and F-DAT (5) dramatically reduced H. pylori's ability to synthesize glycoproteins and led to diminished growth, motility, and biofilm formation. In contrast, these analogues had no effect on glycan biosynthesis or fitness in C. jejuni, while BnFucNAc (3) and an entirely different analogue, BnDAT (2), led to subtle changes in B. fragilis glycosylation. These data indicate that the choice of metabolic inhibitor can be tailored to elicit strain-selective effects in bacteria of interest. Thus, these compounds are useful probes and have the potential to form the basis of new glycosylation-based strategies to interfere with bacterial pathogenicity.
The differential effect of BnBac (1), BnDAT (2), BnFucNAc (3), and F-DAT (5) in bacterial strains could reflect underlying variation in glycosylation machinery present within each species. H. pylori, C. jejuni, and B. fragilis use some of the same monosaccharide building blocks, yet the structures of the glycans produced by these bacteria and the glycosyltransferases that stitch these higher order glycans together diverge.79–82 For example, although H. pylori and C. jejuni have analogous dedicated flagellin glycosylation systems, in which pseudaminic acid is installed onto serine and threonine residues on flagellin proteins,1,3 their general glycosylation systems differ.79,82 These differences have manifested in species-selective utilization of azidosugars in metabolic labeling experiments.34 Metabolic inhibitors may exploit inherent differences in glycan biosynthesis, such as glycosyltransferase substrate specificity and relative permissiveness for unnatural substrates, to induce species-selective glycosylation defects. Metabolic inhibitors that impact glycosylation could be leveraged to yield insight into glycan composition and assembly. This information could, in turn, provide information about how glycan structure correlates to function.
Some surprising effects were observed in light of what is documented in the literature. For example, C. jejuni glycoproteins have bacillosamine at the core,78 yet BnBac had no effect on this bacteria's protein glycosylation. Likewise, B. fragilis contains GalNAc in polysaccharide A,80 yet BnGalNAc had no appreciable effect on PSA biosynthesis. These observations reveal unknowns worthy of further exploration, including relative substrate promiscuity of glycosyltransferases and carbohydrate uptake mechanisms.
Small molecule-induced functional defects in growth, motility, and biofilm formation – three attributes which are absolutely critical for establishment and maintenance of infection within a host – suggest the potential functional importance of H. pylori's general protein glycosylation machinery and ensuing suitability as a drug target. It is well established that flagellin glycosylation is a virulence factor in H. pylori, as targeted genetic disruption of the dedicated flagellin protein glycosylation system impedes flagella formation, motility, and colonization of the host.9 In contrast, the link between H. pylori's general protein glycosylation system and virulence is not known, as the general protein glycosylation system is not well characterized. Metabolic inhibitor-induced functional defects, coupled to those induced via genetic interruption of H. pylori's general protein glycosylation system (ΔGT, Fig. 4), provide the first evidence that H. pylori's general protein glycosylation system may be a virulence factor. Metabolic inhibitor-induced functional defects appeared even more striking than those induced via genetic interruption. This observation could stem from the differential glycan biosynthesis disruption caused by metabolic inhibitors relative to genetic glycosyltransferase deletion. Metabolic inhibitors might interrupt glycan biosynthesis at an earlier point in biosynthesis. Thus, metabolic inhibitors have the potential to yield insightful information, complementary to genetics experiments, about the suitability of glycan biosynthesis as a virulence factor and potential drug target.
With metabolic inhibitors as probe compounds, it is now possible to query how changes in glycan structure precipitate altered function. Molecular-level information concerning how glycan structure changes in the presence of metabolic inhibitors will be critical to gain incisive information on the structure–function relationship. Recent reports by Paulson and coworkers44 and Wang et al.,45 for example, indicate successful strategies for characterizing the effects of metabolic inhibitors on cellular glycosylation in mammalian systems that should translate to bacterial systems. Moreover, molecular-level evidence will reveal the mechanism by which these analogues inhibit bacterial glycosylation. Benzyl glycosides likely act as substrate decoys that divert glycan biosynthesis onto mock substrates; buildup of glycans on benzyl glycosides within cells would support this hypothesis. By contrast, fluoro analogues are presumably activated in situ as unnatural nucleotide sugars that then directly (via glycan installation) or indirectly (via feedback inhibition) terminate glycan biosynthesis; an analysis of nucleotide sugar pools and of fluoro-sugar fate within treated cells would yield insight into these possible mechanisms.
Glycan-disrupting analogues have the potential to disarm bacteria, rendering them more vulnerable to host immune defenses, sensitized to existing antibiotics, and susceptible to competition with other microflora.83,84 Based on precedents in mammalian systems, metabolic inhibitors modulate glycosylation on cells in vitro and in animal models.50 Therefore, the compounds developed here could be deployed in animal infection models to perturb glycans in complex microbial communities. Ultimately, this strategy has the potential to lead to the development of a narrow-spectrum glycosylation-based antibacterial strategy that could treat priority pathogen infections while minimizing the deleterious effects on beneficial bacteria.
Conclusions
Rare bacterial sugars and associated biosynthetic machinery have untapped potential as targets of selective interference. Despite their appeal, no general strategy has been developed to block the biosynthesis of elaborated bacterial glycans. This work represents the first demonstration, to our knowledge, of using MOE and unnatural monosaccharides to globally inhibit the bacterial glycome. While this work focused on a small panel of metabolic inhibitors, the general approach presented here will pave the way for the development of a broad array of metabolic inhibitors to study and alter bacterial glycans in diverse species. Indeed, metabolic inhibitors based on the plethora of exclusively bacterial monosaccharides have the potential to form the basis of tailor-made probe compounds that provide insight into the functional importance of bacterial glycans and serve as agents that selectively disarm bacteria within a mixed population. Broadly, this work introduces new approaches to study and harness glycans of priority pathogens to create urgently needed antibiotics.Experimental
Materials and chemical synthesis
Organic chemicals and anti-FLAG antibodies were purchased from Sigma-Aldrich unless otherwise noted. 4F-GlcNAc (9) and 4F-GalNAc (10) were acquired from Sussex Research. H. pylori strain G27 (ref. 85) was a gift of Manuel Amieva (Stanford University) and protein glycosylation mutant (ΔGT), H. pylori G27 insertionally inactivated in HPG27_580 with a chloramphenicol resistance cassette,86 was a gift from Nina Salama (Fred Hutchinson Cancer Research Center). Other bacterial cells (C. jejuni ATCC 33560; B. fragilis ATCC 23745) were purchased from ATCC and grown according to the supplier's instructions. Ac4GlcNAc, Ac4GlcNAz, Ac4GalNAz and Phos-FLAG were synthesized as previously described.26,87 Compounds 1–6 were synthesized using standard organic chemistry procedures and characterized by standard techniques including 1H and 13C NMR spectroscopy and mass spectrometry. Compounds 1–6 were purified using flash silica gel chromatography.Metabolic labeling
Bacterial cells were grown in rich liquid media supplemented with 0.5 mM Ac4GlcNAz, Ac4GalNAz, or the azide-free control Ac4GlcNAc for 2–4 days, depending on the doubling rate of the organism. H. pylori was metabolically labeled for 4 days under microaerophilic conditions (14% CO2, 37 °C), C. jejuni was metabolically labeled for 3 days under microaerophilic conditions, and B. fragilis was metabolically labeled for 2 days under anaerobic conditions (created by a Thermo Scientific Oxoid AnaeroGen Sachet in an airtight container; 37 °C) using 0.1 mM Ac4GalNAz according to previous literature.69 Cells were then harvested, washed with phosphate buffered saline (PBS), and whole cells were analyzed by flow cytometry or lysed for subsequent western blot analysis, as described below.Western blot
Lysates from metabolically labeled cells were standardized to a protein concentration of ∼2 mg mL−1 and reacted 1:1 with 500 μM Phos-FLAG63 overnight at 37 °C for detection of azide-labeled glycans. Reacted lysates were loaded onto a 12% Tris–HCl SDS-PAGE gel, separated by electrophoresis, and transferred to nitrocellulose paper. Anti-FLAG-HRP was employed to visualize FLAG-tagged proteins via chemiluminescence.
Lectin binding
H. pylori treated with 2 mM inhibitor or with no inhibitor (wildtype) for 4 days were probed with Alexa Fluor 488-conjugated Concanavalin A (ConA), then analysed by flow cytometry on a BD Accuri C6 (BD Biosciences) instrument. As a negative control, ConA was pre-incubated with 400 mM mannose (carbo-block) prior to binding to untreated H. pylori.Flow cytometry of B. fragilis
Metabolically labeled intact B. fragilis were reacted with Alexa Fluor Dye 488 DBCO (AF488-DBCO, 20 μM) for 5 hours in the dark for strain-promoted azide–alkyne cycloaddition detection of azides (Click Chemistry Tools; ex.: 488/em.: 519). Cells were analysed by flow cytometry on a BD Accuri C6 (BD Biosciences) instrument, with 30000 live cells gated for each replicate experiment.
Growth curves
Growth was measured over the course of 3–5 days, as indicated. Bacteria treated were inoculated at a starting OD600 of 0.1–0.3 into culture tubes containing 3 mL of rich media along with 1 mM (H. pylori) or 2 mM (C. jejuni) of the indicated inhibitor (1–10). Cultures were kept at 37 °C and 14% CO2 with gentle shaking. The OD600 of each culture was measured using spectrophotometry at the indicated timepoints. We note that the relative change in OD600 depended upon whether cultures were started in the exponential phase of the growth curve or closer to saturation.Motility assays
The ability of the bacteria to swarm in the presence of compounds 1–8 was monitored over the course of 4–8 days. Bacteria were standardized to an OD600 of 1.0, then incubated in rich media containing 1 mM (H. pylori) or 2 mM (C. jejuni) of the indicated inhibitor (1–8) or no inhibitor overnight at 37 °C and 14% CO2 with gentle shaking. An aliquot of the overnight culture was concentrated by centrifugation and resuspension in media, and 10 μL of the concentrated suspension was plated into soft agar plates supplemented with 4% agar and 10% fetal bovine serum. Plates were incubated at 37 °C and 14% CO2, and colony diameter was measured daily.Biofilm formation assays
Biofilm formation for bacteria cultured in the presence versus absence of 1 mM (H. pylori) or 2 mM (C. jejuni) compounds 1–8 was measured after 4 days following a literature protocol.65 Briefly, bacteria were standardized to an OD600 of 1.0 in rich media containing 2 mM compounds 1–8 or no inhibitor, plated in triplicate in the side wells of a 96 well plate, and incubated at 37 °C and 14% CO2. Following 4 days of growth, media was removed from the wells and the remaining biofilm was stained with 0.15% crystal violet. Sideview images of the triplicate wells were taken after staining.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge insightful conversations with A. McBride and B. Kohorn, and members of our research laboratories for support and guidance. Research reported in this publication was supported by the National Institutes of Health under grant number R15GM109397 to D. H. D., an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103423, and by awards to D. A. W. from the James Stacy Coles Fellowship. S. S. K. thanks the Science and Engineering Research Board (grant No. CRG/2019/000025), the ISF-University Grants Commission (UGC) joint research program framework (grant No. 2253) and Department of Biotechnology (BT/INF/22/SP23026/2017) for financial support. K. P. and A. P. thank Indian Institute of Technology for fellowships.Notes and references
- D. H. Dube, K. Champasa and B. Wang, Chem. Commun., 2011, 47, 87–101 RSC.
- S. A. Longwell and D. H. Dube, Curr. Opin. Chem. Biol., 2013, 17, 41–48 CrossRef CAS PubMed.
- V. N. Tra and D. H. Dube, Chem. Commun., 2014, 50, 4659–4673 RSC.
- J. T. Park and J. L. Strominger, Science, 1957, 125, 99–101 CrossRef CAS PubMed.
- H. R. Perkins, Biochem. J., 1969, 111, 195–205 CrossRef CAS PubMed.
- D. R. Storm and J. L. Strominger, J. Biol. Chem., 1973, 248, 3940–3945 CAS.
- E. Tacconelli and N. Magrini, Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics, World Health Organization, 2017 Search PubMed.
- S. Herget, P. V. Toukach, R. Ranzinger, W. E. Hull, Y. A. Knirel and C. W. von der Lieth, BMC Struct. Biol., 2008, 8, 35 CrossRef PubMed.
- M. Schirm, E. C. Soo, A. J. Aubry, J. Austin, P. Thibault and S. M. Logan, Mol. Microbiol., 2003, 48, 1579–1592 CrossRef CAS PubMed.
- M. J. Morrison and B. Imperiali, Biochemistry, 2014, 53, 624–638 CrossRef CAS PubMed.
- M. D. Hartley, M. J. Morrison, F. E. Aas, B. Borud, M. Koomey and B. Imperiali, Biochemistry, 2011, 50, 4936–4948 CrossRef CAS PubMed.
- J. Horzempa, T. K. Held, A. S. Cross, D. Furst, M. Qutyan, A. N. Neely and P. Castric, Clin. Vaccine Immunol., 2008, 15, 590–597 CrossRef CAS PubMed.
- S. Grass, C. F. Lichti, R. R. Townsend, J. Gross and J. W. St. Geme III, PLoS Pathog., 2010, 6, e1000919 CrossRef PubMed.
- J. G. Smedley, E. Jewell, J. Roguskie, J. Horzempa, A. Syboldt, D. B. Stolz and P. Castric, Infect. Immun., 2005, 73, 7922–7931 CrossRef CAS PubMed.
- I. Bleiziffer, J. Eikmeier, G. Pohlentz, K. McAulay, G. Xia, M. Hussain, A. Peschel, S. Foster, G. Peters and C. Heilmann, PLoS Pathog., 2017, 13, e1006110 CrossRef PubMed.
- S. Goon, J. F. Kelly, S. M. Logan, C. P. Ewing and P. Guerry, Mol. Microbiol., 2003, 50, 659–671 CrossRef CAS PubMed.
- T. M. Allison, S. Conrad and P. Castric, Microbiology, 2015, 161, 1780–1789 CrossRef CAS PubMed.
- M. Blaser, Nature, 2011, 476, 393–394 CrossRef CAS PubMed.
- C. A. Lozupone, J. I. Stombaugh, J. I. Gordon, J. K. Jansson and R. Knight, Nature, 2012, 489, 220–230 CrossRef CAS PubMed.
- K. M. Ng, J. A. Ferreyra, S. K. Higginbottom, J. B. Lynch, P. C. Kashyap, S. Gopinath, N. Naidu, B. Choudhury, B. C. Weimer, D. M. Monack and J. L. Sonnenburg, Nature, 2013, 502, 96–99 CrossRef CAS PubMed.
- C. R. Bertozzi, J. D. Esko and R. L. Schnaar, Chemical tools for inhibiting glycosylation, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, A. G. Darvill, T. Kinosita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 2009 Search PubMed.
- Y. Yuan, S. Fuse, B. Ostash, P. Sliz, D. Kahne and S. Walker, ACS Chem. Biol., 2008, 3, 429–436 CrossRef CAS.
- J. W. De Schutter, J. P. Morrison, M. J. Morrison, A. Ciulli and B. Imperiali, J. Med. Chem., 2017, 60, 2099–2118 CrossRef CAS PubMed.
- R. Ménard, I. C. Schoenhofen, L. Tao, A. Aubry, P. Bouchard, C. W. Reid, P. Lachance, S. M. Twine, K. M. Fulton, Q. Cui, H. Hogues, E. O. Purisima, T. Sulea and S. M. Logan, Antimicrob. Agents Chemother., 2014, 58, 7430–7440 CrossRef PubMed.
- D. H. Dube and C. R. Bertozzi, Curr. Opin. Chem. Biol., 2003, 7, 616–625 CrossRef CAS PubMed.
- S. T. Laughlin and C. R. Bertozzi, Nat. Protoc., 2007, 2, 2930–2944 CrossRef CAS PubMed.
- O. T. Keppler, R. Horstkorte, M. Pawlita, C. Schmidts and W. Reutter, Glycobiology, 2001, 11, 11R–18R CrossRef CAS PubMed.
- P. A. Gilormini, A. R. Batt, M. R. Pratt and C. Biot, Chem. Sci., 2018, 9, 7585–7595 RSC.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- R. Sadamoto, K. Niikura, P. S. Sears, H. T. Liu, C. H. Wong, A. Suksomcheep, F. Tomita, K. Monde and S. I. Nishimura, J. Am. Chem. Soc., 2002, 124, 9018–9019 CrossRef CAS PubMed.
- E. Memmel, A. Homann, T. A. Oelschlaeger and J. Seibel, Chem. Commun., 2013, 49, 7301–7303 RSC.
- M. B. Koenigs, E. A. Richardson and D. H. Dube, Mol. BioSyst., 2009, 5, 909–912 RSC.
- K. Champasa, S. A. Longwell, A. M. Eldridge, E. A. Stemmler and D. H. Dube, Mol. Cell. Proteomics, 2013, 12, 2568–2586 CrossRef CAS PubMed.
- E. L. Clark, M. Emmadi, K. L. Krupp, A. R. Podilapu, J. D. Helble, S. S. Kulkarni and D. H. Dube, ACS Chem. Biol., 2016, 11, 3365–3373 CrossRef CAS.
- G. Andolina, R. Wei, H. Liu, Q. Zhang, X. Yang, H. Cao, S. Chen, A. Yan, X. D. Li and X. Li, ACS Chem. Biol., 2018, 13, 3030–3037 CrossRef CAS.
- B. L. Urbanek, D. C. Wing, K. S. Haislop, C. J. Hamel, R. Kalscheuer, P. J. Woodruff and B. M. Swarts, ChemBioChem, 2014, 15, 2066–2070 CrossRef CAS PubMed.
- K. M. Backus, H. I. Boshoff, C. S. Barry, O. Boutureira, M. K. Patel, F. D'Hooge, S. S. Lee, L. E. Via, K. Tahlan, C. E. Barry 3rd and B. G. Davis, Nat. Chem. Biol., 2011, 7, 228–235 CrossRef CAS PubMed.
- F. Liu, A. J. Aubry, I. C. Schoenhofen, S. M. Logan and M. E. Tanner, ChemBioChem, 2009, 10, 1317–1320 CrossRef CAS PubMed.
- H. Liang, K. E. DeMeester, C. W. Hou, M. A. Parent, J. L. Caplan and C. L. Grimes, Nat. Commun., 2017, 8, 15015 CrossRef CAS PubMed.
- K. E. DeMeester, H. Liang, M. R. Jensen, Z. S. Jones, E. A. D'Ambrosio, S. L. Scinto, J. Zhou and C. L. Grimes, J. Am. Chem. Soc., 2018, 140, 9458–9465 CrossRef CAS PubMed.
- A. Dumont, A. Malleron, M. Awwad, S. Dukan and B. Vauzeilles, Angew. Chem., Int. Ed., 2012, 51, 3143–3146 CrossRef CAS PubMed.
- T. M. Gloster, W. F. Zandberg, J. E. Heinonen, D. L. Shen, L. Deng and D. J. Vocadlo, Nat. Chem. Biol., 2011, 7, 174–181 CrossRef CAS PubMed.
- T. M. Gloster and D. J. Vocadlo, Nat. Chem. Biol., 2012, 8, 683–694 CrossRef CAS PubMed.
- C. D. Rillahan, A. Antonopoulos, C. T. Lefort, R. Sonon, P. Azadi, K. Ley, A. Dell, S. M. Haslam and J. C. Paulson, Nat. Chem. Biol., 2012, 8, 661–668 CrossRef CAS PubMed.
- S.-S. Wang, X. Gao, V. d. Solar, X. Yu, A. Antonopoulos, A. E. Friedman, E. K. Matich, G. E. Atilla-Gokcumen, M. Nasirikenari, J. T. Lau, A. Dell, S. M. Haslam, R. A. Laine, K. L. Matta and S. Neelamegham, Cell Chem. Biol., 2018, 25, 1519–1532.e1515 CrossRef CAS PubMed.
- S. F. Kuan, J. C. Byrd, C. Basbaum and Y. S. Kim, J. Biol. Chem., 1989, 264, 19271–19277 CAS.
- A. K. Sarkar, T. A. Fritz, W. H. Taylor and J. D. Esko, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3323–3327 CrossRef CAS PubMed.
- J. R. Brown, M. M. Fuster, R. Li, N. Varki, C. A. Glass and J. D. Esko, Clin. Cancer Res., 2006, 12, 2894–2901 CrossRef CAS PubMed.
- J. R. Brown, F. Yang, A. Sinha, B. Ramakrishnan, Y. Tor, P. K. Qasba and J. D. Esko, J. Biol. Chem., 2009, 284, 4952–4959 CrossRef CAS PubMed.
- C. J. Dimitroff, T. S. Kupper and R. Sackstein, J. Clin. Invest., 2003, 112, 1008–1018 CrossRef CAS PubMed.
- J. A. Villalobos, B. R. Yi and I. S. Wallace, PLoS One, 2015, 10, e0139091 CrossRef PubMed.
- S. Nishimura, M. Hato, S. Hyugaji, F. Feng and M. Amano, Angew. Chem., Int. Ed., 2012, 51, 3386–3390 CrossRef CAS PubMed.
- S. R. Barthel, A. Antonopoulos, F. Cedeno-Laurent, L. Schaffer, G. Hernandez, S. A. Patil, S. J. North, A. Dell, K. L. Matta, S. Neelamegham, S. M. Haslam and C. J. Dimitroff, J. Biol. Chem., 2011, 286, 21717–21731 CrossRef CAS PubMed.
- F. Megraud, F. Bonnet, M. Garnier and H. Lamouliatte, J. Clin. Microbiol., 1985, 22, 1007–1010 CrossRef CAS PubMed.
- H. Bi, L. Zhu, J. Jia and J. E. Cronan, Sci. Rep., 2016, 6, 21162 CrossRef CAS.
- M. Emmadi and S. S. Kulkarni, Nat. Protoc., 2013, 8, 1870–1889 CrossRef CAS.
- M. Emmadi and S. S. Kulkarni, Org. Biomol. Chem., 2013, 11, 3098–3102 RSC.
- S. R. Sanapala and S. S. Kulkarni, J. Am. Chem. Soc., 2016, 138, 4938–4947 CrossRef CAS.
- J. Karban, I. Cisarova, T. Strasak, L. C. Stastna and J. Sykora, Org. Biomol. Chem., 2012, 10, 394–403 RSC.
- L. Quiquempoix, Z. Wang, J. Graton, P. G. Latchem, M. Light, J. Y. Le Questel and B. Linclau, J. Org. Chem., 2019, 84, 5899–5906 CrossRef CAS.
- M. L. Uhrig, B. Lantano and A. Postigo, Org. Biomol. Chem., 2019, 17, 5173–5189 RSC.
- D. J. Vocadlo, H. C. Hang, E.-J. Kim, J. A. Hanover and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9116–9121 CrossRef CAS PubMed.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS PubMed.
- M. Schirm, E. C. Soo, A. J. Aubry, J. Austin, P. Thibault and S. M. Logan, Mol. Microbiol., 2003, 48, 1579–1592 CrossRef CAS PubMed.
- G. A. O'Toole, J. Visualized Exp., 2011, 47, e2437 Search PubMed.
- H. Li, M. Marceau, T. Yang, T. Liao, X. Tang, R. Hu, Y. Xie, H. Tang, A. Tay, Y. Shi, Y. Shen, T. Yang, X. Pi, B. Lamichhane, Y. Luo, A. W. Debowski, H.-O. Nilsson, S. M. Haslam, B. Mulloy, A. Dell, K. A. Stubbs, B. J. Marshall and M. Benghezal, PLoS Genet., 2019, 15, e1008497 CrossRef PubMed.
- F. Liu, A. J. Aubry, I. C. Schoenhofen, S. M. Logan and M. E. Tanner, ChemBioChem, 2009, 10, 1317–1320 CrossRef CAS PubMed.
- H. C. Hang, C. Yu, D. L. Kato and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14846–14851 CrossRef CAS PubMed.
- N. Geva-Zatorsky, D. Alvarez, J. E. Hudak, N. C. Reading, D. Erturk-Hasdemir, S. Dasgupta, U. H. von Andrian and D. L. Kasper, Nat. Med., 2015, 21, 1091–1100 CrossRef CAS PubMed.
- S. Herget, P. V. Toukach, R. Ranzinger, W. E. Hull, Y. A. Knirel and C.-W. von der Lieth, BMC Struct. Biol., 2008, 8, 35 CrossRef PubMed.
- L. V. Lee, M. L. Mitchell, S. J. Huang, V. V. Fokin, K. B. Sharpless and C. H. Wong, J. Am. Chem. Soc., 2003, 125, 9588–9589 CrossRef CAS.
- C. D. Rillahan, S. J. Brown, A. C. Register, H. Rosen and J. C. Paulson, Angew. Chem., Int. Ed., 2011, 50, 12534–12537 CrossRef CAS.
- B. J. Gross, J. G. Swoboda and S. Walker, J. Am. Chem. Soc., 2008, 130, 440–441 CrossRef CAS.
- Y. Hu, J. S. Helm, L. Chen, C. Ginsberg, B. Gross, B. Kraybill, K. Tiyanont, X. Fang, T. Wu and S. Walker, Chem. Biol., 2004, 11, 703–711 CrossRef CAS.
- S. El Qaidi, C. Zhu, P. McDonald, A. Roy, P. K. Maity, D. Rane, C. Perera and P. R. Hardwidge, Front. Cell. Infect. Microbiol., 2018, 8, 435 CrossRef CAS.
- A. G. E. Madec, N. S. Schocker, S. Sanchini, G. Myratgeldiyev, D. Das and B. Imperiali, ACS Chem. Biol., 2018, 13, 2542–2550 CrossRef CAS.
- S. Nayyab, M. O'Connor, J. Brewster, J. Gravier, M. Jamieson, E. Magno, R. D. Miller, D. Phelan, K. Roohani, P. Williard, A. Basu and C. W. Reid, ACS Infect. Dis., 2017, 3, 421–427 CrossRef CAS PubMed.
- B. Ostash and S. Walker, Curr. Opin. Chem. Biol., 2005, 9, 459–466 CrossRef CAS PubMed.
- N. M. Young, J.-R. Brisson, J. Kelly, D. C. Watson, L. Tessier, P. H. Lanthier, H. C. Jarrell, N. Cadotte, F. St. Michael, E. Aberg and C. M. Szymanski, J. Biol. Chem., 2002, 277, 42530–42539 CrossRef CAS PubMed.
- D. Linton, N. Dorrell, P. G. Hitchen, S. Amber, A. V. Karlyshev, H. R. Morris, A. Dell, M. A. Valvano, M. Aebi and B. W. Wren, Mol. Microbiol., 2005, 55, 1695–1703 CrossRef CAS.
- S. Sharma, K. M. Erickson and J. M. Troutman, ACS Chem. Biol., 2017, 12, 92–101 CrossRef CAS PubMed.
- P. S. Hopf, R. S. Ford, N. Zebian, A. Merkx-Jacques, S. Vijayakumar, D. Ratnayake, J. Hayworth and C. Creuzenet, PLoS One, 2011, 6, e25722 CrossRef CAS PubMed.
- M. Erhardt, Curr. Top. Microbiol. Immunol., 2016, 398, 185–205 CAS.
- S. Hathroubi, S. L. Servetas, I. Windham, D. S. Merrell and K. M. Ottemann, Microbiol. Mol. Biol. Rev., 2018, 82, e00001-18 CrossRef PubMed.
- D. A. Baltrus, M. R. Amieva, A. Covacci, T. M. Lowe, D. S. Merrell, K. M. Ottemann, M. Stein, N. R. Salama and K. Guillemin, J. Bacteriol., 2009, 191, 447–448 CrossRef CAS PubMed.
- N. R. Salama, B. Shepherd and S. Falkow, J. Bacteriol., 2004, 186, 7926–7935 CrossRef CAS PubMed.
- S. J. Luchansky, H. C. Hang, E. Saxon, J. R. Grunwell, C. Yu, D. H. Dube and C. R. Bertozzi, Methods Enzymol., 2003, 362, 249–272 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S5 and full experimental details. See DOI: 10.1039/c9sc05955e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |