
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral cis-iron(II) complexes with metal- and ligand-centered chirality for highly regio- and enantioselective alkylation of N-heteroaromatics†
Jinhu
Wei
 a,
Bei
Cao
a,
Chun-Wai
Tse
a,
Xiao-Yong
Chang
b,
Cong-Ying
Zhou
a and
Chi-Ming
Che
*abc
a,
Bei
Cao
a,
Chun-Wai
Tse
a,
Xiao-Yong
Chang
b,
Cong-Ying
Zhou
a and
Chi-Ming
Che
*abc
aState Key Laboratory of Synthetic Chemistry, Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong SAR, China. E-mail: cmche@hku.hk
bDepartment of Chemistry, Southern University of Science and Technology, Shenzhen, Guangdong 518055, China
cHKU Shenzhen Institute of Research & Innovation, Shenzhen 518055, China
First published on 25th November 2019
Abstract
Iron-catalyzed highly regio- and enantioselective organic transformations with generality and broad substrate scope have profound applications in modern synthetic chemistry; an example is herein described based on cis-FeII complexes having metal- and ligand-centered chirality. The cis-β FeII(N4) complex [FeII(L)(OTf)2] (L = N,N′-bis(2,3-dihydro-1H-cyclopenta-[b]quinoline-5-yl)-N,N′-dimethylcyclohexane-1,2-diamine) is an effective chiral catalyst for highly regio- and enantioselective alkylation of N-heteroaromatics with α,β-unsaturated 2-acyl imidazoles, including asymmetric N1, C2, C3 alkylations of a broad range of indoles (34 examples) and alkylation of pyrroles and anilines (14 examples), all with high product yields (up to 98%), high enantioselectivity (up to >99% ee) and high regioselectivity. DFT calculations revealed that the “chiral-at-metal” cis-β configuration of the iron complex and a secondary π–π interaction are responsible for the high enantioselectivity.
Introduction
The development of highly enantioselective metal-catalyzed reactions usually relies on the design of robust ligand scaffolds containing tetrahedral chiral, axial chiral or planar chiral centers positioned close to the metal ion for effective stereo-differentiation of incoming/coordinated substrate(s). Recently, Meggers and co-workers reported that optically active complexes with metal-centered chirality including iron complexes are potent catalysts for enantioselective organic transformation reactions.1 A challenge in developing these complexes for asymmetric catalysis, particularly over first-row transition metal complexes that are relatively substitutionally labile, is the potential racemization of the stereogenic metal center.2b In this regard, the use of multidentate ligands such as the tetradentate N4 ligands used in this work could make the resulting complexes more configurationally stable.2a,c Acyclic tetradentate ligands form stable octahedral complexes with three possible topologies: trans, cis-α, and cis-β configurations.2a,b Complexes with the cis-α and cis-β configuration feature a stereogenic metal center and a pair of cis-oriented vacant sites for binding the incoming substrate in a η2 fashion. Evans and co-workers reported a chiral cis-β AlIII-salen complex as an effective catalyst for enantioselective aldol reactions of aldehydes and 5-alkoxyoxazoles.3 Yamamoto and co-workers developed a cis-β AlIII catalyst with a tethered bis(8-quinolinolato) (N2O2) ligand for various highly enantioselective organic transformations.4 Chiral FeII complexes supported by tetradentate N4 ligands have been shown to exhibit remarkable catalytic activities in asymmetric hydrocarbon oxidations.5 In a recent study, we showed that the chiral Fe(N4) complexes with bis(quinolyl)diamine ligands can efficiently catalyze highly enantioselective cis-dihydroxylation of alkenes with H2O2 (up to 99.8% ee).6 These FeII complexes possess metal- and ligand-centered chirality with a rigid quinoline-based ligand scaffold and therefore a robust chiral pocket, which is in close proximity to the iron center, for discriminative activation/binding of the incoming reagent/substrate and subsequent reactions. We envision that the chiral pocket together with the Lewis acidity of the FeII/FeIII ion would make chiral Fe(N4) complexes potent chiral Lewis acid catalysts.7,8Chiral indoles are ubiquitous structural motifs in bioactive natural products and pharmaceuticals.9 Over the past few decades, numerous efforts have been dedicated to developing catalytic asymmetric reactions for the synthesis of these compounds.10 As the C3 position of indoles is more reactive than the C2 and N1 positions, many elegant methodologies have been reported for the construction of chiral C3-alkylated indoles.11 In contrast, enantioselective alkylations at the C2 and N1 positions of indoles are limited. For asymmetric C2 alkylation of indoles, the literature examples were realized through the Pictet–Spengler reaction,12 alkylation/oxidation of 4,7-dihydroindoles,11c,13 Michael addition14,15 and other methods.16 So far, examples of highly enantioselective N1 alkylation of indoles have been sparse.17 In view of the prevalence of chiral indoles in pharmaceuticals and the stringent constraint on trace noble metal levels in the pharmaceutical industry, the development of catalysts based on earth-abundant, biocompatible iron metal is appealing. Herein is described highly regio- and enantioselective C3, C2 and N1 alkylation of indoles, as well as alkylation of pyrroles and anilines, with α,β-unsaturated 2-acyl imidazoles catalyzed by chiral cis-iron(II) complexes supported by tetradentate bis(quinolyl)diamine ligands.
Results and discussion
Synthesis and characterization of the catalyst
Following a previously reported method for the synthesis of complexes 1a,6,18a1b and 1c,6 chiral Fe(II) complexes 1a–1f were synthesized, by the reaction of FeII(OTf)2·(CH3CN)2 with a series of chiral bis(quinolyl)diamine N4 ligands 2a–2f, in 80–95% yields (Scheme 1). The new complexes 1d–1f were characterized by ESI-MS, elemental analysis and circular dichroism spectroscopy (ESI†). The structures of 1e and 1f, featuring fused cyclopentane and cyclohexane rings on the quinoline moiety, respectively, were determined by X-ray crystallography, revealing a cis-α configuration for 1f, similar to that for 1b·(H2O)2,6 but, unexpectedly, a cis-β configuration for 1e. As shown in Fig. 1, 1e adopts a cis-β-Δ coordination geometry, where the Fe–N distances of 2.163(2)–2.242(2) Å are comparable to those of cis-β-[FeII(BQCN)(OTf)2] (BQCN = 2a in Scheme 1) reported by Nam and co-workers (Fe–N 2.125(2)–2.224(2) Å).18b In contrast, 1f adopts a cis-α-Δ configuration (Fig. 1); its Fe–N distances of 2.220(6)–2.292(5) Å are comparable to those of 1b·(H2O)2 (cis-α-Λ; Fe–N 2.195(5)–2.261(5) Å),6 while considerably longer than those of cis-α-[FeII(BQCN)(CH3CN)2](ClO4)2 (Fe–N 2.112(3)–2.192(3) Å),18b attributed to the steric repulsion between the quinolyl substituents and the cis-coordinated ligands. Notably, the two methyl groups on the cyclohexane-1,2-diamine nitrogen atoms are oriented syn to each other in 1e but anti to each other in 1b and 1f. Consistent with the relatively long Fe–N distances, both 1e (μeff = 4.92 μB) and 1f (μeff = 5.44 μB) are high-spin with S = 2 as determined by the Evans method. These complexes are configurationally stable; no change in the circular dichroism (CD) signal was observed upon standing an acetonitrile solution of 1e or 1f at room temperature for up to 4 days (Fig. S1, ESI†). The 1H NMR spectrum of 1e in CD3CN at room temperature showed no significant change after the solution was left standing for 8 days (Fig. S2, ESI†).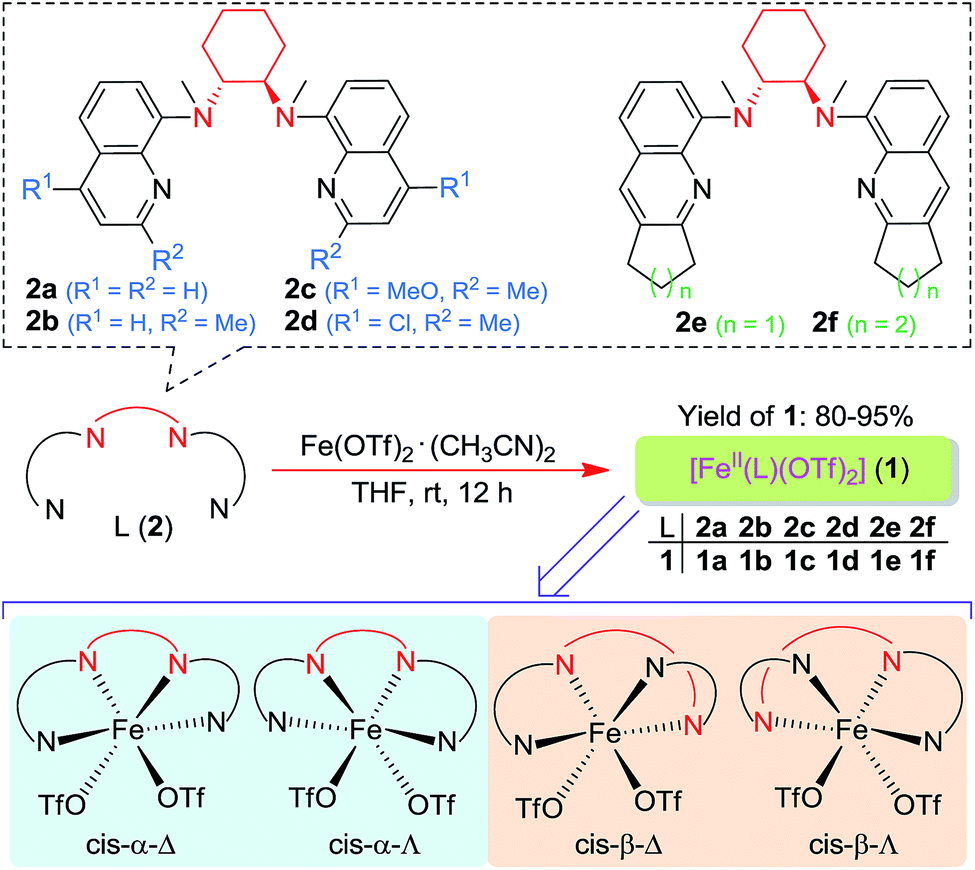 | ||
| Scheme 1 Chiral N4 ligands L (2a–2f) and FeII complexes [FeII(L)(OTf)2] (1a–1f) employed in this work. The bottom shows four possible configurations for such complexes. | ||
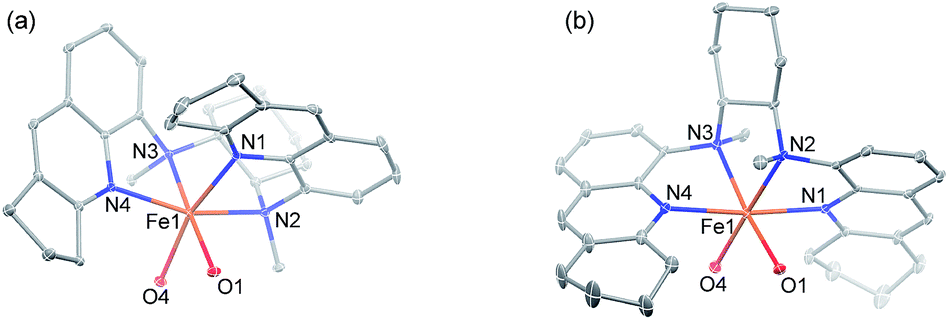 | ||
| Fig. 1 ORTEP drawings of (a) 1e and (b) 1f (30% probability ellipsoids) with the omission of H atoms and OTf− ligands except O atoms bound to FeII. | ||
Catalyst screening
At the outset, the catalytic activity of Fe(II) complexes 1a–1f was examined for C3 alkylation of N-methylindole 3a with α,β-unsaturated 2-acyl imidazole 4a in the presence of molecular sieves (MS) and under argon (under air and/or without MS, the reaction time increased and enantioselectivity reduced, cf. entries 1–3 in Table 1). Complex [FeII(6-Me2-BPMCN)(OTf)2],5a bearing a bis(pyridylmethyl)diamine N4 ligand, was also tested; this complex did not catalyze the alkylation reaction under similar conditions. Revealingly, the reaction of 3a and 4a in the presence of 5 mol% Fe(II) catalysts 1a–1f in CH2Cl2 at 5 °C for 10–15 h (entries 4–9, Table 1) afforded the alkylation product 5a in high yields (87–99%) and with moderate-to-good enantioselectivity (57–85% ee). Complex 1e was found to exhibit the highest catalytic activity and enantioselectivity (entry 8). With 1e as the catalyst, lowering the reaction temperature to −15 °C improved the enantioselectivity to 90% (entry 10). A screening of solvents at this temperature (entries 10–13) revealed that CH3CN gave the highest enantioselectivity of 95% ee. Further lowering the reaction temperature to −25 °C afforded 5a in 91% yield and 97.5% ee (entry 14).|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Solvent/T (°C) | T (h) | % Yb | % eec |
| a Reaction conditions: 3a (0.45 mmol), 4a (0.3 mmol), 1 (5 mol%), 4 Å MS (100 mg) and solvent (3 mL), under argon. b Isolated yield. c Determined by chiral-phase HPLC. d Without 4 Å MS. e Without 4 Å MS, under air. | |||||
| 1 | 1a | CH2Cl2/25 | 3 | 90 | 65 |
| 2d | 1a | CH2Cl2/25 | 4 | 92 | 60 |
| 3e | 1a | CH2Cl2/25 | 5 | 88 | 58 |
| 4 | 1a | CH2Cl2/5 | 10 | 91 | 73 |
| 5 | 1b | CH2Cl2/5 | 10 | 98 | 79 |
| 6 | 1c | CH2Cl2/5 | 15 | 90 | 69 |
| 7 | 1d | CH2Cl2/5 | 12 | 87 | 57 |
| 8 | 1e | CH2Cl2/5 | 10 | 99 | 85 |
| 9 | 1f | CH2Cl2/5 | 10 | 94 | 71 |
| 10 | 1e | CH2Cl2/−15 | 24 | 95 | 90 |
| 11 | 1e | ClCH2CH2Cl/−15 | 24 | 97 | 93 |
| 12 | 1e | CH3Cl/−15 | 24 | 96 | 91 |
| 13 | 1e | CH3CN/−15 | 20 | 92 | 95 |
| 14 | 1e | CH3CN/−25 | 48 | 91 | 97.5 |
Reaction/substrate scope
Under the optimized conditions, we examined the substrate scope of the Fe(II)-catalyzed C3 alkylation of various indoles with α,β-unsaturated 2-acyl imidazoles. As shown in Table 2, in the presence of 5 mol% of 1e, a series of substituted indoles reacted with α,β-unsaturated 2-acyl imidazole 4a and the desired alkylation products 5b–5i were obtained in high yields (87–98%) and with excellent enantioselectivities (95–99% ee). No C2 alkylation product was observed. The sterically hindered 1,2-dimethylindole 3i could also undergo C3 alkylation, giving 5i in 98% yield and 95% ee. The absolute configuration at the stereogenic carbon center of 5i was determined to be R by X-ray crystallography (Fig. 2). The C3 alkylation of N-methyl-5-bromoindole proceeded smoothly at −25 °C to give 5f in 90% yield and 95% ee. However, the presence of the more electron-withdrawing substituent, Cl, on the 5-position of indole was found to deactivate the reaction. This issue was addressed by increasing the temperature to 0 °C, which led to alkylation product 5g in 87% yield and 95% ee. The scope of α,β-unsaturated 2-acyl imidazoles was also examined. As shown in Table 2, various α,β-unsaturated 2-acyl imidazoles are reactive for the C3 alkylation of indoles to give the corresponding products 5j–5m in high yields (87–97%) and enantioselectivities (91–96% ee). Neither β- nor N-substitution of α,β-unsaturated 2-acyl imidazoles was found to have significant impact on the product enantioselectivity.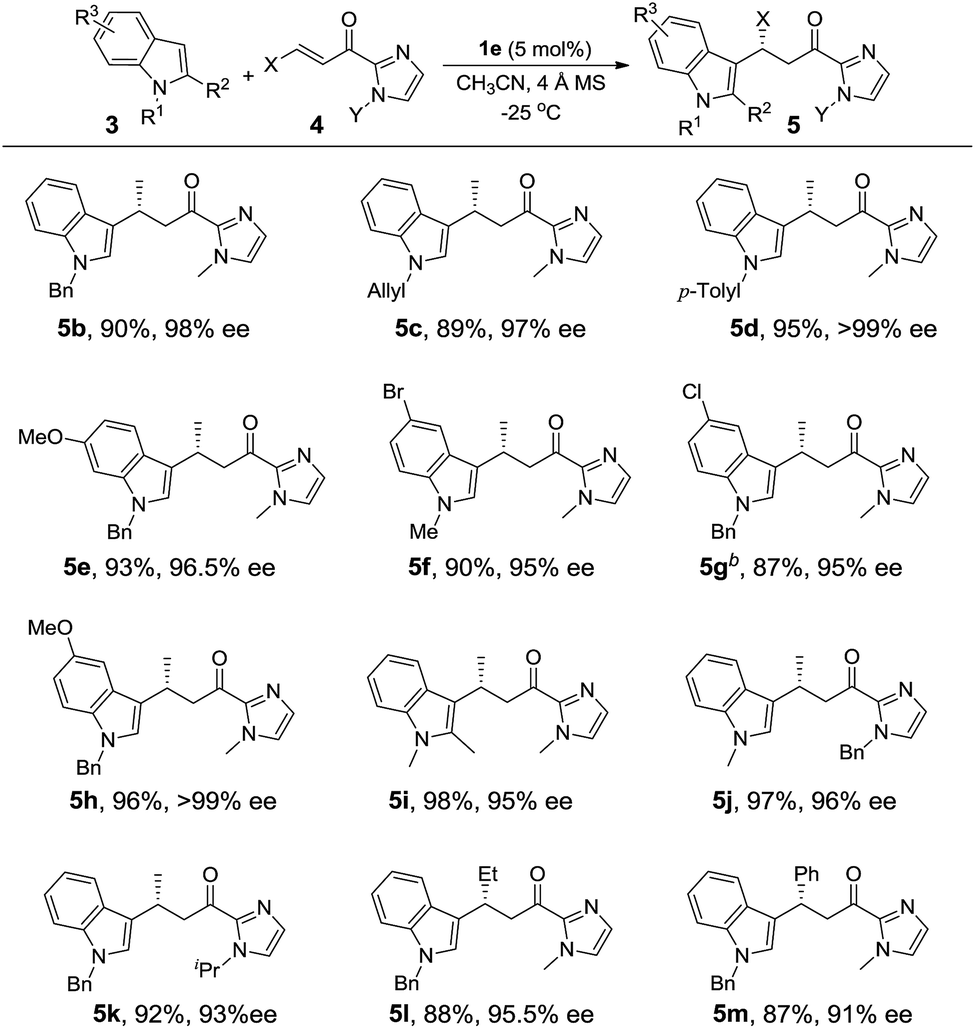
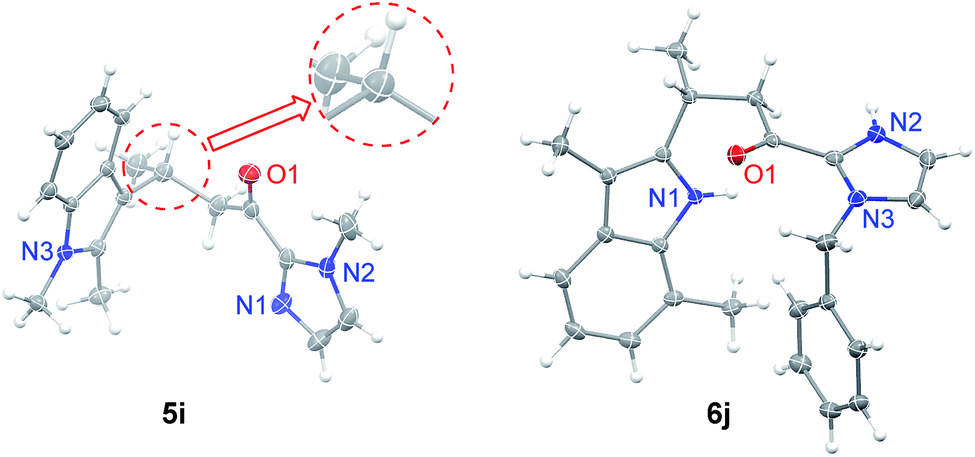 | ||
| Fig. 2 X-ray crystal structures of 5i and 6j. Thermal ellipsoids are drawn at the 50% probability level. | ||
We next investigated the C2 alkylation of 3-substituted indoles. The reaction of 3-methylindole and α,β-unsaturated 2-acyl imidazole 4a with 5 mol% of 1e in CH2Cl2 at room temperature led to the corresponding C2-alkylated product 6a in 83% yield and 87% ee along with the N1 alkylation product 7a′ (6a/7a′ = 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15, Table S2, ESI†). CH3CN as solvent was found to disfavor the C2 alkylation reaction (Table S2, ESI†). Decreasing the reaction temperature to −40 °C further improved the enantioselectivity to 96% ee, although with an extended reaction time of 60 h. Under the optimized conditions, a variety of 3-substituted indoles reacted with α,β-unsaturated 2-acyl imidazoles to give the C2 alkylation products in 76–96% yields and ∼72–98% ee (Table 3). It is noteworthy that the reaction of 3,7-dimethylindole with 4b bearing a bulkier N-substituent afforded 6j in 94% yield and 96.5% ee, without detectable N1 alkylation product. This is attributed to the presence of the 7-methyl substitution which makes N1 alkylation sterically unfavorable. When β-ester substituted α,β-unsaturated 2-acyl imidazole was used as the alkylating agent, in which both the α and β carbons are electrophilic, the nucleophilic addition of indoles took place at the β position (6f–6h). X-ray crystallography of 6j revealed an R configuration at the stereogenic carbon center (Fig. 2), which is the same as that of 5i.
:15, Table S2, ESI†). CH3CN as solvent was found to disfavor the C2 alkylation reaction (Table S2, ESI†). Decreasing the reaction temperature to −40 °C further improved the enantioselectivity to 96% ee, although with an extended reaction time of 60 h. Under the optimized conditions, a variety of 3-substituted indoles reacted with α,β-unsaturated 2-acyl imidazoles to give the C2 alkylation products in 76–96% yields and ∼72–98% ee (Table 3). It is noteworthy that the reaction of 3,7-dimethylindole with 4b bearing a bulkier N-substituent afforded 6j in 94% yield and 96.5% ee, without detectable N1 alkylation product. This is attributed to the presence of the 7-methyl substitution which makes N1 alkylation sterically unfavorable. When β-ester substituted α,β-unsaturated 2-acyl imidazole was used as the alkylating agent, in which both the α and β carbons are electrophilic, the nucleophilic addition of indoles took place at the β position (6f–6h). X-ray crystallography of 6j revealed an R configuration at the stereogenic carbon center (Fig. 2), which is the same as that of 5i.
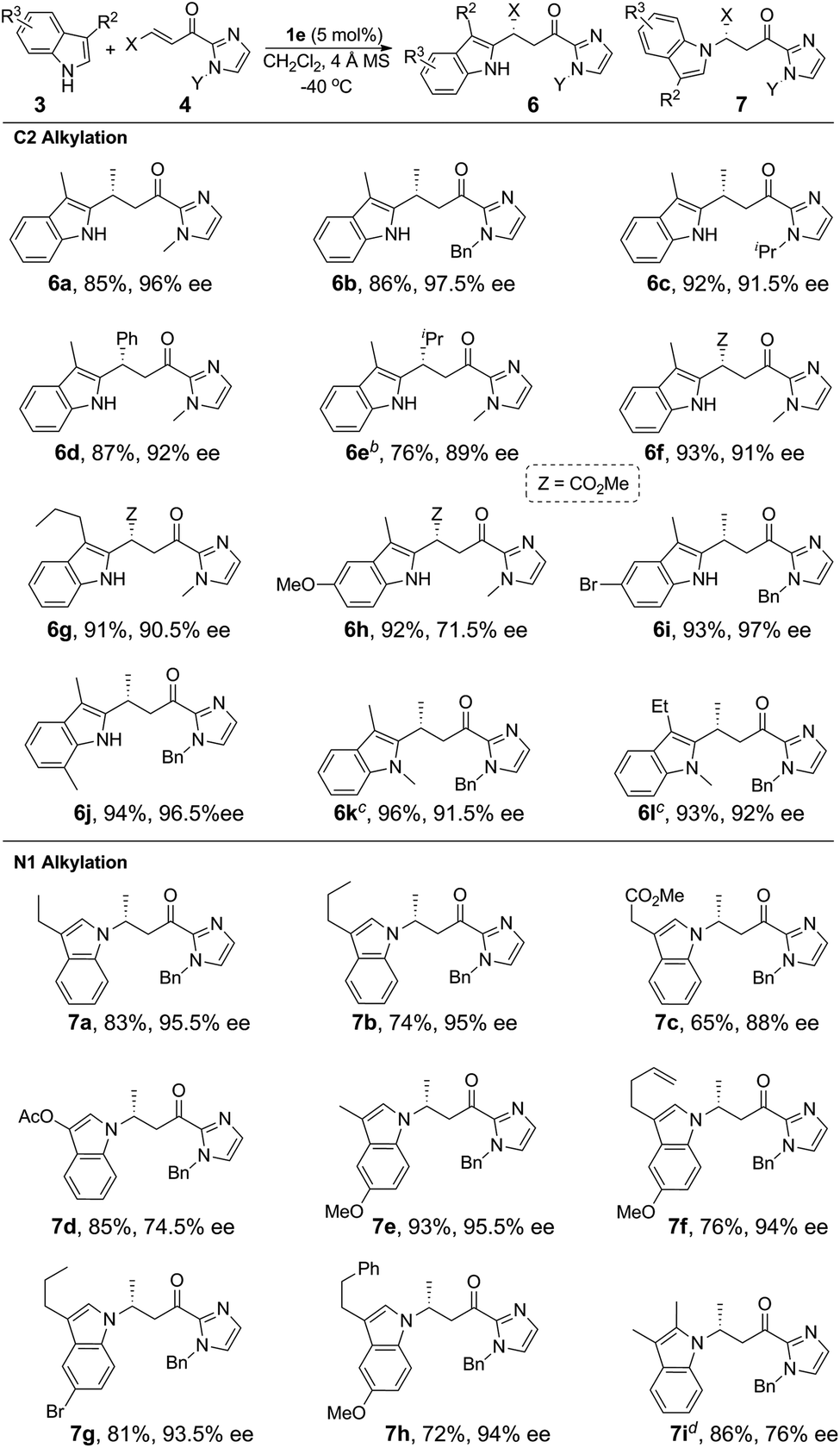
For the reaction of 3-methylindole with α,β-unsaturated 2-acyl imidazoles, the N1 alkylation product was obtained in ∼10% yield, implying a competition between C2 and N1 positions as nucleophiles on the olefinic electrophile. We envisioned that the alkylation of indoles at the nitrogen atom would be favored by increasing the bulkiness of the C3 substituent. To validate this hypothesis, we examined the reaction of 3-ethylindole with α,β-unsaturated 2-acyl imidazole 4b catalyzed by complex 1e at −40 °C. Gratifyingly, the reaction afforded the N1 alkylation product in 83% yield and 95.5% ee (Table 3, 7a). The C2 alkylation product in this case was obtained in less than 10% yield. As shown in Table 3, a range of 3-substituted indoles are reactive for the N1 alkylation, giving the desired products in good yields (65–93%) and with high enantioselectivity (74.5–95.5% ee). It was found that the electron-donating substituent on the indole makes N1 alkylation more favorable. For example, the reaction of 5-methoxy-3-methylindole with 4b gave the N1 alkylation product in 93% yield and 95.5% ee (Table 3, 7e). This is in contrast to the dominating C2 alkylation in the reaction of 3-methylindole and 4b (Table 3, 6b), suggesting that the electronic effect, in addition to the steric effect, of the substituent on the indoles also plays a significant role in directing the regioselectivity. The substituents of α,β-unsaturated 2-acyl imidazoles were also found to have a significant impact on the regioselectivity of the Fe(II)-catalyzed reaction. For example, when β-ester substituted α,β-unsaturated 2-acyl imidazole was used as the substrate, C2 alkylation is favored over N1 alkylation irrespective of the choice of indoles (compare 7bvs.6g and 7evs.6h, Table 3), suggesting that the electronic effect of the β-ester group of α,β-unsaturated 2-acyl imidazoles outweighs the electronic and steric effects of substituents on the indoles on determining the regioselectivity. Sterically hindered 2,3-dimethylindole is also reactive for the N1 alkylation at −20 °C to give 7i in 86% yield and 76% ee.
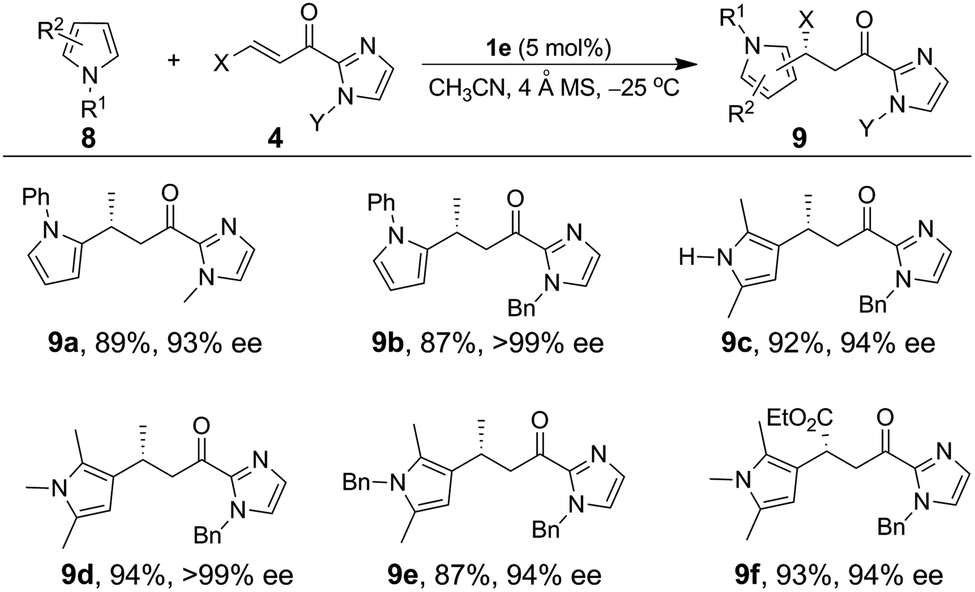
The Fe(II)-catalyzed asymmetric addition reaction is also applicable to the alkylation of pyrroles.11a,c,e,i,19 The reaction of N-phenyl pyrrole with α,β-unsaturated 2-acyl imidazole 4a and 5 mol% of 1e in CH3CN at −25 °C gave the C2 alkylation product 9a (Table 4) in 89% yield and 93% ee. The ee value reached 99% (9b) when a bulkier N-substituent is present on the α,β-unsaturated 2-acyl imidazole 4b. In both cases, the C3 alkylation product was not detected; this is consistent with the C2 position of pyrroles being more nucleophilic than the C3 one. The C3 alkylation of pyrroles was achieved with 2,5-substituted pyrroles, which led to the formation of the C3-alkylated products (9c–9f) in high yields (87–94%) and with excellent enantioselectivity (94–99% ee).
| a Reaction conditions: 8 (0.45 mmol), 4 (0.3 mmol), 1e (5 mol%), 4 Å MS (100 mg), CH3CN (3 mL), under argon. |
|---|
|
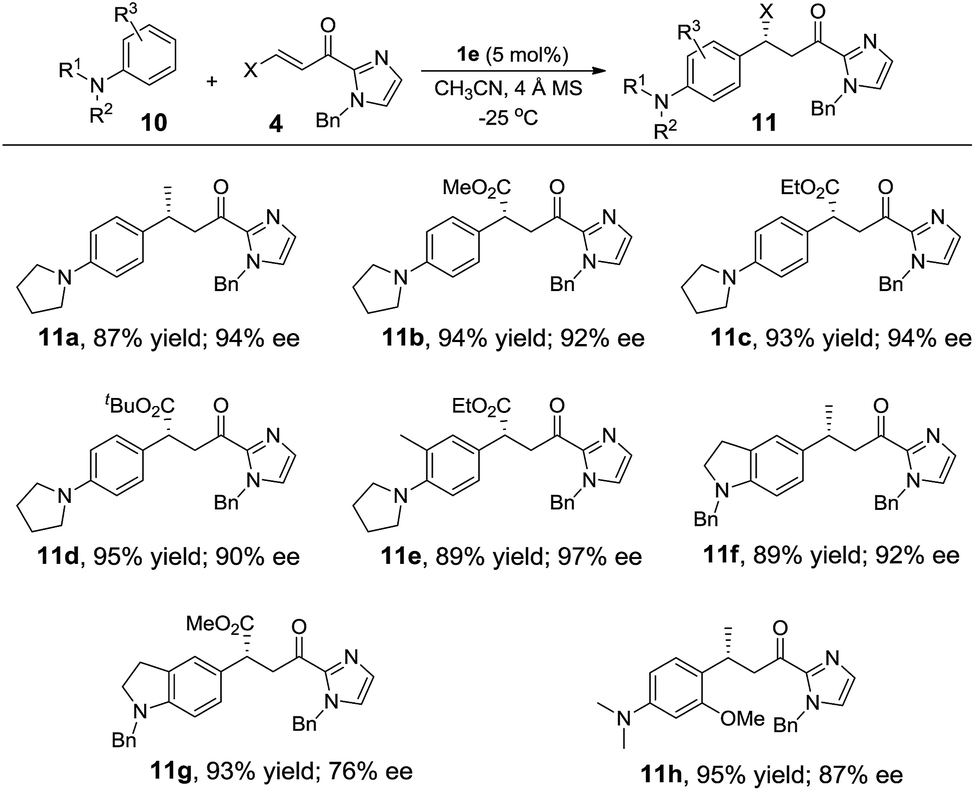
Alkylation of electron-rich arenes such as anilines with α,β-unsaturated 2-acyl imidazoles11c,l could also be achieved using the Fe(II) complex 1e as the catalyst. The 1e-catalyzed alkylation reaction of 10 with 4 gave para-alkylated 11a–11g in 87–95% yields and 76–97% ee (Table 5). When 3-methoxy-N,N-dimethylaniline was used as the substrate, in which two strongly electron-donating groups are present, the reaction proceeded in high regioselectivity giving 11h in 95% yield and 87% ee (Table 5).
| a Reaction conditions: 10 (0.45 mmol), 4 (0.3 mmol), 1e (5 mol%), 4 Å MS (100 mg), CH3CN (3 mL), under argon. |
|---|
|
Synthetic applications
The indolyl 2-acyl imidazoles obtained in this work could be readily converted to a range of indole compounds11a,c which are of interest in medicinal chemistry. For example, methylation of the imidazole moiety of N1 alkylation product 7e with iodomethane in DMF, followed by hydrolysis with water and DBU, gave the indolyl carboxylic acid 12 in 81% yield (Scheme 2), the analogues of which have been reported to be potent inhibitors of integrin αvβ3.20 In a similar fashion, the ester and amide derivatives 13 and 14 were obtained in 95% and 88% yield, respectively (Scheme 2). The amide 14 can be reduced to an analogue of 3-(1H-indol-1-yl)-3-arylpropan-1-amine;21 the latter was reported to display inhibition to norepinephrine and serotonin reuptake.22 The pyrrolo[1,2-α]indole scaffold is a basic structure of many bioactive natural products.23 Methylation and subsequent intramolecular acylation of the C2 alkylation product 6b afforded pyrrolo[1,2-α]indole 15 in 75% yield (Scheme 2). Notably, these organic transformations are accompanied by retention of ee values, indicating preservation of the stereogenic carbon centers.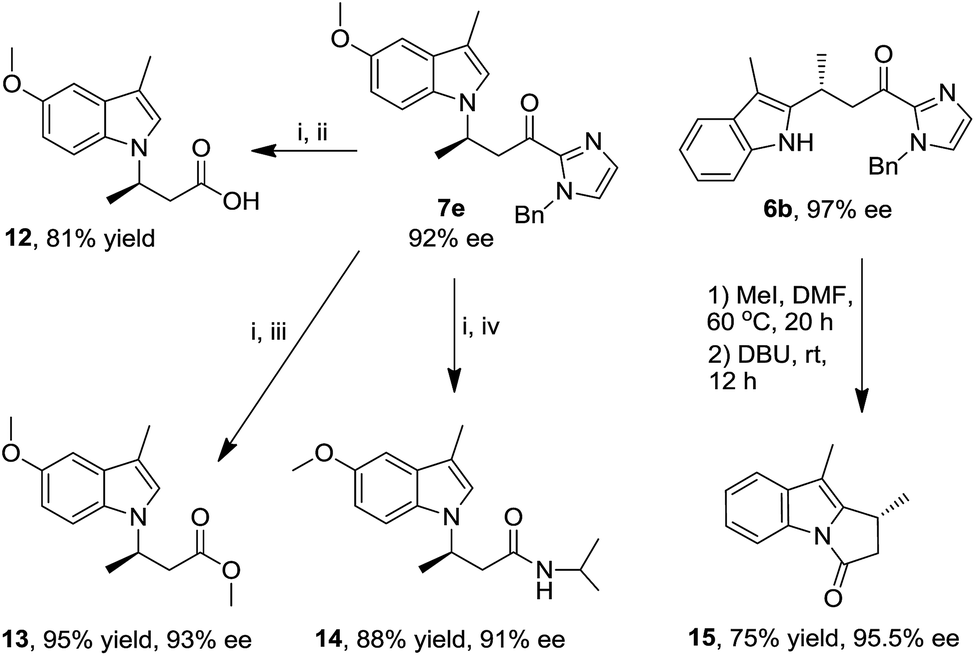 | ||
| Scheme 2 Synthetic elaboration studies. Reaction conditions: (i) MeI, DMF, 60 °C, 20 h; (ii) DBU, H2O, rt, 4 h; (iii) DBU, CH3OH, CH2Cl2, rt, 6 h; (iv) DBU, iPrNH2, rt, 12 h. | ||
Mechanistic studies
A proposed reaction mechanism for the 1e-catalyzed alkylation of indoles is presented in Scheme 3. Binding of α,β-unsaturated 2-acyl imidazoles (e.g.4b) via the imidazolyl nitrogen and carbonyl oxygen atoms to the chiral Fe(II) center generates an intermediate denoted as 1e-(N^O). The Fe(II) catalyst acts as a Lewis acid to activate the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond for nucleophilic attack by indoles (e.g.3a) to give the addition product (e.g.5j). The enantioselectivity is governed by the chiral pocket environment around the Fe(II) center which shields the Si face from indolyl nucleophile attack.
C bond for nucleophilic attack by indoles (e.g.3a) to give the addition product (e.g.5j). The enantioselectivity is governed by the chiral pocket environment around the Fe(II) center which shields the Si face from indolyl nucleophile attack.
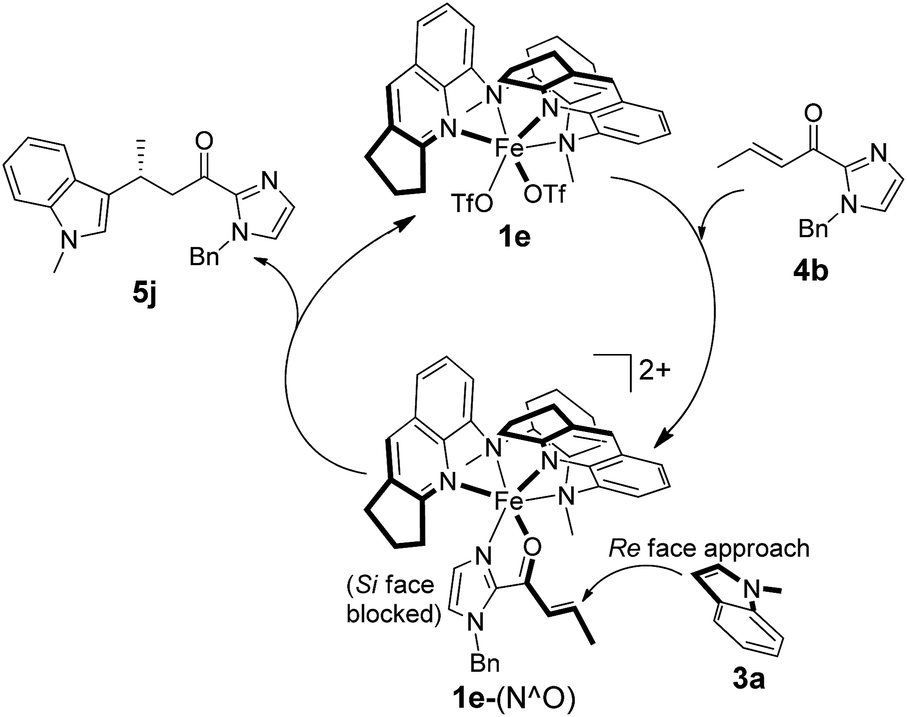 | ||
| Scheme 3 Proposed catalytic cycle for asymmetric alkylation of indoles catalyzed by 1e. | ||
To gain support for the proposed reaction mechanism, the binding between 1e and 4b was examined by high-resolution ESI-MS. Treatment of an acetonitrile solution of 1e with 2 equiv. of 4b resulted in a predominant ion peak at m/z 379.1707 (Fig. 3), where the isotopic distribution pattern (Fig. S3, ESI†) and collision-induced dissociation analysis (Fig. S4, ESI†) are well consistent with the proposed formulation of 1e-(N^O).
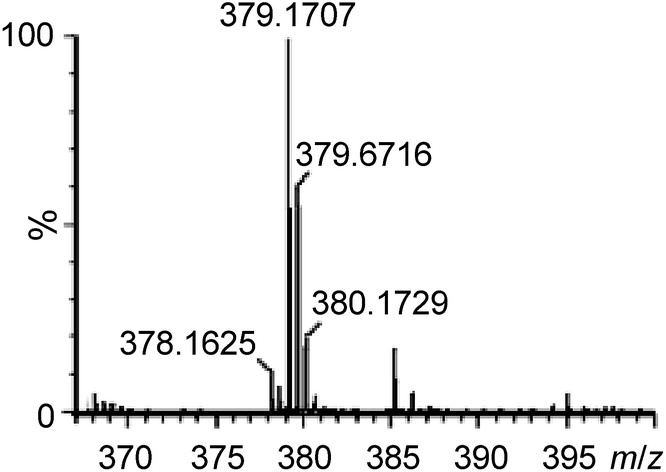 | ||
| Fig. 3 High-resolution ESI-MS spectrum of a reaction mixture of 1e with 4b in CH3CN. | ||
DFT calculations were performed to examine the reaction details of 1e-catalyzed C3 alkylation of N-methylindole (3a) with α,β-unsaturated 2-acyl imidazole 4b and to elucidate the origin of the enantioselectivity (Scheme 4). Since the two cis-oriented coordination sites of 1e are chemically inequivalent, there are two possible binding interactions between 1e and 4b (IntA1 and IntB1). Both binding modes were calculated to form stable intermediates with a decrease in free energy by more than 10 kcal mol−1 as a result of replacement of two coordinated solvent molecules (acetonitrile) by imidazole 4b as a bidentate ligand. In addition, IntB1 is 3.8 kcal mol−1 more stable than IntA1. Accordingly, there are four possible transition states (TSA2R, TSA2S, TSB2R and TSB2S) regarding the two approach directions of 3a to the CC bond of the 1e-(N^O) intermediate (Scheme 4). Among the four transition states, TSA2R was found to have the lowest transition barrier with a free energy of 17.3 kcal mol−1 (TSA2S: 21.4 kcal mol−1; TSB2R: 23.4 kcal mol−1; TSB2S: 23.1 kcal mol−1). There is π–π stacking interaction between the quinoline ring of 1e and the indole ring of 3a in TSA2R (Fig. S5, ESI†) with a distance of about 3.4 Å, which possibly accounts for the lower energy barrier. Accordingly, the indole ring resides closer to the quinoline ring in TSA2R and the additional π–π interaction leads to the comparatively restrained position of the indole, and thus its C1–C3 bond (2.08 Å) is slightly longer than those in the other three transition states (2.03 or 2.04 Å). The R-enantiomer of the C3 alkylation product is mainly obtained viaTSA2R, which is consistent with the configuration observed experimentally in the isolated products (e.g.5i, Table 2). The free energy differences between the four transition states (TSA/B2R/S) lead to a calculated ee of 96.3% at −25 °C, which is in excellent agreement with the experimental result of 96% ee. The subsequent steps proceed feasibly with much lower energy barriers or great energy release. When the C1–C3 bond is formed (IntA3R, IntA3S, IntB3R and IntB3S), the most stable intermediate is IntA3S, which probably stems from its strong hydrogen bond between H1 and O (1.87 Å), compared with other intermediates (2.16, 2.22, and 2.59 Å for IntA3R, IntB3R and IntB3S, respectively). After forming the C1–C3 bond, intramolecular proton transfer takes place from C3 to O by forming a six-membered ring (TSA4R, TSA4S, TSB4R and TSB4S). In these transition states, the Fe–O bonds are weakened (from 2.01–2.04 in IntA/B3R/S to 2.08–2.17 Å in TSA/B4R/S) in association with the shortening of the C1–C3 bond (from 1.58–1.61 Å in IntA/B3R/S to 1.54 Å in TSA/B4R/S). Both TSA4R and TSA4S hold the π–π stacking between the quinoline and indole, which could stabilize TSA4R and TSA4S and therefore lead to the free energy difference between TSA4R/S and TSB4R/S. After proton transfer, four possible enol intermediates are generated (IntA5R, IntA5S, IntB5R and IntB5S) with significant energy release (5.8–16.0 kcal mol−1). Comparatively, the lower free energy of IntB5R and IntB5S possibly results from their stronger Fe–N1 coordination than those in IntA5R and IntA5S. The subsequent exothermic tautomerization leads to the final alkylation product coordinated to the catalyst (IntA6R, IntA6S, IntB6R and IntB6S), which would possibly result in some extent of product inhibition of the catalyst. Indeed, time-course studies revealed that addition of 25 mol% of product 5j to the initial reaction mixture of the 1e-catalyzed reaction of 3a and 4b appreciably lowered the product yield (Fig. S6, ESI†).
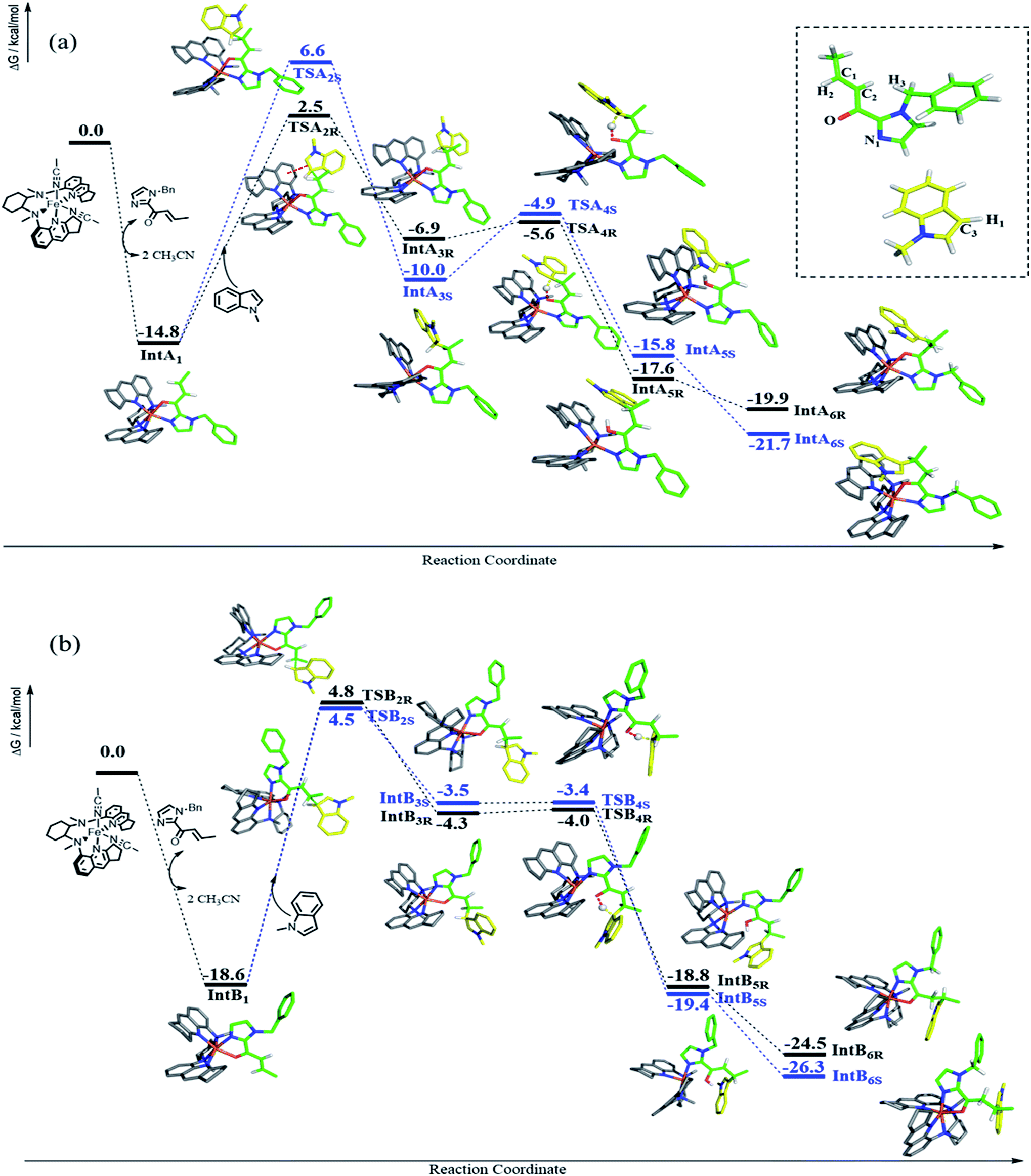 | ||
| Scheme 4 The reaction details of the Fe(II) complex 1e-catalyzed C3 alkylation of N-methylindole 3a by α,β-unsaturated 2-acyl imidazole 4b. Owing to the inequivalent cis-oriented binding sites of 1e, two pathways are presented in panel a and panel b, representing coordination pattern A and B, respectively. Because of the stereoselectivity of the attacked alkene, the reaction pathways of both A and B possess both R and S configurations, which are displayed in black and blue, respectively. In each transition state, the breaking and forming bonds are highlighted by dashed lines. Color code: iron, orange; oxygen, red; nitrogen, blue; carbon, gray for 1e, yellow for 3a and green for 4b; hydrogen (white). For simplicity, only critical hydrogen atoms are displayed. Atomic labels referred to in the text are denoted in the inset of panel a. | ||
General remarks
The Fe(II) catalysis reported herein is reminiscent of an Ir(III) system developed by Meggers and co-workers for the same type of reaction,1a where the “chiral-at-metal” property of the catalyst is responsible for the excellent enantioselectivity as the reaction occurs in close proximity to the metal bearing “intense” chiral information. These Ir(III) catalytic systems composed of achiral ligands are very potent in effecting a wide range of enantioselective reactions and could be accessed in relatively facile pathways.1b However, it should be noted that such systems rely heavily on specific heavy metal ions, i.e. Ir(III) and Rh(III), and/or specific ligand sets, i.e., cyclometalating ligands, to maintain a high coordination stability to prevent catalyst racemization. These features are generally not compatible with the development of base metal catalysts, e.g. Fe complexes, when it comes to harnessing their non-toxicity and earth-abundance for chemical synthesis. In this regard, we demonstrate here, along with other reported examples,2,3 that efficient “chiral-at-metal” catalysis could be achieved with base metals through rational ligand design, e.g. using chiral tetradentate N4 ligands, installing rigidity, and fine-tuning the sterics on the ligand fragments.In recent years, chiral Fe(N4) complexes have been extensively studied in oxidative organic catalysis such as asymmetric alkene epoxidation,5c–g asymmetric alkene cis-dihydroxylation5a,b,6 and asymmetric C–H hydroxylation.5h The class of [FeII(N4)(OTf)2] complexes presented in this work, as well as those in the literature,5,6 features some of the fundamental defining properties of an appealing chiral transition-metal asymmetric catalyst, including (1) ease of preparation and modification, (2) configurationally stable chirality, (3) possession of labile coordination sites for reagent/substrate activation, and (4) high sustainability. The chiral, linear N4 ligands could be prepared on the multigram scale with good yields from commercially available optically active diamine backbones. The steric and electronic properties of the pyridyl/quinolyl donors are highly flexible for manipulation, as have been demonstrated by us6 and by Costas and co-workers.5e Upon coordination to Fe(II) ions, under mild conditions, the N4 ligands form stable cis-α or cis-β chelates depending on the ligand structure. These complexes possess metal-centered chirality without further needs of chiral resolution or purification. Thanks to the large barrier possibly resulting from the different orientations of the N-methyl groups in the cis-α and cis-β isomers, these chelates do not undergo isomerization in solution;6,18b thus the stereogenic configuration is robust. The weakly coordinating triflate (OTf−) ligands, in addition to the fast ligand substitution rate of Fe(II), are favorable for substrate binding/activation. Beyond that, iron is readily available, relatively inexpensive and non-toxic which makes the utilization of iron complexes in catalysis particularly attractive.24 In our continuing efforts to develop synthetically useful catalysis mediated by chiral Fe(N4) complexes, we were interested to examine their potential as Lewis acid catalysts which has not been documented to the best of our knowledge.
As there are many organic reactions of synthetic interest that can be catalyzed by Lewis acids, the development of effective chiral Lewis acid catalysts for asymmetric organic transformations has been receiving considerable interest in organic chemistry. The N4 ligands employed in this work are quinolylamines. Due to the conjugation with the quinoline moiety, the amino group of bis(quinolyl)diamines is a less effective donor to the metal ion than that of bis(pyridylmethyl)diamine ligands, thereby making the corresponding Fe(II) complexes more Lewis acidic and capable of catalyzing a wide range of addition reactions, even at low temperature. In addition, due to the lack of a methylene moiety, tetradentate quinolylamine ligands have a more rigid scaffold than pyridylmethylamine analogues, which would provide a robust chiral pocket around the reaction center for reactions.
In the literature, reports on asymmetric C3 addition of indoles to α,β-unsaturated 2-acyl imidazoles are confined to the use of the Sc(III) complex,11a Cu(II)-DNA/protein adducts,11e,l and Ir(III) complex1a as Lewis acid catalysts. In this work, we report for the first time that chiral FeII(N4) complexes could catalyze similar reactions.
Due to the inherently low nucleophilicity of the nitrogen atom, asymmetric N1 alkylation of indoles with α,β-unsaturated 2-acyl imidazoles remains a formidable challenge. There are only a few examples of highly enantioselective N1 alkylation of indoles reported in the literature, including intramolecular aza-Michael additions,17b,e cinchona alkaloid-catalyzed reaction with Morita–Baylis–Hillman carbonates,17c Pd(II) and Ir(I)-catalyzed N-allylations17d,f,i and Ir(I)-catalyzed allylation/oxidation of indolines.17h Our present method provides an alternative access to N-alkyl indoles in good yields and high enantiomeric excess via Fe(II)-catalyzed intermolecular alkylation which is unprecedented in the literature. To the best of our knowledge, 1e also represents the first example of a single chiral catalyst capable of effecting intermolecular alkylation of various indoles at the C3, C2 and N1 positions, respectively, with high enantio- and regioselectivities.
DFT calculations provide some insight into the origin of high enantioselectivities in the 1e-catalyzed reactions. The two possible binding interactions between 1e and α,β-unsaturated 2-acyl imidazole 4b, in addition to the two possible approach faces (Si or Re) of indole 3a to the 1e-(N^O) adduct, generate four possible reaction pathways. In all the four pathways, nucleophilic attack of 3a on 1e-(N^O) was found to have the largest barrier (17.3–23.4 kcal mol−1) and logically this is the enantioselectivity-determining step. Among them, TSA2R was computed to have the lowest transition barrier (17.3 kcal mol−1), being 4.1 kcal mol−1 lower than that of the second-lowest TSA2S; the calculated ee at −25 °C derived from this energy difference is in excellent agreement with the experimental value. In the DFT-optimized structure of TSA2R, interestingly, there is substantial π–π stacking between the quinoline moiety of 1e and indole 3a. This suggests that the enantioselectivity is not only determined by how well the steric environment around the cis-β Fe(II) center can guide/shield the approach of indoles, but also assisted by secondary π–π interaction provided by the quinolyl ligand. The DFT-calculation findings are insightful for the rational design of future generation of FeII(N4) Lewis acid catalysts with more effective chiral pockets for the enantio-differentiation of aromatic substrates.
Conclusions
In summary, we have developed C3, C2 and N1 alkylations of indoles catalyzed by a chiral cis-β Fe(II) complex supported by a (R,R)-tetradentate bis(quinolyl)diamine ligand. A range of indoles reacted with various α,β-unsaturated 2-acyl imidazoles to give diverse chiral indoles in high yields with high enantio- and regioselectivity. The catalysis is also applicable to the alkylation of pyrroles and anilines. The thus obtained products can be converted to a variety of chiral indole derivatives with potential synthetic and biological values. The stable configuration of the iron(II) complex and a secondary π–π interaction in the transition state are suggested to be the origin of the high enantioselectivity. Our results suggest that chiral FeII(N4) complexes are potentially a new and efficient class of Lewis acid catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Hong Kong Research Grants Council General Research Fund (17303815, 17306714, and 17301817), National Natural Science Foundation of China (NSFC 91856203), and Basic Research Program-Shenzhen Fund (JCYJ20170412140251576, JCYJ20180508162429786).Notes and references
- (a) H. Huo, C. Fu, K. Harms and E. Meggers, J. Am. Chem. Soc., 2014, 136, 2990 CrossRef CAS PubMed; (b) L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320 CrossRef CAS PubMed; (c) Y. Hong, L. Jarrige, K. Harms and E. Meggers, J. Am. Chem. Soc., 2019, 141, 4569 CrossRef CAS PubMed.
- (a) P. D. Knight and P. Scott, Coord. Chem. Rev., 2003, 242, 125 CrossRef CAS; (b) E. B. Bauer, Chem. Soc. Rev., 2012, 41, 3153 RSC; (c) T. Cruchter and V. A. Larionov, Coord. Chem. Rev., 2018, 376, 95 CrossRef CAS.
- D. A. Evans, J. M. Janey, N. Magomedov and J. S. Tedrow, Angew. Chem., Int. Ed., 2001, 40, 1884 CrossRef CAS.
- J. P. Abell and H. Yamamoto, Chem. Soc. Rev., 2010, 39, 61 RSC.
- Examples of Fe(N4)-catalyzed asymmetric cis-dihydroxylation or epoxidation of alkenes or asymmetric oxidation of C–H bonds: (a) M. Costas, A. K. Tipton, K. Chen, D.-H. Jo and L. Que Jr, J. Am. Chem. Soc., 2001, 123, 6722 CrossRef CAS PubMed; (b) K. Suzuki, P. D. Oldenburg and L. Que Jr, Angew. Chem., Int. Ed., 2008, 47, 1887 CrossRef CAS PubMed; (c) M. Wu, C.-X. Miao, S. Wang, X. Hu, C. Xia, F. E. Kühn and W. Sun, Adv. Synth. Catal., 2011, 353, 3014 CrossRef CAS; (d) O. Y. Lyakin, R. V. Ottenbacher, K. P. Bryliakov and E. P. Talsi, ACS Catal., 2012, 2, 1196 CrossRef CAS; (e) O. Cussó, I. Garcia-Bosch, X. Ribas, J. Lloret-Fillol and M. Costas, J. Am. Chem. Soc., 2013, 135, 14871 CrossRef PubMed; (f) O. Cussó, X. Ribas, J. Lloret-Fillol and M. Costas, Angew. Chem., Int. Ed., 2015, 54, 2729 CrossRef PubMed; (g) O. Cussó, M. Cianfanelli, X. Ribas, R. J. M. Klein Gebbink and M. Costas, J. Am. Chem. Soc., 2016, 138, 2732 CrossRef PubMed; (h) M. Milan, M. Bietti and M. Costas, ACS Cent. Sci., 2017, 3, 196 CrossRef CAS PubMed.
- C. Zang, Y. Liu, Z.-J. Xu, C.-W. Tse, X. Guan, J. Wei, J.-S. Huang and C.-M. Che, Angew. Chem., Int. Ed., 2016, 55, 10253 CrossRef CAS PubMed.
- Selected reviews on iron asymmetric catalysis: (a) R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282 RSC; (b) M. Darwish and M. Wills, Catal. Sci. Technol., 2012, 2, 243 RSC; (c) K. Gopalaiah, Chem. Rev., 2013, 113, 3248 CrossRef CAS PubMed; (d) P. E. Sues, K. Z. Demmans and R. H. Morris, Dalton Trans., 2014, 43, 7650 RSC; (e) A. Fingerhut, O. V. Serdyuk and S. B. Tsogoeva, Green Chem., 2015, 17, 2042 RSC; (f) T. Ollevier, Catal. Sci. Technol., 2016, 6, 41 RSC.
- Examples on iron asymmetric catalysis not using N4 ligands: (a) Y. Yamashita, M. Ueno, Y. Kuriyama and S. Kobayashi, Adv. Synth. Catal., 2002, 344, 929 CrossRef CAS; (b) L. Yang, Q. Zhu, S. Guo, B. Qian, C. Xia and H. Huang, Chem.–Eur. J., 2010, 16, 1638 CrossRef CAS PubMed; (c) S.-F. Zhu, Y. Cai, H.-X. Mao, J.-H. Xie and Q.-L. Zhou, Nat. Chem., 2010, 2, 546 CrossRef CAS PubMed; (d) T. Ollevier and B. Plancq, Chem. Commun., 2012, 48, 2289 RSC; (e) Q.-H. Deng, T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2013, 135, 5356 CrossRef CAS PubMed; (f) K. S. Williamson, J. W. Sawicki and T. P. Yoon, Chem. Sci., 2014, 5, 3524 RSC; (g) X. Gu, Y. Zhang, Z.-J. Xu and C.-M. Che, Chem. Commun., 2014, 50, 7870 RSC; (h) J. Loup, D. Zell, J. C. A. Oliveira, H. Keil, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 14197 CrossRef CAS PubMed; (i) H. Xu, Y.-P. Li, Y. Cai, G.-P. Wang, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 7697 CrossRef CAS PubMed; (j) P. Zhou, L. Lin, L. Chen, X. Zhong, X. Liu and X. Feng, J. Am. Chem. Soc., 2017, 139, 13414 CrossRef CAS PubMed.
- (a) J. Bonjoch and D. Solé, Chem. Rev., 2000, 100, 3455 CrossRef CAS PubMed; (b) D. J. Faulkner, Nat. Prod. Rep., 2002, 19, 1 CAS; (c) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed; (d) M. Somei and F. Yamada, Nat. Prod. Rep., 2004, 21, 278 RSC; (e) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef CAS PubMed; (f) S. Agarwal, S. Caemmerer, S. Filali, W. Froehner, J. Knoell, M. P. Krahl, K. R. Reddy and H.-J. Knolker, Curr. Org. Chem., 2005, 9, 1601 CrossRef CAS; (g) S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532 RSC.
- Selected reviews: (a) M. Bandini, A. Melloni and A. Umani-Ronchi, Angew. Chem., Int. Ed., 2004, 43, 550 CrossRef CAS PubMed; (b) T. B. Poulsen and K. A. Jøgensen, Chem. Rev., 2008, 108, 2903 CrossRef CAS PubMed; (c) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CrossRef CAS PubMed; (d) S.-L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190 RSC; (e) M. Zeng and S.-L. You, Synlett, 2010, 9, 1289 Search PubMed; (f) G. Bartoli, G. Bencivenni and R. Dalpozzo, Chem. Soc. Rev., 2010, 39, 4449 RSC; (g) P. Chauhan and S. S. Chimni, RSC Adv., 2012, 2, 6117 RSC; (h) M. Shiri, Chem. Rev., 2012, 112, 3508 CrossRef CAS PubMed; (i) S. Lancianesi, A. Palmieri and M. Petrini, Chem. Rev., 2014, 114, 7108 CrossRef CAS PubMed; (j) R. Dalpozzo, Chem. Soc. Rev., 2015, 44, 742 RSC.
- Examples of asymmetric C3 alkylation of indoles: (a) D. A. Evans, K. R. Fandrick and H.-J. Song, J. Am. Chem. Soc., 2005, 127, 8942 CrossRef CAS PubMed; (b) Y.-X. Jia, J. Zhong, S.-F. Zhu, C.-M. Zhang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2007, 46, 5565 CrossRef CAS PubMed; (c) D. A. Evans, K. R. Fandrick, H.-J. Song, K. A. Scheidt and R. Xu, J. Am. Chem. Soc., 2007, 129, 10029 CrossRef CAS PubMed; (d) J. Itoh, K. Fuchibe and T. Akiyama, Angew. Chem., Int. Ed., 2008, 47, 4016 CrossRef CAS PubMed; (e) A. J. Boersma, B. L. Feringa and G. Roelfes, Angew. Chem., Int. Ed., 2009, 48, 3346 CrossRef CAS PubMed; (f) H. Y. Kim, S. Kim and K. Oh, Angew. Chem., Int. Ed., 2010, 49, 4476 CrossRef CAS PubMed; (g) T. P. Pathak, K. M. Gligorich, B. E. Welm and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 7870 CrossRef CAS PubMed; (h) J. Lv, L. Zhang, Y. Zhou, Z. Nie, S. Luo and J.-P. Cheng, Angew. Chem., Int. Ed., 2011, 50, 6610 CrossRef CAS PubMed; (i) J.-R. Gao, H. Wu, B. Xiang, W.-B. Yu, L. Han and Y.-X. Jia, J. Am. Chem. Soc., 2013, 135, 2983 CrossRef CAS PubMed; (j) S. Raja, M. Nakajima and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 2762 CrossRef CAS PubMed; (k) C. Wang, L.-A. Chen, H. Huo, X. Shen, K. Harms, L. Gong and E. Meggers, Chem. Sci., 2015, 6, 1094 RSC; (l) X. Shen, H. Huo, C. Wang, B. Zhang, K. Harms and E. Meggers, Chem.–Eur. J., 2015, 21, 9720 CrossRef CAS PubMed; (m) R.-R. Liu, S.-C. Ye, C.-J. Lu, G.-L. Zhuang, J.-R. Gao and Y.-X. Jia, Angew. Chem., Int. Ed., 2015, 54, 11205 CrossRef CAS PubMed; (n) J. Bos, W. R. Browne, A. J. M. Driessen and G. Roelfes, J. Am. Chem. Soc., 2015, 137, 9796 CrossRef CAS PubMed; (o) M. M. Heravi, V. Zadsirjan, B. Masoumi and M. Heydari, J. Organomet. Chem., 2019, 879, 78 CrossRef CAS.
- For examples of asymmetric Pictet–Spengler reactions of indoles see: (a) M. S. Taylor and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 10558 CrossRef CAS PubMed; (b) J. Seayad, A. M. Seayad and B. List, J. Am. Chem. Soc., 2006, 128, 1086 CrossRef CAS PubMed; (c) M. J. Wanner, R. N. S. van der Haas, K. R. de Cuba, J. H. van Maarseveen and H. Hiemstra, Angew. Chem., Int. Ed., 2007, 46, 7485 CrossRef CAS PubMed; (d) I. T. Raheem, P. S. Thiara, E. A. Peterson and E. N. Jacobsen, J. Am. Chem. Soc., 2007, 129, 13404 CrossRef CAS PubMed; (e) M. E. Muratore, C. A. Holloway, A. W. Pilling, R. I. Storer, G. Trevitt and D. J. Dixon, J. Am. Chem. Soc., 2009, 131, 10796 CrossRef CAS PubMed; (f) Q. Cai, X.-W. Liang, S.-G. Wang, J.-W. Zhang, X. Zhang and S.-L. You, Org. Lett., 2012, 14, 5022 CrossRef CAS PubMed; (g) A. W. Gregory, P. Jakubec, P. Turner and D. J. Dixon, Org. Lett., 2013, 15, 4330 CrossRef CAS PubMed.
- (a) Q. Kang, X.-J. Zheng and S.-L. You, Chem.–Eur. J., 2008, 14, 3539 CrossRef CAS PubMed; (b) M. Zeng, Q. Kang, Q.-L. He and S.-L. You, Adv. Synth. Catal., 2008, 350, 2169 CrossRef CAS; (c) Y.-F. Sheng, G.-Q. Li, Q. Kang, A.-J. Zhang and S.-L. You, Chem.–Eur. J., 2009, 15, 3351 CrossRef CAS PubMed; (d) L. Hong, C. Liu, W. Sun, L. Wang, K. Wong and R. Wang, Org. Lett., 2009, 11, 2177 CrossRef CAS PubMed; (e) L. Hong, W. Sun, C. Liu, L. Wang, K. Wong and R. Wang, Chem.–Eur. J., 2009, 15, 11105 CrossRef CAS PubMed; (f) T. Wang, G.-W. Zhang, Y. Teng, J. Nie, Y. Zheng and J.-A. Ma, Adv. Synth. Catal., 2010, 352, 2773 CrossRef CAS; (g) H. Wu, R.-R. Liu, C. Shen, M.-D. Zhang, J. Gao and Y.-X. Jia, Org. Chem. Front., 2015, 2, 124 RSC.
- (a) H.-G. Cheng, L.-Q. Lu, T. Wang, Q.-Q. Yang, X.-P. Liu, Y. Li, Q.-H. Deng, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2013, 52, 3250 CrossRef CAS PubMed; (b) Y. Zhang, X. Liu, X. Zhao, J. Zhang, L. Zhou, L. Lin and X. Feng, Chem. Commun., 2013, 49, 11311 RSC; (c) B. Bi, Q.-X. Lou, Y.-Y. Ding, S.-W. Chen, S.-S. Zhang, W.-H. Hu and J.-L. Zhao, Org. Lett., 2015, 17, 540 CrossRef CAS PubMed; (d) J.-Q. Weng, R.-J. Fan, Q.-M. Deng, R.-R. Liu, J.-R. Gao and Y.-X. Jia, J. Org. Chem., 2016, 81, 3023 CrossRef CAS PubMed.
- Z. Zhou, Y. Li, L. Gong and E. Meggers, Org. Lett., 2017, 19, 222 CrossRef CAS PubMed.
- Other types of asymmetric C2 alkylations: (a) S. Lee and D. W. C. MacMillan, J. Am. Chem. Soc., 2007, 129, 15438 CrossRef CAS PubMed; (b) M. Bandini, A. Bottoni, M. Chiarucci, G. Cera and G. P. Miscione, J. Am. Chem. Soc., 2012, 134, 20690 CrossRef CAS PubMed; (c) P. Maity, R. P. Pemberton, D. J. Tantillo and U. K. Tambar, J. Am. Chem. Soc., 2013, 135, 16380 CrossRef CAS PubMed.
- Examples of asymmetric N1 alkylation of indoles: (a) B. M. Trost, M. J. Krische, V. Berl and E. M. Grenzer, Org. Lett., 2002, 4, 2005 CrossRef CAS PubMed; (b) M. Bandini, A. Eichholzer, M. Tragni and A. Umani-Ronchi, Angew. Chem., Int. Ed., 2008, 47, 3238 CrossRef CAS PubMed; (c) H.-L. Cui, X. Feng, J. Peng, J. Lei, K. Jiang and Y.-C. Chen, Angew. Chem., Int. Ed., 2009, 48, 5737 CrossRef CAS PubMed; (d) L. M. Stanley and J. E. Hartwig, Angew. Chem., Int. Ed., 2009, 48, 7841 CrossRef CAS PubMed; (e) Q. Cai, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2010, 49, 8666 CrossRef CAS PubMed; (f) B. M. Trost, M. Osipov and G. Dong, J. Am. Chem. Soc., 2010, 132, 15800 CrossRef CAS PubMed; (g) Y. Xie, Y. Zhao, B. Qian, L. Yang, C. Xia and H. Huang, Angew. Chem., Int. Ed., 2011, 50, 5682 CrossRef CAS PubMed; (h) W.-B. Liu, X. Zhang, L.-X. Dai and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 5183 CrossRef CAS PubMed; (i) L.-Y. Chen, X.-Y. Yu, J.-R. Chen, B. Feng, H. Zhang, Y.-H. Qi and W.-J. Xiao, Org. Lett., 2015, 17, 1381 CrossRef CAS PubMed.
- (a) J. England, G. J. P. Britovsek, N. Rabadia and A. J. P. White, Inorg. Chem., 2007, 46, 3752 CrossRef CAS PubMed; (b) S. Hong, Y.-M. Lee, K.-B. Cho, K. Sundaravel, J. Cho, M. J. Kim, W. Shin and W. Nam, J. Am. Chem. Soc., 2011, 133, 11876 CrossRef CAS PubMed.
- Examples of asymmetric C3 alkylation of pyrroles: (a) C.-X. Zhuo, Q.-F. Wu, Q. Zhao, Q.-L. Xu and S.-L. You, J. Am. Chem. Soc., 2013, 135, 8169 CrossRef CAS PubMed; (b) Y. Zhang, N. Yang, X. Liu, J. Guo, X. Zhang, L. Lin, C. Hua and X. Feng, Chem. Commun., 2015, 51, 8432 RSC.
- K. Leonard, W. Pan, B. Anaclerio, J. M. Gushue, Z. Guo, R. L. DesJarlais, M. A. Chaikin, J. Lattanze, C. Crysler, C. L. Manthey, B. E. Tomczuk and J. J. Marugan, Bioorg. Med. Chem. Lett., 2005, 15, 2679 CrossRef CAS PubMed.
- D. Seebach, H.-O. Kalinowski, W. Langer, G. Crass and E.-M. Wilka, Org. Synth., 1983, 61, 24 CrossRef CAS.
- P. E. Mahaney, A. T. Vu, C. C. McComas, P. Zhang, L. M. Nogle, W. L. Watts, A. Sarkahian, L. Leventhal, N. R. Sullivan, A. J. Uveges and E. J. Trybulski, Bioorg. Med. Chem., 2006, 14, 8455 CrossRef CAS PubMed.
- (a) U. Galm, M. H. Hager, S. G. Van Lanen, J. Ju, J. S. Thorson and B. Shen, Chem. Rev., 2005, 105, 739 CrossRef CAS PubMed; (b) G. A. Elmegeed, A. R. Baiuomy and O. M. E. Abdel-Salam, Eur. J. Med. Chem., 2007, 42, 1285 CrossRef CAS PubMed; (c) L. S. Fernandez, M. L. Sykes, K. T. Andrews and V. M. Avery, Int. J. Antimicrob. Agents, 2010, 36, 275 CrossRef CAS PubMed; (d) I. Ngantchou, B. Nyasse, C. Denier, C. Blonski, V. Hannaert and B. Schneider, Bioorg. Med. Chem. Lett., 2010, 20, 3495 CrossRef CAS PubMed.
- A. Fürstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Data for new compounds and experimental procedures. CCDC 1513960 (1e), 1513961 (1f), 1513962 (5i), and 1513963 (6j). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04858h |
| This journal is © The Royal Society of Chemistry 2020 |