
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTryptophan scanning mutagenesis as a way to mimic the compound-bound state and probe the selectivity of allosteric inhibitors in cells†
Isabelle R.
Taylor‡
a,
Victoria A.
Assimon‡
a,
Szu Yu
Kuo‡
a,
Silvia
Rinaldi
b,
Xiaokai
Li
a,
Zapporah T.
Young
a,
Giulia
Morra
b,
Keith
Green
d,
Daniel
Nguyen
a,
Hao
Shao
 a,
Sylvie
Garneau-Tsodikova
d,
Giorgio
Colombo
*bc and
Jason E.
Gestwicki
*a
a,
Sylvie
Garneau-Tsodikova
d,
Giorgio
Colombo
*bc and
Jason E.
Gestwicki
*a
aDepartment of Pharmaceutical Chemistry, University of California at San Francisco, 675 Nelson Rising Lane, San Francisco, CA 94158, USA. E-mail: jason.gestwicki@ucsf.edu
bIstituto di Chimica del Riconoscimento Molecolare, CNR, Via Mario Bianco, 9 20131 Milano, Italy
cDepartment of Chemistry, University of Pavia, V.le Taramelli, 12 27100, Pavia, Italy. E-mail: g.colombo@unipv.it
dDepartment of Pharmaceutical Sciences, University of Kentucky, Lexington, KY 40536-0596, USA
First published on 10th January 2020
Abstract
Understanding the selectivity of a small molecule for its target(s) in cells is an important goal in chemical biology and drug discovery. One powerful way to address this question is with dominant negative (DN) mutants, in which an active site residue in the putative target is mutated. While powerful, this approach is less straightforward for allosteric sites. Here, we introduce tryptophan scanning mutagenesis as an expansion of this idea. As a test case, we focused on the challenging drug target, heat shock cognate protein 70 (Hsc70), and its allosteric inhibitor JG-98. Structure-based modelling predicted that mutating Y149W in human Hsc70 or Y145W in the bacterial ortholog DnaK would place an indole side chain into the allosteric pocket normally occupied by the compound. Indeed, we found that the tryptophan mutants acted as if they were engaged with JG-98. We then used DnaK Y145W to suggest that this protein may be an anti-bacterial target. Indeed, we found that DnaK inhibitors have minimum inhibitory concentration (MIC) values <0.125 μg mL−1 against several pathogens, including multidrug-resistant Staphylococcus aureus (MRSA) strains. We propose that tryptophan scanning mutagenesis may provide a distinct way to address the important problem of target engagement.
Introduction
It is often challenging to discern whether a small molecule exerts its biological response through the intended target protein(s) or whether it is acting through polypharmacology.1–7 Accordingly, multiple methods to address this question have emerged,8,9 including pull-downs,10,11 partial proteolysis,12,13 genetic screens for resistance,14,15 techniques such as shRNA16 or CRISPRi,17,18 sequencing-based correlations with transcriptomics,19–22 and variations of cellular thermal shift assays (CETSA).10,23,24 Another common approach is to show that treatment of cells with an inhibitor produces a phenotype that is similar to expression of a dominant negative (DN), a variant of the target enzyme in which a key, catalytic residue is removed.25 Because each of these methods has its own strengths, it often takes more than one experiment (e.g. pulldowns combined with a DN) to ultimately conclude that a compound is sufficiently selective to be used as a chemical probe.3,4 However, achieving this preponderance of evidence can be challenging, especially when the inhibitor acts through a cryptic, allosteric binding site.In an effort to expand the scope of target exploration methods, we envisioned that tryptophan scanning mutagenesis might be a complementary approach. This proposed use of scanning mutagenesis has clear origins in alanine scanning26 and other types of tryptophan scanning,27–29 which, to date, have been primarily used for exploring questions in the fields of protein folding, protein–protein interactions and studying the structure of membrane proteins. Put simply, we thought that another way to use tryptophan mutants would be to strategically place bulky, indole rings into a region that might, in some cases, partially mimic the compound-bound state. This approach would be expected to create a version of the target protein that is allosterically inhibited.
To test this idea, we turned to the molecular chaperone: heat shock protein 70 (Hsp70). Members of the Hsp70 family of chaperones, including the constitutively expressed, human Hsc70 and the prokaryotic DnaK ortholog, are emerging as drug target for a number of diseases.30,31 However, this protein class is a particularly challenging system for probing target selectivity using DNs because of its complex allosteric motions and numerous compound-binding sites.32 In both prokaryotes and eukaryotes, members of the Hsp70 family are composed of two domains: a ∼50 kDa nucleotide binding domain (NBD) and a ∼25 kDa substrate binding domain (SBD).33 The NBD has ATPase activity and, during nucleotide cycling, it switches between a “closed”, ATP-bound state and an “open”, ADP-bound state.34–36 More specifically, the identity of the bound nucleotide controls a scissoring motion between the two lobes (I and II) of the NBD, resulting in a relatively acute angle (∼40°) in the ATP-bound state and a more open angle (∼50°) in the ADP-bound state.37,38 In turn, these lobe motions in the NBD regulate the strength of inter-domain interactions with the SBD and overall quaternary structure of Hsp70s.39 Many inhibitors of Hsp70s have been developed and these molecules bind to different locations within the NBD and SBD.40,41 For example, JG-98 binds to a cryptic, allosteric site between the two lobes of the NBD, stabilizing the ADP-like state by acting as a “door jamb” to scissoring motions.37,42 Despite progress in characterizing their molecular MoAs, it is often difficult to ascribe the cellular activity of Hsp70 inhibitors, such as JG-98, to a specific structural state of the protein in cells, owing to the Hsp70's complex series of nucleotide-driven motions. Moreover, there are thirteen hsp70 genes in humans and it can be challenging to ascribe an inhibitor's activity to a specific one of these isoforms because of the potential for feedback mechanisms and compensation.43 These issues, and others, often contribute to a disconnect between the detailed knowledge of an inhibitor's MoA from in vitro experiments and the results of compound treatment in cells or animals.
Herein, we leverage knowledge of the JG-98 binding site, combined with molecular dynamic simulations and tryptophan scanning mutagenesis, to create a genetic tool for exploring these questions. Simulations predicted that installing a tryptophan at residue 149 in human Hsc70 (Y149W Hsc70) or 145 in DnaK (Y145W DnaK) would place steric bulk into the same allosteric site that is normally occupied by JG-98, at the hinge between lobes I and II. Indeed, we found that the tryptophan mutant acted as if it was bound to the inhibitor in a series of biochemical and structural assays. We also used this approach to show that DnaK is a potential new drug target in important human pathogens. Thus, we envision that tryptophan scanning mutagenesis might supplement existing methodologies. This method might be particularly useful for studying allosteric inhibitors, where traditional, enzyme-dead DN mutants fail to fully replicate the compound's MoA.
Results
In silico tryptophan scanning mutagenesis
JG-98 is predicted to bind an allosteric site between lobes I and II in the NBD of human Hsc70 (Fig. 1A).42,44 This site is highly conserved in prokaryotic isoforms of the Hsp70 family, including E. coli DnaK (ESI Fig. 1†). Moreover, this binding site is adjacent to, but not overlapping with, the nucleotide-binding cleft in the NBD (Fig. 1A), suggesting an allosteric mechanism. Indeed, recent studies have shown that JG-98 restricts the nucleotide-driven, “scissoring” motions of lobes I and II.37 In this way, JG-98 partially interrupts nucleotide cycling by stabilizing a conformation of the NBD that resembles the ADP-bound state, even if ATP is present.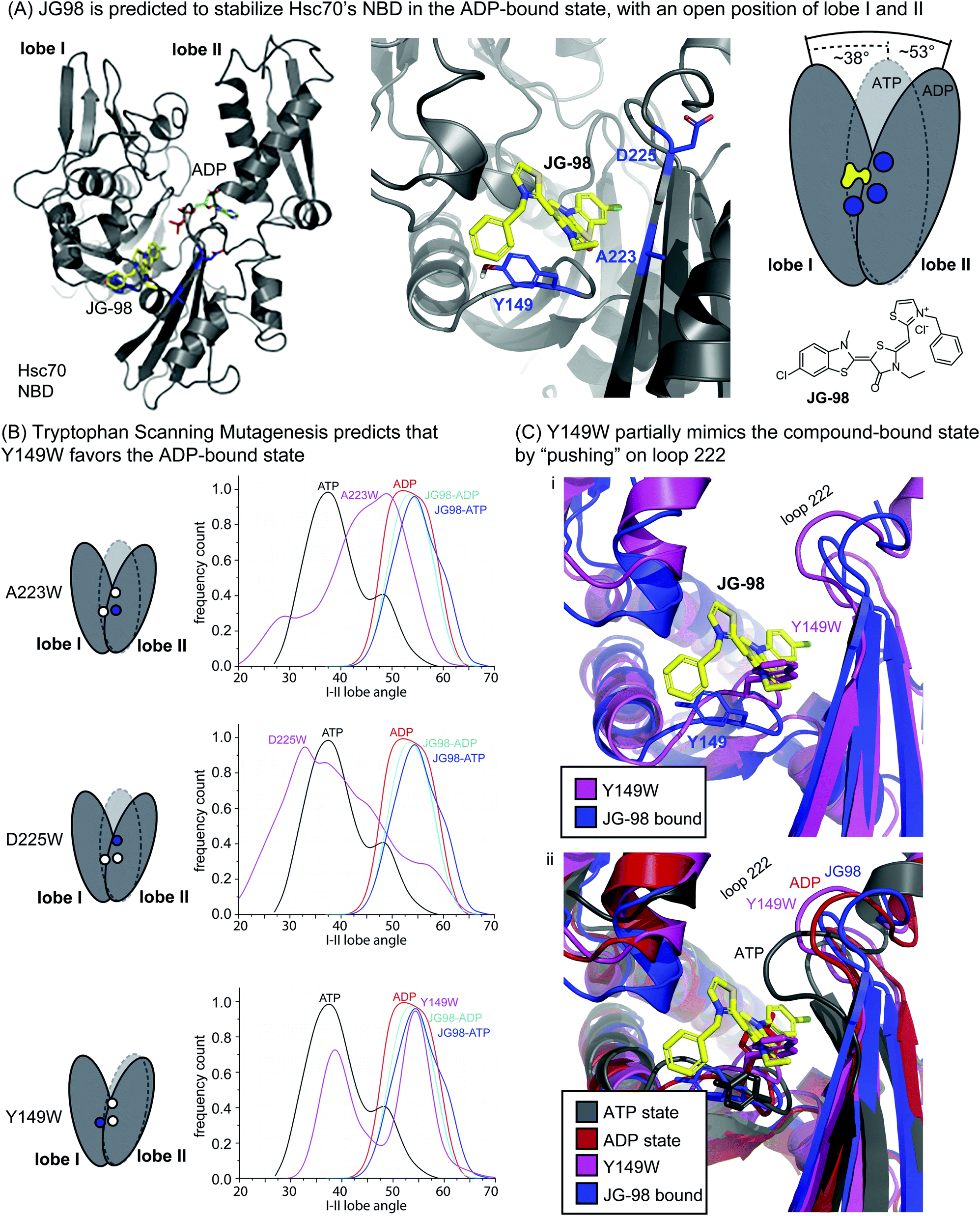 | ||
| Fig. 1 Tryptophan scanning mutagenesis predicts that Y149W may partially mimic inhibitor binding by limiting motion of lobe I/II in Hsc70's NBD. (A) Docked structure of Hsc70's NBD in the ADP-bound state (pdb 3C7N), with a closeup of residues A223, D225 and Y149 (blue) at the interface of lobes I and II. JG-98 is known to limit “scissoring” motions of lobes I and II. (B) Summary of how in silico mutants A223W, D225W and Y149W sample the ATP-bound (closed) or ADP-bound (open) orientations of lobes I/II during MD simulations. Results are representative of two independent simulations. See Methods. (C) Comparison of the Y149W and ATP-, ADP- and JG98-bound structures. Based on the position of loop 222, Y149W, is predicted to partially mimic JG-98. | ||
Guided by this MoA, we focused on mutating residues at the interface between lobes I and II, such as Y149, A223 and D225 (Fig. 1A). The hypothesis was that bulky tryptophan residues at this key, crux point might, like JG-98, partially restrict scissoring of lobes I and II. To test this idea, we performed Molecular Dynamics (MD) simulations on the NBD of wild-type (WT) Hsc70 (PDB 3C7N)45 and compared the nucleotide-dependent motions of its lobes to those of the in silico point mutants: A223W, D225W, and Y149W. In the MD trajectories, we monitored the relative frequency at which the NBD occupied either the ATP-like, closed conformation or the ADP-like, open conformation. First, we confirmed that, for WT Hsc70, the angle between lobes I and II was primarily predicted to be in the closed state (∼38°) in the presence of ATP, while it became relatively open (∼53°) in the presence of bound ADP (Fig. 1B). Moreover, we confirmed that JG-98 favored the open angle between the lobes (∼54 to 55°), regardless of whether ATP or ADP was present. Then, we screened the candidate tryptophan mutants for those that might mimic JG-98. We reasoned that, if the tryptophan mutants mirrored the MoA of JG-98, they might be expected to also favor the ADP-like state. Two of the tryptophan scanning mutants, A223W and D225W Hsc70 NBD, failed to show stabilization of the open state; rather, they showed a broad distribution of predicted angles (Fig. 1B). However, the third mutant, Y149W Hsc70 NBD, was predicted to stabilize the ADP-like lobe angle (∼55°), at the expense of the ATP-like, closed state (Fig. 1B). Thus, these results suggested that mutation of Y149W might partially recapitulate the effects of JG-98, presumably by structural mimicry of the compound's MoA.
To examine this possibility, we looked at snapshots from the MD simulations of Hsc70 Y149W. This analysis suggested that the indole ring is preferentially placed into the cavity between the lobes, partially limiting their closure (Fig. 1C(i)). Further, the positioning of the indole ring closely traced the predicted position of the thioindole ring of JG-98. The global effect of the Y149W mutation on NBD structure was perhaps best observed by examining the motions of loop 222, which is known to undergo a diagnostic shift during the switch from the ATP- to ADP-state.37 Specifically, crystal structures and modelling have shown that loop 222 is relatively compact in the ATP-bound state, but it is more extended in the ADP-bound state (Fig. 1C(ii)).35,46 We found that Y149W favored positioning of loop 222 into a partially ADP-like state, much like JG-98. Neither of the other mutants, A223W or D225W, were able to do this (ESI Fig. 2†). It is worth noting that the extent of loop 222 motion induced by Y149W was not as dramatic as that caused by JG-98 (see Fig. 1C(i)), but we were encouraged that a single mutation could even partially replicate a complex, sub-domain motion.
The purified tryptophan mutant behaves as if it is inhibitor-bound in both the prokaryotic and eukaryotic systems
As a next step in understanding whether tryptophan mutants might be useful tools, we expressed and purified the Y149W mutant of human Hsc70, along with the corresponding Y145W mutant of E. coli DnaK. In this manuscript, we will refer to the Hsc70 variant as Y149W Hsc70 and the DnaK variant as Y145W DnaK, but these residue numbers signify the same structural position (see ESI Fig. 1†). In addition, we created the corresponding alanine mutants at these positions: Y149A Hsc70 and Y145A DnaK. These mutants were created to explore whether the traditional alanine-scanning approach might also be useful for this allosteric site. The purified WT and mutant proteins were generally well-folded, as judged by differential scanning fluorimetry (ESI Fig. 3A†). With these proteins in hand, we turned to studying them in a battery of biochemical and structural tests.First, we measured the ability of the Hsc70 and DnaK variants to hydrolyze ATP using malachite green (MG) assays.47 In these experiments, we added a J-domain protein (JDP) to the Hsp70s because these factors promote their very low, intrinsic ATP hydrolysis rates, increasing the signal-to-noise ratio of the assay.48 Specifically, we chose human DnaJA2 as the JDP to activate Hsc70s, while E. coli DnaJ was used for the corresponding DnaKs. Then, we tested the effects of mutations or inhibitor on the JDP-stimulated ATPase activity. As an inhibitor, we used a close analog of JG-98, JG-48 (see Fig. 2), because of a known, fluorescence spectral overlap between MG and JG-98 which prevents use of that particular analog in MG-based, ATPase assays.49 When we measured ATP hydrolysis in this way, we found that the apparent Km values for nucleotide turnover were not significantly different for any of the untreated proteins and, likewise, that this value was unaffected by treatment of Hsc70 WT with JG-48 (Fig. 2). Because the Km value is a rough approximation of how tightly JDPs bind, this result suggests that physical interactions with JDPs were not dramatically affected by inhibitors or mutations. Indeed, this result was expected because the binding site of JDPs is far from residue Y149 or Y145.35,46 However, we found that the Vmax values for Y149W Hsc70 and Y145W DnaK mutants were reduced (approximately 2- to 4-fold). The decreased Vmax values are consistent with partial stabilization of the ADP-state and a slower rate of nucleotide cycling. Strikingly, we found that JG-48 reduced Vmax by roughly this same extent (∼4-fold) as the point mutants. In addition, we found that the ATPase activity of the Y145W DnaK mutant was relatively resistant to JG-48 (ESI Fig. 3B†), consistent with occlusion of the binding site by the indole side chain. Together, these results suggest that the tryptophan mutants of Hsc70 and DnaK partially mimic the inhibitor-bound state. It is worth noting that traditional DN mutants of Hsc70, in which the catalytic lysine residue is mutated, lack any hydrolysis activity (Vmax and Km values ∼zero).50 Thus, the Y149W and Y145W mutants appear to be a better mimic of allosteric inhibitors than the typical DN.
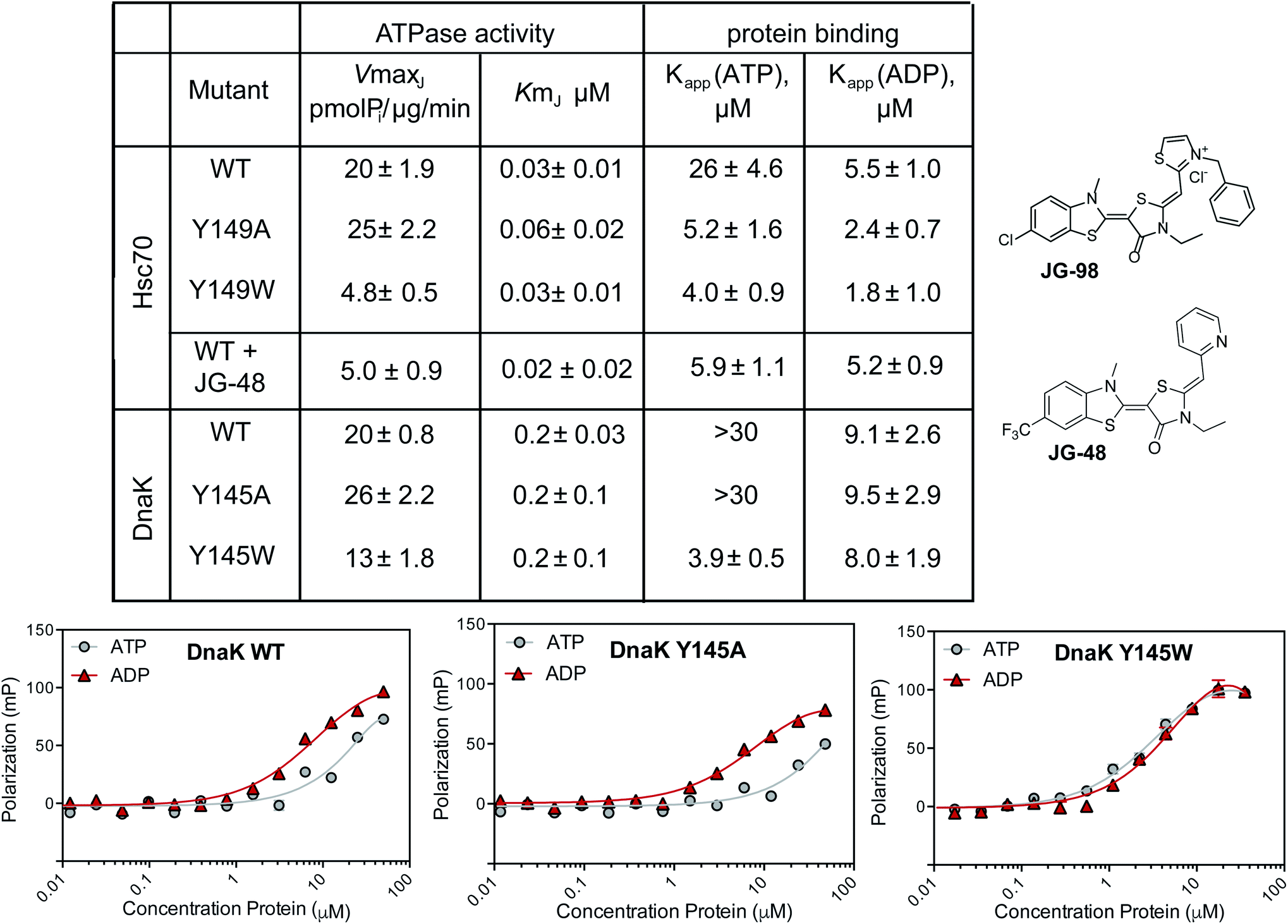 | ||
| Fig. 2 Y149W and Y145W partially mimic the biochemical behavior of inhibitor treatment. Summary of ATPase activity and peptide-binding assays, using purified human Hsc70 or E. coli DnaK and the corresponding mutants. Steady state ATPase activity was measured using malachite green (MG) and binding to a fluorescent FAM-HLA tracer was measured using fluorescence polarization (FP). In the ATPase assays, a J-domain protein (JDP; either DnaJA2 or DnaJ) was added to increase signal intensity. Examples of the FP results for DnaK and its mutants are shown (bottom). Error bars represent SEM of two independent, biological replicates, each performed in technical triplicate. See the Methods for details. | ||
Another sensitive indicator of nucleotide status in Hsp70 family members is their affinity for peptides.47,51 This important function is commonly measured by using fluorescence polarization (FP) in which model peptides are attached to fluorophores and used as tracers. Briefly, Hsp70s bind more tightly to these tracer peptides in the ADP-bound state, when compared to the ATP-bound state.51 JG-48 is known to enhance WT Hsc70's affinity for FAM-HLA in the FP experiment (∼5 μM with either nucleotide).49 Thus, we hypothesized that the Y149W and Y145W mutants might also have a tighter apparent affinity (Kapp) in the presence of either nucleotide. To test this idea, we used model peptide, HLA, attached to a FAM fluorophore (termed FAM-HLA) (see Methods). We predicted that addition of ATP would be unable to weaken the interaction of Y149W Hsc70 with FAM-HLA, because the mutants would be relatively “locked” into the tight-binding, ADP-state. Before asking this question, we first confirmed that FAM-HLA binds tighter to WT Hsc70 in the presence of ADP (Kapp ∼ 5 μM) than ATP (Kapp ∼ 26 μM). Similar results were obtained with WT DnaK, which had an affinity of 9 μM in the ADP state and >30 μM in the ATP state (Fig. 2A). Consistent with the design, we found that Y145W DnaK bound to FAM-HLA, with relatively tight affinity in the presence of either ATP (Kapp ∼ 4 μM) or ADP (Kapp ∼ 8 μM) (Fig. 2). Likewise, the Y149W Hsc70 mutant bound relatively tightly in the presence of either nucleotide (∼4 to 5 μM). In contrast, the control Y145A DnaK behaved similarly to WT DnaK, only binding tightly in the ADP-state (∼9 μM), but not the ATP state (>30 μM). In the human system, the Hsc70 Y149A control still bound relatively tightly in the presence of ATP (∼4 μM; Fig. 2). The reasons for this result are not clear at this time, but it is possible that this mutant may somehow affect NBD-SBD coupling.
Partial proteolysis is another diagnostic way to discern whether Hsp70s are in the ATP-like or ADP-like conformer.52 Briefly, short treatments with trypsin cleave Hsc70 at the linker between the NDB and SBD (Fig. 3A), giving rise to a truncated product of ∼55 kDa on a denaturing polyacrylamide gel (termed band 2). In the ADP-like state, this band is favored because the NBD and SBD are apart from each other.46 In contrast, a mixture of products is formed between 50 to 60 kDa (termed bands 1, 2 and 3) in the ATP-bound state and the overall intensity of band 2 is typically reduced because the system is in a more compact state. Thus, the relative ratio of the intensity of band 2 to band 1 (band 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) can broadly diagnose whether the protein's structure is relatively more ATP- or ADP-like. Consistent with the literature, we found that proteolysis of WT Hsc70 yielded the predicted pattern of products (Fig. 3A). Specifically, treatment of the apo-protein with ADP (1 mM) stabilized band 2 by ∼3- to 4-fold, while ATP did not. In addition, we confirmed that JG-98 could produce the ADP-like banding pattern, even if ATP was added (ESI Fig. 3C†). Finally, we found that Y149W Hsc70 but not the alanine mutant, Y149A Hsc70, could also, like JG-98, favor the ADP-like conformation (Fig. 3A), regardless of whether ADP or ATP was added. Moreover, similar results were observed using DnaK and its variants, although the bands 1, 2 and 3 are located around 45 to 50 kDa in that protein system (Fig. 3B). Specifically, WT DnaK and Y145A DnaK gave the predicted pattern of three bands in the presence of ATP, but Y145W was able to resist this conformational change, staying in the ADP-like state (Fig. 3B). Together, these results support the idea that Y149W Hsc70 and Y145W DnaK behave as if bound to JG-98.
:1) can broadly diagnose whether the protein's structure is relatively more ATP- or ADP-like. Consistent with the literature, we found that proteolysis of WT Hsc70 yielded the predicted pattern of products (Fig. 3A). Specifically, treatment of the apo-protein with ADP (1 mM) stabilized band 2 by ∼3- to 4-fold, while ATP did not. In addition, we confirmed that JG-98 could produce the ADP-like banding pattern, even if ATP was added (ESI Fig. 3C†). Finally, we found that Y149W Hsc70 but not the alanine mutant, Y149A Hsc70, could also, like JG-98, favor the ADP-like conformation (Fig. 3A), regardless of whether ADP or ATP was added. Moreover, similar results were observed using DnaK and its variants, although the bands 1, 2 and 3 are located around 45 to 50 kDa in that protein system (Fig. 3B). Specifically, WT DnaK and Y145A DnaK gave the predicted pattern of three bands in the presence of ATP, but Y145W was able to resist this conformational change, staying in the ADP-like state (Fig. 3B). Together, these results support the idea that Y149W Hsc70 and Y145W DnaK behave as if bound to JG-98.
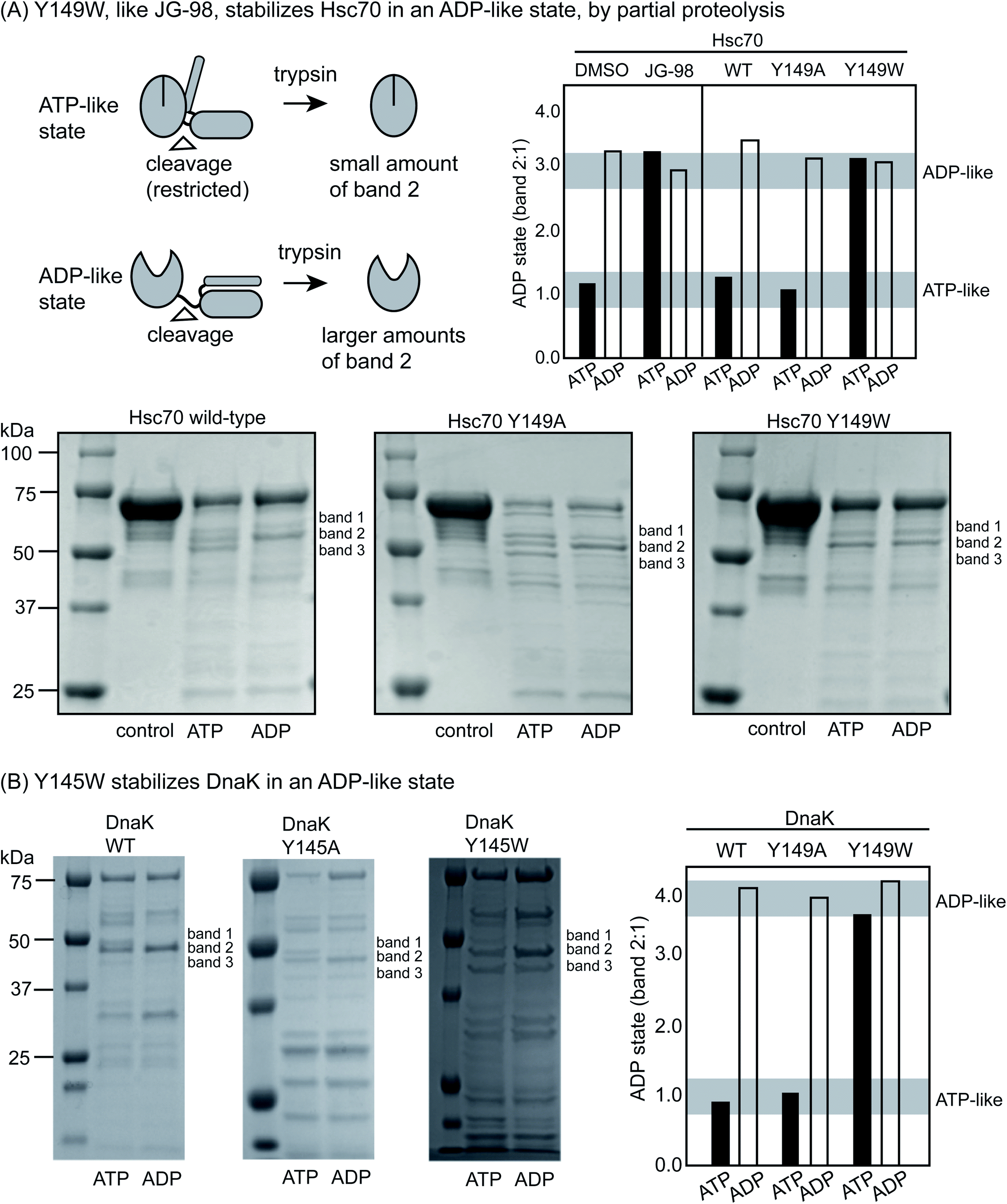 | ||
| Fig. 3 Y149W stabilizes the ADP-like state of Hsc70 and DnaK. (A) Treatment with trypsin is known to produce relatively more band 2 (composed of the NBD and a short section of the linker domain) in the ADP-like state because of the relative availability of the linker to proteolysis. Conversely, this cleavage site is relatively obscured in the ATP-like state. WT Hsc70 gives the expected banding pattern in the ATP- and ADP-like states. Y145W, but not Y145A, stabilizes the ADP-like state, even when ATP (1 mM) is added. Treatment of WT Hsc70 with JG-98 (100 μM) mirrors the Y145W mutant. Band 2 intensity is plotted as a ratio of band 2:1. Results are representative of experiments performed in duplicate. (B) Similar results were observed with DnaK, with Y145W stabilizing band 2 and the ADP-like state. | ||
Expression of Y149W DnaK phenocopies treatment with JG-98 in E. coli
One important way we anticipated using the Y145W mutant was to explore DnaK as a putative drug target in pathogens. This idea is based on studies in Δdnak E. coli strains, which fail to grow at elevated temperature,53,54 owing to an increase in aggregated protein.55 Moreover, the infectivity of Δdnak strains of S. aureus are significantly reduced in mouse models56 and these strains are more sensitive to antibiotics.57 Together, these findings broadly suggest that DnaK might be a new target for the development of anti-infectives. However, there are multiple ways of inhibiting DnaK; for example, one might block nucleotide binding,58 collaborations with DnaJ59 or allostery.60,61 Which of these approaches would be predicted to be anti-bacterial? Answering this question is made more difficult by observations that DnaK's ATPase activity does not correlate directly with its function in vivo,62,63 so traditional DN mutants do not produce informative results.We reasoned that Y145W DnaK could be a useful tool for addressing this issue. Accordingly, we introduced WT, Y145W and Y145A DnaK into a Δdnak strain of E. coli. Each of the proteins was expressed at similar levels (ESI Fig. 3D†). At permissive temperature, all of the strains were viable (Fig. 4A). However, Y145W was not able to complement growth at 40 °C. This result suggested that stabilizing the ADP-like state, as accomplished by Y145W, might be important for growth under these conditions. Indeed, we found that JG-319 (a more permeable analog of JG-98; see below), like Y145W, could partially suppress growth of E. coli in liquid cultures, especially at elevated temperature (Fig. 4B). Together, these studies support the idea that DnaK is a putative drug target.
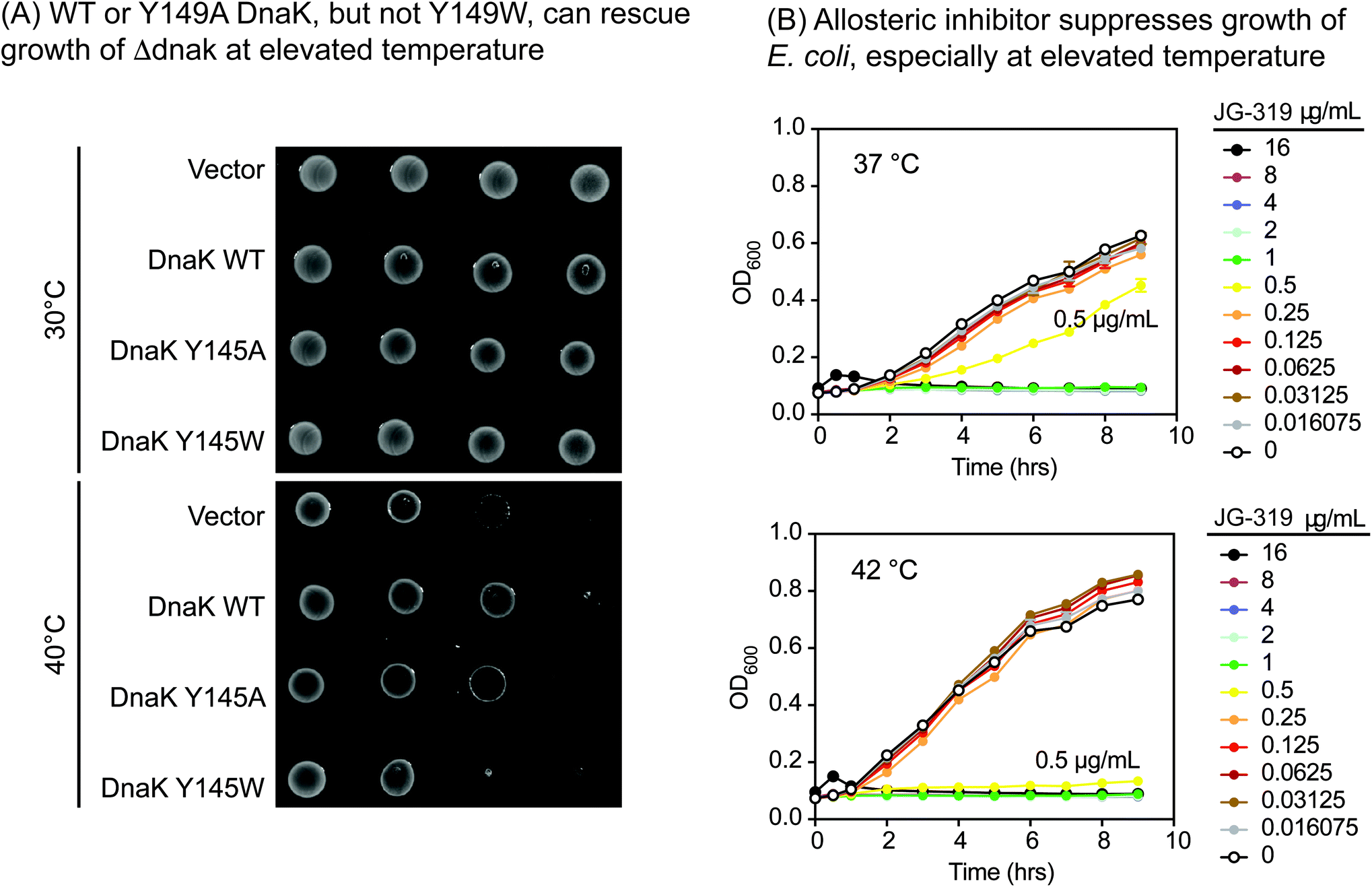 | ||
| Fig. 4 Exploration of DnaK as a potential anti-bacterial target. (A) WT and Y145A DnaK, but not Y145W DnaK, complement for loss of DnaK in E. coli, as judged by spot dilution assays. These results are representative of experiments performed in duplicate. (B) Treatment with a representative allosteric inhibitor, JG-319, partially suppresses growth of E. coli (K12) in liquid media at elevated temperature (42 °C). The position of the JG-319 (0.5 μg mL−1; yellow) results are highlighted. Results are the average of experiments performed in duplicate. | ||
Analogs of JG-98 have anti-bacterial activity
Inspired by these findings, we then tested a panel of ∼54 recently reported analogs of JG-98 (ref. 17) at a single concentrations (16 μg mL−1) for the ability to suppress growth of six common strains, representing Gram-positive (Bacillus subtilis, B. anthracis, B. cereus and S. aureus) and Gram-negative (E. coli and H. influenza) bacteria. In these initial screens, we measured OD600 in liquid cultures at 0, 3 and 6 hours at 37 °C and used tetracycline as a positive control (ESI Fig. 4A†). We found that 31/54 compounds (∼57%) had measurable activity and 24/54 compounds (44%) had minimum inhibitory concentration (MIC) values below 16 μg mL−1 in dose response curves (ESI Fig. 4B†). Of the active molecules, 18 suppressed growth of both Gram-negative and Gram-positive bacteria (ESI Fig. 4C†). Thus, DnaK does indeed seem to be a promising anti-infective target, at least for compounds with the MoA of JG-98. Guided by these results, we synthesized 29 additional analogs of JG-98 (ESI Fig. 5†). These analogs were synthesized through a reported route17 and screened in dose response curves to calculate MIC values against a broader collection of clinically relevant, human pathogens, including A. baumanni ATCC 19606, E. coli MC1061, P. aeruginosa ATCC 27853, L. monocytogenes ATCC 19115, M. smegmatis MC122, multidrug-resistant S. aureus MRSA1, MRSA2, USA100, USA200, USA300, USA600 and ATCC 29213 and a vancomycin resistant enterococcal (VRE) strain.In the first 15 compounds, we retained the pendant benzyl moiety in JG-98 and varied the identity of substituents at R1 and R2 (structure I; Fig. 5). Although there were clearly strain-specific differences in sensitivity, we found that halogens at R1 (compounds 12, 13 and 15) were generally less active than modest-sized alkyl groups, such as 5-methyl, 5-ethyl and 5-isopropyl. For example, a 5-isopropyl group (JG-325) was generally more active than a 5-ethyl (5) or 5-bromo group (12). Substitutions at R2 had less influence on activity; for example, analogs with o-Cl (3), o-Me (4) or o-OCF3 (5) at R2 had comparable MIC values. Because R2 seemed to have relatively minimal effect in this context, we decided to explore distinct ring systems on that side of the molecule (structure II, Fig. 5). Although hetero-atom containing rings (furans 16–19, thiophenes 25–27) were not substantially better than the benzyl group in structure I, we found that adding a methyl group to the linker (JG-319, JG-320, 22–23) produced some of the most potent analogs in the series (MIC values of <0.125 μg mL−1 against multiple strains). Importantly, JG-273, which is unable to bind DnaK because it is missing the critical benzothiazole, was inactive (MIC values >64 μg mL−1). Together, these results suggest that DnaK may be an under-explored target, because the best molecules had promising MIC values (∼0.125 μg mL−1).
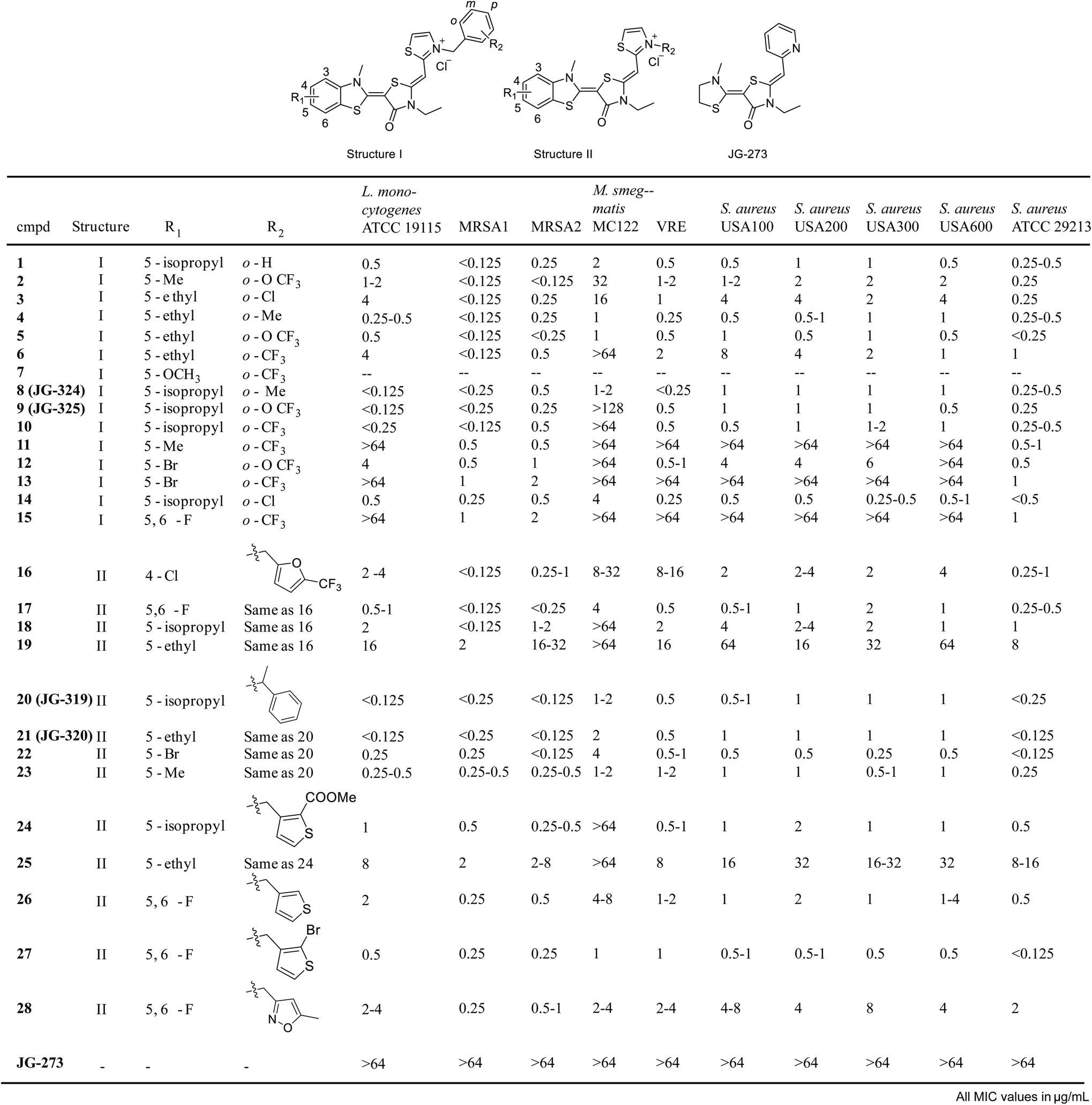 | ||
| Fig. 5 Summary of minimum inhibitory concentration (MIC) values from growth assays in a panel of human pathogens. JG-98 analogs were tested as described in the Methods. Compound 7 was not soluble enough for testing (--). All MIC values are in μg mL−1. | ||
Discussion
It is often difficult to compare knockdown phenotypes to the effects of chemical inhibitors, because many proteins serve scaffolding functions in addition to enzymatic activities.64 Indeed, this is why DN mutants can be powerful probes, because they strategically remove catalytic activity without impacting other protein functions. However, DN mutants completely ablate enzymatic activity (in most cases), making it harder to compare to treatment with allosteric inhibitors that might only tune Kcat or Km. Indeed, during our ongoing efforts to develop inhibitors of Hsp70s, we found that it was difficult to satisfyingly link JG-98 to Hsp70s using DNs. Accordingly, we envisioned an adaptation of tryptophan scanning mutagenesis to create DN-like point mutants that act as if bound to the allosteric inhibitor. This approach clearly requires substantial, pre-existing knowledge of the compound's binding site and MoA; therefore, it is likely less amenable to systems in which these features are not yet defined. However, we reasoned that tryptophan scanning mutagenesis may still find use in exploring targets, such as Hsp70s, that have available structures and multiple, allosteric binding sites. In those cases, traditional DN approaches, such as enzyme-dead mutants lacking catalytic residues, would not be expected to fully recapitulate the inhibitor MoA. Indeed, this is what we observed with Hsc70 Y149W and DnaK Y145W, which behaved more similarly to JG-98 than a typical DN in a series of biochemical and functional assays. Thus, we suggest that tryptophan scanning mutagenesis might be especially useful for studying the selectivity of allosteric inhibitors and their targets.To explore the approach's possible utility, we used the DnaK Y149W mutant to implicate DnaK as a putative drug target in bacteria. Perhaps more pointedly, our results suggested that compounds able to stabilize the ADP-like state of DnaK would be promising to explore. Indeed, this prediction led us to design, synthesize and screen JG-98 analogs, revealing promising MIC values (<0.125 μg mL−1) against a number of important, human pathogens. Although the conservation of the JG-98 binding site with mammalian orthologs likely makes DnaK a challenging target for developing clinical candidates, our studies suggest that JG-98 analogs might still be useful chemical probes for studying DnaK's role(s) in virulence. It will also be interesting to see if minor differences between DnaKs (see ESI Fig. 1†) could be exploited to develop selective inhibitors. It will also be interesting to confirm that JG-98 analogs are selective for DnaK, using complementary approaches, such as CETSA and/or screening for resistance mutants.
For the human Hsp70 system, our ongoing inquiries suggest that it will be important to knockdown or knockout the WT Hsp70 isoform in each experiment, much like what was done with the ΔdnaK strain. Advances in CRISPR and shRNA technologies may make this objective more tractable. Despite these hurdles, we posit that tryptophan scanning mutagenesis might serve as an effective way to build tools for human Hsp70 family members and maybe other chaperones. More broadly, this approach might be useful in making DN variants for a wider range of protein targets. We predict that it may be especially useful for those targets with allosteric binding sites, where traditional DNs are less effective.
Methods
Molecular dynamics (MD) simulations
MD simulations of Hsc70 NBD (PDB code: 3C7N) and its mutants were performed using the protocol previously described in Rinaldi et al.37 using the AMBER 16.0 package65 with the AMBER ff99SB force field.66 Mutants described in the text were generated using the Pymol mutation tool, selecting the best possible rotamer for the corresponding structure. The simulations were run in the presence or in the absence of ATP or ADP in the orthosteric site. All the structures were solvated with TIP3P water molecules,67 counter-ions were randomly added to ensure overall charge neutrality. The system was first minimized using the steepest descendent scheme. Secondly a 0.4 ns simulation was run in an NVT (constant Number, Volume and Temperature) ensemble in which the positions of alpha carbons were weakly restrained (force constant 10 kcal mol−1 Å−2) and slowly increasing the temperature up to 300 K. Later a 0.2 ns simulation in an NPT (constant Number, Pressure and Temperature) ensemble maintaining the restraints on alpha carbons was performed. Finally, for each system we ran two independent MD replicas for each system under NPT conditions, each simulation being 500 ns long. All the simulations were performed in periodic boundary condition (PBC). Short-range electrostatic and van der Waals interactions were calculated within a 10 Å cutoff, whereas long-range electrostatic interactions were assessed using the particle Ewald method.68 The Shake algorithm was used to treat all bonds involving hydrogen atoms.69 All simulations were run in two independent replicates.The docked co-structures of JG-98 were generated by docking the molecule into the representative conformation of the most populated structural cluster of the allosteric site induced by MKT077, described previously.36 This strategy was shown to be efficient and effective in the design allosteric modulators of chaperone proteins.70,71 Briefly, JG-98 was docked using both Induced Fit and Glide docking software (Software Suite 2017-3, Schrodinger Inc).
Calculation of predicted lobe I/II angle
To calculate the angle between lobes I and II, we defined two axes of inertia, one for each lobe. The angle defined by such axes is referred to as the I/II angle. Axes and angles were calculated using the program Chimera (UCSF). All the trajectories were sampled every 500 ps starting at 50 ns. On each extracted structure, to determine the axes, we used only residues forming the conserved secondary structures along the simulation time. Next, we calculated the distribution of the values of the I/II angle for each simulated system.Protein expression and purification
Human Hsc70 (HSPA8) WT, Y149W and Y149A were expressed in E. coli BL21 (DE3) cells. Cultures were grown at 37 °C until an OD600 of 0.6, then the temperature was adjusted to 25 °C and expression was induced with isopropyl β-D-1-thiogalactopyranoside (IPTG; 500 μM). Induced cultures were grown overnight at 25 °C before sonication and centrifugation, then the pellets were re-suspended in His-binding buffer (50 mM Tris, 10 mM imidazole, 500 mM NaCl, pH 8.0) supplemented with protease inhibitor cocktail. The supernatant was then applied to NiII-NTA His-Bind Resin (Novagen, Darmstadt, Germany) and washed twice before elution with His-elution buffer (50 mM Tris, 300 mM imidazole, 300 mM NaCl, pH 8). A nearly identical protocol was used for human DnaJA2 and E. coli DnaJ and DnaK (WT, Y145A, Y145W), as described.47,62 Eluted proteins were concentrated and stored in 50 mM Tris, 300 mM NaCl, pH 7.4 buffer.ATPase assays
Nucleotide turnover was measured using malachite green (MG) assays, as previously described.72 Briefly, the steady state ATPase activity of Hsc70 or DnaK (1 μM) was measured in the presence of increasing concentrations of DnaJA2 or DnaJ (0 to 10 μM), respectively. The background signal from non-specific ATP hydrolysis was subtracted and a phosphate standard curve (potassium dibasic phosphate) was used to calculate pmol Pi per μM protein per min. Experimental data were analyzed using GraphPad Prism 6 software, fitting to the Michaelis–Menton equation. Here, the Vmax is defined as the maximum turnover at saturating DnaJA2/DnaJ, while the Km is the apparent half maximum amount of DnaJA2/DnaJ that is needed for stimulation. The effects of JG-48 (10 μM) on turnover by Hsc70 and DnaJA2 was previously reported.49 As mentioned above, JG-98 has a fluorescence overlap with MG, so the close analog JG-48 is used in these experiments.Fluorescence polarization
Peptide binding to Hsc70 or DnaK was measured using a fluorescent peptide FAM-HLA (AnaSpec), as described.47 Briefly, the HLA peptide is known to bind the canonical, binding cleft in the SBD of Hsp70 family members. The addition of the FAM fluorophore is commonly used to convert this peptide into a FP tracer. In these experiments, apo-Hsc70 (0 to 50 μM) and FAM-HLA (25 nM) were mixed with nucleotide (1 mM, ATP or ADP) and incubated for 30 minutes at room temperature in assay buffer (100 mM Tris, 20 mM KCl, 6 mM MgCl2 pH 7.4). After incubation, fluorescence polarization was measured (excitation: 485 nm emission: 535 nm) using a SpectraMax M5 plate reader. The apparent half-maximum is reported as Kapp.Partial proteolysis
Partial proteolysis experiments were performed using the procedure described.42 Briefly, samples of Hsc70 or DnaK were prepared in a partial proteolysis buffer (40 mM HEPES, 20 mM NaCl, 8 mM MgCl2, 20 mM KCl, 0.3 mM EDTA, pH 8.0) with either 1 mM ATP or ADP. Samples were incubated at room temperature for 30 minutes prior to addition of trypsin (EC 3.4.21.4, Sigma) at a 1:4 molar ratio (trypsin:Hsc70). Proteolysis was carried out for 40 minutes and quenched with 25 μL of SDS loading buffer (240 mM Tris, 6% (w/v) SDS, 30% (v/v) glycerol, 16% (v/v) β-mercaptoethanol, 0.6 mg mL−1 bromophenol blue, pH 6.8) and then heated at 95 °C for 3 minutes. Samples were separated by SDS-PAGE using 10% Mini-PROTEAN TGX Precast Gels and stained with Coomassie blue.
DnaK growth complementation
ΔdnaK E. coli35 were transformed with plasmids containing L-arabinose-inducible 6xHis-tagged dnaK variants or the empty vector. A single colony was cultured overnight in Luria Broth at 30 °C. The OD value of each culture was then adjusted to 0.5 and the expression of DnaK variants was induced by 20 μM L-arabinose (Sigma Aldrich) for 2 hours at 30 °C. 3-fold serial dilutions were spotted onto LB-agar plates. Plates were then incubated at 30 °C or 40 °C overnight. DnaK expression levels after L-arabinose induction for 2 hours were verified by immunoblotting using anti-Tetra-His antibody (QIAGEN).Synthesis of JG-98 analogs
Synthesis of JG-98 analogs was carried out as reported.17,44 For each purified compound, 1H-NMR spectra were obtained using a Bruker 400 or 500 Ultrashield™ spectrometer and analyzed using Bruker TOPSPIN. Active analogs were soluble in aqueous solutions at 50 μM, as determined by DLS and turbidity measurements.Anti-bacterial MIC assays
MIC experiments were performed using the double dilution method. Briefly, inoculum of each strain was prepared by diluting a fresh culture of an OD600 of ∼0.4 1:1000 and 100 μL was added compound (100 μL) to generate a 2-fold dilution series (final concentrations in the range of 64 to 0.125 μg mL−1). Plates were covered and incubated at 37 °C 16–24 hours before MIC values were determined visual inspection or staining with MTT when needed. For pathogens, 37 °C was considered a mild temperature stress.
Conflicts of interest
The authors claim no competing financial interests.Acknowledgements
The authors thank Oren Rosenberg (UCSF) for supplying the PBAD vector and Mathias Meyer for supplying the ΔdnaK strain BB1994. We also thank Oliver Gestwicki for editing the figures. This work was supported by grants from the NIH (R01NS059690; J. E. G.) and AIRC (IG20019; G. C.).References
- M. Schurmann, P. Janning, S. Ziegler and H. Waldmann, Small-molecule target engagement in cells, Cell Chem. Biol., 2016, 23, 435–441 CrossRef PubMed.
- G. C. Terstappen, C. Schlupen, R. Raggiaschi and G. Gaviraghi, Target deconvolution strategies in drug discovery, Nat. Rev. Drug Discovery, 2007, 6, 891–903 CrossRef CAS PubMed.
- S. V. Frye, The art of the chemical probe, Nat. Chem. Biol., 2010, 6, 159–161 CrossRef CAS PubMed.
- C. H. Arrowsmith, J. E. Audia, C. Austin, J. Baell, J. Bennett, J. Blagg, C. Bountra, P. E. Brennan, P. J. Brown, M. E. Bunnage, C. Buser-Doepner, R. M. Campbell, A. J. Carter, P. Cohen, R. A. Copeland, B. Cravatt, J. L. Dahlin, D. Dhanak, A. M. Edwards, M. Frederiksen, S. V. Frye, N. Gray, C. E. Grimshaw, D. Hepworth, T. Howe, K. V. Huber, J. Jin, S. Knapp, J. D. Kotz, R. G. Kruger, D. Lowe, M. M. Mader, B. Marsden, A. Mueller-Fahrnow, S. Muller, R. C. O'Hagan, J. P. Overington, D. R. Owen, S. H. Rosenberg, B. Roth, R. Ross, M. Schapira, S. L. Schreiber, B. Shoichet, M. Sundstrom, G. Superti-Furga, J. Taunton, L. Toledo-Sherman, C. Walpole, M. A. Walters, T. M. Willson, P. Workman, R. N. Young and W. J. Zuercher, The promise and peril of chemical probes, Nat. Chem. Biol., 2015, 11, 536–541 CrossRef CAS PubMed , PMC4706458.
- M. Schenone, V. Dancik, B. K. Wagner and P. A. Clemons, Target identification and mechanism of action in chemical biology and drug discovery, Nat. Chem. Biol., 2013, 9, 232–240 CrossRef CAS PubMed.
- Z. A. Knight, H. Lin and K. M. Shokat, Targeting the cancer kinome through polypharmacology, Nat. Rev. Cancer, 2010, 10, 130–137 CrossRef CAS PubMed , PMC2880454.
- J. U. Peters, Polypharmacology – foe or friend?, J. Med. Chem., 2013, 56, 8955–8971 CrossRef CAS PubMed.
- R. A. McClure and J. D. Williams, Impact of mass spectrometry-based technologies and strategies on chemoproteomics as a tool for drug discovery, ACS Med. Chem. Lett., 2018, 9, 785–791 CrossRef CAS PubMed , PMC6088350.
- Y. Feng, T. J. Mitchison, A. Bender, D. W. Young and J. A. Tallarico, Multi-parameter phenotypic profiling: Using cellular effects to characterize small-molecule compounds, Nat. Rev. Drug Discovery, 2009, 8, 567–578 CrossRef CAS PubMed.
- S. E. Ong, M. Schenone, A. A. Margolin, X. Li, K. Do, M. K. Doud, D. R. Mani, L. Kuai, X. Wang, J. L. Wood, N. J. Tolliday, A. N. Koehler, L. A. Marcaurelle, T. R. Golub, R. J. Gould, S. L. Schreiber and S. A. Carr, Identifying the proteins to which small-molecule probes and drugs bind in cells, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4617–4622 CrossRef CAS PubMed , PMC2649954.
- K. S. Yang, G. Budin, C. Tassa, O. Kister and R. Weissleder, Bioorthogonal approach to identify unsuspected drug targets in live cells, Angew. Chem., Int. Ed. Engl., 2013, 52, 10593–10597 CrossRef CAS PubMed , PMC3856564.
- P. F. Liu, D. Kihara and C. Park, Energetics-based discovery of protein-ligand interactions on a proteomic scale, J. Mol. Biol., 2011, 408, 147–162 CrossRef CAS PubMed , PMC3073411.
- B. Lomenick, R. Hao, N. Jonai, R. M. Chin, M. Aghajan, S. Warburton, J. Wang, R. P. Wu, F. Gomez, J. A. Loo, J. A. Wohlschlegel, T. M. Vondriska, J. Pelletier, H. R. Herschman, J. Clardy, C. F. Clarke and J. Huang, Target identification using drug affinity responsive target stability (darts), Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21984–21989 CrossRef CAS PubMed , 2789755.
- S. French, C. Mangat, A. Bharat, J. P. Cote, H. Mori and E. D. Brown, A robust platform for chemical genomics in bacterial systems, Mol. Biol. Cell, 2016, 27, 1015–1025 CrossRef CAS PubMed , PMC4791123.
- T. M. Kapoor and R. M. Miller, Leveraging chemotype-specific resistance for drug target identification and chemical biology, Trends Pharmacol. Sci., 2017, 38, 1100–1109 CrossRef CAS PubMed , PMC5708298.
- C. J. Matheny, M. C. Wei, M. C. Bassik, A. J. Donnelly, M. Kampmann, M. Iwasaki, O. Piloto, D. E. Solow-Cordero, D. M. Bouley, R. Rau, P. Brown, M. T. McManus, J. S. Weissman and M. L. Cleary, Next-generation nampt inhibitors identified by sequential high-throughput phenotypic chemical and functional genomic screens, Chem. Biol., 2013, 20, 1352–1363 CrossRef CAS PubMed , 3881547.
- H. Shao, X. Li, M. A. Moses, L. A. Gilbert, C. Kalyanaraman, Z. T. Young, M. Chernova, S. N. Journey, J. S. Weissman, B. Hann, M. P. Jacobson, L. Neckers and J. E. Gestwicki, Exploration of benzothiazole-rhodacyanines as allosteric inhibitors of protein-protein interactions with heat shock protein 70 (hsp70), J. Med. Chem., 2018, 61, 6163–6177 CrossRef CAS PubMed.
- R. M. Deans, D. W. Morgens, A. Okesli, S. Pillay, M. A. Horlbeck, M. Kampmann, L. A. Gilbert, A. Li, R. Mateo, M. Smith, J. S. Glenn, J. E. Carette, C. Khosla and M. C. Bassik, Parallel shrna and crispr-cas9 screens enable antiviral drug target identification, Nat. Chem. Biol., 2016, 12, 361–366 CrossRef CAS PubMed , PMC4836973.
- M. P. Patricelli, T. K. Nomanbhoy, J. Wu, H. Brown, D. Zhou, J. Zhang, S. Jagannathan, A. Aban, E. Okerberg, C. Herring, B. Nordin, H. Weissig, Q. Yang, J. D. Lee, N. S. Gray and J. W. Kozarich, In situ kinase profiling reveals functionally relevant properties of native kinases, Chem. Biol., 2011, 18, 699–710 CrossRef CAS PubMed , PMC3142620.
- C. Ye, D. J. Ho, M. Neri, C. Yang, T. Kulkarni, R. Randhawa, M. Henault, N. Mostacci, P. Farmer, S. Renner, R. Ihry, L. Mansur, C. G. Keller, G. McAllister, M. Hild, J. Jenkins and A. Kaykas, Drug-seq for miniaturized high-throughput transcriptome profiling in drug discovery, Nat. Commun., 2018, 9, 4307 CrossRef PubMed , PMC6192987.
- M. G. Rees, B. Seashore-Ludlow, J. H. Cheah, D. J. Adams, E. V. Price, S. Gill, S. Javaid, M. E. Coletti, V. L. Jones, N. E. Bodycombe, C. K. Soule, B. Alexander, A. Li, P. Montgomery, J. D. Kotz, C. S. Hon, B. Munoz, T. Liefeld, V. Dancik, D. A. Haber, C. B. Clish, J. A. Bittker, M. Palmer, B. K. Wagner, P. A. Clemons, A. F. Shamji and S. L. Schreiber, Correlating chemical sensitivity and basal gene expression reveals mechanism of action, Nat. Chem. Biol., 2016, 12, 109–116 CrossRef CAS PubMed , PMC4718762.
- N. A. Pabon, Y. Xia, S. K. Estabrooks, Z. Ye, A. K. Herbrand, E. Suss, R. M. Biondi, V. A. Assimon, J. E. Gestwicki, J. L. Brodsky, C. J. Camacho and Z. Bar-Joseph, Predicting protein targets for drug-like compounds using transcriptomics, PLoS Comput. Biol., 2018, 14, e1006651 CrossRef PubMed , PMC6300300.
- N. J. Martinez, R. R. Asawa, M. G. Cyr, A. Zakharov, D. J. Urban, J. S. Roth, E. Wallgren, C. Klumpp-Thomas, N. P. Coussens, G. Rai, S. M. Yang, M. D. Hall, J. J. Marugan, A. Simeonov and M. J. Henderson, A widely-applicable high-throughput cellular thermal shift assay (cetsa) using split nano luciferase, Sci. Rep., 2018, 8, 9472 CrossRef PubMed , PMC6013488.
- D. Martinez Molina and P. Nordlund, The cellular thermal shift assay: A novel biophysical assay for in situ drug target engagement and mechanistic biomarker studies, Annu. Rev. Pharmacol. Toxicol., 2016, 56, 141–161 CrossRef CAS PubMed.
- P. G. Wyatt, I. H. Gilbert, K. D. Read and A. H. Fairlamb, Target validation: Linking target and chemical properties to desired product profile, Curr. Top. Med. Chem., 2011, 11, 1275–1283 CrossRef CAS PubMed , PMC3182078.
- B. C. Cunningham and J. A. Wells, High-resolution epitope mapping of hgh-receptor interactions by alanine-scanning mutagenesis, Science, 1989, 244, 1081–1085 CrossRef CAS PubMed.
- A. Depriest, P. Phelan and I. Martha Skerrett, Tryptophan scanning mutagenesis of the first transmembrane domain of the innexin shaking-b(lethal), Biophys. J., 2011, 101, 2408–2416 CrossRef CAS PubMed , PMC3218331.
- L. L. Sharp, J. Zhou and D. F. Blair, Tryptophan-scanning mutagenesis of motb, an integral membrane protein essential for flagellar rotation in escherichia coli, Biochemistry, 1995, 34, 9166–9171 CrossRef CAS PubMed.
- A. Vallee-Belisle and S. W. Michnick, Visualizing transient protein-folding intermediates by tryptophan-scanning mutagenesis, Nat. Struct. Mol. Biol., 2012, 19, 731–736 CrossRef CAS PubMed.
- V. A. Assimon, A. T. Gillies, J. N. Rauch and J. E. Gestwicki, Hsp70 protein complexes as drug targets, Curr. Pharm. Des., 2013, 19, 404–417 CrossRef CAS PubMed , 3593251.
- M. Ferraro, I. D'Annessa, E. Moroni, G. Morra, A. Paladino, S. Rinaldi, F. Compostella and G. Colombo, Allosteric modulators of hsp90 and hsp70: Dynamics meets function through structure-based drug design, J. Med. Chem., 2019, 62, 60–87 CrossRef CAS PubMed.
- E. R. P. Zuiderweg, E. B. Bertelsen, A. Rousaki, M. P. Mayer, J. E. Gestwicki and A. Ahmad, Allostery in the hsp70 chaperone proteins, Top. Curr. Chem., 2012, 1–55 CAS.
- M. P. Mayer and B. Bukau, Hsp70 chaperones: Cellular functions and molecular mechanism, Cell. Mol. Life Sci., 2005, 62, 670–684 CrossRef CAS PubMed.
- A. Zhuravleva and L. M. Gierasch, Allosteric signal transmission in the nucleotide-binding domain of 70-kda heat shock protein (hsp70) molecular chaperones, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6987–6992 CrossRef CAS PubMed , 3084084.
- R. Kityk, J. Kopp and M. P. Mayer, Molecular mechanism of j-domain-triggered atp hydrolysis by hsp70 chaperones, Mol. Cell, 2018, 69, 227–237 CrossRef CAS PubMed , e224.
- C. J. Harrison, M. Hayer-Hartl, M. Di Liberto, F. Hartl and J. Kuriyan, Crystal structure of the nucleotide exchange factor grpe bound to the atpase domain of the molecular chaperone dnak, Science, 1997, 276, 431–435 CrossRef CAS PubMed.
- S. Rinaldi, V. A. Assimon, Z. T. Young, G. Morra, H. Shao, I. R. Taylor, J. E. Gestwicki and G. Colombo, A local allosteric network in heat shock protein 70 (hsp70) links inhibitor binding to enzyme activity and distal protein-protein interactions, ACS Chem. Biol., 2018, 13, 3142–3152 CrossRef CAS PubMed , PMC6377810.
- A. Bhattacharya, A. V. Kurochkin, G. N. Yip, Y. Zhang, E. B. Bertelsen and E. R. Zuiderweg, Allostery in hsp70 chaperones is transduced by subdomain rotations, J. Mol. Biol., 2009, 388, 475–490 CrossRef CAS PubMed , 2693909.
- J. F. Swain, G. Dinler, R. Sivendran, D. L. Montgomery, M. Stotz and L. M. Gierasch, Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker, Mol. Cell, 2007, 26, 27–39 CrossRef CAS PubMed.
- X. Li, H. Shao, I. R. Taylor and J. E. Gestwicki, Targeting allosteric control mechanisms in heat shock protein 70 (hsp70), Curr. Top. Med. Chem., 2016, 16, 2729–2740 CrossRef CAS PubMed , PMC5502483.
- L. Shrestha, H. J. Patel and G. Chiosis, Chemical tools to investigate mechanisms associated with hsp90 and hsp70 in disease, Cell Chem. Biol., 2016, 23, 158–172 CrossRef CAS PubMed , PMC4779498.
- A. Rousaki, Y. Miyata, U. K. Jinwal, C. A. Dickey, J. E. Gestwicki and E. R. Zuiderweg, Allosteric drugs: The interaction of antitumor compound mkt-077 with human hsp70 chaperones, J. Mol. Biol., 2011, 411, 614–632 CrossRef CAS PubMed , 3146629.
- J. Hageman, M. A. van Waarde, A. Zylicz, D. Walerych and H. H. Kampinga, The diverse members of the mammalian hsp70 machine show distinct chaperone-like activities, Biochem. J., 2011, 435, 127–142 CrossRef CAS PubMed.
- X. Li, S. R. Srinivasan, J. Connarn, A. Ahmad, Z. T. Young, A. M. Kabza, E. R. Zuiderweg, D. Sun and J. E. Gestwicki, Analogs of the allosteric heat shock protein 70 (hsp70) inhibitor, mkt-077, as anti-cancer agents, ACS Med. Chem. Lett., 2013, 4, 1042–1047 CrossRef CAS PubMed , PMC3845967.
- J. P. Schuermann, J. Jiang, J. Cuellar, O. Llorca, L. Wang, L. E. Gimenez, S. Jin, A. B. Taylor, B. Demeler, K. A. Morano, P. J. Hart, J. M. Valpuesta, E. M. Lafer and R. Sousa, Structure of the hsp110:Hsc70 nucleotide exchange machine, Mol. Cell, 2008, 31, 232–243 CrossRef CAS PubMed.
- E. B. Bertelsen, L. Chang, J. E. Gestwicki and E. R. Zuiderweg, Solution conformation of wild-type E. Coli hsp70 (dnak) chaperone complexed with adp and substrate, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8471–8476 CrossRef CAS PubMed.
- J. N. Rauch and J. E. Gestwicki, Binding of human nucleotide exchange factors to heat shock protein 70 (hsp70) generates functionally distinct complexes in vitro, J. Biol. Chem., 2014, 289, 1402–1414 CrossRef CAS PubMed , 3894324.
- H. H. Kampinga and E. A. Craig, The hsp70 chaperone machinery: J proteins as drivers of functional specificity, Nat. Rev. Mol. Cell Biol., 2010, 11, 579–592 CrossRef CAS PubMed , 3003299.
- Z. T. Young, J. N. Rauch, V. A. Assimon, U. K. Jinwal, M. Ahn, X. Li, B. M. Dunyak, A. Ahmad, G. A. Carlson, S. R. Srinivasan, E. R. Zuiderweg, C. A. Dickey and J. E. Gestwicki, Stabilizing the hsp70-tau complex promotes turnover in models of tauopathy, Cell Chem. Biol., 2016, 23, 992–1001 CrossRef CAS PubMed , 4992411.
- J. Wild, A. Kamath-Loeb, E. Ziegelhoffer, M. Lonetto, Y. Kawasaki and C. A. Gross, Partial loss of function mutations in dnak, the escherichia coli homologue of the 70-kda heat shock proteins, affect highly conserved amino acids implicated in atp binding and hydrolysis, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 7139–7143 CrossRef CAS PubMed , 49661.
- S. V. Slepenkov and S. N. Witt, Peptide-induced conformational changes in the molecular chaperone dnak, Biochemistry, 1998, 37, 16749–16756 CrossRef CAS PubMed.
- P. G. Needham, H. J. Patel, G. Chiosis, P. H. Thibodeau and J. L. Brodsky, Mutations in the yeast hsp70, ssa1, at p417 alter atp cycling, interdomain coupling, and specific chaperone functions, J. Mol. Biol., 2015, 427, 2948–2965 CrossRef CAS PubMed , PMC4569534.
- B. Bukau and G. C. Walker, Cellular defects caused by deletion of the escherichia coli dnak gene indicate roles for heat shock protein in normal metabolism, J. Bacteriol., 1989, 171, 2337–2346 CrossRef CAS PubMed , PMC209906.
- J. C. Bardwell and E. A. Craig, Major heat shock gene of drosophila and the escherichia coli heat-inducible dnak gene are homologous, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 848–852 CrossRef CAS PubMed.
- E. Deuerling, H. Patzelt, S. Vorderwulbecke, T. Rauch, G. Kramer, E. Schaffitzel, A. Mogk, A. Schulze-Specking, H. Langen and B. Bukau, Trigger factor and dnak possess overlapping substrate pools and binding specificities, Mol. Microbiol., 2003, 47, 1317–1328 CrossRef CAS PubMed.
- V. K. Singh, S. Utaida, L. S. Jackson, R. K. Jayaswal, B. J. Wilkinson and N. R. Chamberlain, Role for dnak locus in tolerance of multiple stresses in staphylococcus aureus, Microbiology, 2007, 153, 3162–3173 CrossRef CAS PubMed.
- K. I. Wolska, E. Bugajska, D. Jurkiewicz, M. Kuc and A. Jozwik, Antibiotic susceptibility of escherichia coli dnak and dnaj mutants, Microb. Drug Resist., 2000, 6, 119–126 CrossRef CAS PubMed.
- D. S. Williamson, J. Borgognoni, A. Clay, Z. Daniels, P. Dokurno, M. J. Drysdale, N. Foloppe, G. L. Francis, C. J. Graham, R. Howes, A. T. Macias, J. B. Murray, R. Parsons, T. Shaw, A. E. Surgenor, L. Terry, Y. Wang, M. Wood and A. J. Massey, Novel adenosine-derived inhibitors of 70 kda heat shock protein, discovered through structure-based design, J. Med. Chem., 2009, 52, 1510–1513 CrossRef CAS PubMed.
- S. Wisen, E. B. Bertelsen, A. D. Thompson, S. Patury, P. Ung, L. Chang, C. G. Evans, G. M. Walter, P. Wipf, H. A. Carlson, J. L. Brodsky, E. R. Zuiderweg and J. E. Gestwicki, Binding of a small molecule at a protein-protein interface regulates the chaperone activity of hsp70-hsp40, ACS Chem. Biol., 2010, 5, 611–622 CrossRef CAS PubMed , 2950966.
- T. Taldone, Y. Kang, H. J. Patel, M. R. Patel, P. D. Patel, A. Rodina, Y. Patel, A. Gozman, R. Maharaj, C. C. Clement, A. Lu, J. C. Young and G. Chiosis, Heat shock protein 70 inhibitors. 2. 2,5'-thiodipyrimidines, 5-(phenylthio)pyrimidines, 2-(pyridin-3-ylthio)pyrimidines, and 3-(phenylthio)pyridines as reversible binders to an allosteric site on heat shock protein 70, J. Med. Chem., 2014, 57, 1208–1224 CrossRef CAS PubMed , 3983364.
- A. Q. Hassan, C. A. Kirby, W. Zhou, T. Schuhmann, R. Kityk, D. R. Kipp, J. Baird, J. Chen, Y. Chen, F. Chung, D. Hoepfner, N. R. Movva, R. Pagliarini, F. Petersen, C. Quinn, D. Quinn, R. Riedl, E. K. Schmitt, A. Schitter, T. Stams, C. Studer, P. D. Fortin, M. P. Mayer and H. Sadlish, The novolactone natural product disrupts the allosteric regulation of hsp70, Chem. Biol., 2015, 22, 87–97 CrossRef CAS PubMed.
- L. Chang, A. D. Thompson, P. Ung, H. A. Carlson and J. E. Gestwicki, Mutagenesis reveals the complex relationships between atpase rate and the chaperone activities of escherichia coli heat shock protein 70 (hsp70/dnak), J. Biol. Chem., 2010, 285, 21282–21291 CrossRef CAS PubMed , 2898401.
- T. K. Barthel, J. Zhang and G. C. Walker, Atpase-defective derivatives of escherichia coli dnak that behave differently with respect to atp-induced conformational change and peptide release, J. Bacteriol., 2001, 183, 5482–5490 CrossRef CAS PubMed.
- W. A. Weiss, S. S. Taylor and K. M. Shokat, Recognizing and exploiting differences between rnai and small-molecule inhibitors, Nat. Chem. Biol., 2007, 3, 739–744 CrossRef CAS PubMed , PMC2924165.
- D. A. Case, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, D. Ghoreishi, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, N. Homeyer, S. Izadi, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, H. Nguyen, I. Omelyan, A. Onufriev, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Smith, R. Salomon-Ferrer, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, L. Xiao, D. M. York and P. A. Kollman, AMBER 2018, University of California, San Francisco, 2018 Search PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Improved side-chain torsion potentials for the amber ff99sb protein force field, Proteins, 2010, 78, 1950–1958 CAS , PMC2970904.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, W. Impey and M. L. Klein, Tip3p water model: Comparison of simple potential functions for simulating water, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- R. Salomon-Ferrer, A. W. Gotz, D. Poole, S. Le Grand and R. C. Walker, Routine microsecond molecular dynamics simulations with amber on gpus. 2. Explicit solvent particle mesh ewald, J. Chem. Theory Comput., 2013, 9, 3878–3888 CrossRef CAS PubMed.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- S. Sattin, J. Tao, G. Vettoretti, E. Moroni, M. Pennati, A. Lopergolo, L. Morelli, A. Bugatti, A. Zuehlke, M. Moses, T. Prince, T. Kijima, K. Beebe, M. Rusnati, L. Neckers, N. Zaffaroni, D. A. Agard, A. Bernardi and G. Colombo, Activation of hsp90 enzymatic activity and conformational dynamics through rationally designed allosteric ligands, Chemistry, 2015, 21, 13598–13608 CrossRef CAS PubMed.
- I. D'Annessa, S. Sattin, J. Tao, M. Pennati, C. Sanchez-Martin, E. Moroni, A. Rasola, N. Zaffaroni, D. A. Agard, A. Bernardi and G. Colombo, Design of allosteric stimulators of the hsp90 atpase as new anticancer leads, Chemistry, 2017, 23, 5188–5192 CrossRef PubMed , PMC5927549.
- L. Chang, E. B. Bertelsen, S. Wisén, E. M. Larsen, E. R. Zuiderweg and J. E. Gestwicki, High-throughput screen for small molecules that modulate the atpase activity of the molecular chaperone dnak, Anal. Biochem., 2008, 372, 167–176 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04284a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |