
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conformational control of Pd2L4 assemblies with unsymmetrical ligands†
James E. M.
Lewis
 *,
Andrew
Tarzia
,
Andrew J. P.
White
and
Kim E.
Jelfs
*,
Andrew
Tarzia
,
Andrew J. P.
White
and
Kim E.
Jelfs
Department of Chemistry, Imperial College London, Molecular Sciences Research Hub, 80 Wood Lane, London W12 0BZ, UK. E-mail: james.lewis@imperial.ac.uk
First published on 28th November 2019
Abstract
With increasing interest in the potential utility of metallo-supramolecular architectures for applications as diverse as catalysis and drug delivery, the ability to develop more complex assemblies is keenly sought after. Despite this, symmetrical ligands have been utilised almost exclusively to simplify the self-assembly process as without a significant driving foa mixture of isomeric products will be obtained. Although a small number of unsymmetrical ligands have been shown to serendipitously form well-defined metallo-supramolecular assemblies, a more systematic study could provide generally applicable information to assist in the design of lower symmetry architectures. Pd2L4 cages are a popular class of metallo-supramolecular assembly; research seeking to introduce added complexity into their structure to further their functionality has resulted in a handful of examples of heteroleptic structures, whilst the use of unsymmetrical ligands remains underexplored. Herein we show that it is possible to design unsymmetrical ligands in which either steric or geometric constraints, or both, can be incorporated into ligand frameworks to ensure exclusive formation of single isomers of three-dimensional Pd2L4 metallo-supramolecular assemblies with high fidelity. In this manner it is possible to access Pd2L4 cage architectures of reduced symmetry, a concept that could allow for the controlled spatial segregation of different functionalities within these systems. The introduction of steric directing groups was also seen to have a profound effect on the cage structures, suggesting that simple ligand modifications could be used to engineer structural properties.
Introduction
Non-covalent interactions are utilised by Nature to meticulously control self-assembly processes, ensuring that components combine in specific ratios and orientations to function effectively. For example, proteins can assemble with high fidelity into homo- or hetero-multimeric quaternary structures to carry out a range of biological functions.1 Over the last few decades chemists in various fields have attempted to emulate these principles, applying them to artificial systems.Metallo-supramolecular chemistry entails the self-assembly of a combination of metal ions and ligands to form architectures of defined geometry.2 Following early seminal work by Lehn,3 Fujita4 and Stang5 amongst others, metallo-supramolecular architectures of myriad geometries have been prepared. To promote efficient self-assembly the majority of ligands used are symmetrical and give rise to highly symmetrical self-assembled structures.6
Since their first report by McMorran and Steel over two decades ago,7 Pd2L4 molecular cages8 – assembled from ditopic ligands and “naked” palladium(II) ions – have been examined for a range of functions, including biomedical applications,9 catalysis10 and gas storage.11 Recently several examples have emerged in which two different ligands have been incorporated into cage structures in a controlled manner, resulting in heteroleptic complexes,12 either as a result of steric interactions,13 or through geometric design.14 This approach enables the preparation of cages with cavities that deviate from the standard approximately spherical shape generally associated with these complexes, and could potentially allow different segments of the cage to contain separate functionalities.15 In this manner cages could be prepared that, like enzymes, contain multiple binding sites.
However, functional group segregation could theoretically also be achieved with homoleptic assemblies through the controlled self-assembly of unsymmetrical ligands. Reports on the use of unsymmetrical ligands in metallo-supramolecular constructs,16 and three-dimensional polyhedra in particular,17 are rare. This is likely due to the fact that without a significant bias being built into the system, a statistical mixture of isomeric products will be obtained (Fig. 1). The selective formation of specific assemblies may be particularly challenging without resorting to ligand scaffolds incorporating coordinating moieties of different denticities, or the use of capping ligands to limit effective metal ion coordination numbers. However, it should be possible to design unsymmetrical ligands such that, upon self-assembly with metal ions, certain isomers of the resultant architectures are sufficiently energetically favourable to allow effective self-sorting into a single product. Indeed, whilst this manuscript was in preparation, Ogata and Yuasa reported an investigation into a stimuli-responsive [Pd2L4]4+ cage, capable of binding ReO4− anions, that formed as a single isomer from an unsymmetrical carbazole-based ditopic ligand framework.18
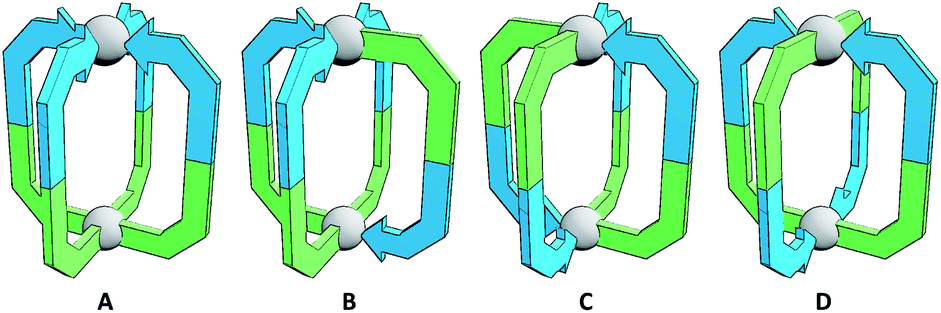 | ||
| Fig. 1 Cartoon representations of the four possible isomers of homoleptic Pd2L4 cages that can be formed with an unsymmetrical ditopic ligand: “all-up” (A), “three-up-one-down” (B), cis (C) and trans (D). Blue and green colours indicate non-equivalent ligand fragments. | ||
Herein we report an initial exploration of unsymmetrical bis-monodentate ligands and their self-assembly with ‘naked’ Pd(II) ions to form Pd2L4 cages. Despite the potential of forming a mixture of different isomers it is shown that, through careful design, trans C2v and cis C2h-symmetry complexes can be formed preferentially due to steric or geometric control, or a combination of the two.
Results and discussion
The simplest assemblies that can be formed from the coordination of unsymmetrical ditopic ligands, incapable of chelation, to square planar palladium(II) ions are Pd2L4 architectures. Due to the lack of bilateral symmetry, the formation of four different isomers is possible (Fig. 1). We hypothesised that by using steric or geometric constraints, or a combination of the two, it should be possible to design systems in which one isomer in particular is sufficiently energetically favourable to be formed to the exclusion of the others, allowing predictable formation of specific cage isomers.Steric constraints
Initially we sought to direct the self-assembly of unsymmetrical ligands using steric control, inspired by recent work on heteroleptic metallo-assemblies. Crowley and co-workers have reported the displacement of unsubstituted di-pyridyl ligands from pre-formed homoleptic Pd2L4 cages with ortho-aminopyridyl ligands, resulting in Pd2L2L′2 architectures.13a Similar Pd(II) cages were prepared by Clever and co-workers using a combination of picolyl-derived ligands with exo- or endo-hedral methyl groups to sterically enforce the formation of heteroleptic structures.13b Ligand 1 was designed around the well-known m-bis(pyridin-3-ylethynyl)aryl ligand motif, with a 6-methyl substituent appended to one of the pyridine rings, and prepared using successive Sonogashira reactions.19Initially combining 1 with [Pd(CH3CN)4](BF4)2 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in CD3CN at room temperature resulted in a mixture of products (Fig. S88†). Pleasingly, heating at 60 °C for 24 h resulted in coalescence to a single major set of signals (Fig. 2a).20 DOSY NMR (D = 8.10 × 10−10 m2 s−1; calculated hydrodynamic radius, RH, of 7.0 Å) and mass spectrometry (MS; m/z = 781 [Pd2(1)4(BF4)2]2+, Fig. S86†) indicated that a Pd2L4 assembly had been formed. To confirm that the observed simple 1H NMR spectrum was the result of a single cage isomer, rather than multiple rapidly interconverting species, a variable temperature NMR experiment was performed with 1H NMR spectra obtained between 20 and −30 °C (Fig. S89†). Even at −30 °C no significant broadening or splitting of the NMR signals could be observed, supporting the conclusion that a single thermodynamically stable cage had been formed.
:1 ratio in CD3CN at room temperature resulted in a mixture of products (Fig. S88†). Pleasingly, heating at 60 °C for 24 h resulted in coalescence to a single major set of signals (Fig. 2a).20 DOSY NMR (D = 8.10 × 10−10 m2 s−1; calculated hydrodynamic radius, RH, of 7.0 Å) and mass spectrometry (MS; m/z = 781 [Pd2(1)4(BF4)2]2+, Fig. S86†) indicated that a Pd2L4 assembly had been formed. To confirm that the observed simple 1H NMR spectrum was the result of a single cage isomer, rather than multiple rapidly interconverting species, a variable temperature NMR experiment was performed with 1H NMR spectra obtained between 20 and −30 °C (Fig. S89†). Even at −30 °C no significant broadening or splitting of the NMR signals could be observed, supporting the conclusion that a single thermodynamically stable cage had been formed.
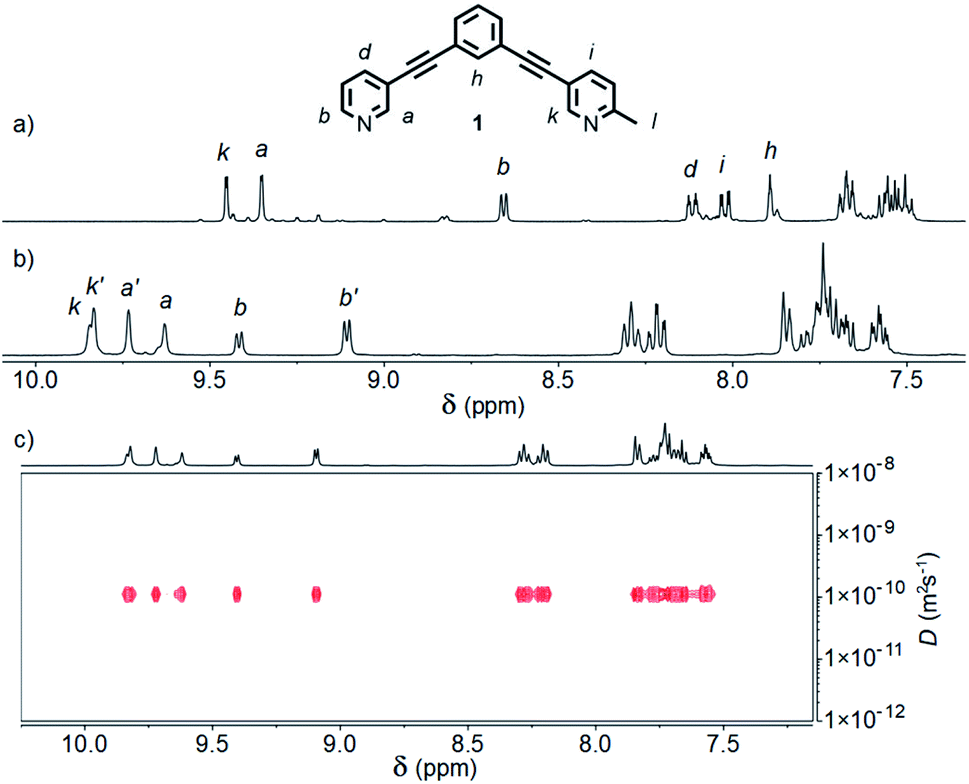 | ||
| Fig. 2 Partial 1H NMR spectra (400 MHz, 298 K) of cages [Pd2(1)4](BF4)4 formed in (a) CD3CN, and (b) d6-DMSO; (c) 1H DOSY spectrum (500 MHz, d6-DMSO) of cis- and trans-[Pd2(1)4](BF4)4 cage mixture. | ||
The symmetry of the 1H NMR spectrum suggested that the product could not be isomer B, whilst cross-peaks observed in the ROESY NMR spectrum (Fig. S84†) between the methyl group (Hl) of one pyridine ring and the exohedral proton ortho to the pyridyl nitrogen atom of the other (Hb) ruled out isomer A. Unfortunately, despite multiple attempts, we were unable to generate X-ray quality crystals of the complex to confirm its structure in the solid state. We therefore turned to density functional theory (DFT) calculations (carried out at the B3LYP/6-31G(d) level of theory using the D3 empirical dispersion correction and the Stuttgart–Dresden (SDD) effective core potentials for Pd(II); see ESI† for details) which were performed to rank the cis (C) and trans (D) isomers in terms of their relative energies (Table S1†). These calculations suggested that the trans-Pd2L4 cage was the thermodynamically favoured species by 6.1 kJ mol−1 (Fig. 3), and thus the likely product. The calculated structures revealed significant differences in the C⋯C distance between methyl groups in the two isomers (3.7 Å for the cis, 4.6 Å for the trans), corroborating the hypothesis that steric interactions between the methyl groups drive the observed isomeric bias.
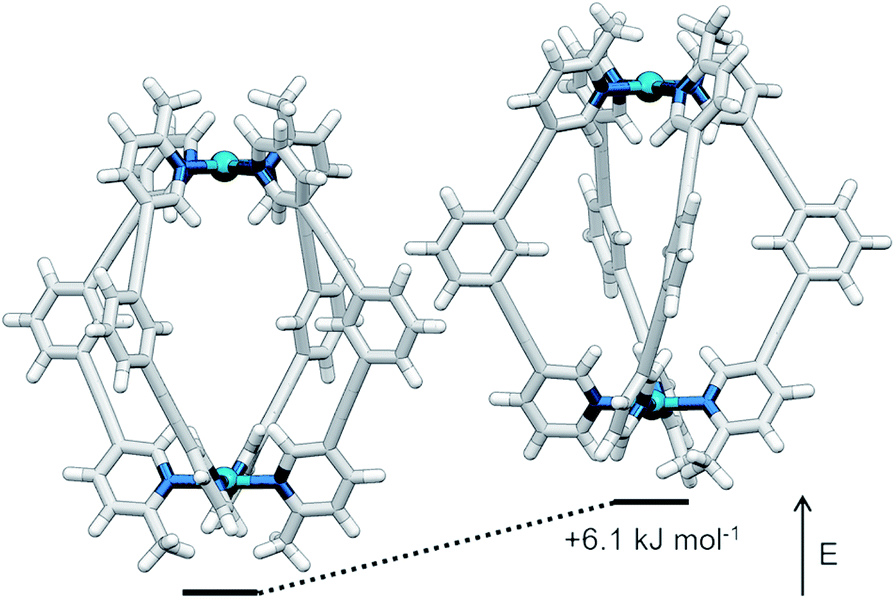 | ||
| Fig. 3 Calculated structures (B3LYP/6-31G(d)/SDD) and relative energies of trans-[Pd2(1)4]4+ (left) and cis-[Pd2(1)4]4+ (right). | ||
Interestingly, when the self-assembly was repeated in d6-DMSO, two species were formed in an approximately 2:3 ratio (Fig. 2b). Continued heating at 60 °C for a prolonged period failed to further simplify the spectrum and ultimately led to degradation of the sample. Both species displayed the same diffusion coefficient (Fig. 2c) which, combined with MS data, indicated exclusive formation of Pd2L4 assemblies. In this instance, however, a mixture of two major isomers appeared to have formed. ROESY and TOCSY NMR (Fig. S74 and S75,† respectively) were used to assign signals as much as possible to the individual species; the former revealed cross-peaks between Hb and Hl for both, indicating that the mixture was composed of the cis (C) and trans (D) cages.
DFT calculations in implicit DMSO and MeCN (Table S1†) showed no difference in the relative energies of the cis and trans isomers in the two solvents, with the trans architecture remaining lower in energy. Unsurprisingly the calculations did show the higher energy cis structure to be the more polar of the two (based on a larger dipole moment from the DFT optimised structure; Table S1†), and it seems plausible that the difference in speciation observed between the two solvents could be related to this. In less polar acetonitrile, the more energetically favourable of the two cage isomers, i.e. trans-[Pd2(1)4](BF4)4, is formed exclusively; in DMSO the more polar cis-[Pd2(1)4](BF4)4 is stabilised by the increased polarity of the solvent.21 In the absence of more persuasive evidence, however, we remain cautious in our explanation of the observed effect on speciation in different solvents.
Thus we have shown that it is possible to use simple steric constraints, in this instance the inclusion of a single methyl group, to control conformational bias in the self-assembly of an unsymmetrical ligand. It was also discovered that this particular system is susceptible to the environment in which it is placed. In this instance, changing the solvent had a dramatic effect on the ratio of cage isomers observed at equilibrium. This sort of solvent-responsive ligand rearrangement in metallo-supramolecular systems is relatively uncommon22 and, once better understood, could potentially be exploited for adaptable constructs that change conformation upon exposure to different environments.23
Geometric complementarity
Subsequently we looked to assemble lower symmetry systems in which the design of the ligand geometry would enforce the assembly of a cis-Pd2L4 cage. To this end ligand 2 was prepared incorporating one isoquinoline and one pyridyl donor (Fig. 4a). This ligand was designed such that the planes orthogonal to the donor nitrogen atoms were no longer coincident with one another. Thus formation of a Pd2L4 species, entropically more favourable than assemblies of higher nuclearity, should occur with ligands arranged in a cis fashion (Fig. 4a).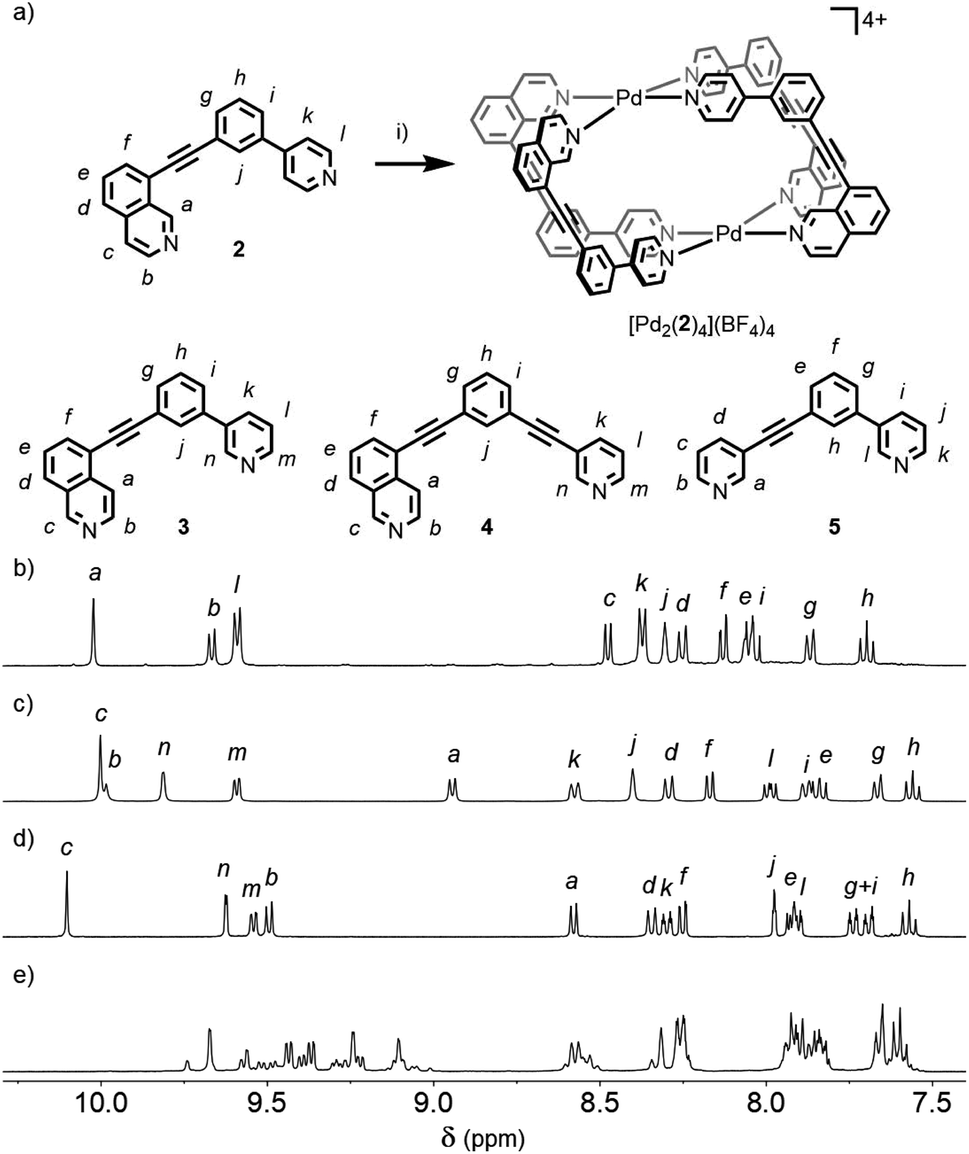 | ||
| Fig. 4 (a) Formation of the cis-Pd2L4 cage [Pd2(2)4](BF4)4 and structures of ligands 3, 4 and 5. Reagents and conditions: (i) [Pd(CH3CN)4](BF4)2, d6-DMSO, rt, 2 h. 1H NMR spectra (400 MHz, 298 K, d6-DMSO) of (b) [Pd2(2)4](BF4)4, (c) [Pd2(3)4](BF4)4, (d) [Pd2(4)4](BF4)4 and (e) equilibrated mixture of 5 and [Pd(CH3CN)4](BF4)2. | ||
Mixing 2 in d6-DMSO with [Pd(CH3CN)4](BF4)2 resulted in clean formation of a single cage, [Pd2(2)4](BF4)4, within 2 h at room temperature. The identity of the assembly was confirmed by DOSY NMR (RH = 8.7 Å) and MS, and specific formation of the cis isomer (C) demonstrated in the solid state by single-crystal X-ray diffraction (SCXRD) (Fig. 5a). In the solid state the cage cavity was found to encapsulate two BF4− anions (Fig. 5b), with the remaining counterions occupying exohedral sites adjacent to the Pd(II) ions of the cage. To probe how significant the deviation from co-planarity the donor atoms needed to be to ensure quantitative assembly to a single cis-Pd2L4 species, the series of ligands 3, 4 and 5 (Fig. 4a) was synthesised for comparison.
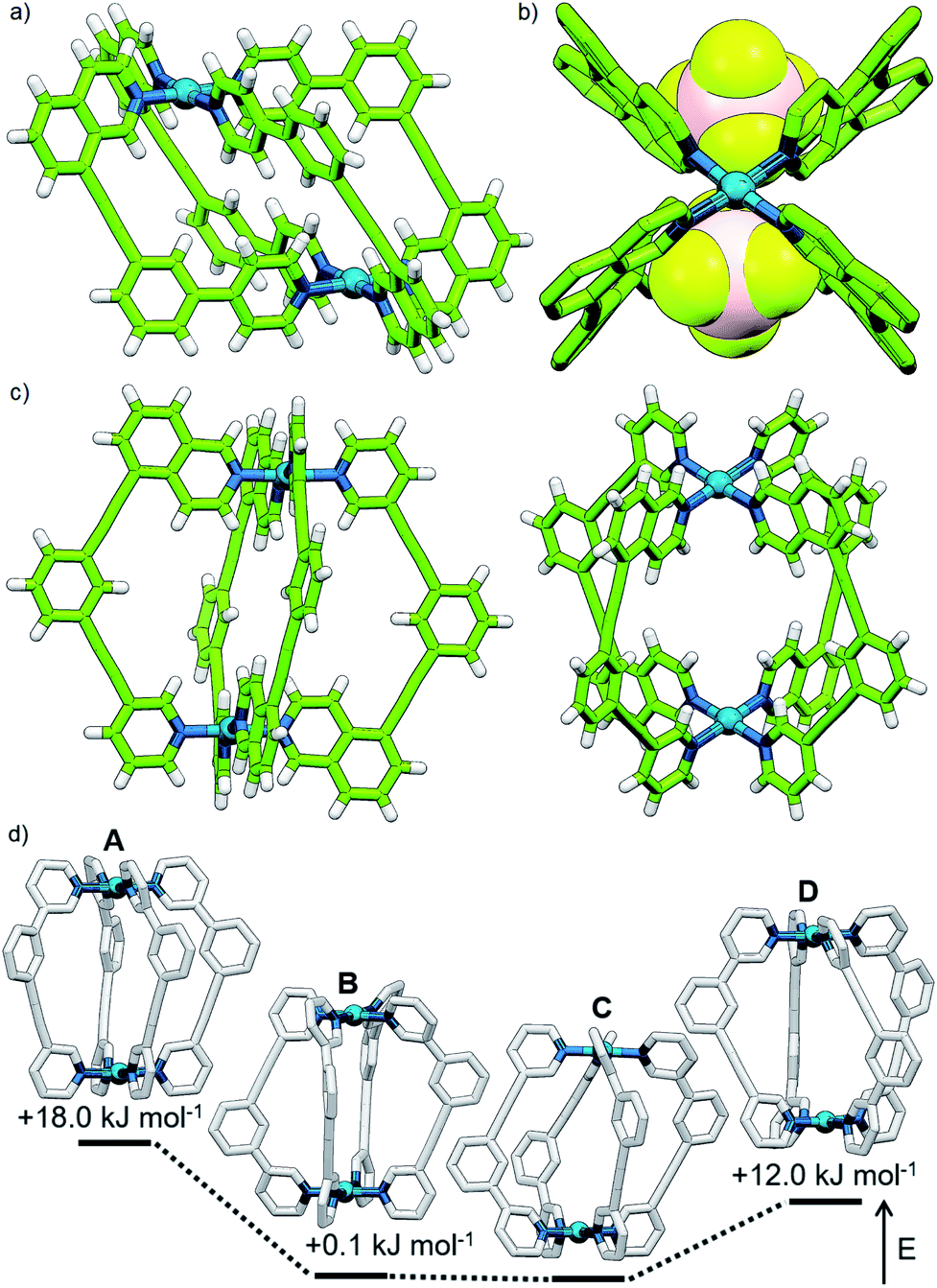 | ||
| Fig. 5 Solid state structure of cis-[Pd2(2)4](BF4)4 viewed (a) from the side and (b) down the Pd–Pd axis with encapsulated BF4− anions shown (hydrogen atoms omitted for clarity). Bond lengths (Å): Pd–Npyridine 1.990(17)/2.069(2); Pd–Nisoquinoline 2.029(3)/2.040(3). (c) Solid state structure of cis-[Pd2(4)4](BF4)4. Bond lengths (Å): Pd–Npyridine 2.044(8)/2.056(8); Pd–Nisoquinoline 2.006(9)/2.007(8). (d) Calculated structures (B3LYP/6-31G(d)/SDD) of the A, B, C and D isomers of [Pd2(5)4]4+ with energies relative to C. Hydrogen atoms have been omitted for clarity. | ||
In the case of ligand 3, an isomer of 2, addition of 0.5 equivalents of [Pd(CH3CN)4](BF4)2 in d6-DMSO also resulted in clean formation of a single Pd2L4 species within 2 h at room temperature as observed by NMR (Fig. 4c) and MS. For ligand 4 equilibration was slower; however, within 24 h a single species was formed cleanly (Fig. 4d) and the expected Pd2L4 structure with cis ligand arrangement confirmed in the solid state by SCXRD (Fig. 5c). DFT calculations (Table S2†) supported the cis-[Pd2(4)4]4+ isomer as being the lowest energy species by 4.4–26.8 kJ mol−1 compared to the other three potential isomers. The “all-up” A configuration was determined to be the highest energy isomer, followed by the transD assembly, with isomer B, in which three of the ligands are orientated in the same direction, the second lowest energy structure.
For 5, however, even after prolonged heating at 60 °C in d6-DMSO,24 the 1H NMR spectrum (Fig. 4e) failed to converge into a single set of signals. DOSY NMR (Fig. S117†) indicated that the assemblies formed were of a similar size, with the calculated RH congruent with Pd2L4 architectures (8.6 Å), suggesting the formation of a mixture of dinuclear cage isomers.
The calculated structural isomers of [Pd2(5)4]4+ (Fig. 5d; Table S2†) exhibited the same trend in relative energies as with [Pd2(4)4]4+, i.e.C < B < D < A. In contrast, however, the D isomer was higher in relative energy (12.0 kJ mol−1) whilst the difference in energies between the B and C isomers was not found to be significant (0.1 kJ mol−1), indicating that a mixture of at least the B and C isomers would potentially result from the equilibrium mixture of 5 and Pd(II). Isomer B would also give the most complicated NMR spectrum due to possessing three different ligand environments. As such a mixture of isomers B and C would be expected to give a complex spectrum in accord with the observed NMR data; unfortunately severely overlapping signals prohibited detailed analysis of the equilibrated mixture.
Combined steric and geometric control
Although the geometric constraint of ligand 5 was insufficient to give clean formation of a single cage isomer, it was hypothesised that in combination with a steric factor the B isomer could become sufficiently raised in energy to allow selective formation of the cis (C) assembly. To this end, ligand 6 (Fig. 6a) was synthesised in which one of the pyridyl moieties was replaced with a 2-picolyl unit; pleasingly, upon combination with Pd(II) ions, a single species was observed to form by 1H NMR in d6-DMSO within 24 h (Fig. 6b). 1H, ROESY and DOSY (RH = 8.7 Å) NMR (Fig. 6b, S128 and S129,† respectively) and MS data all indicated that either the cis- or trans-[Pd2(6)4](BF4)4 cage was formed as the sole product of self-assembly. As the cis (C) and trans (D) isomers could not be distinguished spectroscopically, their relative energies were calculated by DFT (Table S2†), with the cis assembly (Fig. 6c and d) determined to be the lower energy isomer by 12.2 kJ mol−1. We have therefore shown that it is possible to augment a particular geometric ligand design with a sterically influencing group to drive selective formation of a single species when one factor exerts insufficient bias by itself.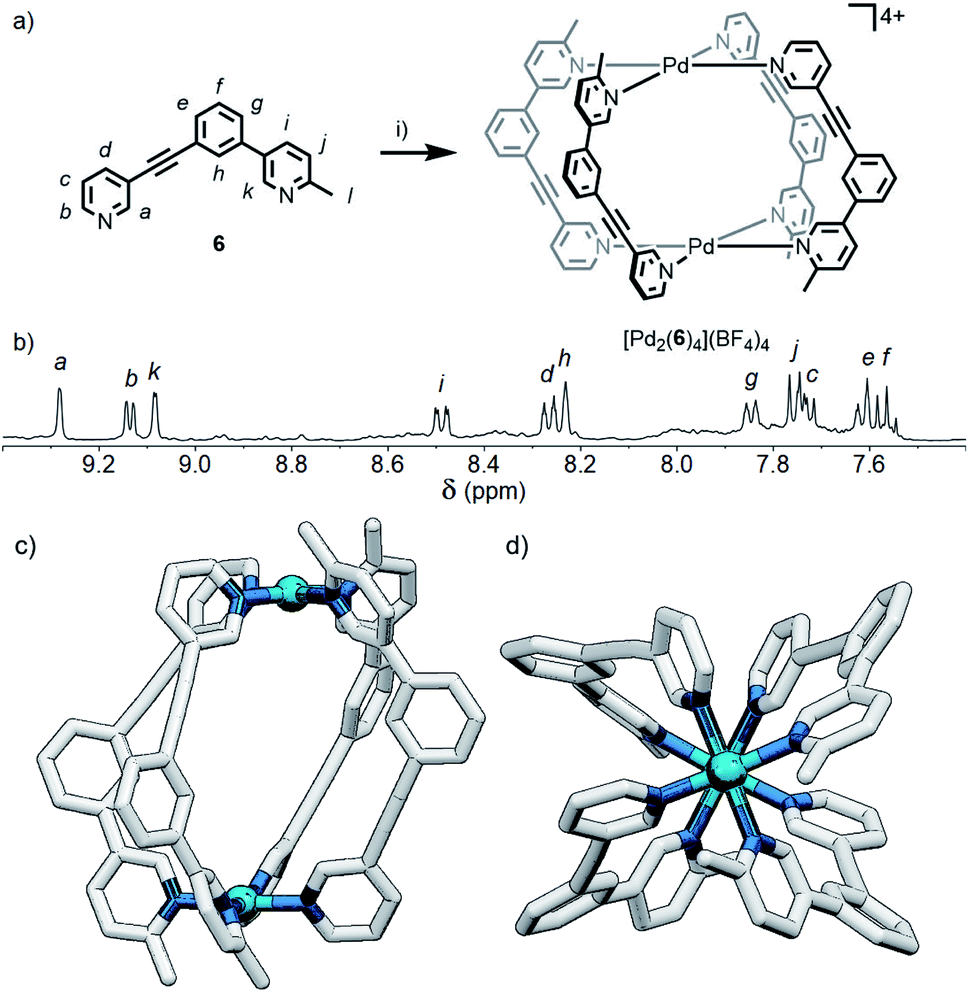 | ||
| Fig. 6 (a) Formation of the cis-Pd2L4 cage [Pd2(6)4](BF4)4. Reagents and conditions: (i) [Pd(CH3CN)4](BF4)2, d6-DMSO, rt, 24 h. (b) Partial 1H NMR spectrum (400 MHz, 298 K, d6-DMSO) of [Pd2(6)4](BF4)4. Calculated structure (B3LYP/6-31G(d)/SDD) of cis-[Pd2(6)4]4+ viewed from (c) the side, and (d) down the Pd–Pd axis; hydrogen atoms have been omitted for clarity. | ||
Effect of steric directing groups on cage conformation
Ligands 5 and 6 differ only in the presence of a single methyl group, yet this small change meant the difference between formation of a single cage isomer or a mixture of isomers upon complexation with Pd(II). A closer examination of the calculated structures of cis-[Pd2(5)4]4+ and cis-[Pd2(6)4]4+ revealed that the steric bulk of the methyl groups also affected the conformation of the ligands and of the cages as a whole (see ESI; Table S3†).25 The twist of the alkyne units (ϕ, the torsion angle across the alkyne bond; Fig. S137a and b†) was found to be increased between cis-[Pd2(5)4]4+ and cis-[Pd2(6)4]4+, and is at least partially responsible for the observed difference in the average ligand twist (θ; defined as the torsion angle between the two Pd–N bonds of an individual ligand, Fig. S137c†): ∼27° for cis-[Pd2(5)4]4+ and ∼41° for cis-[Pd2(6)4]4+, resulting in a larger helical twist. Surprisingly, the increased helicity of cis-[Pd2(6)4]4+ compared to cis-[Pd2(5)4]4+ does not significantly alter the diameter of the largest theoretical sphere able to fit in the cavity of the cage26 (Table S3;† this result may be a limitation of using a spherical probe to analyse intrinsic porosity as pore shapes become more anisotropic).A similar effect on cage conformation could be seen by comparing the calculated structure of trans-[Pd2(1)4]4+ with previously reported27 SCXRD structures of the Pd2L4 complex of the unsubstituted ligand (1,3-bis(pyridin-3-ylethynyl)benzene, L). The latter [Pd2(L)4]4+ species adopt a pseudo-D4h symmetry with minimal twisting of the alkyne units (ϕaverage ≈ 9°) and of the ligands (θ ≤ 1°) which contrasts with the greater distortion seen in the calculated structure of trans-[Pd2(1)4]4+ (ϕ ≈ 17–30°; θ ≈ 42°) (Table S3†). Again, no significant reduction in the calculated cavity size of trans-[Pd2(1)4]4+ was observed.
These structural distortions induced by the steric encumbrance of the methyl groups suggests that relatively minor modifications to the ligand framework could be used to dramatically alter the assembly conformation, allowing precision engineering of the internal cavity space without any significant loss in intrinsic porosity, a concept we are currently exploring.
Conclusions
Metallo-supramolecular systems have evolved in complexity since their initial realisation; heteroleptic, functionalised and even interlocked assemblies28 have been reported. Despite the significant advances made in the chemists' toolbox of techniques for designing these systems, symmetrical ligands still tend to be employed to simplify the self-assembly process. In this work we have shown that it is possible, through careful ligand design, to exploit steric or geometric factors, or a combination of the two, to assemble unsymmetrical ditopic ligands with “naked” palladium(II) ions, forming single Pd2L4 cage isomers with high fidelity. This approach does not require the incorporation of ligand motifs of different denticity, nor the use of substantially bulky moieties, both of which can introduce exorbitant complexity into the ligand framework and make them synthetically very taxing, inhibiting general utility.Structure calculations have been invaluable in helping to explain the outcomes of the self-assembly processes examined. Following on from this work we expect to be able to exploit these as a predictive tool to forecast successful outcomes, defined as the self-assembly of a single cage isomer that is sufficiently lower in energy than other possible structural isomers as to be the only spectroscopically detectable product.
Ongoing work in our lab will also look to utilise the self-assembly principles delineated through this work to prepare multi-functional systems in which moieties are held in specific arrangements relative to each other. It is hoped that in this manner functionalities within the cage cavities may be spatially segregated, a concept we envisage exploiting for a variety of applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
JEML thanks Imperial College London for an Imperial College Research Fellowship, and the Royal Society for a Research Grant (RG170321). AT thanks the Royal Society Enhancement Award 2018 and KEJ thanks the Royal Society for a University Research Fellowship and the ERC through Agreement Number 758370 (ERC-StG-PE5-CoMMaD). Peter Haycock is thanked for assistance with the collection of NMR data. Dr Lisa Haigh is thanked for the collection of MS data. Professor Erin R. Johnson is thanked for useful discussions. Professor Matthew J. Fuchter is thanked for useful discussions and access to equipment and resources.References
- S. E. Ahnert, J. A. Marsh, H. Hernández, C. V. Robinson and S. A. Teichmann, Science, 2015, 350, aaa2245 CrossRef PubMed.
- M. Fujita, Chem. Soc. Rev., 1998, 27, 417 RSC; B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef CAS PubMed; S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2002, 100, 853–908 CrossRef PubMed; R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef PubMed; M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC; T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef PubMed.
- J.-M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed.
- M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 509–518 RSC; M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed; K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS PubMed; B. H. Northrop, Y. Zheng, K. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554–1563 CrossRef PubMed.
- A notable exception includes the work of Schmittel and co-workers, who have extensively studied the self-assembly of unsymmetrical ligands incorporating orthogonal metal-coordination sites to prepare a range of metallo-macrocycles. For select examples, see: K. Mahata and M. Schmittel, J. Am. Chem. Soc., 2009, 131, 16544–16554 CrossRef CAS PubMed; K. Mahata, M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2010, 132, 15933–15935 CrossRef PubMed; M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2013, 135, 17743–17746 CrossRef PubMed; M. L. Saha, N. Mittal, J. W. Bats and M. Schmittel, Chem. Commun., 2014, 50, 12189–12192 RSC; N. Mittal, M. L. Saha and M. Schmittel, Chem. Commun., 2015, 51, 15514–15517 RSC . For reviews, see: S. De, K. Mahata and M. Schmittel, Chem. Soc. Rev., 2010, 39, 1555–1575 RSC; M. L. Saha, S. Neogi and M. Schmittel, Dalton Trans., 2014, 43, 3815–3834 RSC.
- D. A. McMorran and P. J. Steel, Angew. Chem., Int. Ed., 1998, 37, 3295–3297 CrossRef CAS; P. J. Steel and D. A. McMorran, Chem.–Asian J., 2019, 14, 1098–1101 CrossRef PubMed.
- For reviews, see: M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC; A. Schmidt, A. Casini and F. E. Kühn, Coord. Chem. Rev., 2014, 275, 19–36 CrossRef CAS; S. Saha, I. Regeni and G. H. Clever, Coord. Chem. Rev., 2018, 374, 1–14 CrossRef . For select examples, see: D. K. Chand, K. Biradha and M. Fujita, Chem. Commun., 2001, 1, 1652–1653 RSC; G. H. Clever, S. Tashiro and M. Shionoya, Angew. Chem., Int. Ed., 2009, 48, 7010–7012 CrossRef PubMed; J. D. Crowley and E. L. Gavey, Dalton Trans., 2010, 39, 4035–4037 RSC; J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC; N. Kishi, Z. Li, K. Yoza, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2011, 133, 11438–11441 CrossRef PubMed; C. Desmarets, G. Gontard, A. L. Cooksy, M. N. Rager and H. Amouri, Inorg. Chem., 2014, 53, 4287–4294 CrossRef PubMed; S. M. Jansze, M. D. Wise, A. V. Vologzhanina, R. Scopelliti and K. Severin, Chem. Sci., 2017, 8, 1901–1908 RSC; K. Matsumoto, S. Kusaba, Y. Tanaka, Y. Sei, M. Akita, K. Aritani, M. Haga and M. Yoshizawa, Angew. Chem., Int. Ed., 2019, 8463–8467 CrossRef PubMed; T. R. Schulte, J. J. Holstein and G. H. Clever, Angew. Chem., Int. Ed., 2019, 58, 5562–5566 CrossRef PubMed.
- A. Casini, B. Woods and M. Wenzel, Inorg. Chem., 2017, 56, 14715–14729 CrossRef CAS PubMed; S. M. McNeill, D. Preston, J. E. M. Lewis, A. Robert, K. Knerr-Rupp, D. O. Graham, J. R. Wright, G. I. Giles and J. D. Crowley, Dalton Trans., 2015, 44, 11129–11136 RSC; D. Preston, S. M. McNeill, J. E. M. Lewis, G. I. Giles and J. D. Crowley, Dalton Trans., 2016, 45, 8050–8060 RSC; A. Ahmedova, D. Momekova, M. Yamashina, P. Shestakova, G. Momekov, M. Akita and M. Yoshizawa, Chem.–Asian J., 2016, 11, 474–477 CrossRef PubMed; A. Ahmedova, R. Mihaylova, D. Momekova, P. Shestakova, S. Stoykova, J. Zaharieva, M. Yamashina, G. Momekov, M. Akita and M. Yoshizawa, Dalton Trans., 2016, 45, 13214–13221 RSC; R. A. S. Vasdev, L. F. Gaudin, D. Preston, J. P. Jogy, G. I. Giles and J. D. Crowley, Front. Chem., 2018, 6, 563 CrossRef PubMed.
- V. Martí-Centelles, A. L. Lawrence and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 2862–2868 CrossRef PubMed.
- D. Preston, K. F. White, J. E. M. Lewis, R. A. S. Vasdev, B. F. Abrahams and J. D. Crowley, Chem.–Eur. J., 2017, 23, 10559–10567 CrossRef CAS PubMed.
- W. M. Bloch and G. H. Clever, Chem. Commun., 2017, 53, 8506–8516 RSC; S. Pullen and G. H. Clever, Acc. Chem. Res., 2018, 51, 3052–3064 CrossRef CAS; D. Bardhan and D. K. Chand, Chem.–Eur. J., 2019, 25, 12241–12269 CrossRef PubMed.
- (a) D. Preston, J. E. Barnsley, K. C. Gordon and J. D. Crowley, J. Am. Chem. Soc., 2016, 138, 10578–10585 CrossRef CAS PubMed; (b) R. Zhu, W. M. Bloch, J. J. Holstein, S. Mandal, L. V. Schäfer and G. H. Clever, Chem.–Eur. J., 2018, 24, 12976–12982 CrossRef CAS PubMed.
- W. M. Bloch, Y. Abe, J. J. Holstein, C. M. Wandtke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2016, 138, 13750–13755 CrossRef CAS PubMed; W. M. Bloch, J. J. Holstein, W. Hiller and G. H. Clever, Angew. Chem., Int. Ed., 2017, 56, 8285–8289 CrossRef PubMed.
- S. Saha, B. Holzapfel, Y. T. Chen, K. Terlinden, P. Lill, C. Gatsogiannis, H. Rehage and G. H. Clever, J. Am. Chem. Soc., 2018, 140, 17384–17388 CrossRef CAS PubMed.
- K. W. Chi, C. Addicott, A. M. Arif and P. J. Stang, J. Am. Chem. Soc., 2004, 126, 16569–16574 CrossRef CAS PubMed; K. W. Chi, C. Addicott, M. E. Moon, H. J. Lee, S. C. Yoon and P. J. Stang, J. Org. Chem., 2006, 71, 6662–6665 CrossRef; S. Ghosh, D. R. Turner, S. R. Batten and P. S. Mukherjee, J. Chem. Soc., Dalton Trans., 2007, 1869–1871 RSC; L. Zhao, B. H. Northrop, Y. R. Zheng, H. B. Yang, J. L. Hyo, M. L. Young, Y. P. Joo, K. W. Chi and P. J. Stang, J. Org. Chem., 2008, 73, 6580–6586 CrossRef PubMed; Y. Zheng, B. H. Northrop, H. Yang, L. Zhao and P. J. Stang, J. Org. Chem., 2009, 74, 3554–3557 CrossRef PubMed; A. K. Bar, R. Chakrabarty, K. W. Chi, S. R. Batten and P. S. Mukherjee, J. Chem. Soc., Dalton Trans., 2009, 3222–3229 RSC; S. Ghosh and P. S. Mukherjee, Inorg. Chem., 2009, 48, 2605–2613 CrossRef PubMed; C. Reactions, H. Wang, R. Zhong, X. Q. Guo, X. Y. Feng and X. F. Hou, Eur. J. Inorg. Chem., 2010, 174–178 Search PubMed; A. K. Bar, R. Chakrabarty, H. M. Lee and P. S. Mukherjee, Inorg. Chim. Acta, 2011, 372, 313–320 CrossRef; S. Ghosh, O. Mendoza, L. Cubo, F. Rosu, V. Gabelica, A. J. P. White and R. Vilar, Chem.–Eur. J., 2014, 20, 4772–4779 CrossRef PubMed; O. Jurček, P. Bonakdarzadeh, E. Kalenius, J. M. Linnanto, M. Groessl, R. Knochenmuss, J. A. Ihalainen and K. Rissanen, Angew. Chem., Int. Ed., 2015, 54, 15462–15467 CrossRef; A. Torres-Huerta, J. Cruz-Huerta, H. Höpfl, L. G. Hernández-Vázquez, J. Escalante-García, A. Jiménez-Sánchez, R. Santillan, I. F. Hernández-Ahuactzi and M. Sánchez, Inorg. Chem., 2016, 55, 12451–12469 CrossRef PubMed; X. Gao, A. Y. Fu and Y. Y. Wang, Inorg. Chem., 2016, 55, 4330–4334 CrossRef PubMed; E. H. Wi, J. Y. Ryu, S. G. Lee, U. Farwa, M. Pait, S. Lee, S. Cho and J. Lee, Inorg. Chem., 2019, 58, 11493 CrossRef PubMed; D. Preston, A. R. Inglis, A. L. Garden and P. E. Kruger, Chem. Commun., 2019, 55, 13271–13274 RSC.
- S. Hiraoka and M. Fujita, J. Am. Chem. Soc., 1999, 121, 10239–10240 CrossRef CAS; X. P. Zhou, J. Liu, S. Z. Zhan, J. R. Yang, D. Li, K. M. Ng, R. W. Y. Sun and C. M. Che, J. Am. Chem. Soc., 2012, 134, 8042–8045 CrossRef PubMed; X. P. Zhou, Y. Wu and D. Li, J. Am. Chem. Soc., 2013, 135, 16062–16065 CrossRef; S. Bala, A. Goswami, S. Sengupta, S. Ganguly, S. Bhattacharya, S. Khanra and R. Mondal, Cryst. Growth Des., 2013, 13, 5068–5075 CrossRef; S. Gaikwad, M. Lal Saha, D. Samanta and M. Schmittel, Chem. Commun., 2017, 53, 8034–8037 RSC; D. Preston, J. J. Sutton, K. C. Gordon and J. D. Crowley, Angew. Chem., Int. Ed., 2018, 57, 8659–8663 CrossRef PubMed; S. K. Sen and R. Natarajan, Inorg. Chem., 2019, 58, 7180–7188 CrossRef PubMed; X. Zhang, X. Dong, W. Lu, D. Luo, X.-W. Zhu, X. Li, X.-P. Zhou and D. Li, J. Am. Chem. Soc., 2019, 141, 11621–11627 CrossRef PubMed; D. Preston, A. R. Inglis, J. D. Crowley and P. E. Kruger, Chem.–Asian J., 2019, 14, 3404–3408 CrossRef PubMed.
- D. Ogata and J. Yuasa, Angew. Chem., Int. Ed. DOI:10.1002/anie.201911097.
- Due to the volatility of 2-methyl-5-ethynylpyridine, Sonogashira reactions with non-commercially available alkynes were performed with in situ deprotection of trimethylsilylacetylenes based on a reported literature procedure: M. J. Mio, L. C. Kopel, J. B. Braun, T. L. Gadzikwa, K. L. Hull, R. G. Brisbois, C. J. Markworth and P. A. Grieco, Org. Lett., 2002, 4, 3199–3202 CrossRef CAS PubMed.
- Small additional peaks in the NMR spectra are assumed to arise from the presence of minor cage isomers at equilibrium.
- M. Hutin, G. Bernardinelli and J. R. Nitschke, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17655–17660 CrossRef CAS PubMed.
- M. Fujita, F. Ibukuro, H. Hagihara and K. Ogura, Nature, 1994, 367, 720–723 CrossRef CAS; P. N. W. Baxter, R. G. Khoury, J. M. Lehn, G. Baum and D. Fenske, Chem.–Eur. J., 2000, 6, 4140–4148 CrossRef; S. J. Park, D. M. Shin, S. Sakamoto, K. Yamaguchi, Y. K. Chung, M. S. Lah and J. I. Hong, Chem.–Eur. J., 2005, 11, 235–241 CrossRef; K. Suzuki, M. Kawano and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 2819–2822 CrossRef; J. Ramírez, A. M. Stadler, N. Kyritsakas and J. M. Lehn, Chem. Commun., 2007, 237–239 RSC; J. Heo, Y. M. Jeon and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 7712–7713 CrossRef; B. Kilbas, S. Mirtschin, R. Scopelliti and K. Severin, Chem. Sci., 2012, 3, 701–704 RSC; D. M. Weekes, C. Diebold, P. Mobian, C. Huguenard, L. Allouche and M. Henry, Chem.–Eur. J., 2014, 20, 5092–5101 CrossRef; O. Gidron, M. Jirásek, N. Trapp, M. O. Ebert, X. Zhang and F. Diederich, J. Am. Chem. Soc., 2015, 137, 12502–12505 CrossRef PubMed.
- W. Wang, Y. X. Wang and H. B. Yang, Chem. Soc. Rev., 2016, 45, 2656–2693 RSC.
- Similar results were observed in CD3CN: see ESI† for details.
- Ligand distortion parameters were adapted from: T. A. Young, V. Martí-Centelles, J. Wang, P. J. Lusby and F. Duarte, ChemRxiv Preprint, 2019, DOI:10.26434/chemrxiv.9576644.
- M. Miklitz and K. E. Jelfs, J. Chem. Inf. Model., 2018, 58, 2387–2391 CrossRef CAS PubMed.
- P. Liao, B. W. Langloss, A. M. Johnson, E. R. Knudsen, F. S. Tham, R. R. Julian and R. J. Hooley, Chem. Commun., 2010, 46, 4932–4934 RSC; D. P. August, G. S. Nichol and P. J. Lusby, Angew. Chem., Int. Ed., 2016, 55, 15022–15026 CrossRef CAS.
- M. Frank, M. D. Johnstone and G. H. Clever, Chem.–Eur. J., 2016, 22, 14104–14125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data, NMR spectra, crystallographic details and computational methods. CCDC 1946272 and 1946273. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05534g |
| This journal is © The Royal Society of Chemistry 2020 |