DOI:
10.1039/C9SC04569D
(Edge Article)
Chem. Sci., 2020,
11, 1912-1917
From 1,2-difunctionalisation to cyanide-transfer cascades – Pd-catalysed cyanosulfenylation of internal (oligo)alkynes†
Received
10th September 2019
, Accepted 22nd December 2019
First published on 23rd December 2019
Abstract
Internal alkynes substituted by aliphatic or aromatic moieties or by heteroatoms were converted into sulphur-substituted acrylonitrile derivatives. Key is the use of Pd catalysis, which allows the addition of aromatic and aliphatic thiocyanates in an intra- and intermolecular manner. Substrates with several alkyne units underwent further carbopalladation steps after the initial thiopalladation step, thus generating in a cascade-like fashion an oligoene unit with sulphur at one terminus and the cyano group at the other.
Introduction
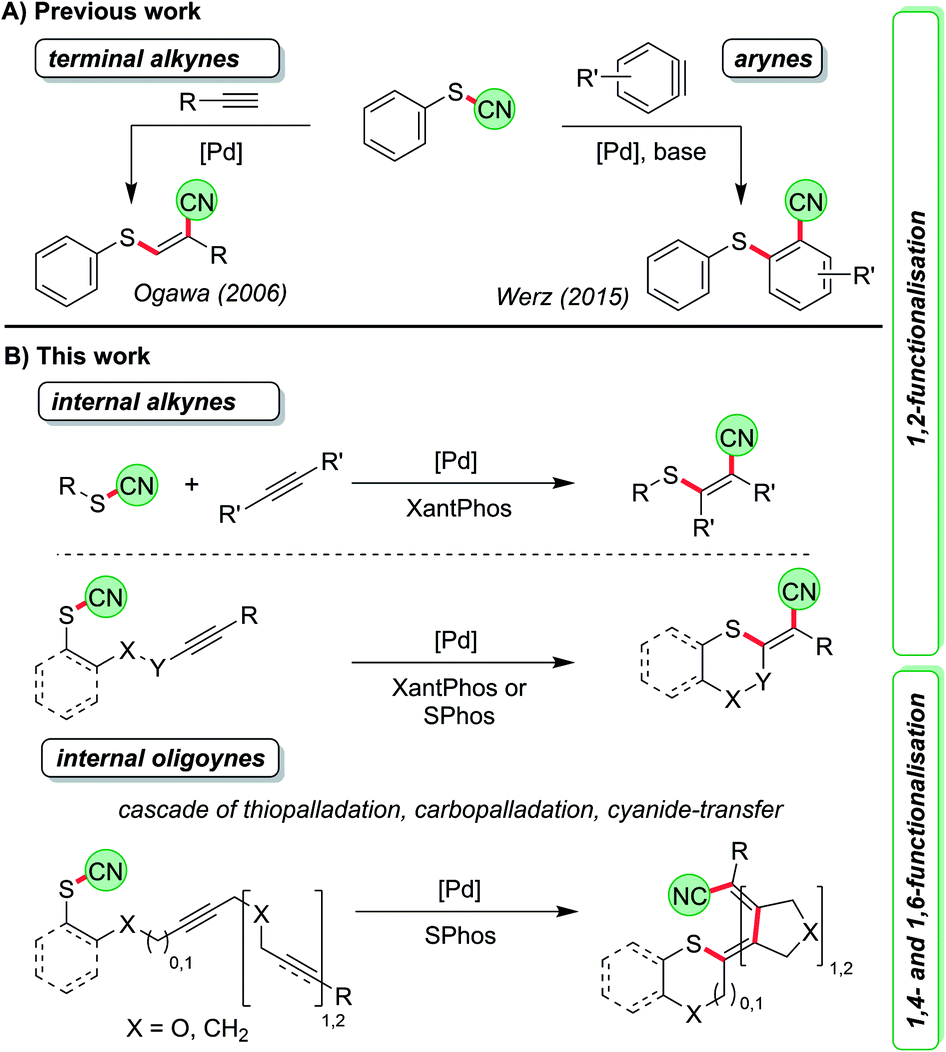
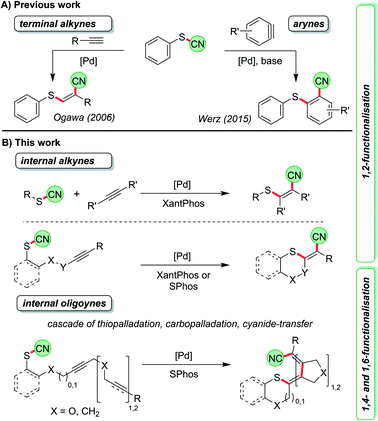
An important method of accessing tetrasubstituted double bonds is the dual functionalisation of internal alkynes.1 In its ideal and atom-economic way a substrate X–Y is attached to the C–C triple bond in a completely regio- and diastereoselective fashion. Depending on the type of substituents, either specially designed reagents are required or transition-metal catalysts are employed.2–4 As a prime example for the first scenario a syn-chlorocyanation using imidazolium thiocyanate in combination with BCl3 was recently reported by Alcarazo et al.2a Morandi developed a Pd-catalysed intermolecular syn-aryliodination of internal alkynes5 while Lautens and co-workers disclosed a series of intramolecular reactions leading to syn- and anti-carbohalogenations of triple bonds.6 The key step of the latter reaction is a carbopalladation of the C–C triple bond followed by reductive elimination of the Pd to generate the carbon–halogen bond. Such a step is rare and can only be triggered by specific ligands.5,6 The more common scenario is further reaction in a carbopalladation cascade7 when other alkyne or alkene units are present.8 In most cases a final elimination or cross-coupling reaction is employed to terminate the process. By such protocols, structures of astonishing complexity9 such as fenestranes,10 cyclooctatetraenes,11 helicenes,12 and several natural products13 have been prepared from appropriately designed starting materials in just one synthetic step. The groups of Lautens, Cook and others have precisely designed intramolecular iodine-transfer reactions along these lines.14 With other moieties such transfer reactions over several carbon atoms have only rarely been investigated.
Cyanosulfenylation reactions of terminal alkynes have been developed to access sulphur-substituted acrylonitrile derivatives starting from thiocyanates (Scheme 1A, left).15 In these transformations the S–CN bond is broken and added across the C–C triple bond, which is comparable to reactions where O–CN or N–CN-bonds are activated to react with alkenes.16 Later, we found that a similar Pd-catalysed process allows the reaction with arynes, which contain highly reactive formal triple bonds, leading to o-thiobenzonitriles (Scheme 1A, right).17
 |
| | Scheme 1 Inter- and intramolecular cyanosulfenylation reactions. | |
Thus, we were first interested in seeking conditions to transform internal non-activated triple bonds to generate tetrasubstituted alkenes with sulphur and cyano in 1,2-position (Scheme 1B), and to determine whether this reaction might be also the starting point for a longer cascade involving – besides the thiopalladation step – carbopalladation steps of additional C–C triple bonds. The termination of the cascade was envisioned as a reductive elimination of the Pd catalyst to form the C–CN bond. Such an approach would lead to formal 1,4- and 1,6-cyanosulfenylation reactions (Scheme 1B).
Results and discussions
Optimisation of the reaction conditions
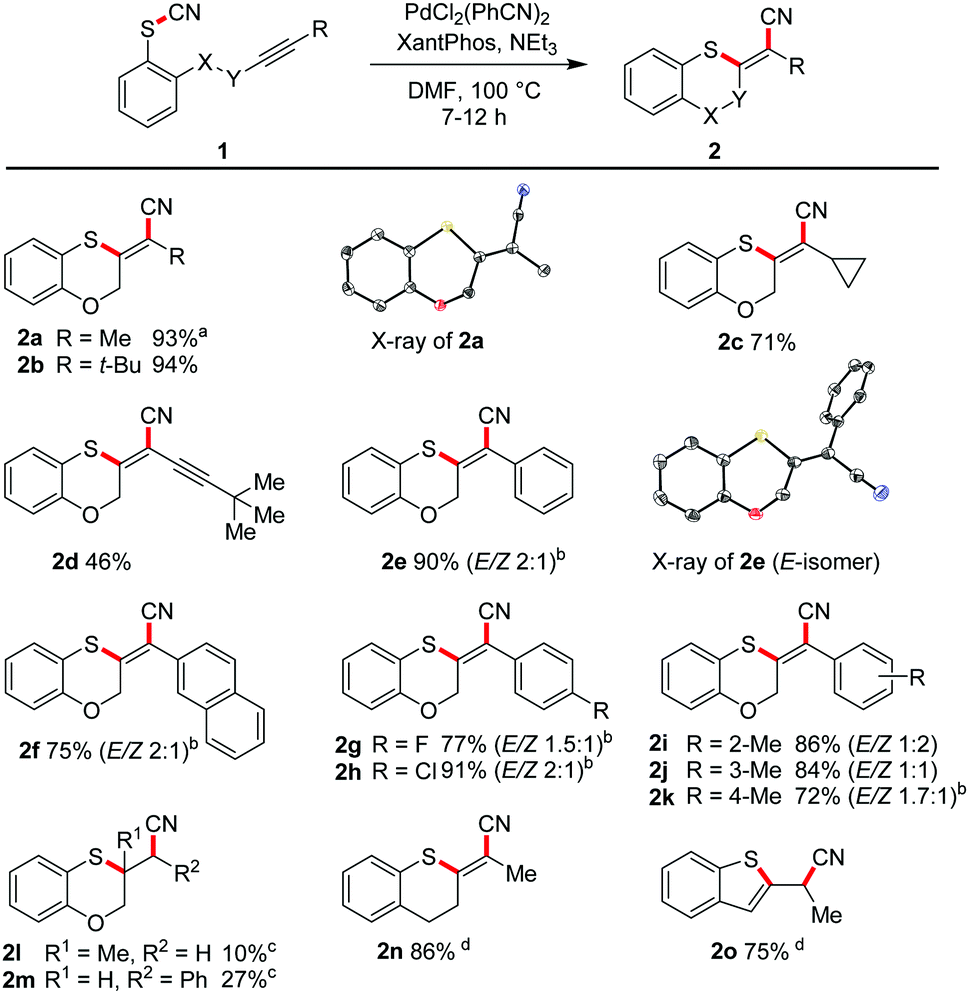
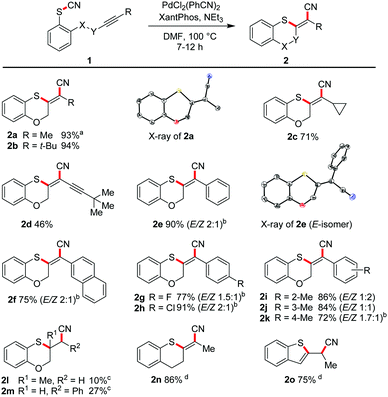
Initially, we explored the Pd-catalysed reaction of the aromatic thiocyanate unit across the C–C triple bond in 1a. PdCl2(PhCN)2, tris(t-butyl)phosphine as ligand (derived from Fu's salt18) and triethylamine in DMF at 100 °C generated the benzoxathiin 2a in 4 h with 35% yield. Various other ligands gave no better results. A breakthrough was achieved using XantPhos, which led to an excellent yield of 93% (for full optimisation details, see ESI†).
Scope of aromatic thiocyanates
With these conditions in hand, we explored the scope and limitations of this transformation (Scheme 2).
 |
| | Scheme 2 Intramolecular cyanosulfenylation with aromatic thiocyanates. Reaction conditions: 1 (1.00 equiv.), PdCl2(PhCN)2 (10 mol%), XantPhos (20 mol%), NEt3 (5.00 equiv.), DMF (20 mM), 100 °C, 7 h. aLarge scale (1.5 mmol, 93%), Pd(PPh3)4 (5 mol%), SPhos (20 mol%), 160 °C, 3 h, PhMe (20 mM). bZ-isomer is shown even if it is the minor isolated product because that is consistent with the plausible mechanism; E-isomer arises as result of a strong push–pull-system.19cPd(PPh3)4 (5 mol%), SPhos (20 mol%), 160 °C, 6 h, DMF (20 mM). dPd(PPh3)4 (5 mol%), SPhos (20 mol%), 110 °C, 3 h, DMF (20 mM). X = O, (CH2)m; Y = (CH2)n; m, n = 0, 1, 2. | |
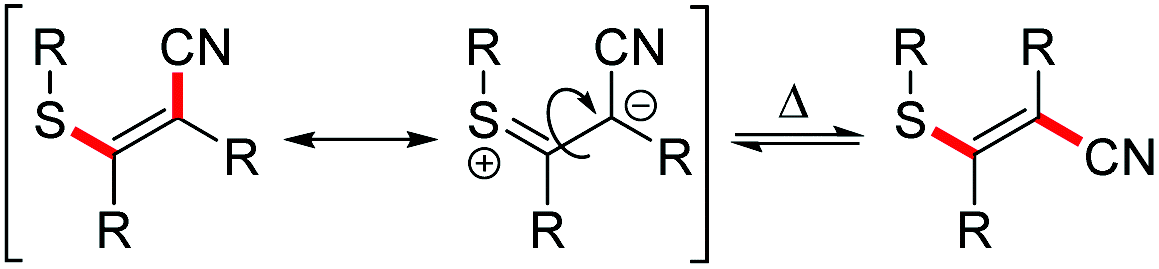
Substrates with aliphatic termini were smoothly transformed and yielded products 2a–2c in 71–94% yield. A 1,3-diyne unit reacted to enyne 2d (46%). Differently substituted aromatic termini were tolerated and furnished products 2e–2k in up to 91% yield. However, mixtures of E/Z-isomers were found because of the highly polarized character of the emerging tetrasubstituted olefin moiety. In these cases, the thioacrylonitrile moiety, as a strong push–pull-system, easily stabilizes a zwitterionic structure.19 The positive charge is well stabilized at the sulphur and the negative charge next to the aryl substituent. This formal single bond character allows rotation leading to isomeric mixtures.20
Furthermore, 2l (10%) and 2m (27%) were synthesised from two alkenes as starting materials to demonstrate that also other π-systems can be employed in our protocol. To address smaller ring sizes the tether between the aromatic ring system and the C–C triple bond was shortened to afford substituted benzothiophene 2o as the result of a subsequent isomerisation to the more stabilized aromatic system. A further shortening led not to an isolable four-membered ring benzothiete, but to a 3-cyano-substituted benzothiophene (2s, see ESI†), as proved by X-ray crystallography.21
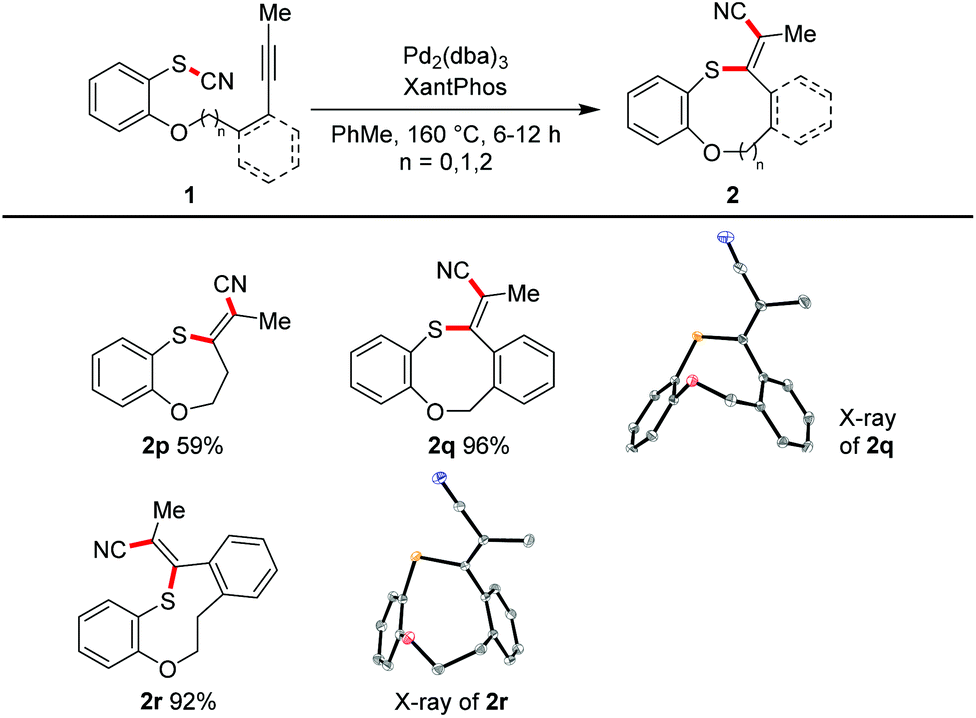
Seven- to nine-membered ring systems
We varied the ring size also in the opposite direction by increasing the tether length. In moderate to excellent yields the seven-membered ring 2p (59%), the eight-membered oxathiocin 2q (96%) and even the nine-membered oxathionin derivative 2r (92%) were obtained. The structures of the latter two compounds were proved by X-ray crystallography (Scheme 3). The formation of a ten- or a twelve-membered ring system was, however, not achieved.22
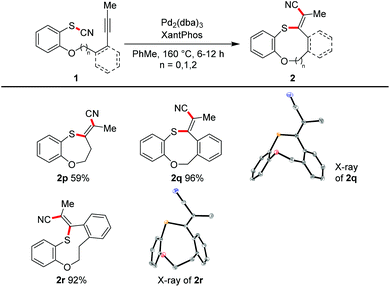 |
| | Scheme 3 Intramolecular cyanosulfenylation for large ring synthesis. Reaction conditions: 1 (1.00 equiv.), Pd2(dba)3 (10 mol%), XantPhos (20 mol%), PhMe (20 mM), 160 °C, 6–12 h. | |
Scope of aliphatic thiocyanates
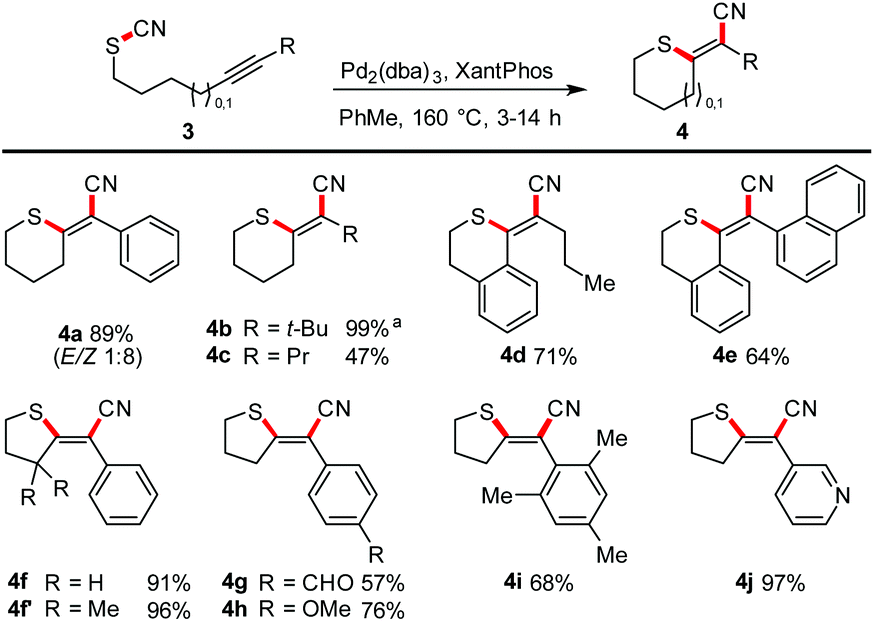
Next, we focused on the scope with respect to aliphatic thiocyanates (Scheme 4). We realized that higher temperatures are required to enable the transformation. This observation might be traced back to a stronger S–CN bond (because of its non-conjugated nature) and/or the lack of preorganisation. Hence, a complete reoptimisation of the reaction conditions was necessary (see ESI†). Finally, Pd2(dba)3 or Pd(PPh3)4 and the use of toluene as solvent and a reaction temperature of 160 °C proved to be the optimal choice. Under these conditions 4a was obtained in 89% yield. The successful synthesis of products 4b–4d shows that the reaction tolerates aliphatic termini, as well as a conjugated triple bond as exemplified in isothiochromene 4d. The motif of a molecular switch was achieved with the synthesis of 4e (64%). The generation of five-membered exocyclic thioenol ethers also proceeded smoothly. Electron-poor (4g), electron-rich (4h), sterically encumbered (4i), aromatic and heteroaromatic (4j) termini also permit the transformation. Compound 4f′ was prepared to investigate whether an acceleration of the reaction by a Thorpe–Ingold effect can be observed; the effect was small.23
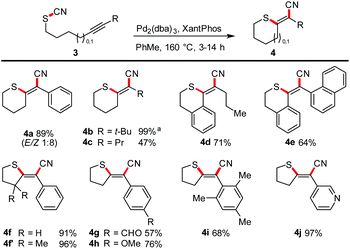 |
| | Scheme 4 Intramolecular cyanosulfenylation with aliphatic thiocyanates. Reaction conditions: 3 (1.00 equiv.), Pd2(dba)3 (10 mol%), XantPhos (20 mol%), PhMe (20 mM), 160 °C, 3–14 h. aPd(PPh3)4 (5 mol%), SPhos (20 mol%), 160 °C, 3 h, PhMe (20 mM). | |
Multiple heteroatom-substitution
Because we have a special interest in multiply heterosubstituted C–C double bonds, we subjected several α- and α,α′-(di)substituted triple bonds to the reaction conditions (Scheme 5).
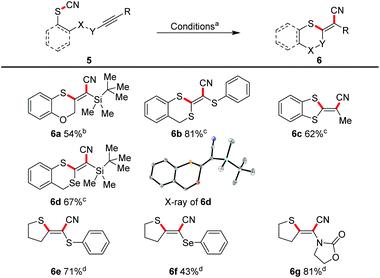 |
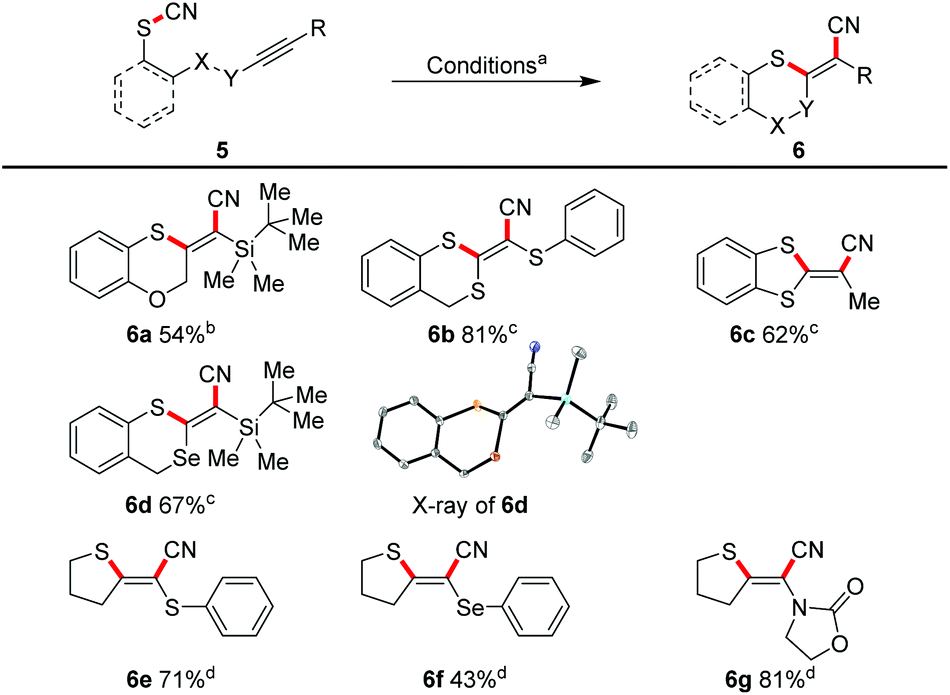
| | Scheme 5 Cyanosulfenylation of heterosubstituted triple bonds. aReaction conditions: 5 (1.00 equiv.). bPd(PPh3)4 (5 mol%), SPhos (20 mol%), 110 °C, 3 h, DMF (20 mM). cPdCl2(PhCN)2 (10 mol%), XantPhos (20 mol%), NEt3 (5.00 equiv.), DMF (20 mM), 100 °C, 3–7 h. dPd2(dba)3 (10 mol%), XantPhos (20 mol%), 160 °C, 3 h, PhMe (20 mM). | |
This allowed a diastereoselective access to the corresponding doubly or triply heterosubstituted olefin moieties in moderate to good yields. TBS-substituted triple bonds (e.g.6a, 6d), and also the very electron-rich mono- or bis-substituted triple bonds with sulphur (6b, 6c, 6e) and selenium (6d, 6f) were successfully transformed. An ynamide furnished the sulphur/nitrogen-substituted (E)-alkene 6g in 81%.
Intermolecular cyanosulfenylation
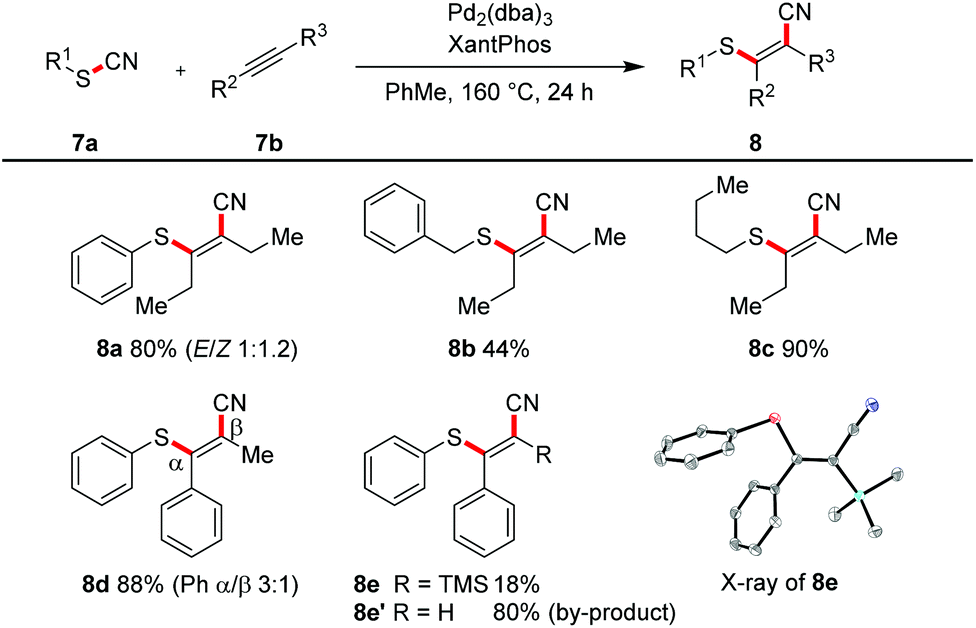
All the transformations described so far proceeded in an intramolecular manner. With an excess of alkyne (2.0 equiv.) and the catalytic system of Pd2(dba)3 and Xantphos in toluene at 160 °C we were able to realize an intermolecular variant (Scheme 6). 3-Hexyne was converted in yields of 44–90% into the tetrasubstituted alkenes 8a–8c. The reaction with methylphenyl acetylene afforded in 88% yield a regioisomeric mixture (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); the steric and electronic differentiation of the two acetylenic carbons seems to be not high enough to lead to a clear preference of the thiopalladation step. In contrast, with phenyl trimethylsilyl acetylene only one regioisomeric product 8e was formed (compare also X-ray crystal structure); however, as the major product desilylated trisubstituted olefin 8e′ was found.
:1); the steric and electronic differentiation of the two acetylenic carbons seems to be not high enough to lead to a clear preference of the thiopalladation step. In contrast, with phenyl trimethylsilyl acetylene only one regioisomeric product 8e was formed (compare also X-ray crystal structure); however, as the major product desilylated trisubstituted olefin 8e′ was found.
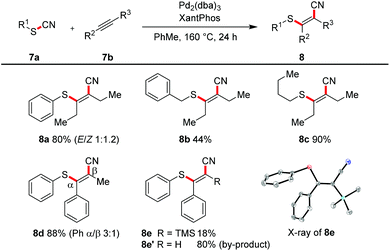 |
| | Scheme 6 Intermolecular cyanosulfenylation with different thiocyanates. Reaction conditions: 7a (1.00 equiv.), 7b (2.00 equiv.), Pd2(dba)3 (10 mol%), XantPhos (20 mol%), PhMe (20 mM), 160 °C, 24 h. | |
Thiopalladation–carbopalladation cascade with cyanide transfer
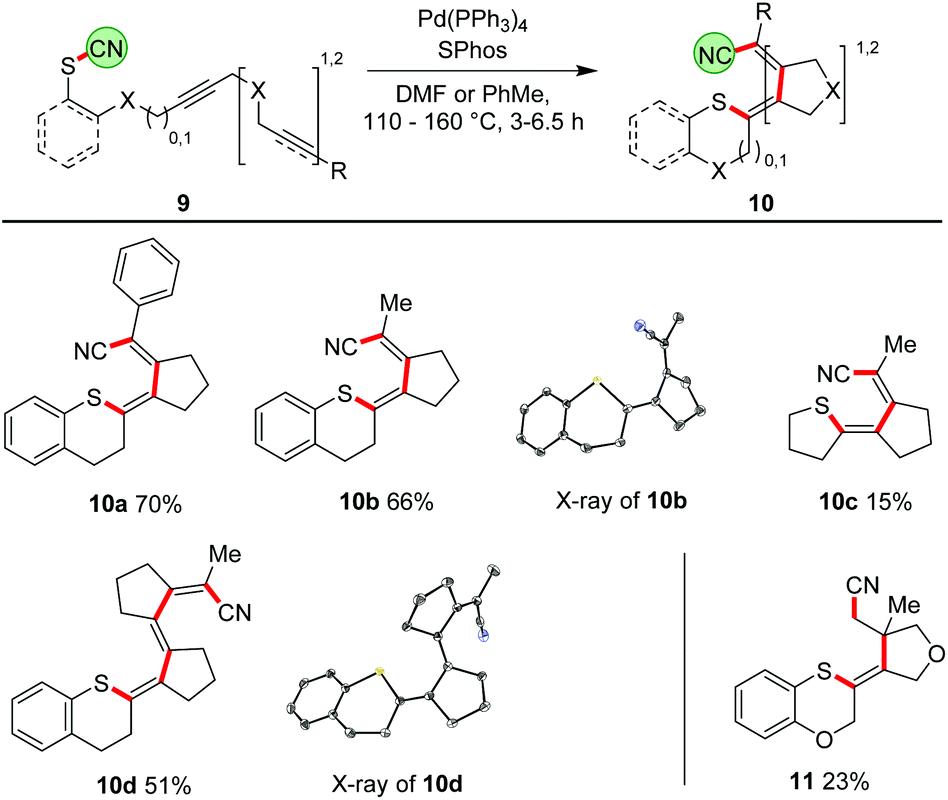
Our ultimate goal was to extend the sequence that starts with the thiopalladation by further carbopalladation steps. The reductive elimination of the Pd should then occur as the final step, generating a C–CN bond. Such an approach using oligoalkynes would lead to a formal 1,4- or 1,6-cyanosulfenylation with a conjugated double bond system in between (Scheme 7). To perform such a cascade successfully, the carbopalladation step must be faster than the reductive elimination of Pd forming the C–CN bond. Initial experiments using model compound 9a and trimethylphosphine as ligand for Pd showed that a thiopalladation–carbopalladation sequence is possible. However, as final step a protodepalladation occurred and thus the cyano moiety was not found in the product (for X-ray analysis of this undesired product 10a′, see ESI†). Reoptimisation studies revealed that the best results are obtained with SPhos and Pd(PPh3)4 as Pd source. The 1,4-cyanosulfenylation products 10a and 10b were obtained in yields of 66–70%. Because the conditions had been optimised for aryl thiocyanates, we expected a much lower yield for cascades starting with alkyl thiocyanates. Indeed, compound 10c was obtained in only 15% yield. The thiopalladation–carbopalladation–carbopalladation cascade, generating one C–S and three C–C bonds in one step, yielded push–pull-substituted triene 10d in 51% yield. X-ray crystallography unequivocally showed the translocation of the cyano group; the central double bond has isomerized to avoid a helical arrangement.24 First unoptimised experiments with an alkyne-alkene system, furnishing 11, demonstrated that, after formation of the tetrasubstituted double bond, the subsequent carbopalladation also allows the generation of a C(sp3)-CN bond.25
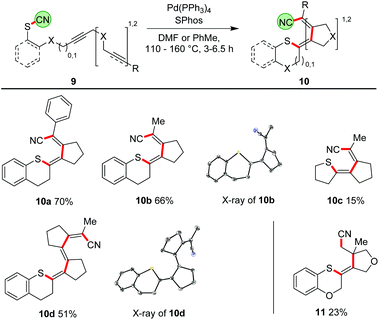 |
| | Scheme 7 Thiopalladation/carbopalladation/(carbopalladation)/cyanide transfer cascades with translocation of the CN group over four or six atoms. Product 9 uses an enyne as starting material. Conditions: 7 (1.00 equiv.), Pd(PPh3)4 (5 mol%), SPhos (20 mol%), 110–160 °C, 3–7 h, DMF or PhMe (20 mM). | |
Proposed mechanism
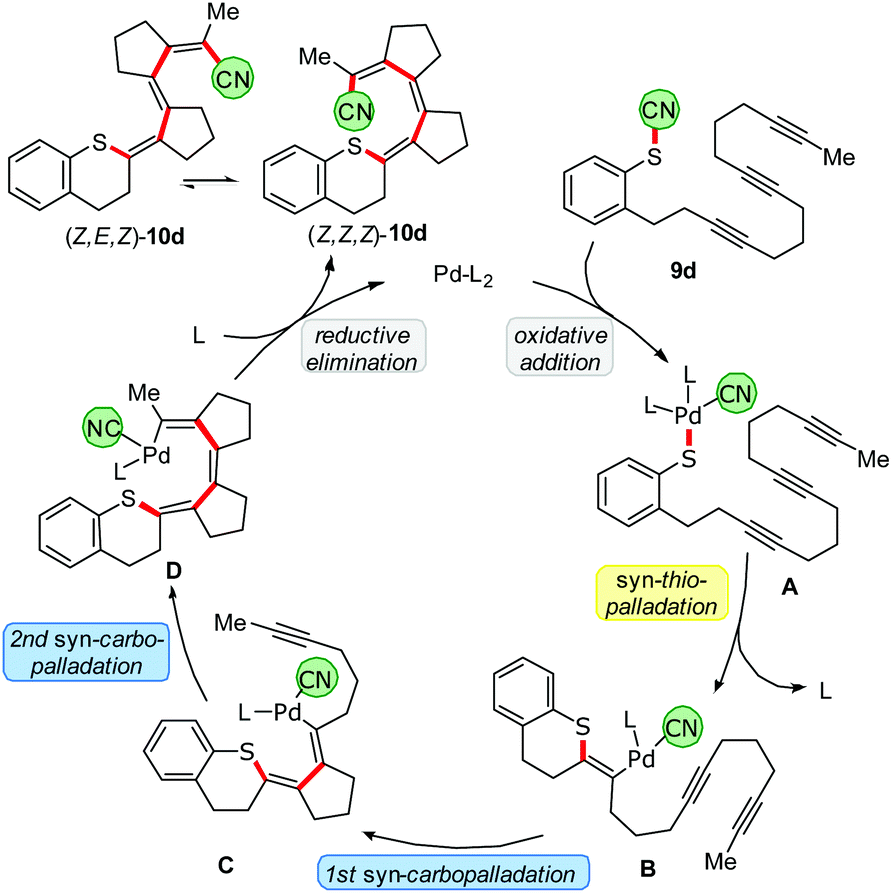
A plausible mechanism of the thiopalladation/carbopalladation/carbopalladation/cyanide transfer cascade to 10d, based on our additional experimental results26 (such as protodepalladation) and previously reported carbopalladation cascades,10–12 is depicted in Scheme 8. After the oxidative addition of the Pd catalyst into the S–CN bond of 9d, a thiopalladation occurs (A → B). Instead of delivering the CN group via reductive elimination of the Pd to the emerging double bond, the sequence continues by a first syn-carbopalladation to yield C. A repetition with another triple bond affords D. As terminating step, the C(sp2)-CN bond is formed by reductive elimination of the Pd to furnish E. Because a strongly push–pull-substituted oligoene is formed, the double bonds are much weaker than in non-push–pull-substituted oligoenes; thus, isomerisation from (Z,Z,Z)-10d to (Z,E,Z)-10d might happen relatively easily.
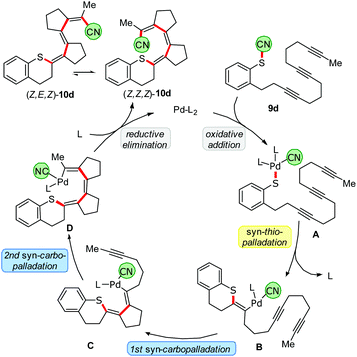 |
| | Scheme 8 Proposed mechanism of the thiopalladation/carbopalladation/carbopalladation/cyanide transfer cascade. | |
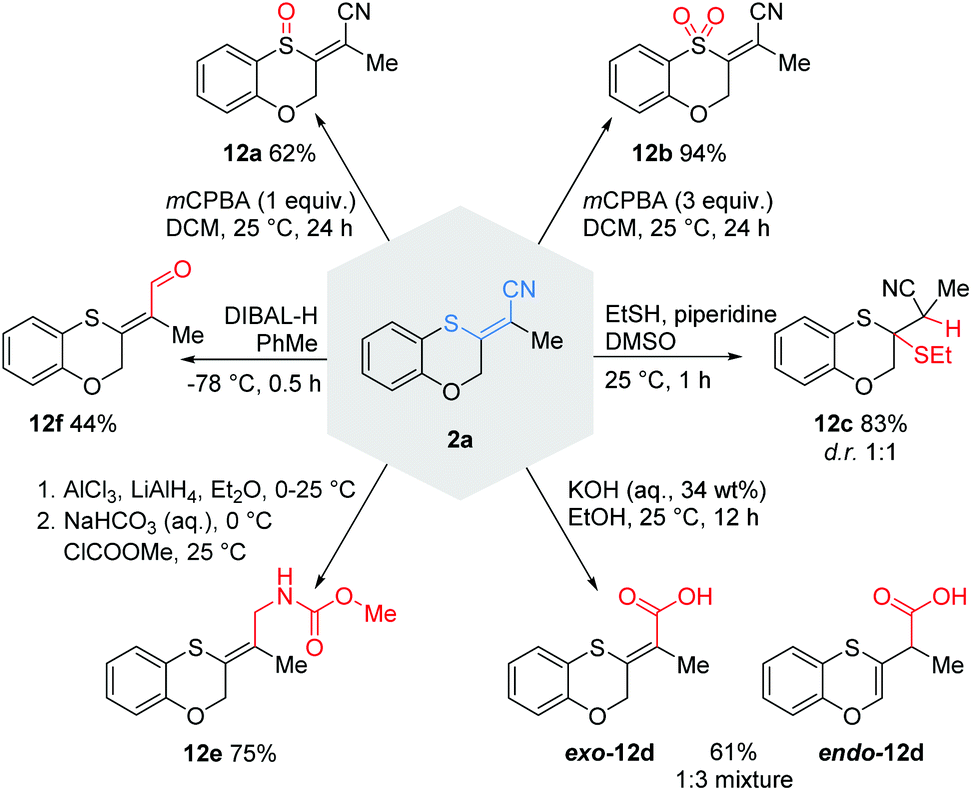
Follow-up chemistry
To demonstrate the versatility of the thioacrylonitrile moiety, some postsynthetic functionalisations with 2a were performed (Scheme 9). Depending on the amount of mCPBA, we were able to prepare either sulfoxide 12a or sulfone 12b in good to excellent yields, thus transforming the strongly polarized olefin into very electron-poor olefins. A Michael addition with ethanethiol led to a diastereomeric mixture of 12c in 83% yield. Furthermore, the hydrolysis of the nitrile with aqueous KOH gave access to the carboxylic acids exo-12d and endo-12d. The partial reduction of the Michael system trapped by methyl chloroformate led to carbamate 12e in a good yield of 75%. Reaction of 2a with DIBAL-H furnished the aldehyde 12f in a moderate yield.
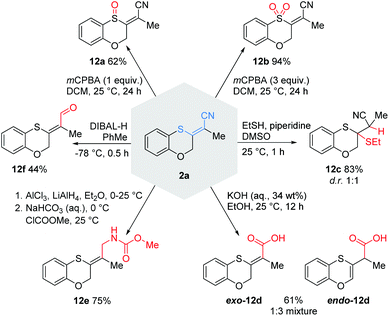 |
| | Scheme 9 Follow-up chemistry. | |
Conclusions
In conclusion, we have developed a Pd-catalysed syn-1,2-cyanosulfenylation of internal alkynes to access tetra-substituted double bonds with sulphur and cyano in adjacent positions. Both aromatic and aliphatic thiocyanates undergo the reaction. Various substitution patterns of the C–C triple bond are tolerated, such as aliphatic and aromatic residues, but also heteroatoms such as sulphur, selenium, silicon and nitrogen. Our methodology facilitates access to tetrasubstituted olefins with four different elements as substituents of the double bond. The reaction works in an intramolecular manner paving the way to five-, six-, seven-, eight, and nine-membered ring systems, but also in an intermolecular way leading to acyclic compounds. By offering further alkyne moieties the transformation was extended to a thiopalladation/carbopalladation/(carbo-palladation) cascade with translocation of the cyano group over four or six carbon atoms, generating up to four new bonds in one step.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the TU Braunschweig for funding. Marcel Bürger thanks Anna Lehmann, Theresa Schitter and Adrian Bauschke for their support.
Notes and references
-
I. Marek and Y. Minko, in Metal-Catalyzed Cross-Coupling Reactions and More, ed. A. de Meijere, M. Oestreich and S. Bräse, Wiley-VCH, Weinheim, 2014 Search PubMed.
- For examples of addition of special reagents see:
(a) A. G. Barrado, A. Zieliński, R. Goddard and M. Alcarazo, Angew. Chem., Int. Ed., 2017, 56, 13401 CrossRef CAS;
(b) X. Wang and A. Studer, J. Am. Chem. Soc., 2016, 138, 2977 CrossRef CAS.
- Selected examples for transition-metal catalysed functionalisations:
(a) P. D. G. Greenwood, E. Grenet and J. Waser, Chem.–Eur. J., 2019, 25, 3010 CrossRef CAS PubMed;
(b) M. Ahmad, A.-C. Gaumont, M. Durandetti and J. Maddaluno, Angew. Chem., Int. Ed., 2017, 56, 2464 CrossRef CAS PubMed;
(c) N. Sakata, K. Sasakura, G. Matsushita, K. Okamoto and K. Ohe, Org. Lett., 2017, 19, 3422 CrossRef CAS PubMed;
(d) Y. Nakao, Bull. Chem. Soc. Jpn., 2012, 85, 731 CrossRef CAS;
(e) N. R. Rondla, S. M. Levi, J. M. Ryss, R. A. Vanden Berg and C. J. Douglas, Org. Lett., 2011, 13, 1940 CrossRef CAS PubMed;
(f) Y. Hirata, A. Yada, E. Morita, Y. Nakao, T. Hiyama, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2010, 132, 10070 CrossRef CAS PubMed;
(g) Y. Nakao, A. Yada, S. Ebata and T. Hiyama, J. Am. Chem. Soc., 2007, 129, 2428 CrossRef CAS PubMed;
(h) M. Suginome, A. Yamamoto and M. Murakami, Angew. Chem., Int. Ed., 2005, 44, 2380 CrossRef CAS PubMed;
(i) Y. Nakao, S. Oda and T. Hiyama, J. Am. Chem. Soc., 2004, 126, 13904 CrossRef CAS;
(j) M. Suginome, A. Yamamoto and M. Murakami, J. Am. Chem. Soc., 2003, 125, 6358 CrossRef CAS PubMed.
- For miscellaneous examples see:
(a) S. Higashimae, D. Kurata, S.-I. Kawaguchi, S. Kodama, M. Sonoda, A. Nomoto and A. Ogawa, J. Org. Chem., 2018, 83, 5267 CrossRef CAS PubMed;
(b) D. Xu, R. Rios, F. Ba, D. Ma, G. Gu, A. Ding, Y. Kuang and H. Guo, Asian J. Org. Chem., 2016, 5, 981 CrossRef CAS;
(c) M. Murai, R. Hatano, S. Kitabata and K. Ohe, Chem. Commun., 2011, 47, 2375 RSC.
- Y. H. Lee and B. Morandi, Angew. Chem., Int. Ed., 2019, 58, 6444 CrossRef CAS PubMed.
-
(a) T. Sperger, C. M. Le, M. Lautens and F. Schoenebeck, Chem. Sci., 2017, 8, 2914 RSC;
(b) C. M. Le, P. J. C. Menzies, D. A. Petrone and M. Lautens, Angew. Chem., Int. Ed., 2015, 54, 254 CrossRef CAS;
(c) C. M. Le, X. Hou, T. Sperger, F. Schoenebeck and M. Lautens, Angew. Chem., Int. Ed., 2015, 54, 15897 CrossRef CAS PubMed.
-
(a)
L.-F. Tietze, G. Brasche and K. M. Gericke, Domino Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 2006 CrossRef;
(b) L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef CAS PubMed.
-
(a) Review: A. Düfert and D. B. Werz, Chem.–Eur. J., 2016, 22, 16718 CrossRef PubMed;
(b) M. Pawliczek, T. F. Schneider, C. Maaß, D. Stalke and D. B. Werz, Angew. Chem., Int. Ed., 2015, 54, 4119 CrossRef CAS PubMed;
(c) M. Leibeling, M. Pawliczek, D. Kratzert, D. Stalke and D. B. Werz, Org. Lett., 2012, 14, 346 CrossRef CAS PubMed.
-
(a) L. Bai, Y. Yuan, J. Liu, J. Wu, L. Han, H. Wang, Y. Wang and X. Luan, Angew. Chem., Int. Ed., 2016, 55, 6946 CrossRef CAS PubMed;
(b) L. F. Tietze, B. Waldecker, D. Ganapathy, C. Eichhorst, T. Lenzer, K. Oum, S. O. Reichmann and D. Stalke, Angew. Chem., Int. Ed., 2015, 54, 10317 CrossRef CAS PubMed;
(c) J. Wallbaum, R. Neufeld, D. Stalke and D. B. Werz, Angew. Chem., Int. Ed., 2013, 52, 13243 CrossRef CAS PubMed;
(d) K. Parthasarathy, H. Han, C. Prakash and C.-H. Cheng, Chem. Commun., 2012, 48, 6580 RSC;
(e) M. Leibeling, D. C. Koester, M. Pawliczek, S. C. Schild and D. B. Werz, Nat. Chem. Biol., 2010, 6, 199 CrossRef CAS PubMed;
(f) G. Blond, C. Bour, B. Salem and J. Suffert, Org. Lett., 2008, 10, 1075 CrossRef CAS PubMed;
(g) Y. Zhang, G. Z. Wu, G. Agnel and E. I. Negishi, J. Am. Chem. Soc., 1990, 112, 8590 CrossRef CAS.
-
(a) M. Charpenay, A. Boudhar, C. Hulot, G. Blond and J. Suffert, Tetrahedron, 2013, 69, 7568 CrossRef CAS;
(b) M. Charpenay, A. Boudhar, G. Blond and J. Suffert, Angew. Chem., Int. Ed., 2012, 51, 4379 CrossRef CAS PubMed.
- S. Blouin, V. Gandon, G. Blond and J. Suffert, Angew. Chem., Int. Ed., 2016, 55, 7208 CrossRef CAS PubMed.
-
(a) B. Milde, M. Leibeling, M. Pawliczek, J. Grunenberg, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2015, 54, 1331 CrossRef CAS PubMed;
(b) B. Milde, M. Leibeling, A. Hecht, P. G. Jones, A. Visscher, D. Stalke, J. Grunenberg and D. B. Werz, Chem.–Eur. J., 2015, 21, 16136 CrossRef CAS PubMed.
-
(a) B. Milde, M. Pawliczek, P. G. Jones and D. B. Werz, Org. Lett., 2017, 19, 1914 CrossRef CAS PubMed;
(b) L. Li, Q. Yang, Y. Wang and Y. Jia, Angew. Chem., Int. Ed., 2015, 54, 6255 CrossRef CAS PubMed;
(c) S. S. Goh, G. Chaubet, B. Gockel, M.-C. A. Cordonnier, H. Baars, A. W. Phillips and E. A. Anderson, Angew. Chem., Int. Ed., 2015, 54, 12618 CrossRef CAS PubMed;
(d) L. F. Tietze, S.-C. Duefert, J. Clerc, M. Bischoff, C. Maaß and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 3191 CrossRef CAS;
(e) D. Shan, Y. Gao and Y. Jia, Angew. Chem., Int. Ed., 2013, 52, 4902 CrossRef CAS.
-
(a) B. M. Monks and S. P. Cook, Angew. Chem., Int. Ed., 2013, 52, 14214 CrossRef CAS PubMed;
(b) X. Jia, D. A. Petrone and M. Lautens, Angew. Chem., Int. Ed., 2012, 51, 9870 CrossRef CAS PubMed;
(c) S. G. Newman and M. Lautens, J. Am. Chem. Soc., 2011, 133, 1778 CrossRef CAS PubMed;
(d) H. Liu, C. Li, D. Qiu and X. Tong, J. Am. Chem. Soc., 2011, 133, 6187 CrossRef CAS.
-
(a) T. Ozaki, A. Nomoto, I. Kamiya, J.-i. Kawakami and A. Ogawa, Bull. Chem. Soc. Jpn., 2011, 84, 155 CrossRef CAS;
(b) W. Zheng, A. Ariafard and Z. Lin, Organometallics, 2008, 27, 246 CrossRef CAS;
(c) M. Wang, L. Cheng and Z. Wu, Dalton Trans., 2008, 3879 RSC;
(d) Y. T. Lee, S. Y. Choi and Y. K. Chung, Tetrahedron Lett., 2007, 48, 5673 CrossRef CAS;
(e) I. Kamiya, J.-i. Kawakami, S. Yano, A. Nomoto and A. Ogawa, Organometallics, 2006, 25, 3562 CrossRef CAS.
-
(a) K. M. Korch and D. A. Watson, Chem. Rev., 2019, 119, 8192 CrossRef CAS PubMed;
(b) Z. Pan, S. Wang, J. T. Brethorst and C. J. Douglas, J. Am. Chem. Soc., 2018, 140, 3331 CrossRef CAS PubMed;
(c) S. V. C. Vummaleti, M. Al-Ghamdi, A. Poater, L. Falivene, J. Scaranto, D. J. Beetstra, J. G. Morton and L. Cavallo, Organometallics, 2015, 34, 5549 CrossRef CAS;
(d) B. Rao and X. Zeng, Org. Lett., 2014, 16, 314 CrossRef CAS PubMed;
(e) Y. Miyazaki, N. Ohta, K. Semba and Y. Nakao, J. Am. Chem. Soc., 2014, 136, 3732 CrossRef CAS PubMed;
(f) D. C. Koester, M. Kobayashi, D. B. Werz and Y. Nakao, J. Am. Chem. Soc., 2012, 134, 6544 CrossRef CAS PubMed;
(g) A. Yada, S. Ebata, H. Idei, D. Zhang, Y. Nakao and T. Hiyama, Bull. Chem. Soc. Jpn., 2010, 83, 1170 CrossRef CAS.
- M. Pawliczek, L. K. B. Garve and D. B. Werz, Org. Lett., 2015, 17, 1716 CrossRef CAS PubMed.
- M. R. Netherton and G. C. Fu, Org. Lett., 2001, 3, 4295 CrossRef CAS PubMed.
-
 .
.
- The configurational stability of E- and Z-isomers of 2e was investigated in additional experiments under the reaction conditions. The E-configured starting material shows the generation of the Z-configured counterpart and vice versa. For more information, see ESI.†.
- CCDC 1948156 (2a), 1948157 (2e), 1969697 (2q), 1969698 (2r), 1948158 (6d), 1996969 (8e), 1948159 (10b), 1948160 (10d), 1948161 (2s, ESI†) and 1948162 (10a′, ESI†) contain the supplementary crystallographic data for this paper.
- For the synthesis of the starting material for larger rings, see ESI.†.
- For respective GC experiments, see ESI.†.
- An HPLC experiment directly after the reaction showed the existence of (Z,Z,Z)-8d, which slowly isomerizes to (Z,E,Z)-8d. For results of thermal and photochemical experiments, see ESI.†.
- The formation of a product from an alkene–alkene system was observed only in traces in GC-MS. For the synthesis of starting material, see ESI.†.
- Experiments in the presence of TEMPO did not significantly decrease the yield (77% of 2a); thus, we rule out the involvement of any radical intermediates.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterisation data. CCDC 1948156–1948162 and 1969696–1969698. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04569d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Maximilian N.
Loch
a,
Maximilian N.
Loch