
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aldehydes and ketones influence reactivity and selectivity in nickel-catalysed Suzuki–Miyaura reactions†‡§
Alasdair K.
Cooper
a,
David K.
Leonard¶
 a,
Sonia
Bajo||
a,
Paul M.
Burton
b and
David J.
Nelson
*a
a,
Sonia
Bajo||
a,
Paul M.
Burton
b and
David J.
Nelson
*a
aWestCHEM Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, Scotland, UK. E-mail: david.nelson@strath.ac.uk
bSyngenta, Jealott's Hill International Research Centre, Bracknell, Berkshire RG42 6EY, UK
First published on 6th January 2020
Abstract
The energetically-favorable coordination of aldehydes and ketones – but not esters or amides – to Ni0 during Suzuki–Miyaura reactions can lead either to exquisite selectivity and enhanced reactivity, or to inhibition of the reaction. Aryl halides where the C–X bond is connected to the same π-system as an aldehyde or ketone undergo unexpectedly rapid oxidative addition to [Ni(COD)(dppf)] (1), and are selectively cross-coupled during competition reactions. When aldehydes and ketones are present in the form of exogenous additives, the cross-coupling reaction is inhibited to an extent that depends on the strength of the coordination of the pendant carbonyl group to Ni0. This work advances our understanding of how common functional groups interact with Ni0 catalysts and how these interactions affect workhorse catalytic reactions in academia and industry.
Introduction
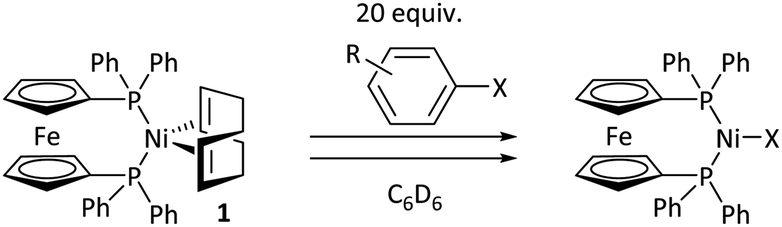
Nickel catalysis has the potential to replace palladium catalysis in some reactions and to enable new reactivity that can be exploited in organic synthesis.1 These reactions include tandem photocatalysis/cross-coupling,2,3 reductive cross-electrophile coupling,4 and the cross-coupling of phenol derivatives,5 aryl fluorides,6,7 and amides.8 Several issues remain to be resolved before the full potential and impact of nickel catalysis can be realised. We must understand how nickel interacts with the functional groups that are present in target molecules in the fine chemicals, agrochemicals, and pharmaceutical industries to understand the scope and limitations of existing methods and the opportunities and challenges to consider when developing new ones. The underlying reaction mechanisms in nickel catalysis, and how these depend on substrate and ligand structure, remain relatively poorly understood compared to palladium catalysis, and so nickel-catalysed reactions are often treated as a ‘black box’. Correspondingly, reaction design and optimisation rely heavily on empirical observations.The mechanisms of nickel-catalysed cross-coupling reactions show a complex dependence on ligand and/or substrate. The mechanistic landscape is further complicated by the often-ambiguous role of nickel(I) species in catalysis.9–12 [Ni(NHC)2] and [Ni(PR3)4] complexes react with aryl halides by oxidative addition or halide abstraction, depending on the ligand and substrate structure.13–17 [Ni(COD)(dppf)] (1)18 undergoes oxidative addition to aryl halides, followed by rapid comproportionation to form [NiX(dppf)] (Fig. 1(a));19 we have established the order of reactivity of a series of aryl (pseudo) halides. It was noted during our previous studies that aryl halides that had aldehyde or ketone substituents underwent unexpectedly fast oxidative addition. Further investigations have revealed that aldehyde and ketone functional groups can act either as directing groups for selective synthesis or as inhibitors of catalytic reactions (Fig. 1(b)). Our initial empirical study showed that various coordinating groups elicit these effects, and that they are considerably more marked for nickel than for palladium.20 Here, we examine the specific case of aldehydes and ketones in depth using a range of experimental and computational techniques.
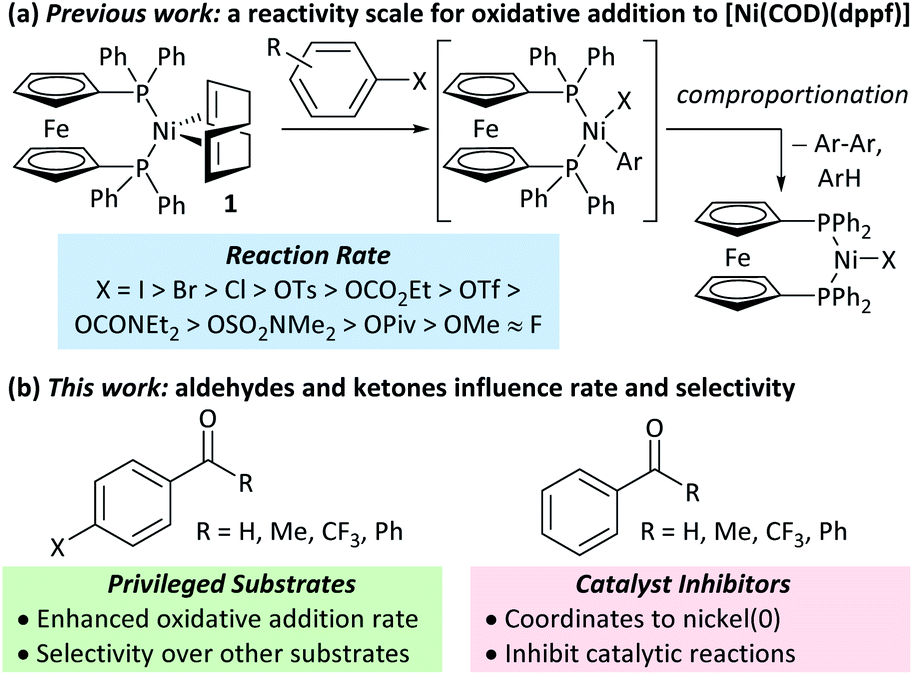 | ||
| Fig. 1 (a) Previous work. (b) This work. | ||
Results and discussion
Kinetic studies of oxidative addition
Aldehyde and ketone-substituted aryl chlorides undergo surprisingly rapid oxidative addition, compared to the oxidative addition rates for a set of other substrates. The consumption of 1 in the presence of an excess of aryl halide was monitored using 31P NMR spectroscopy and exhibited pseudo-first order behaviour; rate data are recorded in Table 1. The ultimate nickel-containing products are [NiX(dppf)] complexes, as confirmed by EPR and 1H NMR spectroscopies.21 In contrast to cross-coupling reactions (vide infra) where transmetalation and reductive elimination can take place, the only pathway available to the arylnickel(II) halide intermediates formed during these kinetic experiments is comproportionation with 1 to form NiI products.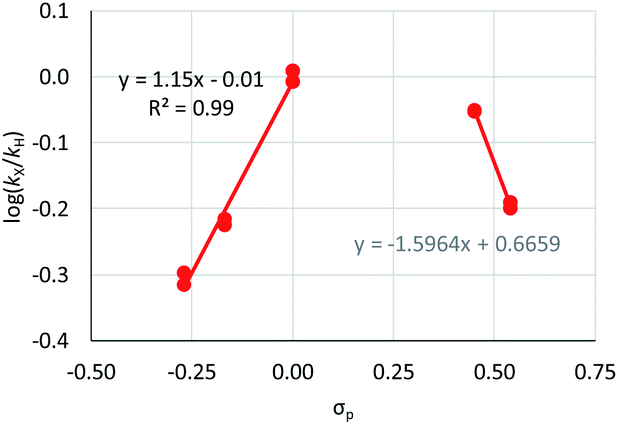 | ||
| Fig. 2 Hammett plot for oxidative addition of aryl chlorides to 1 from reactions at 50 °C in benzene-d6.19 | ||
|
|
|||
|---|---|---|---|
| Substrate | Oxidative addition rate constants | ||
| k obs (20 °C) | k obs (50 °C) | k rel | |
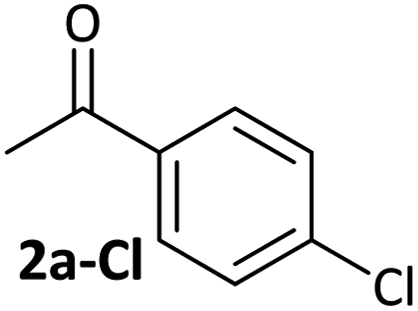
|
2.5(3) × 10−4 s−1 | [nd] | 0.32 |
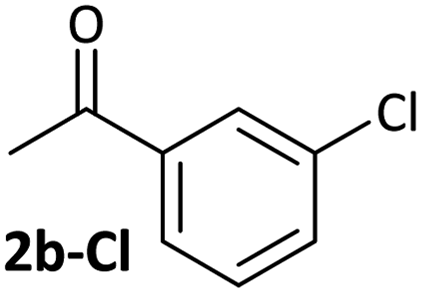
|
4.2(3) × 10−4 s−1 | [nd] | 0.54 |

|
3.0(2) × 10−4 s−1 | [nd] | 0.38 |

|
1.41(1) × 10−4 s−1 | [nd] | 0.18 |
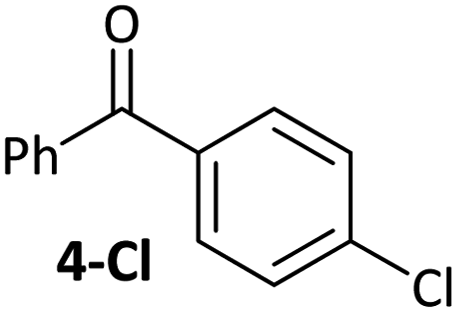
|
9.2(2) × 10−5 s−1 | [nd] | 0.12 |
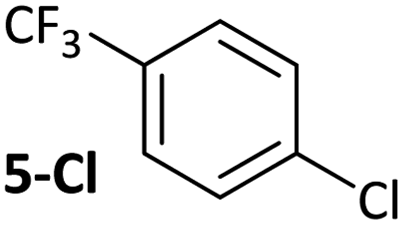
|
[nd] | 2.39(3) × 10−4 s−1 | 0.007 |
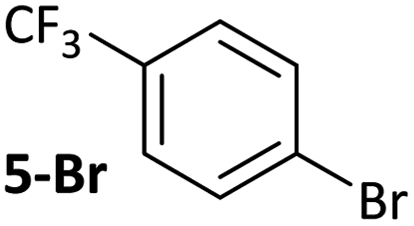
|
2.38(4) × 10−5 s−1 | 1.02(1) × 10−3 s−1 | 0.031 |
|
7.8(1) × 10−4 s−1 | [nd] | 1.0 |
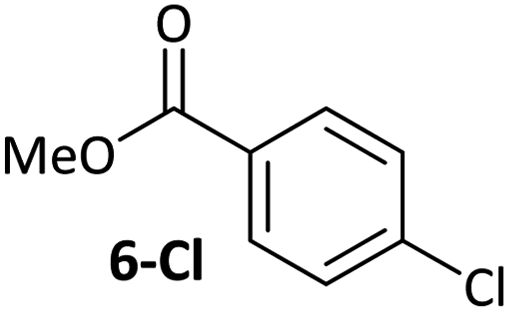
|
[nd] | 1.85(4) × 10−4 s−1 | 0.005 |
Substrates 2-Cl to 4-Cl undergo oxidative addition much more rapidly than electron-deficient aryl chloride 5-Cl or bromide 5-Br. These large rate differences cannot be attributed simply to inductive or mesomeric electronic effects; Hammett σp is 0.4–0.5 for ketones, aldehydes and esters and 0.54 for trifluoromethyl.22 The Hammett plot for oxidative addition of aryl chlorides to 1 is shown in Fig. 2.19
Ligand exchange, where COD is replaced with the aryl halide, and the oxidative addition event that follows cannot be deconvoluted; we propose that the favourable coordination of aldehydes and ketones to Ni0 (vide infra) improves the equilibrium position of the ligand exchange (COD versus aryl halide), leading to a reaction that is more rapid overall. Consistent with this, signals that are tentatively assigned to an η2(CO) complex are observed during the oxidative addition reactions of 3-Cl; various complexes of this type are known,23–30 and have been implicated in nickel-mediated reactions.31 The observed signals in the 31P{1H} NMR spectrum are consistent with a square planar complex (two doublets with 2JPP of 31 Hz). This geometry is a result of electron donation from the bisphosphine ligand into the 3d(x2–y2) orbital, which is also engaged in d to π* back-bonding.32,33 The measured Keq for the displacement of COD from 1 using benzaldehyde (>20), benzophenone (0.65), and acetophenone (0.02) are sufficiently large that under catalytic conditions – i.e. in the presence of a large excess (ca. 10–100 equiv.) of each cross-coupling partner – that this coordination would be expected to occur to the extent that between 2 and 100% of the Ni0 present would be coordinated to a carbonyl group. The lack of a similar effect for esters (e.g.6-Cl) is attributed to n to 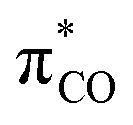 resonance effects between the oxygen lone pair and the ester carbonyl group.
resonance effects between the oxygen lone pair and the ester carbonyl group.
Cross-coupling selectivity
The coordination of aldehydes and ketones to Ni0 can be leveraged to achieve selective cross-coupling. Optimised cross-coupling conditions were developed for the prototypical cross-coupling of 2a-Br with p-tolylboronic acid, catalysed by well-defined [NiCl(o-tol)(dppf)] pre-catalyst 7,34 using factorial experimental design.§ To dissect the contributions of electronic and coordination effects, experiments were performed in which bromobenzene and a functionalized aryl bromide competed for a limiting amount of boronic acid (Fig. 3(a)).** Data were interpreted by quantifying selectivity using eqn (1) (Fig. 3(b)); this has values between −1 and +1 which represent complete selectivity for bromobenzene and functionalised aryl bromide, respectively. The reaction selectivity was plotted versus σ (Fig. 3(c)).35 These data show that selectivity is typically insensitive to the electronic properties of the aryl halide, but significant selectivity is achieved when the aryl halide has a ketone or aldehyde functional group.36 This selectivity trend is observed for both meta- and para-substituted aryl bromides. These data confirm that aldehyde and ketone substituents can be used to induce selective cross-coupling at one substrate present within the reaction.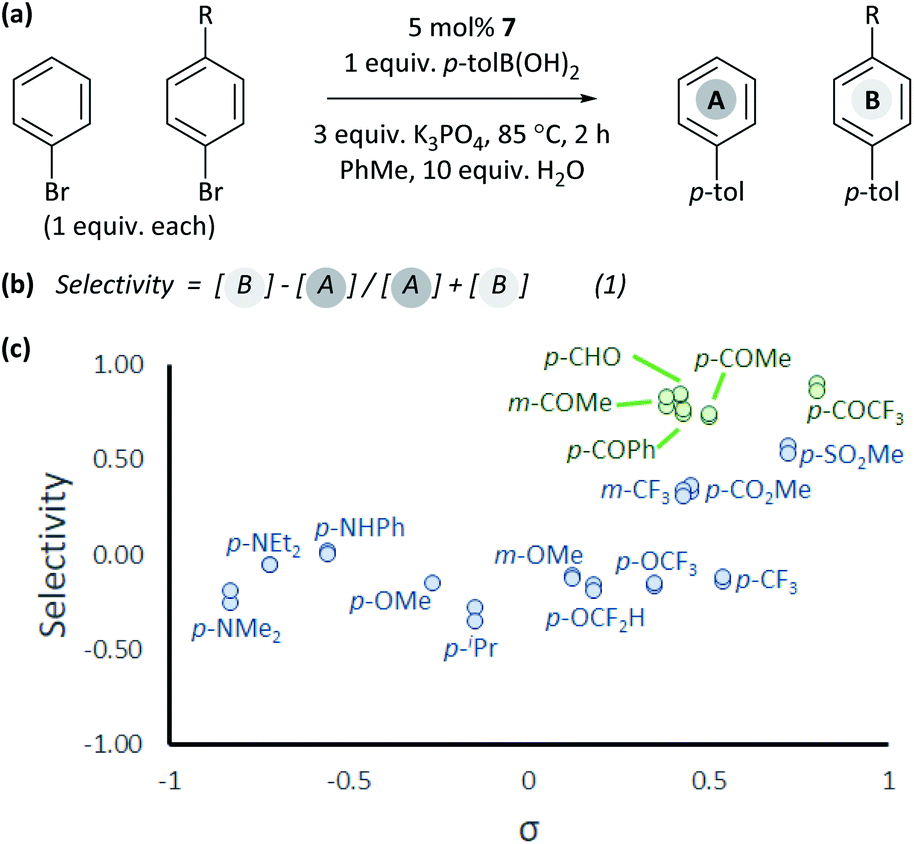 | ||
| Fig. 3 Competition experiments: (a) reaction conditions; (b) quantification of selectivity; and (c) reactions in toluene with 10 equiv. water. Each individual competition experiment is plotted as a separate point. | ||
Computational modelling of ring walking and oxidative addition
DFT calculations give further insights into these reactions.†† [Ni(dppf)(η2-benzene)] (8) was assigned Grel = 0. Consistent with experiment, aldehydes and ketones coordinate [Ni(dppf)] exergonically via the carbonyl group (Fig. 4(a)). Free energy profiles were calculated for the oxidative addition reactions of selected aryl bromides (Fig. 4(b));37 reactions proceed via transition state B to irreversibly form NiII products C. The aldehyde substrate can also form the η2(CO) complex.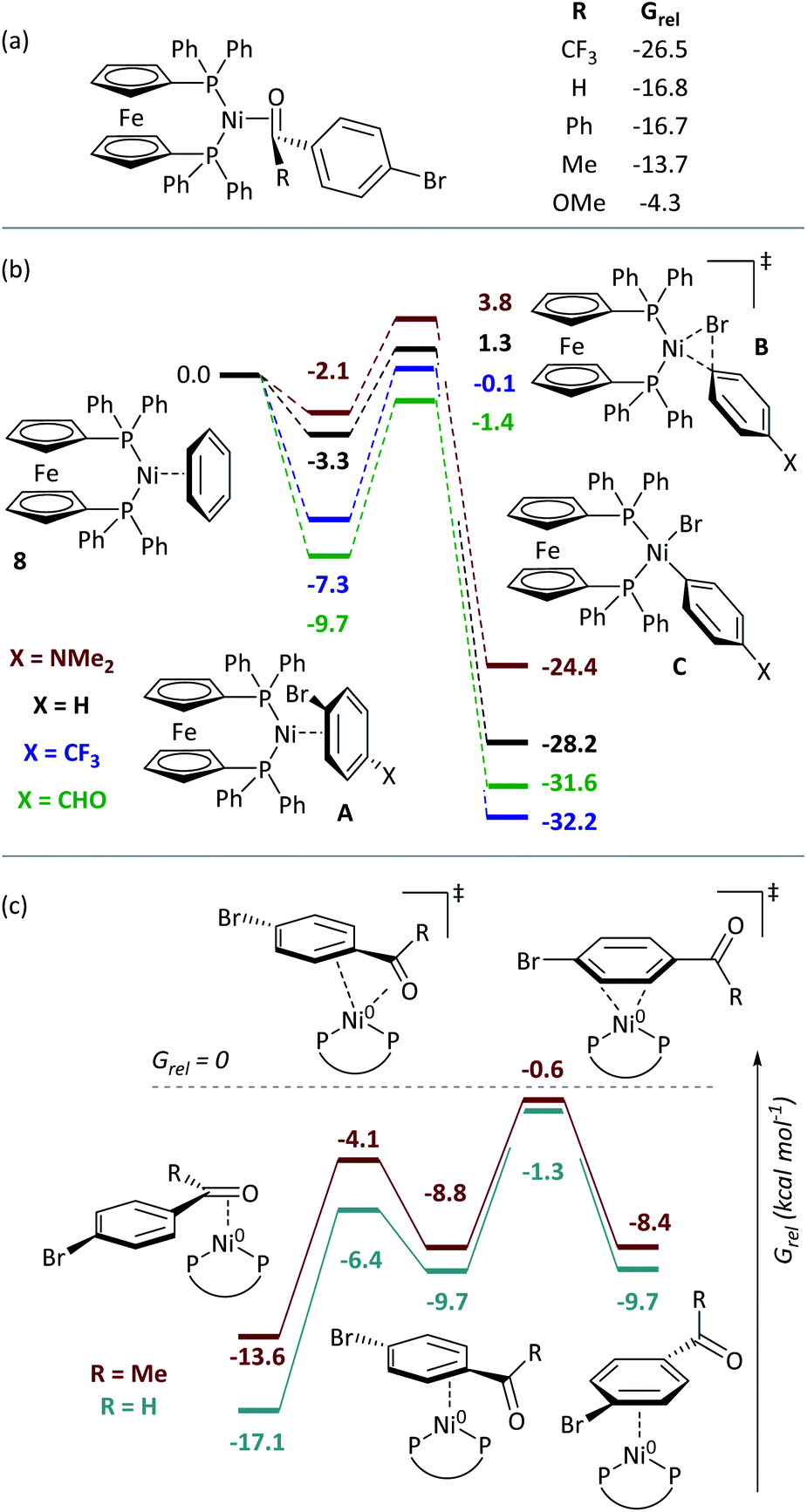 | ||
| Fig. 4 (a) Coordination to aldehydes, ketones, and esters. (b) Free energy profile for oxidative addition. (c) Calculations on the ring-walking process. All energies are free energies in toluene solution and are quoted in kcal mol−1. | ||
Further calculations were undertaken to model the ring-walking38–42 step, using 2a-Br and 3-Br as substrates. In each case, three η2-complexes are linked by two transition states, leading from the carbonyl group to the halide site (Fig. 4(c)). The transition states for these ring-walking steps are all lower in energy than 8 and so this process is more energetically favourable than the dissociation of the aryl halide and coordination of a new aryl halide; once an aldehyde- or ketone-containing substrate coordinates the nickel centre via the carbonyl ligand, it will undergo oxidative addition after ring walking, rather than ligand exchange for an alternative substrate. Substrate 2c-Br undergoes a similar ring-walking process, again with no intermediates or transition states that have Grel > 0.§ These data are consistent with two key experimental observations: (i) the enhanced rate of oxidative addition, because η2(CO) coordination renders aldehyde- and ketone-containing aryl halides competitive ligands (versus COD); and (ii) selective cross-coupling, because aldehyde- and ketone-containing aryl halides are much better ligands for Ni0 than aryl halides without such strongly coordinating groups.
Catalyst inhibition by aldehydes and ketones
The coordination of ketones and aldehydes to Ni0 might be expected to have a detrimental effect on the performance of cross-coupling reactions if this behaviour sequesters the active catalyst. A ‘robustness screen’43,44 was used, in which a model reaction was carried out in the presence of 1 equiv. of each of a series of additives (9–19).** This provides a rapid and quantitative measure of the effect of each additive. A palette of additives was examined (Fig. 5).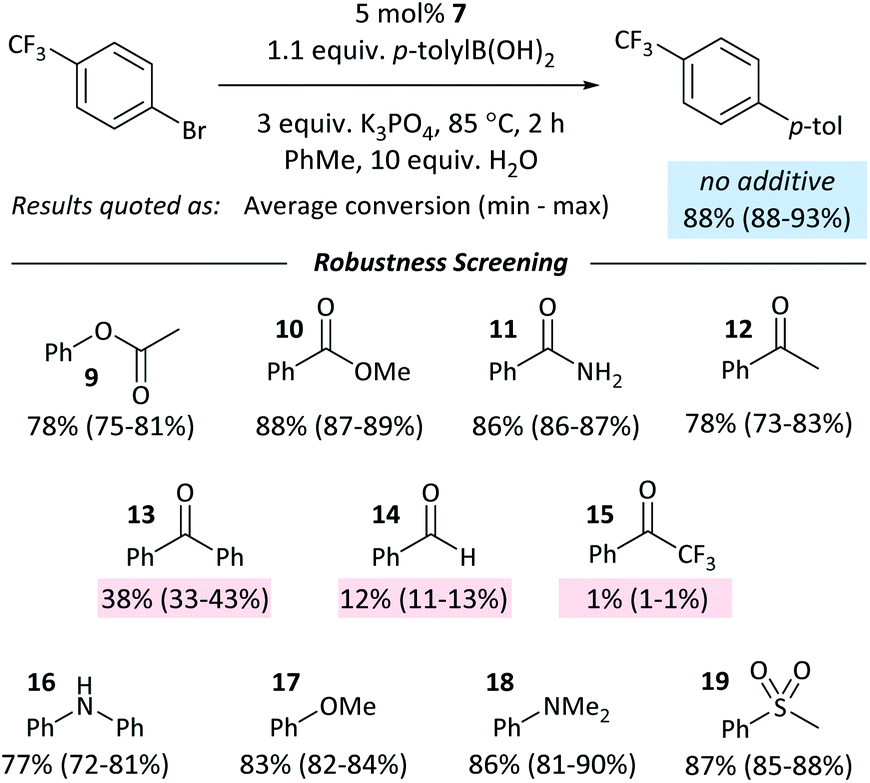 | ||
| Fig. 5 Results from the robustness screen. | ||
Most additives had little effect on the reaction conversion. Aldehyde and ketone additives – except for acetophenone (12) – had a significant and detrimental effect, with the reaction almost completely ceasing when 1 equiv. of 2,2,2-trifluoroacetophenone (15) was present. The degree of inhibition of the reaction is correlated to Keq for the displacement of COD from 1 (vide supra); it may be necessary to protect aldehydes and ketones in some reactions to prevent their coordination to Ni0. If catalyst decomposition was responsible for the decrease in yield then we would expect that at the end of the reaction ≥5% of the additive would have been consumed;45 in the case of 13 and 14, ≤ 2% of the additive is unaccounted for, while 15 hydrates in the presence of water, resulting in the absence of 12–15% of 15 at the end of the reaction. There is no correlation between the degree of inhibition and the amount of additive unaccounted for.
Control experiments where one equivalent of benzaldehyde was added to the cross-coupling of 4-Cl and p-tolylboronic acid showed complete conversion to the cross-coupling product. Aldehyde and ketone additives therefore have less of an effect on the cross-coupling reactions of aldehyde- and ketone-containing aryl halides than they do on the cross-coupling of aryl halides that do not contain these functional groups.
The robustness screening data are consistent with coordination of an exogenous additive to the Ni0 catalyst inhibiting the reaction. No ring-walking is possible from such intermediates to a site for oxidative addition or other exergonic onward reaction.
On the generality of these effects
These observations are not limited to dppf-based (pre)catalysts. [NiCl(o-tol)(L)n] (L = dppe (20), Xantphos (21))34 were applied in: (i) competition reactions between 5-Br and 3-Br, in which 3-Br reacts exclusively; and (ii) robustness screening reactions using 5-Br as the substrate and benzaldehyde as the additive, showing significant reaction inhibition (Fig. 6).†† Complexes 20 and 21 perform significantly less well than 7 but may yield better results if further reaction optimisation was carried out. The results in Fig. 6 illustrate that the effect is general to nickel phosphine complexes, and not limited to dppf-based catalysts. Palladium, specifically [PdCl2(dppf)], shows a far reduced propensity to undergo selective cross-coupling reactions, but has far better functional group tolerance.20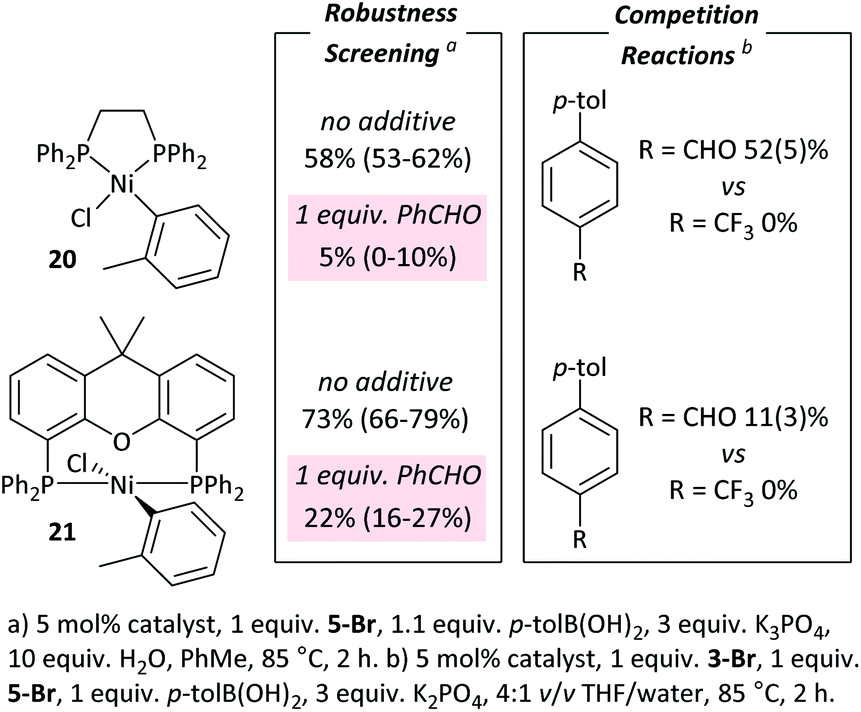 | ||
| Fig. 6 Results with dppe and Xantphos catalysts. | ||
Opportunities for exploitation
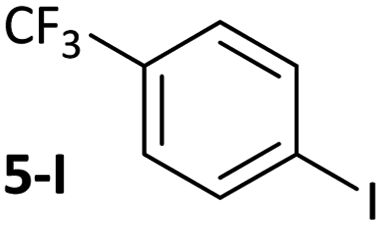
The carbonyl coordination effect was leveraged to achieve site selectivity in cross-coupling reactions, both intermolecularly and intramolecularly (Fig. 7). The aldehyde/ketone effect allows the normal reactivity order of aryl halides (I > Br > Cl)19 to be entirely overridden, to the extent that an aryl chloride (2a-Cl) undergoes selective coupling in the presence of an electron-deficient aryl bromide (5-Br) and even in the presence of an aryl iodide (5-I) (Fig. 7(a)). Reactions are selective for aldehydes over ketones, as judged from further competition experiments.§ Ketones that are not in conjugation with the aryl halide π-system are not selectively coupled (e.g. 22-Br) (Fig. 7(b)). Intramolecular competition experiments were carried out using 23-Cl, which has two aryl chloride sites that are available for cross-coupling but only one is in conjugation with a ketone (Fig. 7(c)); reactions with 23-Cl establish that selective cross-coupling occurs at the site that is in conjugation with the ketone. Intrinsic differences in the reactivities of aryl chlorides, bromides, and iodides have been leveraged in the past for selective cross-coupling reactions,46 but this work shows that the substitution pattern of the aryl halide can significantly change the order of reactivity, opening new avenues for creative organic synthesis using nickel-catalysed cross-coupling.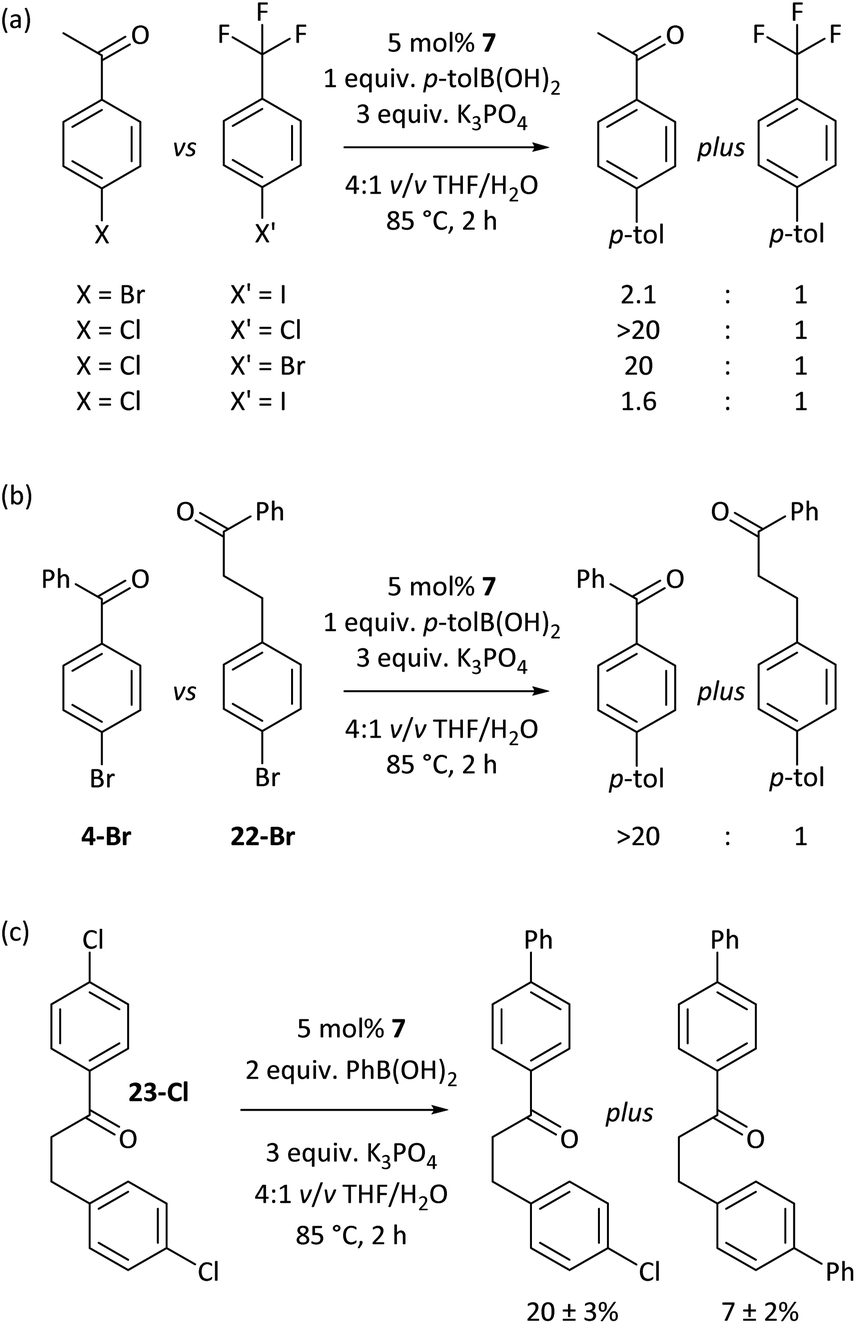 | ||
| Fig. 7 (a) Overriding the intrinsic reactivity differences between aryl halides. (b) Selectivity between different carbonyl-containing substrates. (c) Site-selectivity within a molecule, enforced by carbonyl coordination. | ||
Conclusions
This work establishes how aldehydes and ketones can have positive or negative effects on nickel-catalysed reactions, depending on their location. The formation of η2-complexes has been implicated in some nickel-catalysed reactions30,31,39,47,48 but this work shows that coordination effects can influence even simple and ubiquitous Suzuki–Miyaura reactions.• Aryl halides with aldehyde and ketone functional groups undergo rapid oxidative addition because of favourable ligand exchange and ring-walking processes.
• Selectivity is achieved for aldehyde- or ketone-containing aryl halides over other aryl halide substrates because of this favourable coordination.
• Aldehydes and ketones that are present in cross-coupling reactions but are not in conjugation with the aryl halide act as inhibitors of catalysis.
• Aldehydes and ketone-containing aryl halides undergo successful cross-coupling.
Fig. 8 summarises the conclusions of this study using Ni0/NiII cycles based on literature evidence from studies of nickel/dppf-catalysed reactions.18,19,49–53Fig. 8(a) shows a cycle for a reaction in which an aldehyde or ketone is present as an additive; a low energy η2(CO) complex sequesters the active catalyst and slows the reaction because the η2-complex with the aryl halide is higher in energy. Fig. 8(b) shows a cycle for an aryl halide with an aldehyde or ketone substituent.
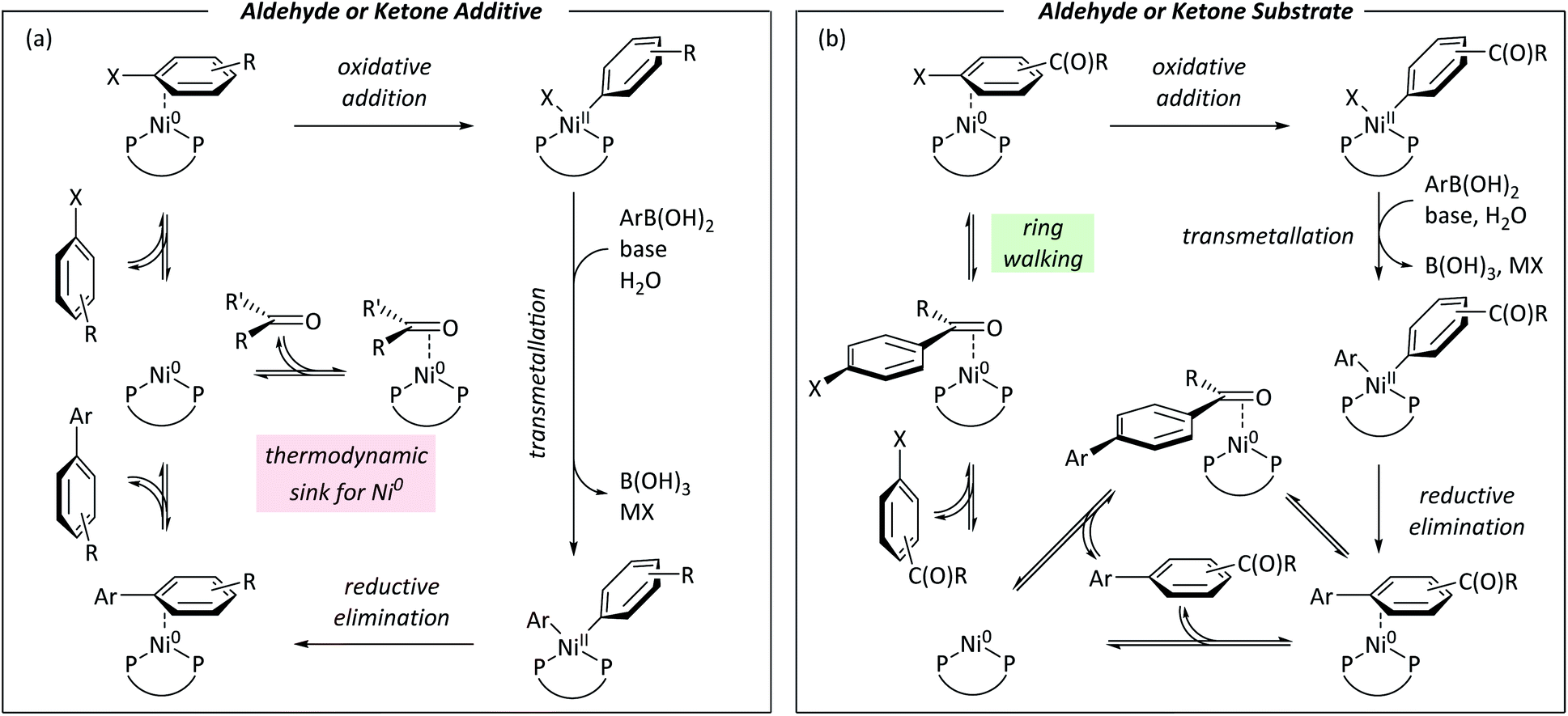 | ||
| Fig. 8 (a) Catalytic cycle for nickel-catalysed Suzuki–Miyaura reactions in the presence of exogenous aldehyde or ketone. (b) Catalytic cycle for nickel-catalysed Suzuki–Miyaura reactions of aldehyde- and ketone-containing aryl halides. | ||
We are currently pursuing further work within our laboratories to understand the effects of a wider range of functional groups on nickel-catalysed reactions and their fundamental steps using kinetic and mechanistic studies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for funding from: a Syngenta/Engineering and Physical Sciences Research Council (EPSRC) Industrial CASE Studentship for AKC (EP/P51066X/1); the EPSRC (EP/M027678/1); the University of Strathclyde for a Chancellor's Fellowship for DJN (2014–2018); and the Carnegie Trust for the Universities of Scotland for a Research Incentive Grant (RIG008165). We thank the Department of Pure and Applied Chemistry at the University of Strathclyde for consumables and facilities funding for DKL. We thank Mr G. Bain, Mr C. Irving, Ms P. Keating, and Dr J. Parkinson for assistance with technical and analytical facilities. We thank Dr J. Sanderson (University of Newcastle) for assistance with initial factorial experimental design studies. Some of the results reported here were obtained using the EPSRC-funded ARCHIE-WeSt high-performance computer (archie-west.ac.uk) (EP/K000586/1); we are grateful to Mr J. Buzzard, Dr K. Kubiak-Ossowska, and Dr R. Martin for their assistance with this facility. We thank Dr S. Sproules for EPR analyses and Dr A. Watson (University of St Andrews) for helpful discussions.Notes and references
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- J. Twilton, P. Zhang, M. H. Shaw, R. W. Evans and D. W. MacMillan, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS PubMed.
- D. J. Weix, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS PubMed.
- T. Mesganaw and N. K. Garg, Org. Process Res. Dev., 2012, 17, 29–39 CrossRef.
- T. Schaub, M. Backes and U. Radius, J. Am. Chem. Soc., 2006, 128, 15964–15965 CrossRef CAS PubMed.
- M. Tobisu, T. Xu, T. Shimasaki and N. Chatani, J. Am. Chem. Soc., 2011, 133, 19505–19511 CrossRef CAS PubMed.
- J. E. Dander and N. K. Garg, ACS Catal., 2017, 7, 1413–1423 CrossRef CAS PubMed.
- C. Y. Lin and P. P. Power, Chem. Soc. Rev., 2017, 46, 5347–5399 RSC.
- J. Jover, Catal. Sci. Technol., 2019, 9, 5962–5970 RSC.
- T. Inatomi, Y. Fukahori, Y. Yamada, R. Ishikawa, S. Kanegawa, Y. Koga and K. Matsubara, Catal. Sci. Technol., 2019, 9, 1784–1793 RSC.
- G. D. Jones, J. L. Martin, C. McFarland, O. R. Allen, R. E. Hall, A. D. Haley, R. J. Brandon, T. Konovalova, P. J. Desrochers, P. Pulay and D. A. Vicic, J. Am. Chem. Soc., 2006, 128, 13175–13183 CrossRef CAS PubMed.
- I. Funes-Ardoiz, D. J. Nelson and F. Maseras, Chem.–Eur. J., 2017, 23, 16728–16733 CrossRef CAS PubMed.
- D. J. Nelson and F. Maseras, Chem. Commun., 2018, 54, 10646–10649 RSC.
- K. Zhang, M. Conda-Sheridan, S. R. Cooke and J. Louie, Organometallics, 2011, 30, 2546–2552 CrossRef CAS PubMed.
- D. S. McGuinness, K. J. Cavell, B. W. Skelton and A. H. White, Organometallics, 1999, 18, 1596–1605 CrossRef CAS.
- A. Manzoor, P. Wienefeld, M. C. Baird and P. H. M. Budzelaar, Organometallics, 2017, 36, 3508–3519 CrossRef CAS.
- G. Yin, I. Kalvet, U. Englert and F. Schoenebeck, J. Am. Chem. Soc., 2015, 137, 4164–4172 CrossRef CAS PubMed.
- S. Bajo, G. Laidlaw, A. R. Kennedy, S. Sproules and D. J. Nelson, Organometallics, 2017, 36, 1662–1672 CrossRef CAS.
- A. K. Cooper, P. M. Burton and D. J. Nelson, Synthesis, 2020 DOI:10.1055/s-0039-1690045.
- We have established the mechanism of this reaction, and showed that this proceeds via oxidative addition to form Ni(II) intermediates – which are only observable when the aryl halide has ortho-substituents – which then undergo comproportionation to Ni(0). Please see ref. 19 for details.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- D. Walther, Z. Anorg. Allg. Chem., 1977, 431, 17–30 CrossRef CAS.
- D. Walther, J. Organomet. Chem., 1980, 190, 393–401 CrossRef CAS.
- J. Kaiser, J. Sieler, D. Walther, E. Dinjus and L. Golic, Acta Crystallogr., Sect. B: Struct. Sci., 1982, 38, 1584–1586 CrossRef.
- R. Countryman and B. R. Penfold, J. Cryst. Mol. Struct., 1972, 2, 281–290 CrossRef CAS.
- T. T. Tsou, J. C. Huffman and J. K. Kochi, Inorg. Chem., 1979, 18, 2311–2317 CrossRef CAS.
- A. Flores-Gaspar, P. Pinedo-González, M. G. Crestani, M. Muñoz-Hernández, D. Morales-Morales, B. A. Warsop, W. D. Jones and J. J. García, J. Mol. Catal. A: Chem., 2009, 309, 1–11 CrossRef CAS.
- R. Doi, K. Kikushima, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2015, 137, 3276–3282 CrossRef CAS PubMed.
- C. Lei, Y. J. Yip and J. S. Zhou, J. Am. Chem. Soc., 2017, 139, 6086–6089 CrossRef CAS PubMed.
- Y. Hoshimoto, M. Ohashi and S. Ogoshi, Acc. Chem. Res., 2015, 48, 1746–1755 CrossRef CAS PubMed.
- A. N. Desnoyer, W. He, S. Behyan, W. Chiu, J. A. Love and P. Kennepohl, Chemistry, 2019, 25, 5259–5268 CrossRef CAS PubMed.
- W. He and P. Kennepohl, Faraday Discuss., 2019, 220, 133–143 RSC.
- E. A. Standley, S. J. Smith, P. Müller and T. F. Jamison, Organometallics, 2014, 33, 2012–2018 CrossRef CAS PubMed.
- Equivalent data are obtained in a THF/water solvent mixture; please see ref. 16.
- Control experiments with ketone-functionalised boronic acids did not lead to selective cross-coupling of these species (see the ESI§). Coordination occurs to Ni(0) only, and not to the more sterically-crowded and less electron-rich Ni(II) intermediates.
- Free energy profiles for the corresponding reactions of a wider range of aryl halides can be found in the ESI.§.
- N. Yoshikai, H. Matsuda and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 15258–15259 CrossRef CAS PubMed.
- L. Guo, S. Dai, X. Sui and C. Chen, ACS Catal., 2016, 6, 428–441 CrossRef CAS.
- O. V. Zenkina, A. Karton, D. Freeman, L. J. W. Shimon, J. M. L. Martin and M. E. van der Boom, Inorg. Chem., 2008, 47, 5114–5121 CrossRef CAS PubMed.
- J. A. Bilbrey, A. N. Bootsma, M. A. Bartlett, J. Locklin, S. E. Wheeler and W. D. Allen, J. Chem. Theory Comput., 2017, 13, 1706–1711 CrossRef CAS PubMed.
- S. K. Sontag, J. A. Bilbrey, N. E. Huddleston, G. R. Sheppard, W. D. Allen and J. Locklin, J. Org. Chem., 2014, 79, 1836–1841 CrossRef CAS PubMed.
- K. D. Collins and F. Glorius, Acc. Chem. Res., 2015, 48, 619–627 CrossRef CAS PubMed.
- K. D. Collins and F. Glorius, Nat. Chem., 2013, 5, 597–601 CrossRef CAS PubMed.
- This assumes a >1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 additive:nickel stoichiometry in such a decomposition reaction, based on the low concentrations of nickel in the reactions and the high concentrations of additive (20 equiv. with respect to nickel)..
:1 additive:nickel stoichiometry in such a decomposition reaction, based on the low concentrations of nickel in the reactions and the high concentrations of additive (20 equiv. with respect to nickel).. - C. P. Seath, J. W. B. Fyfe, J. J. Molloy and A. J. B. Watson, Angew. Chem., Int. Ed., 2015, 54, 9976–9979 CrossRef CAS PubMed.
- E. L. Lanni and A. J. McNeil, J. Am. Chem. Soc., 2009, 131, 16573–16579 CrossRef CAS PubMed.
- W. He, B. O. Patrick and P. Kennepohl, Nat. Commun., 2018, 9, 3866 CrossRef PubMed.
- L. M. Guard, M. Mohadjer Beromi, G. W. Brudvig, N. Hazari and D. J. Vinyard, Angew. Chem., Int. Ed., 2015, 54, 13352–13356 CrossRef CAS PubMed.
- I. Kalvet, Q. Guo, G. J. Tizzard and F. Schoenebeck, ACS Catal., 2017, 7, 2126–2132 CrossRef CAS PubMed.
- M. Mohadjer Beromi, A. Nova, D. Balcells, A. M. Brasacchio, G. W. Brudvig, L. M. Guard, N. Hazari and D. J. Vinyard, J. Am. Chem. Soc., 2017, 139, 922–936 CrossRef CAS PubMed.
- M. Mohadjer Beromi, G. Banerjee, G. W. Brudvig, N. Hazari and B. Q. Mercado, ACS Catal., 2018, 8, 2526–2533 CrossRef CAS PubMed.
- M. Mohadjer Beromi, G. Banerjee, G. W. Brudvig, D. J. Charboneau, N. Hazari, H. M. C. Lant and B. Q. Mercado, Organometallics, 2018, 37, 3943–3955 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnotes |
| † Dedicated to Professor Robert Mulvey on the occasion of his 60th birthday. |
| ‡ All data underpinning this publication are openly available from the University of Strathclyde KnowledgeBase at https://doi.org/10.15129/0e956fd7-939e-4b60-be15-d530ed0b8b2c. |
| § Electronic supplementary information (ESI) available: rate constants, reaction procedures, characterisation data for compounds, and energies and geometries from DFT studies. See DOI: 10.1039/c9sc05444h. |
| ¶ Current address: Leibniz-Institute for Catalysis, Albert-Einstein-Straße 29a, 18059 Rostock, Germany. |
| || Current address: Institute for Chemical Research (IIQ), CSIC-University of Sevilla, Avda. Américo Vespucio 49, 41092 Sevilla, Spain. |
| ** Reaction outcomes were analysed using calibrated GC-FID analyses. In some competition experiments, total yield sometimes slightly exceeded 100% which is likely to be due to errors in weighing out exactly one equivalent of boronic acid. |
| †† Calculations were carried out using Gaussian09 Rev. D01.54 Geometry optimisations were carried out without symmetry constraints using the B3LYP functional with Grimme's D3 dispersion corrections, the LANL2DZ(dp) basis set on Br and I, the LANL2TZ(f) basis set on Ni and Fe, and the 6-31G(d) basis set on all other atoms. The nature of stationary points was confirmed using frequency calculations. Energies were refined using single point calculations in which the 6-31G(d) basis set was exchanged for 6-311+G(d,p). All calculations were carried out in toluene solvent using the SMD solvent model. |
| This journal is © The Royal Society of Chemistry 2020 |