DOI:
10.1039/C9SC05432D
(Edge Article)
Chem. Sci., 2020,
11, 2243-2259
Stable group 8 metal porphyrin mono- and bis(dialkylcarbene) complexes: synthesis, characterization, and catalytic activity†
Received
28th October 2019
, Accepted 30th December 2019
First published on 31st December 2019
Abstract
Alkyl-substituted carbene (CHR or CR2, R = alkyl) complexes have been extensively studied for alkylcarbene (CHR) ligands coordinated with high-valent early transition metal ions (a.k.a. Schrock carbenes or alkylidenes), yet dialkylcarbene (CR2) complexes remain less developed with bis(dialkylcarbene) species being little (if at all) explored. Herein, several group 8 metal porphyrin dialkylcarbene complexes, including Fe- and Ru-mono(dialkylcarbene) complexes [M(Por)(Ad)] (1a,b, M = Fe, Por = porphyrinato dianion, Ad = 2-adamantylidene; 2a,b, M = Ru) and Os-bis(dialkylcarbene) complexes [Os(Por)(Ad)2] (3a–c), are synthesized and crystallographically characterized. Detailed investigations into their electronic structures reveal that these complexes are formally low-valent M(II)-carbene in nature. These complexes display remarkable thermal stability and chemical inertness, which are rationalized by a synergistic effect of strong metal-carbene covalency, hyperconjugation, and a rigid diamondoid carbene skeleton. Various spectroscopic techniques and DFT calculations suggest that the dialkylcarbene Ad ligand is unique compared to other common carbene ligands as it acts as both a potent σ-donor and π-acceptor; its unique electronic and structural features, together with the steric effect of the porphyrin macrocycle, make its Fe porphyrin complex 1a an active and robust catalyst for intermolecular diarylcarbene transfer reactions including cyclopropanation (up to 90% yield) and X–H (X = S, N, O, C) insertion (up to 99% yield) reactions.
Introduction
Since the discovery of a metal-alkylcarbene (M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHR) complex, [Ta(CH2tBu)3(CHtBu)], by Schrock,1 alkyl-substituted carbene ligands (alternative name: alkylidenes), i.e., alkylcarbene (CHR) and dialkylcarbene (CR2), have been mainly studied for MCHR complexes with early transition metals (TMs, groups 4–6), with their roles ranging from reactive synthons in olefin metathesis to supporting ligands (in nucleophilic, dianionic form (CHR)2−) that stabilize high-valent metal ions.2 In contrast, the chemistry of dialkylcarbene complexes of TMs and CHR/CR2 complexes of middle/late TMs remains less developed (Fig. 1a), particularly for mononuclear bis(CHR/CR2) complexes, examples of which are sparse.3 To the best of our knowledge, no mononuclear bis(dialkylcarbene) complexes of TMs have been reported previously.4
CHR) complex, [Ta(CH2tBu)3(CHtBu)], by Schrock,1 alkyl-substituted carbene ligands (alternative name: alkylidenes), i.e., alkylcarbene (CHR) and dialkylcarbene (CR2), have been mainly studied for MCHR complexes with early transition metals (TMs, groups 4–6), with their roles ranging from reactive synthons in olefin metathesis to supporting ligands (in nucleophilic, dianionic form (CHR)2−) that stabilize high-valent metal ions.2 In contrast, the chemistry of dialkylcarbene complexes of TMs and CHR/CR2 complexes of middle/late TMs remains less developed (Fig. 1a), particularly for mononuclear bis(CHR/CR2) complexes, examples of which are sparse.3 To the best of our knowledge, no mononuclear bis(dialkylcarbene) complexes of TMs have been reported previously.4
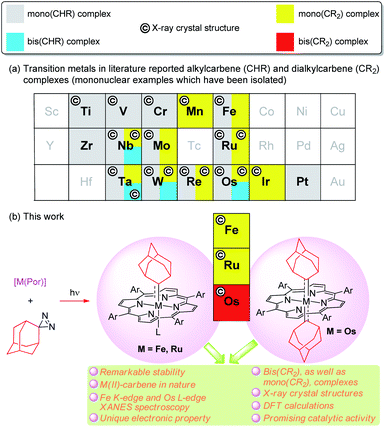 |
| | Fig. 1 (a) Literature examples of isolated mononuclear (di)alkylcarbene complexes of TMs. Mono- and bis(CHR/CR2) complexes are represented by different colors filled. Structurally characterized examples are indicated by ©. TMs for which no isolated examples were found are left blank. (b) Group 8 metal porphyrin dialkylcarbene complexes reported in this work. M![[partial double bond, top dashed]](https://www.rsc.org/images/entities/char_e12f.gif) Ad (M = Fe, Ru, Os) represents an Ad → M σ bonding (–) and a M → Ad π-backbonding ( Ad (M = Fe, Ru, Os) represents an Ad → M σ bonding (–) and a M → Ad π-backbonding ( ). ). | |
We are interested in developing new types of dialkylcarbene complexes of group 8 metals Fe, Ru, and Os. In the literature, Grubbs' Ru-alkylcarbene species have received much attention owing to their essential role in olefin metathesis.5 Related CHR/CR2 species of Fe,6 Ru,7 and Os,3d,8 mostly stabilized by strong-field ligands such as CO/phosphine and Cp (cyclopentadienyl), have also been reported; some Fe-(di)alkylcarbene complexes [Cp(CO)(L)Fe(CHR)]+ (L = PR3, CO)9 or [Cp(CO)2Fe(CR2)]+10 undergo stoichiometric reactions typical of electrophilic carbenes, including cyclopropanation,9a–g,10 Si–H insertion,9h and C–H insertion.9i Interest in Fe-carbene complexes6a,11 also lies in their potential role as next-generation catalysts for olefin metathesis and mechanistic implication of iron porphyrin carbene complexes for artificial metalloenzyme catalysis involving heme-carbenes as key intermediates.12 Recently, we developed catalytic dialkylcarbene transfer reactions13 including C(sp3)–H insertion, cyclopropanation, and Buchner reactions using various metalloporphyrin catalysts including Ru and Fe porphyrins; the active intermediates in these catalytic processes are likely to be the corresponding metalloporphyrin dialkylcarbene complexes [M(Por)(CR2)] (Por = porphyrinato dianion), which, however, have not been directly detected. Unlike the above-mentioned [Cp(CO)(L)Fe(CHR)]+ and [Cp(CO)2Fe(CR2)]+ which bear strong π-acidic CO ligand(s) in favor of low-valent metal centers, complexes [M(Por)(CR2)] can possibly be formulated as either low-valent MII←(:CR2)0 or high-valent MIV(CR2)2− species.14 To clarify this issue, it is highly desirable to isolate and fully characterize stable metalloporphyrin dialkylcarbene complexes for detailed studies on their electronic and molecular structures.
Up to now, metalloporphyrin dialkylcarbene complexes have been rarely reported; the only examples are [Fe(TPP)(CMeR)] (R = nBu, nPr) prepared by treatment of [Fe(TPP)Cl] with terminal alkynes and NaBH4.15 These [Fe(TPP)(CMeR)] complexes were not structurally characterized by X-ray crystallography and were formulated as Fe(II) species by analogy with the 1H NMR, UV-vis, and MS spectra of iron porphyrins bearing other types of carbene ligands. The very limited knowledge on metalloporphyrin dialkylcarbene species contrasts with extensive studies on other types of nonheteroatom-stabilized carbene complexes of Fe,11e,16 Ru,7a,17 and Os18 porphyrins, which were generally prepared using diazo compounds as carbene sources; such a synthetic method is hardly applicable to dialkylcarbene complexes owing to the short lifetime of dialkyldiazomethanes. Moreover, metalloporphyrin dialkylcarbene species could be prone to undergo a 1,2-hydride shift, which is a common side reaction observed in dialkylcarbene transfer catalysis.13 In addition, non-porphyrin group 8 metal dialkylcarbene complexes reported previously were often prepared with a limited choice of ligand sets (e.g., phosphine, Cp, and tris(pyrazolyl)borate) and/or under demanding conditions (e.g., strong acid and reducing medium),5–8 and only a few non-porphyrin Ru-7b–e,19 and Os-dialkylcarbene20 complexes have been structurally characterized by X-ray crystallographic studies and, despite the report of a structurally characterized FeCHMe non-porphyrin complex,21 no crystal structure of an Fe-dialkylcarbene complex has been reported so far.
In quest of a highly stable dialkylcarbene complex of metalloporphyrins, we paid attention to the dialkylcarbene ligand 2-adamantylidene (Ad). This was inspired by the studies of Ad as a model of dialkylcarbene in free carbene chemistry, as it can be photochemically generated from the diazirine compound, aziadamantane, under mild conditions and exhibits great resistance against 1,2-hydride shift due to its rigid diamondoid skeleton,22 coupled with the use of diazirines as carbene sources for the preparation of non-porphyrin metal-C(Ph)X (X = OMe, Br) carbene complexes;23 there is a literature report on the X-ray crystal structure of [Cp(CO)2Mn(Ad)],24 which is hitherto the only example of a known MAd complex, although this MnAd complex was prepared from a metal-borylene precursor rather than using aziadamantane as the Ad source. We envisaged that diazirines could be a new type of carbene source for synthesizing metal-dialkylcarbene species and Ad may also lead to isolable group 8 metal porphyrin dialkylcarbene complexes by reaction of readily accessible metalloporphyrin precursors with aziadamantane under photolysis.
In the present work, we report a simple and mild method of synthesizing dialkylcarbene complexes of TMs using diazirine as the carbene source, leading to the isolation of several MAd complexes of group 8 metal porphyrins including Fe- and Ru-mono(dialkylcarbene) [M(Por)(Ad)] (M = Fe 1, Ru 2) and Os-bis(dialkylcarbene) [Os(Por)(Ad)2] (3) complexes, along with the X-ray crystal structures of these complexes, as well as various spectroscopic studies including XANES (X-ray absorption near edge structure) spectroscopy and also DFT calculations (Fig. 1b), which all lend evidence for the low-valent M(II)-carbene nature of 1–3. Complexes 3a–c contribute unique examples of a bis(dialkylcarbene) complex of TMs. Surprisingly, the metal-dialkylcarbene complexes 1–3 are not reactive toward dialkylcarbene transfer reactions and generally display remarkable stability. Direct comparison is also made among dialkylcarbene Ad and other types of carbene ligands, mainly based on the system of group 8 metal porphyrins. Furthermore, a promising role of dialkylcarbene Ad as a supporting ligand is demonstrated by one of its Fe complexes 1a in homogeneous donor–donor carbene transfer catalysis.
Results
Synthetic procedures
Treatment of [FeII(TPFPP)] and aziadamantane at room temperature under UV irradiation (365 nm) for 15 min afforded 1a in 93% yield as a bright red solid (reaction 1 in Scheme 1a). Alternatively, it could be synthesized directly from commercially available [FeIII(TPFPP)Cl] at 40 °C with a slightly lower yield (85%, reaction 2 in Scheme 1a; the Fe(III) center was presumably reduced in situ by free Ad). Complex [Fe(TTP)(Ad)] (1b) could be prepared by the same two methods yet in much lower isolated yields, probably due to its higher susceptibility to aerobic oxidation as it decomposed immediately to μ-oxo dimer [Fe(TTP)]2O at above 60 °C in air (reaction 4 in Scheme 1a). Six-coordinate iron porphyrin carbene species 1a·Py could be easily accessed by adding pyridine into a solution of 1a (reaction 3 in Scheme 1a). Ruthenium analogues [Ru(Por)(Ad)] (Por = TPFPP: 2a; TTP: 2b) could be similarly prepared from [RuII(Por)(CO)] and aziadamantane in high yields (reaction 5 in Scheme 1b). Besides 2a,b, diazirine complexes 2az and 2bz (Scheme 1b), which are a new class of metalloporphyrin complexes, were isolated as minor species. The coordinated diazirine molecules are less photoactive and 2az could only be sluggishly transformed into 2a under UV irradiation and at elevated temperature (reaction 6 in Scheme 1b); we conceive that the coordinated diazirine ligand was either first converted to the corresponding diazo compound which then reacted with Ru porphyrin, or directly decomposed to free carbene that subsequently coordinated to the Ru center. Complexes 2az and 2bz were also slightly air-sensitive and slowly underwent oxidation to give 2-adamantanone.25
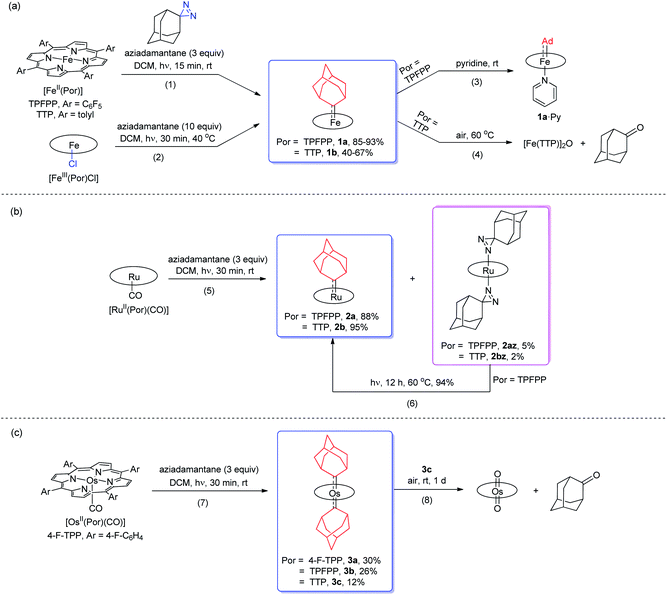 |
| | Scheme 1 Synthesis of Fe (a), Ru (b), and Os (c) dialkylcarbene complexes. | |
Strikingly, by a similar treatment of [Os(Por)(CO)] (Por = 4-F-TPP, TPFPP, and TTP) with aziadamantane, bis(dialkylcarbene) products [Os(Por)(Ad)2] (Por = 4-F-TPP: 3a, TPFPP: 3b, and TTP: 3c) were observed by 1H NMR measurements (reaction 7 in Scheme 1c). Monitoring the reaction of [Os(TPFPP)(CO)] and aziadamantane by 1H NMR showed that 3b was formed immediately after UV irradiation and no mono(dialkylcarbene) species [Os(Por)(Ad)] was detected. Thus it is likely that the bis(dialkylcarbene) complexes 3a–c were formed directly from the Os carbonyl precursors, different from the pre-formation of an isolable mono(diarylcarbene) analogue [Os(TPFPP)(CPh2)] and its conversion to the bis(diarylcarbene) counterpart [Os(TPFPP)(CPh2)2].18e Besides, 3a–c could all be readily purified by using an alumina column in air, in contrast to the analogous bis(diarylcarbene) complexes which could not be isolated in a pure form when supported by simple porphyrin ligands such as TTP.18a,d The lower isolated yields (12–30%) for 3a–c are attributable to the strong absorptivity of the Os precursors at 365 nm; attempts to improve the yields of 3a–c by increasing irradiation time were not successful, as prolonged irradiation caused decomposition of the Os-dialkylcarbene products. The bis(dialkylcarbene) complexes 3a–c were found slightly susceptible to aerobic oxidation, as exemplified by the oxidation of 3c to a trans-dioxo complex [Os(TTP)O2] and 2-adamantanone after standing in aerobic solution for ∼1 d (reaction 8 in Scheme 1c).
X-ray crystallography
The crystal structures of 1b, 1a·Py, 2b·MeOH, and 3a were determined by X-ray crystallography (Fig. 2), and the selected bond distances and angles are listed in Table 1. The sum of angles around Ccarbene atoms (1b: 360.1°; 1a·Py: 359.9°; 2b·MeOH: 359.9°; 3a: 359.8°) is indicative of sp2-hybridization of carbene carbons; the carbene planes are close to bisecting the M–Npyrrole bonds (31.6–44.3°) in all four complexes. The Fe–Ccarbene distance in 1b is 1.770(3) Å, which is typical of five-coordinate iron porphyrin carbene species.11e,16a The TTP macrocycle suffers from saddled distortion and the mean deviation of 24 atoms from the porphyrin mean plane is 0.268 Å, with the iron atom being displaced from this mean plane by 0.269 Å. Coordination of pyridine at the trans axial site causes elongation of the Fe–Ccarbene distance to 1.829(9) Å in 1a·Py, yet this distance is still much shorter than Fe–C single bonds;26 meanwhile, distortion of the porphyrin ligand is greatly suppressed (mean deviation: 0.051 Å) and the iron atom is only marginally deviated (0.074 Å) from the mean porphyrin plane. The crystal structure of 2b·MeOH features a short Ru–Ccarbene distance of 1.856(3) Å, and its porphyrin ring shows a minor doming distortion (mean deviation: 0.037 Å) along with a slightly out-of-plane Ru atom (0.172 Å).
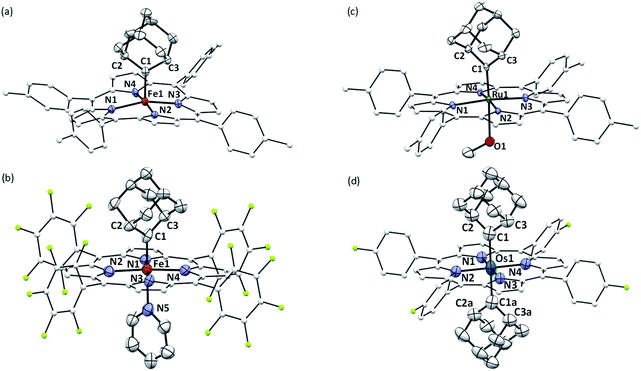 |
| | Fig. 2 ORTEP diagrams of 1b (a), 1a·Py (b), 2b·MeOH (c), and 3a (d) with thermal ellipsoids at the 50% probability level. Hydrogen atoms, disordered atoms/solvent molecules, and ellipsoids of porphyrin ligands (except for pyrrolic nitrogens) are omitted for clarity. | |
Table 1 Selected bond lengths (Å) and angles (o)
| (M = Fe, Ru, Os) |
1b
|
1a·Py |
2b·MeOH |
3a
|
|
All values involving C1–3 are averaged between two Ad groups.
Average distances between M1 and pyrrolic nitrogens (N1–4).
Lax = N5 (for 1a·Py) or O1 (for 2b·MeOH), or C1a (for 3a).
|
| M1–C1 |
1.770(3) |
1.829(9) |
1.856(3) |
2.061(7) |
| C1–C2 |
1.500(4) |
1.50(3) |
1.511(5) |
1.510(7) |
| C1–C3 |
1.488(5) |
1.50(2) |
1.507(5) |
1.510(7) |
| M1–Npyrroleb |
1.978(4) |
1.996(8) |
2.047(3) |
2.065(5) |
| M1–Laxc |
|
2.200(8) |
2.364(3) |
2.061(7) |
| C2–C1–C3 |
112.8(3) |
111.2(12) |
110.7(3) |
106.8(7) |
| C2–C1–M1 |
124.2(2) |
123.2(11) |
124.5(2) |
126.3(6) |
| C3–C1–M1 |
123.1(2) |
125.5(11) |
124.7(2) |
126.7(5) |
| C1–M1–Laxc |
|
178.4(7) |
176.16(12) |
180.0(9) |
The Os–bis(dialkylcarbene) complex 3a features an Os–Ccarbene bond length of 2.061(7) Å, similar to the bis(diphenylcarbene) complex [Os(TPFPP)(CPh2)2] (∼2.03 Å)18e yet apparently longer than that of Os porphyrin mono(carbene) complexes (1.79–1.87 Å),18c,e which is likely due to the strong trans influence exerted by the carbene ligands. A 90° disorder is observed for one of the two Ad ligands, making them either orthogonal or parallel to each other. Notably, the porphyrin plane in 3a is almost planar (mean deviation: 0.025 Å), which is in stark contrast to the strongly ruffled porphyrin ligand observed in [Os(TPFPP)(CPh2)2] (mean deviation: 0.197 Å).18e
Complexes 2az and 2bz represent two of rare examples of metal diazirine complexes25,27 and the X-ray crystal structure of 2az was obtained (Fig. 3). The two diazirine moieties experience slight π-backbonding from the Ru center, as the NN bond distance (1.248(9) Å) is longer than those in non-coordinated diazirines (1.228–1.235 Å)25 while the Ru–Ndiazirine bonds (1.985(6) and 1.996(6) Å) are also shorter than a typical Ru–N single bond (∼2.1 Å). Evidence of such π-backbonding further comes from the NN stretching bands observed in the IR spectrum of 2az (1518 and 1491 cm−1) which appear at appreciably lower frequencies than that of the free ligand (1570 cm−1).28
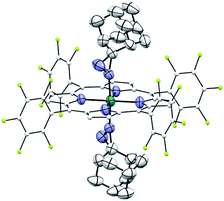 |
| | Fig. 3 ORTEP diagram of 2az with thermal ellipsoids at the 50% probability level. Hydrogen atoms and ellipsoids of porphyrin ligands (except for pyrrolic nitrogens) are omitted for clarity. | |
Spectral features
All the 1H NMR signals of dialkylcarbene complexes 1–3 are in the diamagnetic region, and no significant spectral change was observed up to 60 °C under an inert atmosphere. The Ad protons of these MAd complexes (Scheme 1) are strongly shielded and well separated by the porphyrin ring current, as assigned by COSY spectra (see the ESI†). In the case of 13C NMR spectra, the five-coordinate Fe-dialkylcarbene complexes 1 show characteristic downfield carbene chemical shifts at 394.92–413.57 ppm, and ligation of 1a with an axial ligand such as isocyanide, imidazole, or pyridines results in further downfield shifts of carbene carbons to 433.36–444.78 ppm; less deshielded signals are observed in Ru-mono(dialkylcarbene) complexes 2 as well as Os-bis(dialkylcarbene) complexes 3 (350–370 ppm, Table 2).
Table 2 Carbene chemical shifts δ (ppm)a,b
|
δ(FeC) |
|
δ(RuC) |
|
|
Measured in CDCl3 at 298 K.
ArNC = 2,6-dimethylphenyl isocyanide, CNPy = 4-cyanopyridine, Py = pyridine, NMe2Py = 4-dimethylaminopyridine, and MeIm = 1-methylimidazole.
Measured in C6D6 at 298 K.
|
|
1a
|
413.57 |
2a
|
369.55 |
|
1b
|
394.92 |
2b
|
353.72 |
|
1a·ArNC |
444.78 |
|
|
|
1a·CNPy |
433.85 |
|
|
|
δ(FeC) |
|
δ(OsC) |
|
|
1a·Py |
433.58 |
3a
|
351.28c |
|
1a·NMe2Py |
433.39 |
3b
|
367.04c |
|
1a·MeIm |
433.36 |
3c
|
350.39 |
Several other spectroscopic techniques were employed to probe the oxidation state of metal centers in these metalloporphyrin dialkylcarbene complexes. The Fe K-edge XANES spectrum of 1a was recorded along with that of several reference iron porphyrin complexes. The pre-edge peak of 1a (7113.0 eV) is highly similar to that of [FeII(TPFPP)(CPh2)] (7113.0 eV) yet appreciably lower in energy than that of [FeIII(TPFPP)Cl] (7114.0 eV), thus suggesting an Fe(II) center in the dialkylcarbene complex (Fig. 4, top). These results are also consistent with those obtained previously for Fe porphyrin carbene and related complexes (Table S6†).16a The 57Fe isomer shift (δ = 0.25 mm s−1) of 1a in Mössbauer spectroscopy (Fig. 5) falls in the region of low-spin five-coordinate Fe(II) porphyrin complexes (−0.03 to 0.37 mm s−1);16c the relatively small quadrupole splitting (|ΔEQ| = 1.61 mm s−1) is possibly related to the unique ligand field induced by the dialkylcarbene ligand.29 The resonance Raman spectrum of 1a features a 1356 cm−1 band (Fig. S5†) which is comparable to that of [Fe(TPP)(CCl2)] (1368 cm−1) and indicative of a low spin Fe(II) center (1358–1379 cm−1) rather than other higher valent configurations (>1370 cm−1 for Fe in +3 to +5 oxidation states).16a,30 The diamagnetism of 1a is also suggested by Evans' method where no observable separation of solvent peaks was found. Ruthenium complex 2b displays an oxidation-state marker band at 1009 cm−1 in its IR spectrum, which is within the normal range of Ru(II) porphyrin carbonyl or carbene complexes (1001–1017 cm−1).17g,31
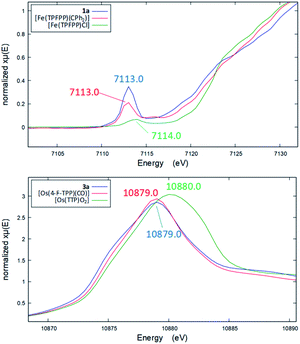 |
| | Fig. 4 Top: Fe pre-edge peaks in the XANES spectra of 1a (blue), [Fe(TPFPP)(CPh2)] (red), and [Fe(TPFPP)Cl] (green). Bottom: Os L3-edge peaks in the XANES spectra of 3a (blue), [Os(4-F-TPP)(CO)] (red), and trans-dioxo complex [Os(TTP)O2] (green). | |
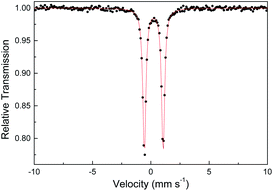 |
| | Fig. 5 Zero-field Mössbauer spectrum of 1a at 298 K. | |
Previously, the oxidation state of metal centers in Os porphyrin carbene complexes remained ambiguous.18c,e,f To clarify this issue, we conducted an Os L-edge XANES study on 3a and two related Os porphyrins (Fig. 4, bottom). Complex 3a exhibits an L3-edge peak at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 879.0 eV in its XANES spectrum, and this was found to be highly similar to that of [OsII(4-F-TPP)(CO)] (10879.0 eV) but different from that of the high-valent trans-dioxo complex [OsVI(TTP)O2] (10880.0 eV); these results are supportive of an OsII-bis(carbene) electronic structure of 3a rather than an OsVI Schrock alkylidene formulation.
879.0 eV in its XANES spectrum, and this was found to be highly similar to that of [OsII(4-F-TPP)(CO)] (10879.0 eV) but different from that of the high-valent trans-dioxo complex [OsVI(TTP)O2] (10880.0 eV); these results are supportive of an OsII-bis(carbene) electronic structure of 3a rather than an OsVI Schrock alkylidene formulation.
UV-vis absorption spectra of Fe complexes 1a,b both exhibit a single Soret band, whereas two separate Soret bands could be observed for Ru congeners (Fig. S1†). Addition of an axial ligand generally leads to a red shift of the Soret band(s), and the extent of the red shift is affected by the electronic properties of the axial ligand (Fig. 6). The binding constants of pyridine with 1a and 2b were measured to be (9.90 ± 0.48) × 102 and (2.375 ± 0.144) × 104 M−1 respectively (Fig. S2†), both being smaller than those of analogous Fe/Ru porphyrin complexes bearing other carbene ligands (3500 M−1 for Fe-dichlorocarbene11a and 1.13 × 105 M−1 for Ru-quinoid carbene complexes17l). Interestingly, no binding between PPh3 and 1a/2b has been observed during UV-vis monitoring. The Soret and Q bands of Os-bis(dialkylcarbene) complexes 3 are generally weakened and broadened, with the Q bands of 3b,c markedly blue-shifted (<500 nm) as compared to those of 1a,b and 2a,b bearing the same porphyrin ligands (Fig. S1†); these spectral features are typically found for Os porphyrin complexes.32
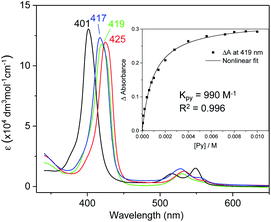 |
| | Fig. 6 UV-vis spectra of 1a (black), and 1a ligated with 4-cyanopyridine (blue), pyridine (green), and 4-dimethylaminopyridine (red) in DCM. Inset: titration of pyridine (5 × 10−5–0.01 M) into a DCM solution of 1a (5 × 10−5 M). | |
Electrochemistry
Electrochemical studies on these group 8 metal dialkylcarbene complexes were conducted to gain insights into their electronic structures (Fig. 7 and S3†). Complexes 1a,b both show one reversible oxidation wave at −0.18 to 0.01 V (vs. Fc+/0, the same as below) in the cyclic voltammograms (Table 3), falling in the region of the FeIII/II couple.33 This assignment complied with the results from XANES, Mössbauer, and resonance Raman spectroscopies (vide supra), and was further verified by the spectroelectrochemical spectrum of 1a in which a broadened Soret band and red-shifted Q bands characteristic of Fe(III) porphyrins appeared after oxidation (Fig. 8, top);34 products derived from porphyrin- or carbene ligand-oxidation could be precluded, since metal-Por+˙ species feature a broad band between 630 and 710 nm while oxidation on the axial ligand generally leads to minor changes in the electronic spectra of [M(Por)] complexes.33,35 Although the oxidation is reversible on the cyclic voltammetry timescale, the oxidized Fe(III) species was found unstable and decomposed within minutes. The first reversible oxidation waves of 2a,b (−0.03 to 0.18 V, Table 3) are also ascribed to be Ru-centered oxidation according to our previous study.17g The Os complexes 3a–c all show a quasi-reversible oxidation wave (−0.17 to 0.04 V) assignable to the OsIII/II couple,36 in agreement with the +2 oxidation state of the Os center suggested by XANES spectroscopy (Fig. 4, bottom). The second oxidation waves in 1b, 2b, 3a, and 3c could be attributed to porphyrin-centered oxidation processes. The first reduction waves of the Fe complexes are strongly influenced by the aryl substituents on porphyrin ligands (Table 3) and are therefore assigned to porphyrin-centered reduction. This was also evidenced by the noticeable spectral changes of Soret and Q bands upon reduction of 1a (Fig. 8, bottom). The first reductions of 2a,b and 3a,b are assigned likewise in view of their similarity in potential and reversibility (Table 3); no reduction peaks are observed for 3c and they are possibly beyond the solvent window of DCM.
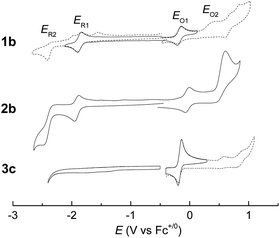 |
| | Fig. 7 Cyclic voltammograms of group 8 metal dialkylcarbene complexes supported by the TTP macrocycle, using 0.1 M (Bu4N)PF6 as the electrolyte and at a scan rate of 0.1 V s−1. Top: 1b and 2b, measured in DMF with SCE as the reference electrode; E(Fc+/0) = 0.49 V. Bottom: 3c, measured in DCM with Ag/AgCl as the reference electrode; E(Fc+/0) = 0.40 V. Dotted lines indicate irreversible redox events. | |
Table 3 Redox potentials V vs. Fc+/0a
|
E (V) |
E
O1
|
E
O2
|
E
R1
|
E
R2
|
|
E
1/2 values. 1a,b and 2a,b were measured in DMF with SCE as the reference electrode; E(Fc+/0) = 0.49 V. 3a–c were measured in DCM with Ag/AgCl as the reference electrode; E(Fc+/0) = 0.40 V.
E
p,a.
E
p,c.
Quasi-reversible.
|
|
1a
|
0.01 |
|
−1.59 |
−2.07 |
|
1b
|
−0.18 |
0.47b |
−1.86 |
−2.38c |
|
2a
|
0.18 |
|
−1.66 |
−2.10 |
|
2b
|
−0.03 |
0.61b |
−1.91 |
−2.50c |
|
3a
|
−0.10d |
0.77b |
−2.05d |
|
|
3b
|
0.04d |
|
−1.71d |
|
|
3c
|
−0.17d |
0.59b |
|
|
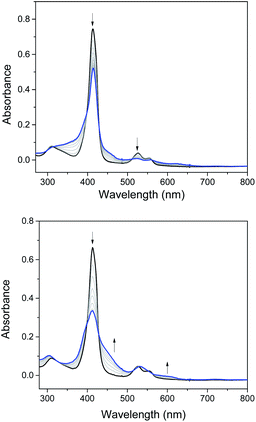 |
| | Fig. 8 Spectroelectrochemistry of 1a in DMF. Electrode potentials: 0.6 V (top) and −1.1 V (bottom) vs. Ag/AgCl. | |
DFT calculations
DFT calculations were performed on 1b and 3a to provide detailed information on the bonding nature of a metalloporphyrin mono(dialkylcarbene) complex and a metal-bis(dialkylcarbene) complex. The optimized structure of 1b and 3a show the M–Ccarbene (M = Fe, Os) bond distances of 1.742 Å and 2.003 Å respectively, which are close to the experimental values (Table 1). A low-spin Fe(II) center with an electronic configuration of (dx2–y2)2(dyz)2(dxz)2(dz2)0(dxy)0 was found for 1b (Fig. 9), consistent with the diamagnetism of this complex and also similar to previous experimental and computational results for iron porphyrin complexes ligated with other types of carbene ligands.16b,37 An Fe-dialkylcarbene π-backbonding interaction is formed between the dyz of Fe and the py orbital of Ccarbene, and the π(FeC) orbital is mostly Fe dπ in character (H-3, 45% Fe, 17% Ccarbene), while the π*(FeC) orbital is more polarized to the carbene carbon (L + 2, 32% Fe, 52% Ccarbene). Therefore, the complex should be best described as a formally FeIIAd0 species with decent Fe-to-carbene π-backbonding. Similar to free Ad in which hyperconjugation plays an essential role in stabilizing the carbene center,22d,e the π*(FeC) he orbital in 1b is also stabilized by such an effect exerted by the flanking alkyl groups. The LUMO is essentially populated on the porphyrin ligand and the HOMO mostly resides at the Fe center, both of which comply with the electrochemical study (vide supra).
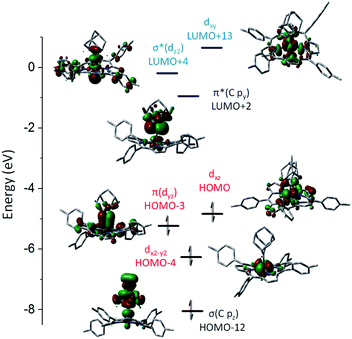 |
| | Fig. 9 Calculated molecular orbitals of [Fe(TTP)(Ad)] (1b). LUMO, which is mainly located on the TTP ligand, is not shown. The z axis is orthogonal to the porphyrin plane and the x and y axes are on the porphyrin plane and bisecting Fe–Npyrrole vectors. | |
A similar M(II) electronic configuration was calculated for Os-bis(dialkylcarbene) complex 3a which features a low-spin d6 Os(II) center and strong metal-carbene covalency (Fig. 10, top). The two perpendicular pπ orbitals of the two Ad ligands interact with the dxz and dyz orbitals of Os, giving rise to two pairs of almost degenerate π(OsC) (H-1 and H-2) and π*(OsC) (L + 2 and L + 3) orbitals. Different from 1b, the dz2 orbital of 3a is higher in energy than dxy as a result of the strongly coordinating dialkylcarbene ligands. TD-DFT calculations could well reproduce the UV-vis spectrum of 3a which consists of two separate Soret bands at 343–395 nm and a weak and broad Q band at 557 nm (Fig. 10, bottom).
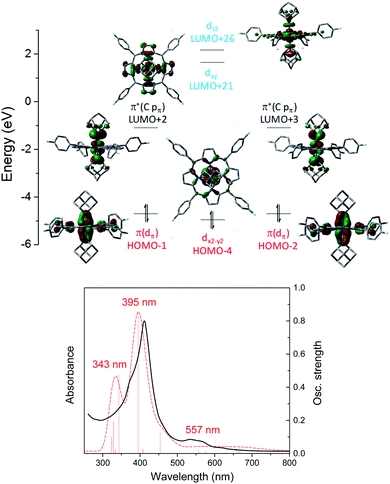 |
| | Fig. 10 Top: Calculated molecular orbitals of [Os(4-F-TPP)(Ad)2] (3a). The z axis is orthogonal to the porphyrin plane and the x and y axes are on the porphyrin plane and bisecting Os–Npyrrole vectors. Bottom: Experimental (black solid line) and TD-DFT simulated (red dashed line) UV-vis spectrum of 3a. | |
Stability and reactivity
Fe(II) porphyrins are usually air-sensitive and could be stabilized by a few π-acceptor ligands (e.g., CS and isocyanides). In this work, the dialkylcarbene Ad ligand was found to confer remarkable stability on Fe(II) porphyrins, especially for 1a bearing the electron-deficient porphyrin ligand TPFPP. No decomposition of 1a could be detected even by treating its benzene solution at 80 °C in air for at least 1 h; its six-coordinate adducts with pyridines and MeIm also stayed intact in solution for at least one week under ambient conditions, which is in sharp contrast to air-sensitive [Fe(TPFPP)(CPh2)(MeIm)].11e Surprisingly, thermogravimetric analysis (TGA) revealed that 1a in the solid state is stable up to ∼230 °C in air (Fig. S6;† the 1H NMR spectrum of 1a, after being heated at 230 °C in the solid state for 5 min in air, remained unchanged). The Ru analogue 2a also exhibits great thermal stability in air with the onset of decomposition appearing at ∼330 °C (Fig. S6†). The Os-bis(dialkylcarbene) complexes are stable below 60 °C under an inert atmosphere in both solution and solid states.
Although Fe porphyrin CPh2 and C(Ph)CO2Et carbene complexes could react stoichiometrically with alkenes and/or C(sp3)–H bonds,11e1a was found resistant toward various organic substrates/nucleophiles (e.g., styrene, 1,4-cyclohexadiene, PhNH2, and PPh3; see the ESI† for details), even in the presence of a trans axial ligand (pyridine and MeIm) and at high temperature (80 °C), nor was it reactive under UV irradiation, which has been reported to trigger reactions between [Fe(TPP)(CX2)] (X = halogens) and alkenes.38 Remarkable stability was also found with the Ru congener 2a which was unreactive under the above conditions. When 1a was treated with anionic nucleophiles such as NaXMe (X = S, O) at 80 °C, SNAr reactions on the TPFPP meso-C6F5 groups slowly took place to afford 1c and 1d respectively (reaction 9 in Scheme 2), which could be traced by the changes in 1H and 19F NMR spectra; the carbene carbons remained intact and still resonated at ∼410 ppm in their 13C NMR spectra, demonstrating the remarkable kinetic inertness of the Ad ligand.
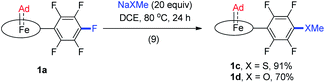 |
| | Scheme 2 Intermolecular reactions of 1a with anionic nucleophiles. | |
Previously, the Os-bis(CPh2) complex has been shown to display stoichiometric diarylcarbene transfer reactivity.18e However, no dialkylcarbene transfer product was detected when treating nucleophilic organic substrates (e.g., styrene, 1,4-cyclohexadiene, PhNH2, PhNO, PPh3, and PhSH) with the bis(dialkylcarbene) complex 3a up to 60 °C; further heating up the reaction mixture to 80 °C only caused decomposition of 3a to complicated mixtures, which was similarly observed in a control experiment in the absence of a substrate. Surprisingly, 3a was also stable against MeLi at −78 °C to room temperature, although MeLi was known to undergo nucleophilic addition or α-deprotonation of metal-carbene complexes. Addition of pyridine into a DCM solution of 3a at room temperature slowly led to carbene ligand dissociation and formation of 2-adamantanone, yet neither a dialkylcarbene transfer product nor an Os-mono(dialkylcarbene) complex could be observed.18f,g In view of the relatively labilized Ad ligands in the Os-bis(dialkylcarbene) complexes 3, we wondered whether 3a would exhibit partial nucleophilic free dialkylcarbene character;22f however, it was found unreactive toward electrophiles such as ethyl acrylate or bis(pinacolato)diboron.
While the studies described above revealed that the dialkylcarbene Ad ligand coordinated to group 8 metal porphyrins is less reactive than diphenylcarbene CPh2, their relative thermodynamic stability was further probed by carbene substitution experiments (Scheme 3). It was found that [Fe(TPFPP)(CPh2)] and [Os(TPFPP)(CPh2)2] could readily react with free Ad, generated from photolysis of aziadamantane, to afford 1a and 3b, respectively (reactions 10 and 12 in Scheme 3), yet treatment of 1a or 3b with excess diazo compound Ph2CN2 was not able to result in replacement of the Ad ligand(s) in the Fe or Os complex by CPh2 even under more forcing conditions (reactions 11 and 13 in Scheme 3). For the reaction of 1a with Ph2CN2 (reaction 11 in Scheme 3), the azine product Ph2CN–NCPh2 was obtained in 90% yield.
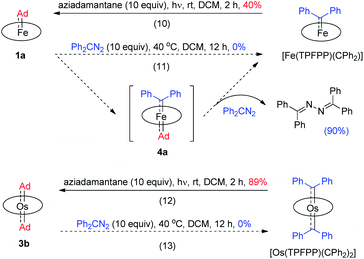 |
| | Scheme 3 Carbene substitution experiments. | |
Catalytic intermolecular donor–donor carbene transfer reactions catalyzed by 1a
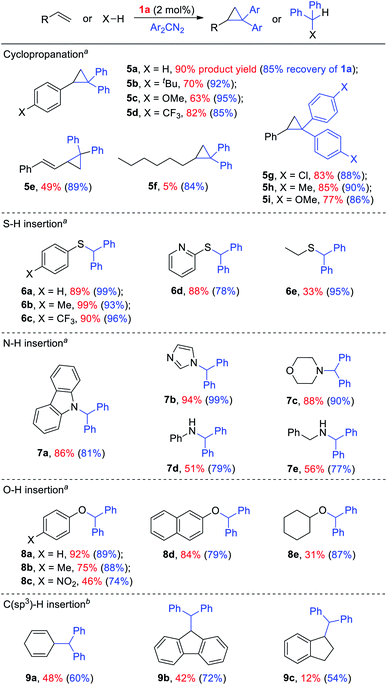
Given the remarkable stability of the Ad ligand, we examined the possibility of using 1a as a robust Fe(II) catalyst for some challenging transformations. Diarylcarbene (CAr2) transfer reactions remain less developed in the literature, which is mainly due to the intrinsic low reactivity of CAr2 carbenes and they are known to form isolable adducts even with the most active catalysts such as Rh39 and Cu.40 Inspired by the carbene substitution experiments (reaction 11 in Scheme 3) revealing the formation of an azine product, we envisioned that 1a might serve as an efficient catalyst for diarylcarbene transfer reactions, since the observed azine product possibly resulted from reaction of Ph2CN2 with a reactive Fe-bis(carbene) intermediate [Fe(TPFPP)(Ad)(CPh2)] (4a, Scheme 3; by drawing an analogy with Os-bis(carbene) analogues), in which the CPh2 ligand was activated with the trans-Ad ligand. Gratifyingly, cyclopropanation of styrene with Ph2CN2 could be catalyzed by 2 mol% of 1a in excellent yield (90%, entry 1 in Table 4). The time course plot of this reaction showed that product 5a was smoothly produced with a very short induction period, and catalyst 1a remained rather stable in the reaction mixture (85% recovery, Fig. 11). Employing [FeII(TPFPP)] as the catalyst led to a poorer yield of 5a (entry 2 in Table 4), which suggests the important role played by the Ad ligand in 1a in this reaction. Complex 1a showed superior catalytic performance and chemoselectivity to other commonly used carbene transfer catalysts (e.g., Rh and Cu catalysts, entries 4–6 in Table 4; see also Table S7†) under the same reaction conditions since the latter catalysts afforded mainly the azine product; the Ru-dialkylcarbene complex 2a gave slightly better results than 1a (entry 3). By using 1a as the catalyst, the scope of alkenes was extended to styrenes bearing electron-donating (5b,c) and -withdrawing (5d) substituents with good to high product yields (Scheme 4). A conjugated alkene was selectively cyclopropanated at the less hindered site in a moderate yield (5e). However, alkyl-substituted alkenes such as 1-octene resulted in a low yield of 5f and mainly the azine by-product. Besides CPh2 carbene, electron-deficient (5g) and -rich (5h,i) diarylcarbenes were also converted to the desired cyclopropanes in high yields.
Table 4 Comparison of catalytic performance among 1a, 2a, and other common carbene transfer catalystsa
| Entry |
Catalyst |
Yield of 5ab (%) |
|
Conditions: Ph2CN2 (0.2 mmol), styrene (2 mmol), catalyst (0.004 mmol), DCM (1 mL), 40 °C, 12 h, and under Ar.
Yield determined by 1H NMR with PhTMS as the internal standard.
|
| 1 |
1a
|
90 |
| 2 |
[FeII(TPFPP)] |
48 |
| 3 |
2a
|
92 |
| 4 |
[Rh2(esp)2] |
10 |
| 5 |
CuI |
1 |
| 6 |
[Ru(TTP)(CO)] |
56 |
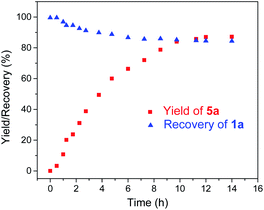 |
| | Fig. 11 Time course plot of the reaction among Ph2CN2, styrene, and 1a during 1H NMR monitoring. | |
 |
| | Scheme 4 Intermolecular diarylcarbene transfer reactions catalyzed by 1a. Product yields in red and recovery of 1a (blue) in parentheses. a Conditions: Ph2CN2 (0.2 mmol), substrate (2 mmol), catalyst (0.004 mmol), DCM (1 mL), 40 °C, 12 h, and under Ar. b Conditions: Ph2CN2 (0.2 mmol, added over 3 h), substrate (2 mmol), catalyst (0.004 mmol), DCE (1 mL), 80 °C, 12 h, and under Ar. | |
Complex 1a could also catalyze intermolecular S–H, N–H, and O–H insertion reactions, thereby introducing benzhydryl groups onto various heteroatoms (Scheme 4). S–H bonds activated by aryl (6a–c) and 2-pyridyl groups (6d) readily underwent carbene insertion in 88–99% yields; alkyl-substituted thiols such as ethanethiol also afforded the desired sulfide product albeit in a lower yield (6e, 33%). Catalytic N–H insertion could be achieved in high yields by using electron-rich N-heterocycles such as carbazole (7a) and imidazole (7b), as well as secondary alkyl amines such as morpholine (7c); primary amines, aryl (7d) and aliphatic (7e), resulted in only moderate product yields. Aryl alcohols including phenols (8a,b) and 2-naphthol (8d) were transformed into O–H insertion products in 75–92% yields, yet electron-poor phenol (8c) and aliphatic alcohol (8e) showed appreciably lower reactivity. Notably, catalytic C(sp3)–H insertion of CPh2 carbene was achieved under more demanding conditions (80 °C and dropwise addition of Ph2CN2), and previously such catalytic transformations were largely confined to intramolecular versions using Rh catalysts.41 C–H insertion of doubly-activated substrates including 1,4-cyclohexadiene (9a) and fluorene (9b) was achieved in moderate yields, while a low yield was observed with the less reactive indane (9c). Moreover, the robustness of catalyst 1a could be demonstrated by the high recovery of 1a after most of the catalytic reactions mentioned above (Scheme 4).
Attempts to isolate or directly detect the proposed Fe-bis(carbene) intermediate 4a have not been successful. We performed DFT calculations on a model complex 4b (Fig. 12), which is identical to 4a except that the meso-aryl groups in 4a were replaced by hydrogen for simplicity.37,42 The calculated structure of 4b features a rather lengthened Fe–CCPh2 bond (1.96 Å) as compared to the Fe–CAd bond (1.89 Å); the LUMO was mainly localized on the pπ of CCPh2 while the pπ of CAd was higher in energy (L + 3); this could account for the relatively high reactivity of CAr2 ligands as well as the stability of 1a and its Ad ligand during catalytic turnover.
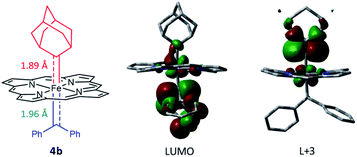 |
| | Fig. 12 DFT-optimized structure of model complex 4b (left) and its LUMO (middle) and L + 3 orbitals (right). | |
Catalytic intermolecular cyclopropanation reactions of other donor–donor (phenyl/alkyl- and dialkyl-) carbenes were also tested by using freshly prepared or in situ generated diazo compounds.43 Complex 1a could catalyze the reactions of N2C(Ph)CF3 and N2C(Ph)Me (the latter was generated in situ from acetophenone hydrazone and Ag2O) with styrene to afford the corresponding cyclopropanation products in moderate to good yields albeit with low diastereomeric ratios (entries 1–2 in Table 5); however, a similar treatment of diazoadamantane (in situ generated from AdNNH2 and PhIO) with styrene using catalyst 1a did not give the cyclopropanation product, and [3 + 2] cycloaddition between styrene and the diazo compound to give 10 was the major reaction observed (entry 3 in Table 5).43c Worthy of note is the high recovery of 1a in all these reactions as this complex showed decent stability toward PhIO which is known to generate Fe-oxo species with common Fe porphyrin complexes.44
Table 5 Intermolecular cyclopropanation reactions between other diazo compounds and styrene catalyzed by 1aa
Discussion
Middle/late transition metal (TM) (di)alkylcarbene species are key intermediates related to olefin metathesis5 and (di)alkylcarbene transfer catalysis,13,45 yet the electronic properties of dialkylcarbene as a ligand, especially in comparison with common carbene ligands including “stable” (NHC and CAAC) and “reactive” (phenyl-/ester-substituted) carbenes, still remain less understood. In this work, by choosing 2-adamantylidene (Ad) as a rigid model, we have synthesized several terminal group 8 metal dialkylcarbene complexes from easily accessible precursors and by a simple and mild method. We have also demonstrated that, different from the well documented reactive features, dialkylcarbene ligands such as Ad can be highly stable upon coordination to TMs including Fe and even act as a robust ancillary ligand for catalytic applications. To gain deeper knowledge, the following aspects of the dialkylcarbene ligand Ad coordinated to TMs are discussed in this section.
Dialkylcarbene vs. common carbene ligands
Although both stable and reactive carbene ligands feature an sp2-hybridized carbene center with a vacant pπ orbital, they have scarcely been linked or discussed together due to the drastic differences in the molecular structure, stability, and/or electronic properties of their metal complexes.46 The uniqueness of dialkylcarbene lies in the fact that it possesses mutual characters from the above two types of carbenes: on the one hand, its carbene transfer reactivity has been well documented in the literature;10,13,45 on the other hand, we have discovered in this work that it confers remarkable thermodynamic and kinetic stability on group 8 metal porphyrin complexes and in particular, it leads to stabilization of an Fe(II) porphyrin species and several Os-bis(carbene) complexes. These findings prompt us to make a closer comparison between dialkylcarbene and stable/reactive carbenes with the aid of theoretical calculations.
(i) Dialkylcarbene vs. stable carbenes (NHC/CAAC).
The carbene substituents for dialkylcarbene and stable carbenes show an interesting pattern ranging from diamino (NHC), amino/alkyl (CAAC) to dialkyl (Ad), and such a variation can directly influence their carbene frontier orbitals, i.e., the σ orbital and pπ orbital, through inductive and mesomeric effects.47 By performing DFT calculations on these free carbenes, we found that the most striking feature of dialkylcarbene is its much lower-lying pπ orbital than NHC and CAAC (ΔE > 1.8 eV, Fig. 13a), likely due to the absence of π-donating atoms adjacent to the carbene center;48 its σ orbital is also slightly higher in energy (ΔE = 0.10–0.52 eV). Therefore, dialkylcarbene acts as a much stronger π-acceptor and a slightly better σ-donor, and thus a stronger-field ligand than these conventional stable carbenes.
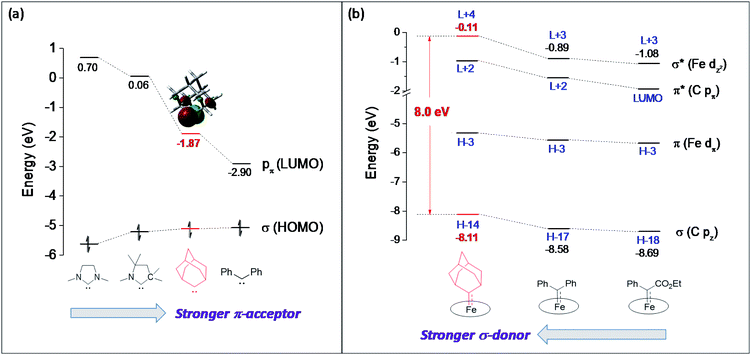 |
| | Fig. 13 (a) Comparison of calculated carbene frontier orbitals of free NHC, CAAC, Ad, and CPh2 carbenes in their singlet states. (b) Comparison of calculated Fe–Ccarbene orbitals in [Fe(Por)(Ad)], [Fe(Por)(CPh2)], and [Fe(Por)(C(Ph)CO2Et)]. | |
The variation in π-character could account for the distinct differences in the Fe/Ru–Ccarbene distances, as the Fe/Ru–CAd bonds are all within double bond regions (Table 1) and are apparently shorter than those in NHC/CAAC complexes (1.87–2.15 Å for Fe and 1.93–2.15 Å for Ru).49 The redox potential of metalloporphyrin carbene complexes, especially the first oxidation potential (EO1), is a good indicator for the electronic properties of carbene ligands,17l and the greater π-acidity of the Ad ligand is also noted by the increased EO1 values of 2a,b as compared to their NHC congeners (Fig. 14 and Table S8†). In addition, NHC often gives bis(carbene) complexes with Fe and Ru porphyrins,49d,j yet only mono(dialkylcarbene) complexes of Fe and Ru porphyrins [MFe/Ru(Por)(Ad)] are isolable, which is likely caused by the prominent trans influence of the Ad ligand. The stronger trans influence of Ad than NHC is also inferred by comparing the solid-state structures of [Ru(TTP)(Ad)(MeOH)] (2b·MeOH) and [Ru(TPP)(IMe2)(THF)] where the axial Ru–O bond is longer in the former (2.36 Å) than in the latter complex (2.22 Å, Fig. S7†).
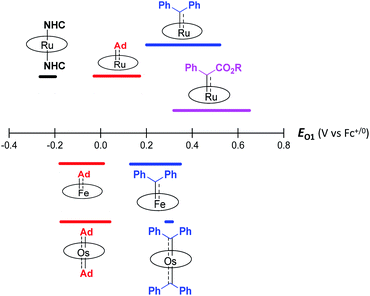 |
| | Fig. 14 First oxidation potential (EO1) range of group 8 metal porphyrin carbene complexes. | |
(ii) Dialkylcarbene vs. reactive carbenes (phenyl-/ester-substituted carbenes).
Besides all being related to carbene transfer catalysis, dialkylcarbene also bears some other resemblance to reactive carbenes. For instance, their complexes of Fe/Ru porphyrins are all mono(carbene) species diaplaying M–Ccarbene double bond character (1.77–1.83 Å for Fe and 1.81–1.88 Å for Ru);16a,17g,i these complexes share similar features in their NMR, UV-vis, and IR spectra and cyclic voltammograms. To further compare their electronic properties, we conducted DFT calculations on Fe-carbene complexes bearing Ad, CPh2, and C(Ph)CO2Et ligands since they have all been isolated in this work or in a previous report;11e the porphyrin meso-substituents are again replaced by hydrogen atoms for simplicity. The most notable difference between dialkylcarbene Ad and reactive carbenes lies in the σ-properties, as the Fe dz2 orbital in the Ad complex is calculated at a much higher energy level (ΔE ≥ 0.78 eV) together with a relatively large energy splitting between σ and σ* orbitals (Fig. 13b), which is attributable to the electron-donating alkyl substituents. Meanwhile, its Fe dπ orbital is also slightly destabilized compared to that of CPh2 and C(Ph)CO2Et carbene complexes (ΔE = 0.25–0.35 eV). Therefore, dialkylcarbene Ad is a stronger σ-donor and slightly weaker π-acceptor, and thus a more electron-donating ligand than the other reactive carbenes.
These computational results are supported by a number of spectroscopic and experimental data. The electron-donating feature of dialkylcarbene can be demonstrated by electrochemical studies where the EO1 values of Ad complexes of group 8 metal porphyrins are appreciably smaller than those of phenyl-/ester-substituted carbene congeners (Table 6 and Fig. 14);17g additionally, a larger isomer shift in the Mössbauer spectrum of 1a (0.25 mm s−1) than that of the analogous CPh2 (0.03–0.19 mm s−1) and CCl2 complexes (0.02–0.10 mm s−1) also reveals a more electron-rich Fe center in the former complex. The much enhanced σ-bonding interaction improves the trans influence, as revealed by smaller binding constants of 1a and 2b (see the Results section), as well as the thermodynamic stability of dialkylcarbene Ad relative to other reactive carbene complexes, which has been demonstrated by the carbene substitution experiments (Scheme 3). The higher carbene pπ orbital in dialkylcarbene complexes is accountable for the kinetic stability of 1a and 3a since they were observed to be unreactive towards stoichiometric carbene transfer reactions while the analogous CPh2 and/or C(Ph)CO2R carbene complexes have been reported to be reactive.11e,18e
Table 6 First oxidation potentials (EO1, V vs. Fc+/0) of group 8 metal carbene complexes supported by the TPFPP macrocycle
| Complex |
E
O1
(V) |
Ref.b |
|
E
1/2 values of reversible oxidation processes.
TW = this work.
Quasi-reversible.
|
|
Fe-mono(carbene) complexes
|
| [Fe(TPFPP)(Ad)] (1a) |
0.01 |
TW |
| [Fe(TPFPP)(CPh2)] |
0.35 |
17g
|
|
|
Ru-mono(carbene) complexes
|
| [Ru(TPFPP)(Ad)] (2a) |
0.18 |
TW |
| [Ru(TPFPP)(CPh2)] |
0.46 |
17g
|
| [Ru(TPFPP)(C(Ph)CO2Me)(MeOH)] |
0.65 |
17g
|
|
|
Os-bis(carbene) complexes
|
| [Os(TPFPP)(Ad)2] (3b) |
0.04c |
TW |
| [Os(TPFPP)(CPh2)2] |
0.30 |
TW |
Ad vs. other dialkylcarbene ligands
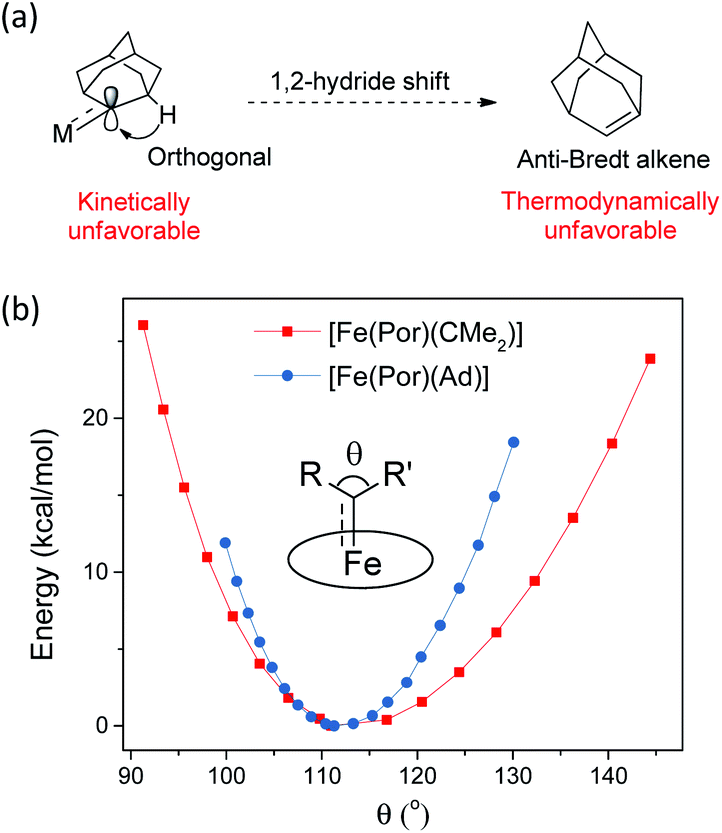
In this work, Ad is used as a model to study the features of dialkylcarbene ligands. To examine the effect of alkyl substituents of dialkylcarbene on the frontier orbitals of the carbene carbon, we performed DFT calculations on two in silico complexes [Fe(Por)(CMe2)] and [Fe(Por)(CiPr2)]; the two calculated complexes show almost identical Fe-carbene bonding structures to the Ad complex (Fig. S8†). However, MAd  complexes (especially the Fe and Ru complexes) are uniquely stable as compared to other examples of Fe/Ru-dialkylcarbene complexes which could undergo a 1,2-hydride/alkyl shift and/or carbene transfer reaction.7e,h,10,19b,20b,50 Such a striking difference is attributable to the rigid structure of Ad. On the one hand, its diamondoid skeleton suppresses the inner-sphere 1,2-hydride shift both thermodynamically and kinetically (Fig. 15a).22c On the other hand, the angle at carbene carbon is relatively fixed in Ad complexes while it is more flexible in other dialkylcarbene such as CMe2, as revealed by DFT calculations (Fig. 15b). Such an angle is closely related to the reactivity of carbene species47 as the energy level of the CMe2 pπ orbital decreases significantly upon increasing this angle (Fig. S9†); in contrast, the Ad complex stays preferentially around the local minimum of its potential energy curve which is apparently steeper than that of the CMe2 complex, thus making it less susceptible to outer-sphere nucleophilic attack.
complexes (especially the Fe and Ru complexes) are uniquely stable as compared to other examples of Fe/Ru-dialkylcarbene complexes which could undergo a 1,2-hydride/alkyl shift and/or carbene transfer reaction.7e,h,10,19b,20b,50 Such a striking difference is attributable to the rigid structure of Ad. On the one hand, its diamondoid skeleton suppresses the inner-sphere 1,2-hydride shift both thermodynamically and kinetically (Fig. 15a).22c On the other hand, the angle at carbene carbon is relatively fixed in Ad complexes while it is more flexible in other dialkylcarbene such as CMe2, as revealed by DFT calculations (Fig. 15b). Such an angle is closely related to the reactivity of carbene species47 as the energy level of the CMe2 pπ orbital decreases significantly upon increasing this angle (Fig. S9†); in contrast, the Ad complex stays preferentially around the local minimum of its potential energy curve which is apparently steeper than that of the CMe2 complex, thus making it less susceptible to outer-sphere nucleophilic attack.
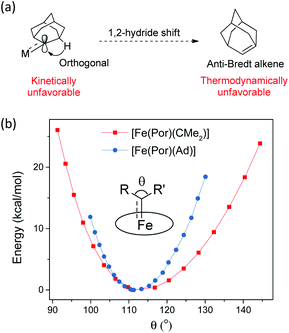 |
| | Fig. 15 (a) Resistance of MAd complexes toward 1,2-hydride shift. (b) Calculated potential energy curves for [Fe(Por)(Ad)] and [Fe(Por)(CMe2)] by changing the angle θ. | |
Ad vs. π-acceptor ligands for Fe(II) porphyrins
Some useful comparisons can also be made among dialkylcarbene Ad and common π-acceptor ligands which are important in stabilizing Fe(II) porphyrins.16b While Ad only forms mono-adducts with the Fe center, bis-adducts of NO,51 CO,52 pyridine,34b and isocyanide53 have all been reported, which implies a stronger ligand field of Ad than other π-acids. Electrochemistry revealed larger EO1 values for CS and NO complexes (≥0.6 V vs. SCE)33,51,54 than the Ad counterparts (0.29–0.50 V vs. SCE) and thus suggested less electron-donating properties of the diatomic ligands; this is consistent with the larger isomer shift of 1a (0.25 mm s−1) in Mössbauer spectroscopy as compared to CS and CO complexes measured at similar temperatures (−0.03 to 0.18 mm s−1).16c
Catalytic diarylcarbene transfer reactivity
Previously, intermolecular diarylcarbene transfer reactions have only been achieved by a limited number of catalytic systems and some of them suffered from a low yield, narrow substrate scope, and/or high catalyst loading.18e,55 The Fe-dialkylcarbene complex 1a is, to the best of our knowledge, the first Fe catalyst that mediates the transfer of diarylcarbene to a panel of substrates with moderate to high yields, while remaining largely intact during turnover. Based on a few reports on the stoichiometric reactivity of Fe-diarylcarbene species with organic substrates,11e,f,h we envision that the plausible intermediates, [Fe(TPFPP)(Ad)(CAr2)], are key to the success in our methodology and thus merit further discussion.
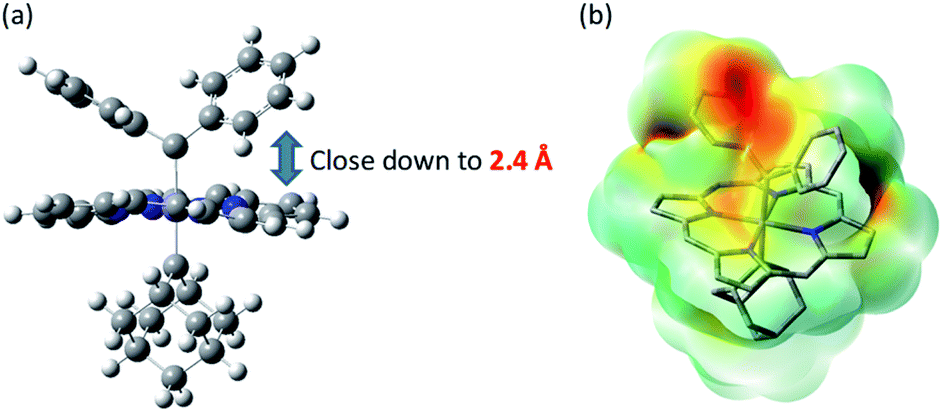
We have not been able to detect the presence of [Fe(TPFPP)(Ad)(CPh2)] species. Nonetheless, according to our calculated structure of 4b (Fig. 12), the CPh2 carbene group is rather labile as indicated by the elongated Fe–CCPh2 bond (1.96 Å), which is also consistent with the high reactivity of 1a involving 4a as a proposed reactive intermediate; meanwhile the Fe–CAd bond is appreciably shorter (1.89 Å) and could account for the stability of the catalyst. However, such a large difference in Fe-carbene distances is in contrast to those found in the corresponding five- or six-coordinate mono(carbene) complexes, i.e., [Fe(Por)(CPh2)] vs. [Fe(Por)(Ad)] or [Fe(Por)(CPh2)(L)] vs. [Fe(Por)(Ad)(L)],11e,16a,c where both pairs of complexes display negligible differences in the Fe–carbene bond distances (<0.02 Å). We attribute this phenomenon to both the trans effect of Ad and the steric effect of CPh2 carbene ligands. The steric repulsion between the bulky CPh2 group and porphyrin ligand has been well documented in the literature, especially in six-coordinate complexes where a strong distortion of the porphyrin ligand has been observed,11e which is distinctly different from the nearly planar porphyrins found in six-coordinate Ad and CCl2 carbene complexes.11a In previous examples with imidazoles as trans-axial ligands, the strong trans effect of CPh2 carbene and the highly distorted porphyrin plane significantly weaken the Fe–imidazole interaction.16c In model complex 4b, however, where the ligand field of Ad is comparable to that of CPh2, the Ad group tends to stay closer to the Fe center and reduces the distortion of porphyrin toward itself; consequently, the steric effect (or Pauli repulsion) between porphyrin and CPh2 is expected to increase, as the distance between them is as short as 2.4 Å (Fig. 16a), which is well within the normal region for π–π interaction,56 and the electrostatic repulsion is also enhanced since both CPh2 and porphyrin carry partial negative charge (Fig. 16b). Elongation of the Fe–CCPh2 bond thus occurs in order to offset these repulsive interactions. It should be noted that such a steric effect is unique to metalloporphyrin systems, since in other common catalysts, such as Rh and Cu catalysts, the coordination environment around CAr2 is much less congested and the aryl ring(s) on CAr2 can even be coplanar with the carbene plane according to their crystal structures.39,40
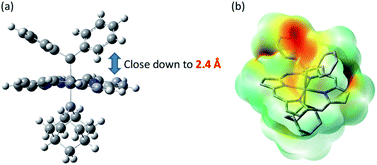 |
| | Fig. 16 (a) Spatial separaton between CPh2 and Por ligands in the optimized structure of 4b. (b) Calculated electrostatic map of 4b. | |
Besides being spatially accessible due to the elongated Fe-carbene distance, CPh2 carbene is also energetically favorable to be transferred to organic substrates in the bis(carbene) intermediate. While the carbene pπ orbitals of CPh2 and Ad ligands in their mono(carbene) porphyrin complexes are both calculated at L + 2 (Fig. 13b), in the bis(carbene) complex 4b the pπ orbital of CPh2 is markedly lowered to the LUMO while that of Ad slightly increases to L + 3 (Fig. 12). This is likely caused by the intrinsic π-characters of the two carbene ligands as the pπ orbital of free Ad lies higher in energy than that of CPh2 (in their singlet states) due to the hyperconjugation effect (Fig. 13a), yet the presence of two π-acceptor ligands on the Fe center decreases the metal-to-carbene backbonding and makes the carbene orbital of CPh2 shift to a lower energy level. Meanwhile, the σ orbital of CPh2 is also strongly destabilized, as it is mainly located at H-17 in the mono(carbene) complex (Fig. 13b), yet in 4b it exists at H-5 and even partially at the HOMO (Fig. S10†). To briefly summarize, the elongated Fe–CCPh2 bond as well as reorganized CPh2 carbene frontier orbitals concomitantly lead to great destabilization of the CPh2 group in the putative bis(carbene) [Fe(Por)(Ad)(CPh2)] intermediate, which could in turn account for the high catalytic activity of 1a in these diarylcarbene transfer reactions.
Conclusions
In this work we have conducted a detailed study on a series of group 8 metal porphyrin dialkylcarbene complexes. With aziadamantane as the carbene source, a new synthetic method is developed toward Fe- and Ru-mono(dialkylcarbene) complexes and also Os-bis(dialkylcarbene) complexes, which, to the best of our knowledge, represent the first examples of transition metal-bis(dialkylcarbene) complexes. Spectroscopic investigations including XANES, NMR, electrochemistry, UV-vis, Mössbauer, resonance Raman, and IR, combined with DFT and TD-DFT calculations, reveal that the electronic structures of these dialkylcarbene complexes are all low-valent d6 metal-carbene MIIAd0 in nature instead of the high-valent Schrock alkylidene formalism. These dialkylcarbene complexes generally exhibit much improved thermodynamic and kinetic stability than other analogous group 8 metal porphyrins bearing nonheteroatom-stabilized carbene ligands such as diphenylcarbene; further comparison with other types of carbene ligands shows that dialkylcarbene Ad is a borderline species in between the common “stable” and “reactive” carbenes and it displays strong σ-donating as well as π-accepting characters. The unique electronic properties and stable features make dialkylcarbene Ad a promising candidate for ligand design in organometallic catalysis, and it has been demonstrated in this work that the Fe(II) complex 1a is highly active and robust in some donor–donor carbene, especially diarylcarbene, transfer reactions where the strong-field trans-Ad ligand is believed to play a crucial role in activating the reactive carbene groups.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the Hong Kong Research Grants Council (HKU 17303815) and Basic Research Program-Shenzhen Fund (JCYJ20170412140251576, JCYJ20170818141858021, and JCYJ20180508162429786). We thank Prof. Ya-Wen Zhang, Mr Haozong Xue, and staff at the Shanghai Synchrotron Radiation Facility (SSRF) for assistance with XANES spectroscopy, Mr Qian Liu from shiyanjia lab (https://www.shiyanjia.com) for support in Mössbauer analysis, and Prof. David L. Phillips, Dr Lili Du, Dr Zhiping Yan, and Ms Xueqin Bai for performing the resonance Raman measurements. We acknowledge helpful discussions with Prof. Michael P. Doyle and Dr Ka-Pan Shing. Assistance in experimental work by Ms Linda Quach and Ms Shuo Xu is also kindly acknowledged.
Notes and references
- R. R. Schrock, J. Am. Chem. Soc., 1974, 96, 6796–6797 CrossRef CAS.
- R. R. Schrock, Chem. Rev., 2002, 102, 145–180 CrossRef CAS PubMed.
-
(a) J. D. Fellmann, G. A. Rupprecht, C. D. Wood and R. R. Schrock, J. Am. Chem. Soc., 1978, 100, 5964–5966 CrossRef CAS;
(b) M. R. Churchill and W. J. Youngs, Inorg. Chem., 1979, 18, 1930–1935 CrossRef CAS;
(c) J. D. Fellmann, R. R. Schrock and G. A. Rupprecht, J. Am. Chem. Soc., 1981, 103, 5752–5758 CrossRef CAS;
(d) A. M. LaPointe, R. R. Schrock and W. M. Davis, J. Am. Chem. Soc., 1995, 117, 4802–4813 CrossRef CAS;
(e) J. B. Diminnie, H. D. Hall and Z. Xue, Chem. Commun., 1996, 2383–2384 RSC;
(f) T. Chen, Z. Wu, L. Li, K. R. Sorasaenee, J. B. Diminnie, H. Pan, I. A. Guzei, A. L. Rheingold and Z. Xue, J. Am. Chem. Soc., 1998, 120, 13519–13520 CrossRef CAS;
(g) T. Chen, X.-H. Zhang, C. Wang, S. Chen, Z. Wu, L. Li, K. R. Sorasaenee, J. B. Diminnie, H. Pan, I. A. Guzei, A. L. Rheingold, Y.-D. Wu and Z.-L. Xue, Organometallics, 2005, 24, 1214–1224 CrossRef CAS;
(h) U. J. Kilgore, J. Tomaszewski, H. Fan, J. C. Huffman and D. J. Mindiola, Organometallics, 2007, 26, 6132–6138 CrossRef CAS;
(i) Z.-L. Xue and L. A. Morton, J. Organomet. Chem., 2011, 696, 3924–3934 CrossRef CAS.
- For a review on Fischer-type multicarbene complexes, see: D. I. Bezuidenhout, S. Lotz, D. C. Liles and B. van der Westhuizen, Coord. Chem. Rev., 2012, 256, 479–524 CrossRef CAS.
- G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS PubMed.
-
(a)
W. Petz, Iron-Carbene Complexes, Springer-Verlag, 1993 CrossRef;
(b) O. Daugulis, A. H. R. MacArthur, F. C. Rix and J. L. Templeton, ACS Catal., 2016, 6, 1518–1532 CrossRef CAS.
-
(a) J. P. Collman, P. J. Brothers, L. McElwee-White, E. Rose and L. J. Wright, J. Am. Chem. Soc., 1985, 107, 4570–4571 CrossRef CAS;
(b) D. G. Gusev, T. Maxwell, F. M. Dolgushin, M. Lyssenko and A. J. Lough, Organometallics, 2002, 21, 1095–1100 CrossRef CAS;
(c) P. Alvarez, E. Lastra, J. Gimeno, M. Bassetti and L. R. Falvello, J. Am. Chem. Soc., 2003, 125, 2386–2387 CrossRef CAS PubMed;
(d) M. Leutzsch, L. M. Wolf, P. Gupta, M. Fuchs, W. Thiel, C. Farès and A. Fürstner, Angew. Chem., Int. Ed., 2015, 54, 12431–12436 CrossRef CAS PubMed;
(e) A. Guthertz, M. Leutzsch, L. M. Wolf, P. Gupta, S. M. Rummelt, R. Goddard, C. Farès, W. Thiel and A. Fürstner, J. Am. Chem. Soc., 2018, 140, 3156–3169 CrossRef CAS PubMed;
(f) A. Fürstner, J. Am. Chem. Soc., 2019, 141, 11–24 CrossRef PubMed;
(g) T. Biberger, C. P. Gordon, M. Leutzsch, S. Peil, A. Guthertz, C. Copéret and A. Fürstner, Angew. Chem., Int. Ed., 2019, 58, 8845–8850 CrossRef CAS PubMed;
(h) S. Peil, A. Guthertz, T. Biberger and A. Fürstner, Angew. Chem., Int. Ed., 2019, 58, 8851–8856 CrossRef CAS PubMed.
-
(a) G. Ferrando, J. N. Coalter III, H. Gérard, D. Huang, O. Eisenstein and K. G. Caulton, New J. Chem., 2003, 27, 1451–1462 RSC;
(b) M. A. Esteruelas, A. M. López and M. Oliván, Coord. Chem. Rev., 2007, 251, 795–840 CrossRef CAS;
(c) V. Martin and S. Blakey, Tetrahedron Lett., 2008, 49, 6800–6803 CrossRef CAS;
(d) T. Bolaño, M. A. Esteruelas and E. Oñate, J. Organomet. Chem., 2011, 696, 3911–3923 CrossRef.
-
(a) M. Brookhart, J. R. Tucker and G. R. Husk, J. Am. Chem. Soc., 1981, 103, 979–981 CrossRef CAS;
(b) K. A. M. Kremer, P. Helquist and R. C. Kerber, J. Am. Chem. Soc., 1981, 103, 1862–1864 CrossRef CAS;
(c) M. Brookhart, J. R. Tucker and G. R. Husk, J. Am. Chem. Soc., 1983, 105, 258–264 CrossRef CAS;
(d) M. Brookhart, D. Timmers, J. R. Tucker, G. D. Williams, G. R. Husk, H. Brunner and B. Hammer, J. Am. Chem. Soc., 1983, 105, 6721–6723 CrossRef CAS;
(e) M. Brookhart, W. B. Studabaker and G. R. Husk, Organometallics, 1985, 4, 943–944 CrossRef CAS;
(f) M. Brookhart, W. B. Studabaker and G. R. Husk, Organometallics, 1987, 6, 1141–1145 CrossRef CAS;
(g) M. Brookhart, Y. Liu, E. W. Goldman, D. A. Timmers and G. D. Williams, J. Am. Chem. Soc., 1991, 113, 927–939 CrossRef CAS;
(h) E. Scharrer and M. Brookhart, J. Organomet. Chem., 1995, 497, 61–71 CrossRef CAS;
(i) S. Ishii, S. Zhao and P. Helquist, J. Am. Chem. Soc., 2000, 122, 5897–5898 CrossRef CAS.
-
(a) K. A. M. Kremer, G.-H. Kuo, E. J. O'Connor, P. Helquist and R. C. Kerber, J. Am. Chem. Soc., 1982, 104, 6119–6121 CrossRef CAS;
(b) C. P. Casey, W. H. Miles, H. Tukada and J. M. O'Connor, J. Am. Chem. Soc., 1982, 104, 3761–3762 CrossRef CAS;
(c) C. P. Casey, W. H. Miles and H. Tukada, J. Am. Chem. Soc., 1985, 107, 2924–2931 CrossRef CAS.
-
(a) D. Mansuy, M. Lange, J. C. Chottard, J. F. Bartoli, B. Chevrier and R. Weiss, Angew. Chem., Int. Ed., 1978, 17, 781–782 CrossRef;
(b) D. Mansuy, Pure Appl. Chem., 1980, 52, 681–690 CAS;
(c) A. Klose, E. Solari, C. Floriani, N. Re, A. Chiesi-Villa and C. Rizzoli, Chem. Commun., 1997, 2297–2298 RSC;
(d) V. Esposito, E. Solari, C. Floriani, N. Re, C. Rizzoli and A. Chiesi-Villa, Inorg. Chem., 2000, 39, 2604–2613 CrossRef CAS PubMed;
(e) Y. Li, J.-S. Huang, Z.-Y. Zhou, C.-M. Che and X.-Z. You, J. Am. Chem. Soc., 2002, 124, 13185–13193 CrossRef CAS PubMed;
(f) S. K. Russell, J. M. Hoyt, S. C. Bart, C. Milsmann, S. C. E. Stieber, S. P. Semproni, S. DeBeer and P. J. Chirik, Chem. Sci., 2014, 5, 1168–1174 RSC;
(g) B. M. Lindley, A. Swidan, E. B. Lobkovsky, P. T. Wolczanski, M. Adelhardt, J. Sutter and K. Meyer, Chem. Sci., 2015, 6, 4730–4736 RSC;
(h) J. Liu, L. Hu, L. Wang, H. Chen and L. Deng, J. Am. Chem. Soc., 2017, 139, 3876–3888 CrossRef CAS PubMed;
(i) J. F. DeJesus and D. M. Jenkins, Chem.–Eur. J., 2019 DOI:10.1002/chem.201905360.
-
(a) H. Renata, Z. J. Wang and F. H. Arnold, Angew. Chem., Int. Ed., 2015, 54, 3351–3367 CrossRef CAS PubMed;
(b) R. D. Lewis, M. Garcia-Borras, M. J. Chalkley, A. R. Buller, K. N. Houk, S. B. J. Kan and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7308–7313 CrossRef CAS PubMed;
(c) T. Hayashi, M. Tinzl, T. Mori, U. Krengel, J. Proppe, J. Soetbeer, D. Klose, G. Jeschke, M. Reiher and D. Hilvert, Nat. Catal., 2018, 1, 578–584 CrossRef CAS.
-
(a) A. R. Reddy, C.-Y. Zhou, Z. Guo, J. Wei and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 14175–14180 CrossRef CAS PubMed;
(b) A. R. Reddy, F. Hao, K. Wu, C.-Y. Zhou and C.-M. Che, Angew. Chem., Int. Ed., 2016, 55, 1810–1815 CrossRef CAS PubMed;
(c) H. Wang, C.-Y. Zhou and C.-M. Che, Adv. Synth. Catal., 2017, 359, 2253–2258 CrossRef CAS;
(d) F. Hao, A. R. Reddy, C.-Y. Zhou and C.-M. Che, Adv. Synth. Catal., 2018, 360, 1433–1438 CrossRef CAS.
-
(a) G. Frenking and N. Fröhlich, Chem. Rev., 2000, 100, 717–774 CrossRef CAS PubMed;
(b)
Y. Jean, Molecular Orbitals of Transition Metal Complexes, OUP Oxford, 2005 Search PubMed;
(c)
J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science BooksSausalito, CA, 2010 Search PubMed;
(d) D. J. Mindiola and J. Scott, Nat. Chem., 2011, 3, 15–17 CrossRef CAS PubMed.
- J.-i. Setsune, Y. Ishimaru and A. Sera, J. Chem. Soc. Chem. Commun., 1992, 328–329 RSC.
-
(a) Y. Liu, W. Xu, J. Zhang, W. Fuller, C. E. Schulz and J. Li, J. Am. Chem. Soc., 2017, 139, 5023–5026 CrossRef CAS PubMed;
(b) Q. Peng, J. T. Sage, Y. Liu, Z. Wang, M. Y. Hu, J. Zhao, E. E. Alp, W. R. Scheidt and J. Li, Inorg. Chem., 2018, 57, 8788–8795 CrossRef CAS PubMed;
(c) H. Wang, C. E. Schulz, X. Wei and J. Li, Inorg. Chem., 2019, 58, 143–151 CrossRef CAS PubMed.
-
(a) N. Rajapakse, B. R. James and D. Dolphin, Can. J. Chem., 1990, 68, 2274–2277 CrossRef CAS;
(b) J. P. Collman, E. Rose and G. D. Venburg, J. Chem. Soc. Chem. Commun., 1993, 934–935 RSC;
(c) E. Galardon, P. L. Maux, L. Toupet and G. Simonneaux, Organometallics, 1998, 17, 565–569 CrossRef CAS;
(d) C.-M. Che, J.-S. Huang, F.-W. Lee, Y. Li, T.-S. Lai, H.-L. Kwong, P.-F. Teng, W.-S. Lee, W.-C. Lo, S.-M. Peng and Z.-Y. Zhou, J. Am. Chem. Soc., 2001, 123, 4119–4129 CrossRef CAS PubMed;
(e) C.-M. Che and J.-S. Huang, Coord. Chem. Rev., 2002, 231, 151–164 CrossRef CAS;
(f) T. Harada, S. Wada, H. Yuge and T. K. Miyamoto, Acta Crystallogr., Sect. C: Struct. Chem., 2003, 59, m37–m39 CrossRef PubMed;
(g) Y. Li, J.-S. Huang, G.-B. Xu, N. Zhu, Z.-Y. Zhou, C.-M. Che and K.-Y. Wong, Chem.–Eur. J., 2004, 10, 3486–3502 CrossRef CAS PubMed;
(h) C.-M. Che, C.-M. Ho and J.-S. Huang, Coord. Chem. Rev., 2007, 251, 2145–2166 CrossRef CAS;
(i) P. Le Maux, T. Roisnel, I. Nicolas and G. Simonneaux, Organometallics, 2008, 27, 3037–3042 CrossRef CAS;
(j) K. Hirasawa, H. Yuge and T. K. Miyamoto, Acta Crystallogr., Sect. C: Struct. Chem., 2008, 64, m97–m100 CrossRef CAS PubMed;
(k) Q.-H. Deng, J. Chen, J.-S. Huang, S. S.-Y. Chui, N. Zhu, G.-Y. Li and C.-M. Che, Chem.–Eur. J., 2009, 15, 10707–10712 CrossRef CAS PubMed;
(l) H.-X. Wang, Q. Wan, K. Wu, K.-H. Low, C. Yang, C.-Y. Zhou, J.-S. Huang and C.-M. Che, J. Am. Chem. Soc., 2019, 141, 9027–9046 CrossRef CAS PubMed.
-
(a) L. K. Woo and D. A. Smith, Organometallics, 1992, 11, 2344–2346 CrossRef CAS;
(b) D. A. Smith, D. N. Reynolds and L. K. Woo, J. Am. Chem. Soc., 1993, 115, 2511–2513 CrossRef CAS;
(c) J.-P. Djukic, D. A. Smith, V. G. Young Jr and L. K. Woo, Organometallics, 1994, 13, 3020–3026 CrossRef CAS;
(d) C. G. Hamaker, J.-P. Djukic, D. A. Smith and L. K. Woo, Organometallics, 2001, 20, 5189–5199 CrossRef CAS;
(e) Y. Li, J.-S. Huang, Z.-Y. Zhou and C.-M. Che, J. Am. Chem. Soc., 2001, 123, 4843–4844 CrossRef CAS PubMed;
(f) Y. Li, J.-S. Huang, Z.-Y. Zhou and C.-M. Che, Chem. Commun., 2003, 1362–1363 RSC;
(g) K. Sawano, H. Yuge and T. K. Miyamoto, Inorg. Chim. Acta, 2005, 358, 1830–1834 CrossRef CAS.
-
(a) Z. Wu, S. T. Nguyen, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1995, 117, 5503–5511 CrossRef CAS;
(b) V. F. Kuznetsov, K. Abdur-Rashid, A. J. Lough and D. G. Gusev, J. Am. Chem. Soc., 2006, 128, 14388–14396 CrossRef CAS PubMed.
-
(a) M. L. Buil, M. A. Esteruelas, C. García-Yebra, E. Gutiérrez-Puebla and M. Oliván, Organometallics, 2000, 19, 2184–2193 CrossRef CAS;
(b) D. G. Gusev and A. J. Lough, Organometallics, 2002, 21, 2601–2603 CrossRef CAS;
(c) R. Castro-Rodrigo, M. A. Esteruelas, S. Fuertes, A. M. López, F. López, J. L. Mascareñas, S. Mozo, E. Oñate, L. Saya and L. Villarino, J. Am. Chem. Soc., 2009, 131, 15572–15573 CrossRef CAS PubMed;
(d) R. Castro-Rodrigo, M. A. Esteruelas, A. M. López, F. López, J. L. Mascareñas, S. Mozo, E. Oñate and L. Saya, Organometallics, 2010, 29, 2372–2376 CrossRef CAS.
- V. Mahias, S. Cron, L. Toupet and C. Lapinte, Organometallics, 1996, 15, 5399–5408 CrossRef CAS.
-
(a) R. A. Moss and M. J. Chang, Tetrahedron Lett., 1981, 22, 3749–3752 CrossRef CAS;
(b) T. Bally, S. Matzinger, L. Truttmann, M. S. Platz and S. Morgan, Angew. Chem., Int. Ed., 1994, 33, 1964–1966 CrossRef;
(c) G. V. Shustov and M. T. H. Liu, Can. J. Chem., 1998, 76, 851–861 CrossRef CAS;
(d) W. Knoll, D. Kaneno, M. M. Bobek, L. Brecker, M. G. Rosenberg, S. Tomoda and U. H. Brinker, J. Org. Chem., 2012, 77, 1340–1360 CrossRef CAS PubMed;
(e) V. C. Rojisha, K. Nijesh, S. De and P. Parameswaran, Chem. Commun., 2013, 49, 8465–8467 RSC;
(f) R. A. Moss, L. Wang and K. Krogh-Jespersen, J. Am. Chem. Soc., 2014, 136, 4885–4888 CrossRef CAS PubMed.
- P. A. Chaloner, G. D. Glick and R. A. Moss, J. Chem. Soc. Chem. Commun., 1983, 880–881 RSC.
- J. Bauer, H. Braunschweig, A. Damme, J. O. C. Jimenez-Halla, T. Kramer, K. Radacki, R. Shang, E. Siedler and Q. Ye, J. Am. Chem. Soc., 2013, 135, 8726–8734 CrossRef CAS PubMed.
- V. B. Shur, I. A. Tikhonova, G. G. Aleksandrov, Y. T. Struchkov, M. E. Vol'pin, E. Schmitz and K. Jähnisch, Inorg. Chim. Acta, 1980, 44, L275–L277 CrossRef.
- Z. Mo and L. Deng, Coord. Chem. Rev., 2017, 350, 285–299 CrossRef CAS.
-
(a) A. Albini and H. Kisch, J. Organomet. Chem., 1975, 94, 75–85 CrossRef CAS;
(b) R. Battaglia, H. Matthäus and H. Kisch, J. Organomet. Chem., 1980, 193, 57–67 CrossRef CAS.
- Q. Ye, I. V. Komarov, A. J. Kirby and M. Jones Jr, J. Org. Chem., 2002, 67, 9288–9294 CrossRef CAS PubMed.
-
A. B. P. Lever and H. B. Gray, Iron Porphyrins, Addison Wesley Publishing Company, 1983 Search PubMed.
-
(a) T. G. Spiro and J. M. Burke, J. Am. Chem. Soc., 1976, 98, 5482–5489 CrossRef CAS PubMed;
(b) G. Chottard and D. Mansuy, J. Chem. Soc. Chem. Commun., 1980, 279–280 RSC;
(c) G. Chottard, P. Battioni, J.-P. Battioni, M. Lange and D. Mansuy, Inorg. Chem., 1981, 20, 1718–1722 CrossRef CAS.
-
(a) J.-S. Huang, C.-M. Che and C.-K. Poon, J. Chem. Soc. Chem. Commun., 1992, 161–163 RSC;
(b) J.-L. Liang, J.-S. Huang, Z.-Y. Zhou, K.-K. Cheung and C.-M. Che, Chem.–Eur. J., 2001, 7, 2306–2317 CrossRef CAS PubMed.
- A. Antipas, J. W. Buchler, M. Gouterman and P. D. Smith, J. Am. Chem. Soc., 1978, 100, 3015–3024 CrossRef CAS.
- D. Lancon and K. M. Kadish, J. Am. Chem. Soc., 1983, 105, 5610–5617 CrossRef CAS.
-
(a) K. M. Kadish, E. Van Caemelbecke, F. D'Souza, M. Lin, D. J. Nurco, C. J. Medforth, T. P. Forsyth, B. Krattinger, K. M. Smith, S. Fukuzumi, I. Nakanishi and J. A. Shelnutt, Inorg. Chem., 1999, 38, 2188–2198 CrossRef CAS PubMed;
(b) K. T. Moore, J. T. Fletcher and M. J. Therien, J. Am. Chem. Soc., 1999, 121, 5196–5209 CrossRef CAS.
-
(a) G. M. Brown, F. R. Hopf, J. A. Ferguson, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1973, 95, 5939–5942 CrossRef CAS PubMed;
(b) T. Malinski, D. Chang, L. A. Bottomley and K. M. Kadish, Inorg. Chem., 1982, 21, 4248–4253 CrossRef CAS;
(c) I.-K. Choi, Y. Liu, D. Feng, K.-J. Paeng and M. D. Ryan, Inorg. Chem., 1991, 30, 1832–1839 CrossRef CAS.
-
(a) G. M. Brown, F. R. Hopf, T. J. Meyer and D. G. Whitten, J. Am. Chem. Soc., 1975, 97, 5385–5390 CrossRef CAS PubMed;
(b) J. P. Collman, J. T. McDevitt, C. R. Leidner, G. T. Yee, J. B. Torrance and W. A. Little, J. Am. Chem. Soc., 1987, 109, 4606–4614 CrossRef CAS.
- R. L. Khade, W. Fan, Y. Ling, L. Yang, E. Oldfield and Y. Zhang, Angew. Chem., Int. Ed., 2014, 53, 7574–7578 CrossRef CAS PubMed.
- C. J. Ziegler and K. S. Suslick, J. Am. Chem. Soc., 1996, 118, 5306–5307 CrossRef CAS.
-
(a) C. Werlé, R. Goddard and A. Fürstner, Angew. Chem., Int. Ed., 2015, 54, 15452–15456 CrossRef PubMed;
(b) C. Werlé, R. Goddard, P. Philipps, C. Farès and A. Fürstner, J. Am. Chem. Soc., 2016, 138, 3797–3805 CrossRef PubMed.
-
(a) X. Dai and T. H. Warren, J. Am. Chem. Soc., 2004, 126, 10085–10094 CrossRef CAS PubMed;
(b) P. Hofmann, I. V. Shishkov and F. Rominger, Inorg. Chem., 2008, 47, 11755–11762 CrossRef CAS PubMed.
-
(a) C. Soldi, K. N. Lamb, R. A. Squitieri, M. González-López, M. J. Di Maso and J. T. Shaw, J. Am. Chem. Soc., 2014, 136, 15142–15145 CrossRef CAS PubMed;
(b) D. Zhu, J. Ma, K. Luo, H. Fu, L. Zhang and S. Zhu, Chem. Commun., 2016, 55, 8452–8456 CAS;
(c) L. W. Souza, R. A. Squitieri, C. A. Dimirjian, B. M. Hodur, L. A. Nickerson, C. N. Penrod, J. Cordova, J. C. Fettinger and J. T. Shaw, Angew. Chem., Int. Ed., 2018, 57, 15213–15216 CrossRef CAS PubMed;
(d) D. Zhu, L. Chen, H. Zhang, Z. Ma, H. Jiang and S. Zhu, Angew. Chem., Int. Ed., 2018, 57, 12405–12409 CrossRef CAS PubMed.
-
(a) D. A. Sharon, D. Mallick, B. Wang and S. Shaik, J. Am. Chem. Soc., 2016, 138, 9597–9610 CrossRef CAS PubMed;
(b) Y. Wei, A. Tinoco, V. Steck, R. Fasan and Y. Zhang, J. Am. Chem. Soc., 2018, 140, 1649–1662 CrossRef CAS PubMed.
-
(a) J. R. Denton, D. Sukumaran and H. M. L. Davies, Org. Lett., 2007, 9, 2625–2628 CrossRef CAS PubMed;
(b) P. Rullière, G. Benoit, E. M. D. Allouche and A. B. Charette, Angew. Chem., Int. Ed., 2018, 57, 5777–5782 CrossRef PubMed;
(c) E. M. D. Allouche and A. B. Charette, Chem. Sci., 2019, 10, 3802–3806 RSC.
- G.-Q. Chen, Z.-J. Xu, C.-Y. Zhou and C.-M. Che, Chem. Commun., 2011, 47, 10963–10965 RSC.
-
(a) P. Q. Le and J. A. May, J. Am. Chem. Soc., 2015, 137, 12219–12222 CrossRef CAS PubMed;
(b) Y. Wang, X. Wen, X. Cui and X. P. Zhang, J. Am. Chem. Soc., 2018, 140, 4792–4796 CrossRef CAS PubMed;
(c) J. Werth and C. Uyeda, Angew. Chem., Int. Ed., 2018, 57, 13902–13906 CrossRef CAS PubMed;
(d) M. Lankelma, A. M. Olivares and B. de Bruin, Chem.–Eur. J., 2019, 25, 5658–5663 CrossRef CAS PubMed;
(e) S. Yu, A. Noble, R. B. Bedford and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 20325–20334 CrossRef CAS PubMed.
- D. Munz, Organometallics, 2018, 37, 275–289 CrossRef CAS.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- C. M. Weinstein, G. P. Junor, D. R. Tolentino, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 9255–9260 CrossRef CAS PubMed.
-
(a) C. Samojłowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708–3742 CrossRef PubMed;
(b) T. Hashimoto, S. Urban, R. Hoshino, Y. Ohki, K. Tatsumi and F. Glorius, Organometallics, 2012, 31, 4474–4479 CrossRef CAS;
(c) P. Stefan and W. Robert, Z. Anorg. Allg. Chem., 2013, 639, 2581–2585 CrossRef;
(d) K.-H. Chan, X. Guan, V. K.-Y. Lo and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 2982–2987 CrossRef CAS PubMed;
(e) P. P. Samuel, K. C. Mondal, N. A. Sk, H. W. Roesky, E. Carl, R. Neufeld, D. Stalke, S. Demeshko, F. Meyer, L. Ungur, L. F. Chibotaru, J. Christian, V. Ramachandran, J. van Tol and N. S. Dalal, J. Am. Chem. Soc., 2014, 136, 11964–11971 CrossRef CAS PubMed;
(f) G. Ung, J. Rittle, M. Soleilhavoup, G. Bertrand and J. C. Peters, Angew. Chem., Int. Ed., 2014, 53, 8427–8431 CrossRef CAS PubMed;
(g) G. Ung and J. C. Peters, Angew. Chem., Int. Ed., 2015, 54, 532–535 CAS;
(h) V. M. Marx, A. H. Sullivan, M. Melaimi, S. C. Virgil, B. K. Keitz, D. S. Weinberger, G. Bertrand and R. H. Grubbs, Angew. Chem., Int. Ed., 2015, 54, 1919–1923 CrossRef CAS PubMed;
(i) L. Wang, L. Hu, H. Zhang, H. Chen and L. Deng, J. Am. Chem. Soc., 2015, 137, 14196–14207 CrossRef CAS PubMed;
(j) K.-P. Shing, Y. Liu, B. Cao, X.-Y. Chang, T. You and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 11947–11951 CrossRef CAS PubMed.
- G. Dazinger and K. Kirchner, Organometallics, 2004, 23, 6281–6287 CrossRef CAS.
- L. W. Olson, D. Schaeper, D. Lancon and K. M. Kadish, J. Am. Chem. Soc., 1982, 104, 2042–2044 CrossRef CAS.
- B. B. Wayland, L. F. Mehne and J. Swartz, J. Am. Chem. Soc., 1978, 100, 2379–2383 CrossRef CAS.
- G. B. Jameson and J. A. Ibers, Inorg. Chem., 1979, 18, 1200–1208 CrossRef CAS.
- L. A. Bottomley, M. R. Deakin and J.-N. Gorce, Inorg. Chem., 1984, 23, 3563–3571 CrossRef CAS.
-
(a) W. Baratta, W. A. Herrmann, R. M. Kratzer and P. Rigo, Organometallics, 2000, 19, 3664–3669 CrossRef CAS;
(b) H. Werner, M. E. Schneider, M. Bosch, J. Wolf, J. H. Teuben, A. Meetsma and S. I. Troyanov, Chem.–Eur. J., 2000, 6, 3052–3059 CrossRef CAS PubMed;
(c) S. Priya, M. S. Balakrishna, S. M. Mobin and R. McDonald, J. Organomet. Chem., 2003, 688, 227–235 CrossRef CAS;
(d) A. Hamze, B. Tréguier, J.-D. Brion and M. Alami, Org. Biomol. Chem., 2011, 9, 6200–6204 RSC;
(e) H. Liu, Y. Wei and C. Cai, New J. Chem., 2016, 40, 674–678 RSC;
(f) Z. Liu, Q. Li, Y. Yang and X. Bi, Chem. Commun., 2017, 53, 2503–2506 RSC.
- R. M. Parrish and C. D. Sherrill, J. Am. Chem. Soc., 2014, 136, 17386–17389 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Abbreviations, experimental procedures, characterization of compounds, computational details, Tables S1–S8, Fig. S1–S11, NMR spectra of compounds, Cartesian coordinates from DFT calculations, and CIF file for the crystal structure of 1b, 1a·Py, 2b·MeOH, 2az, and 3a. CCDC: 1854823, 1854830, 1943318, 1943319, and 1943321. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05432d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Qingyun
Wan
a,
Qingyun
Wan
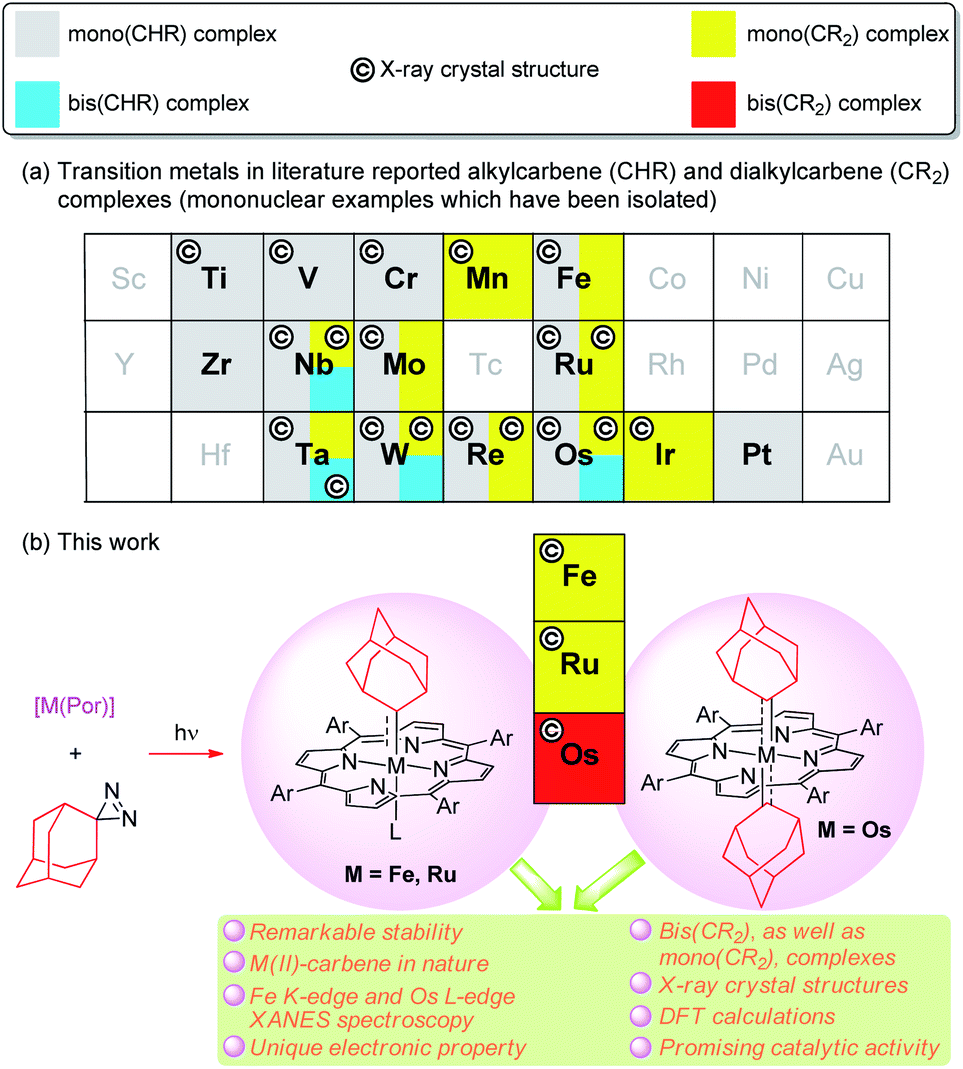
 ).
).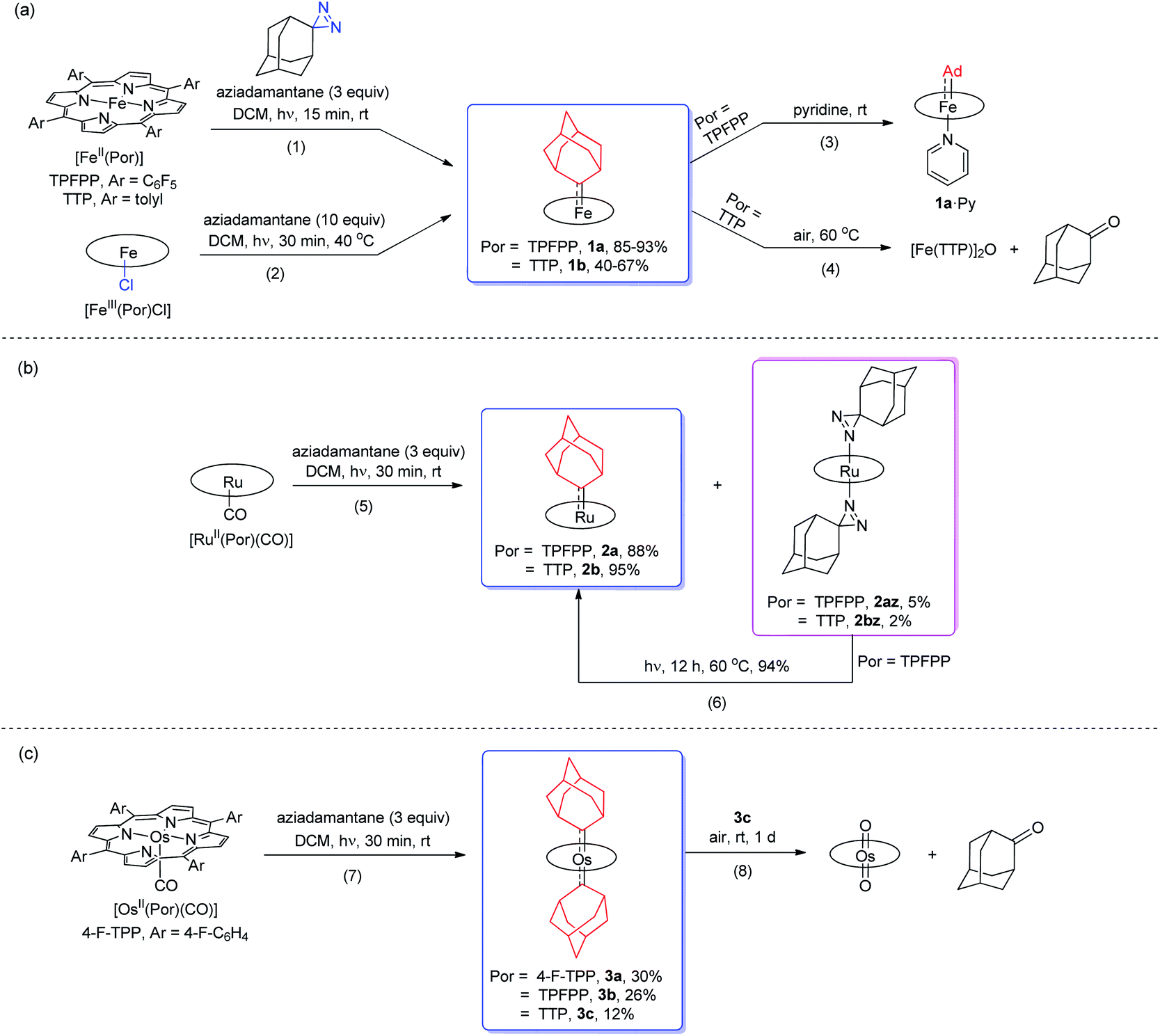
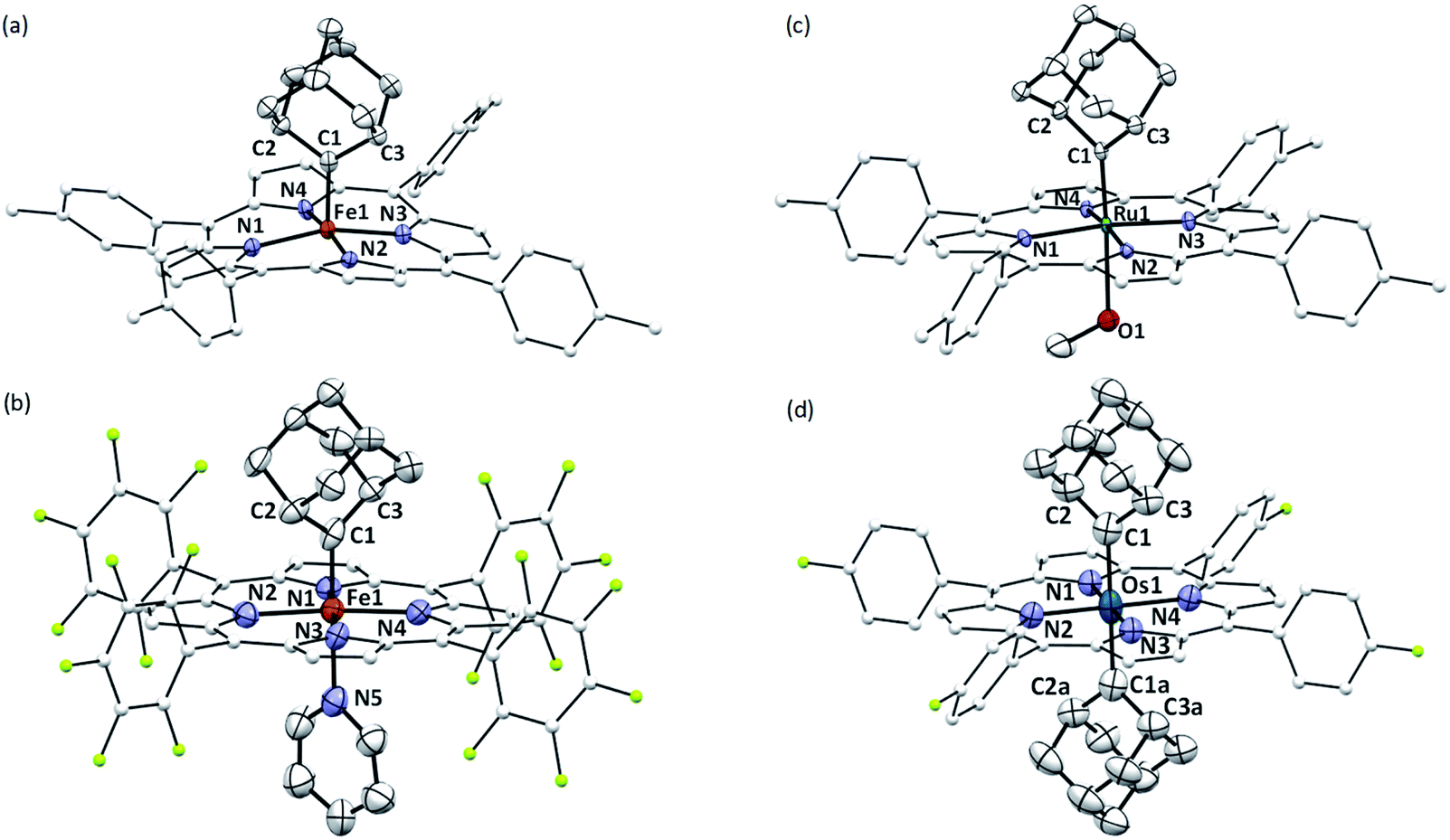
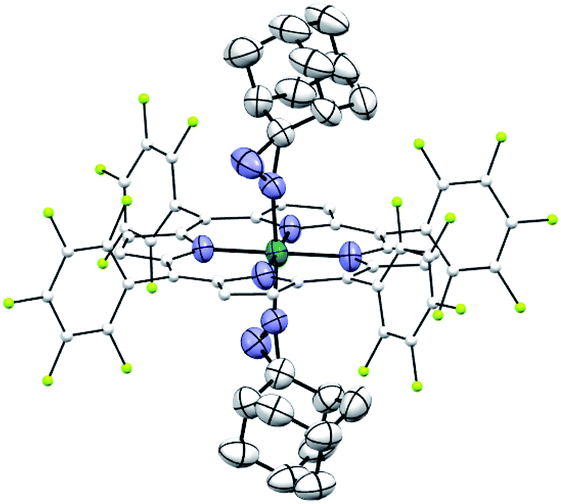
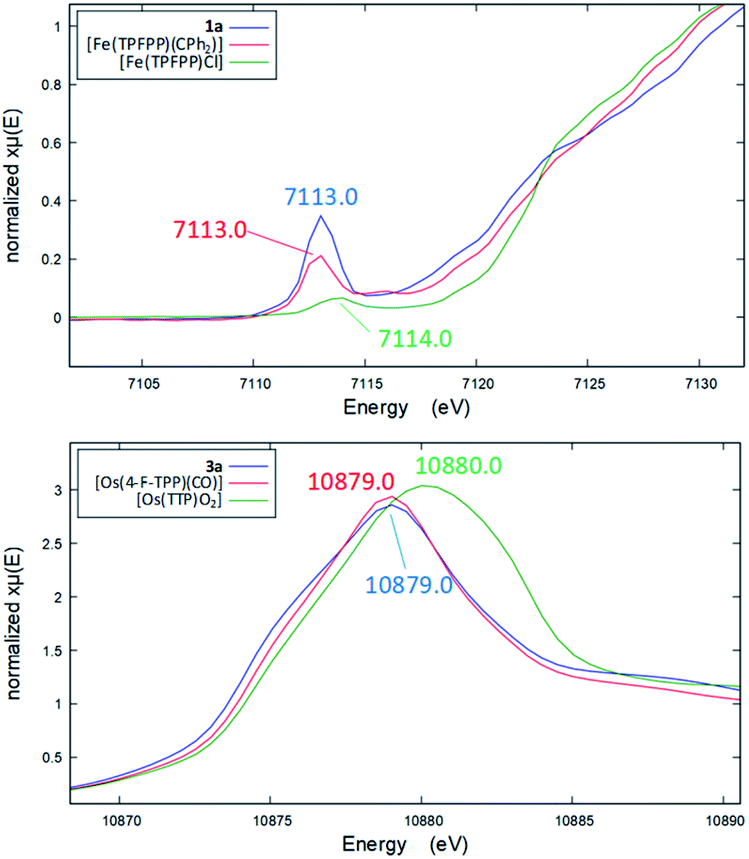
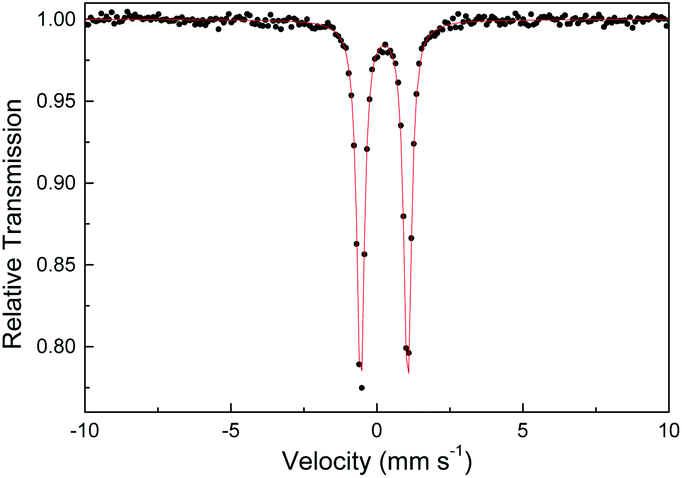
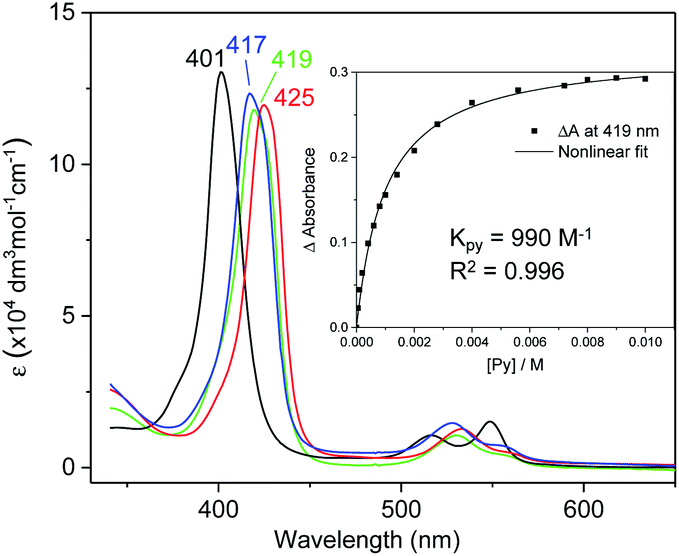
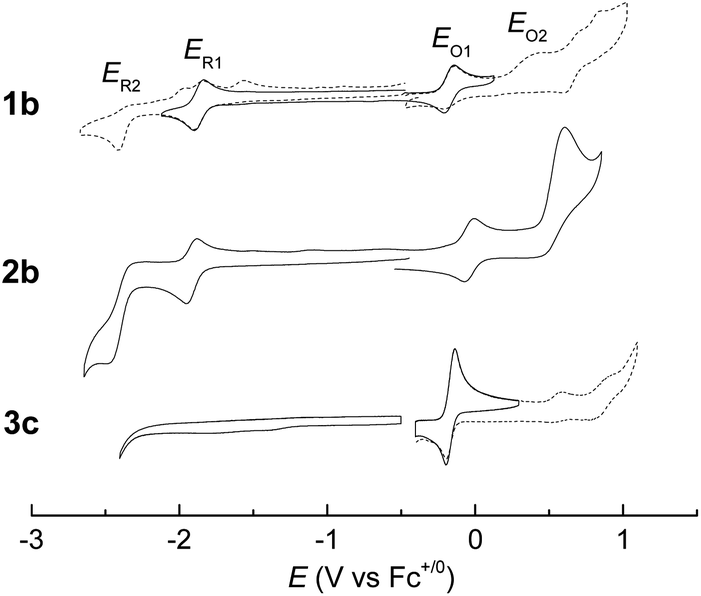

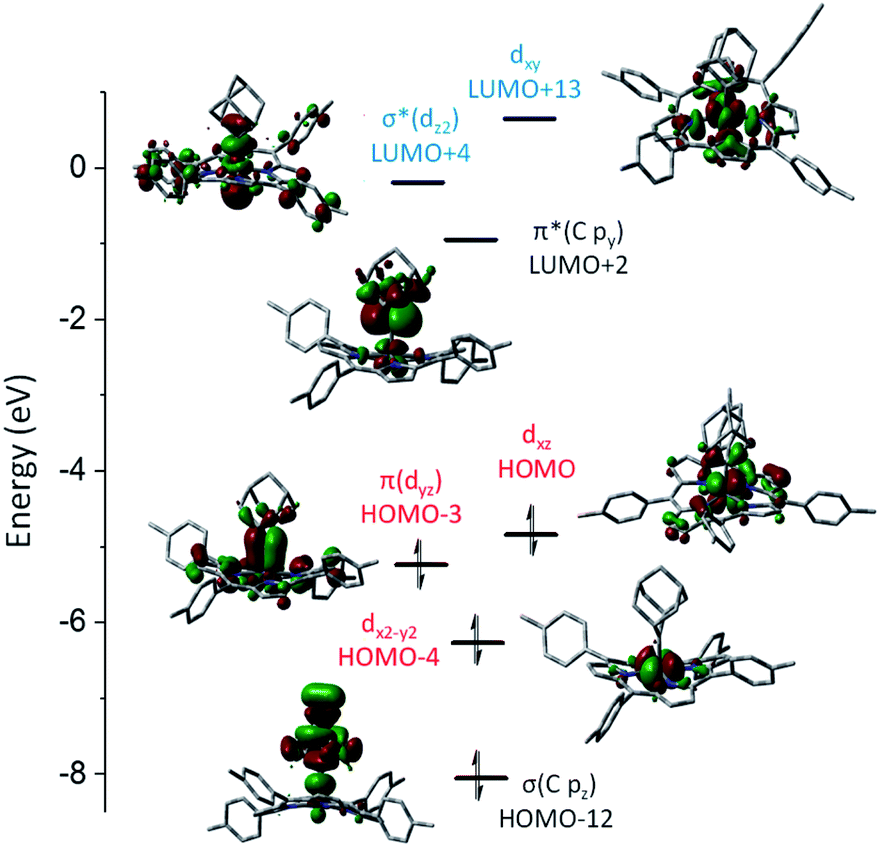
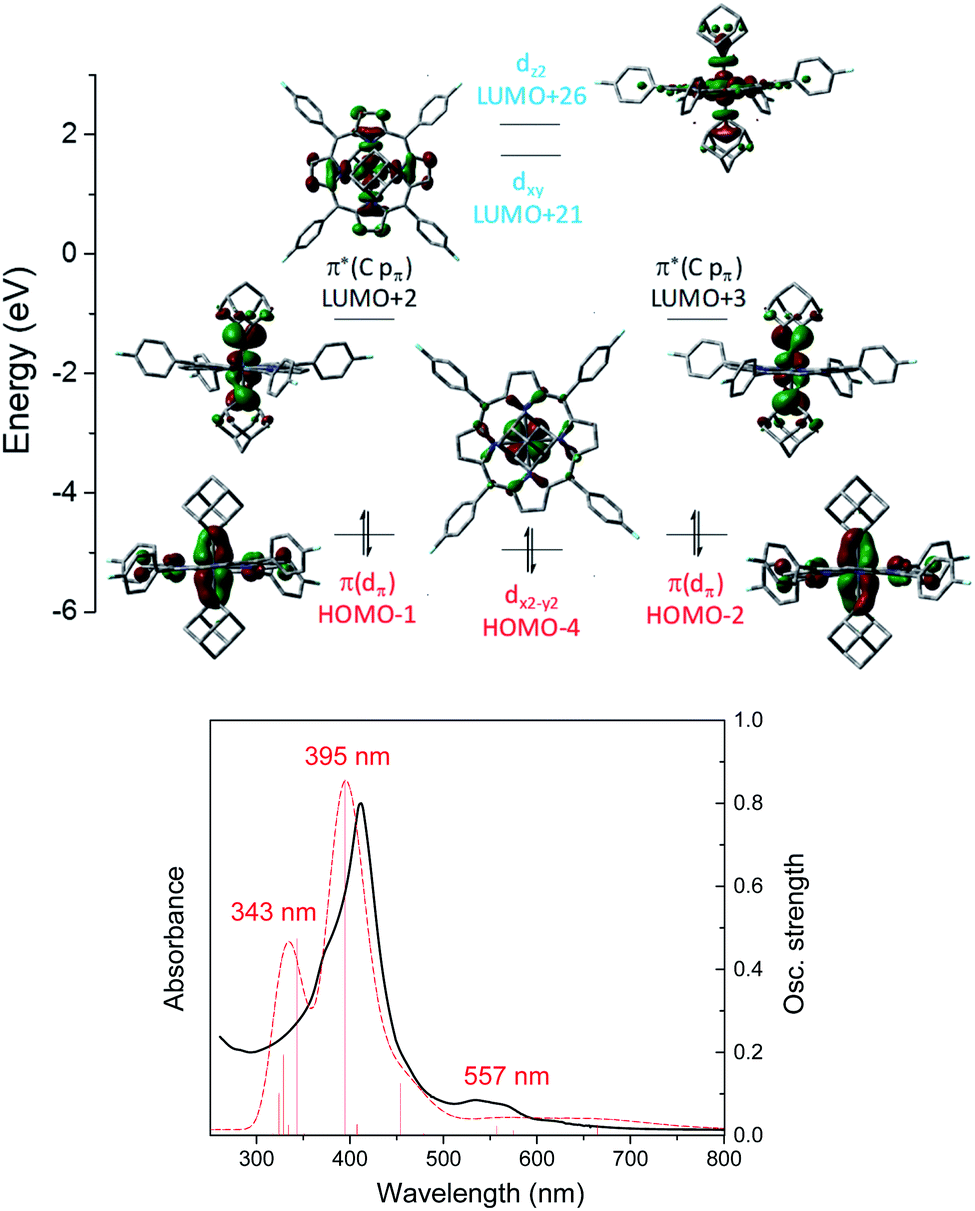
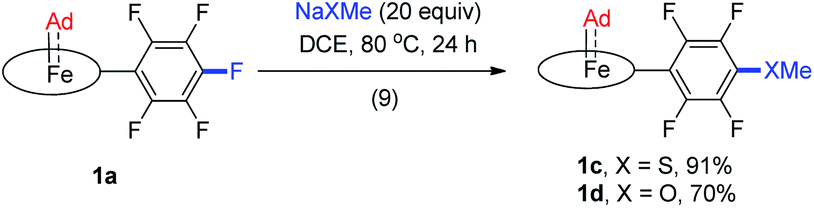
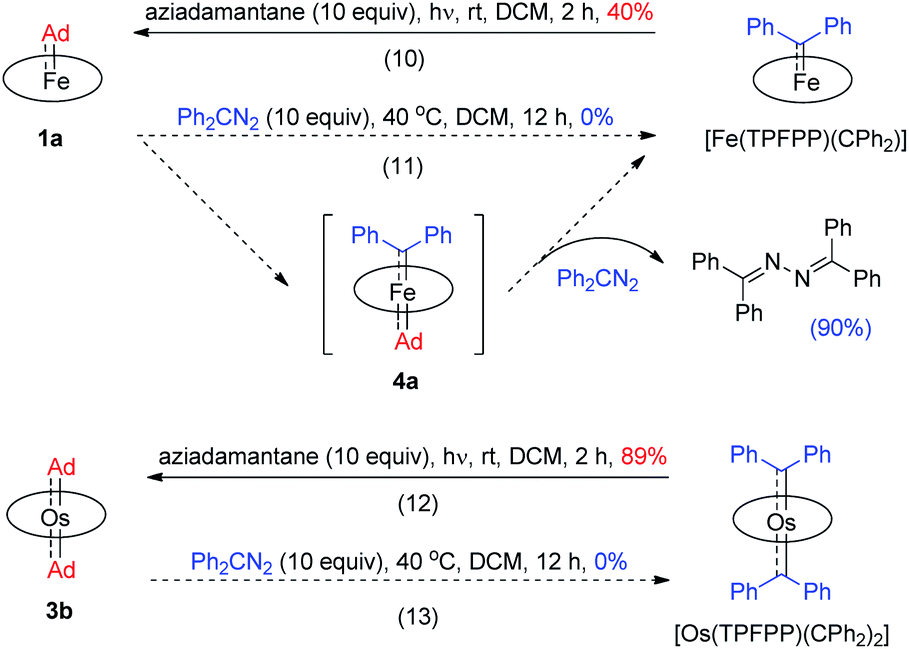
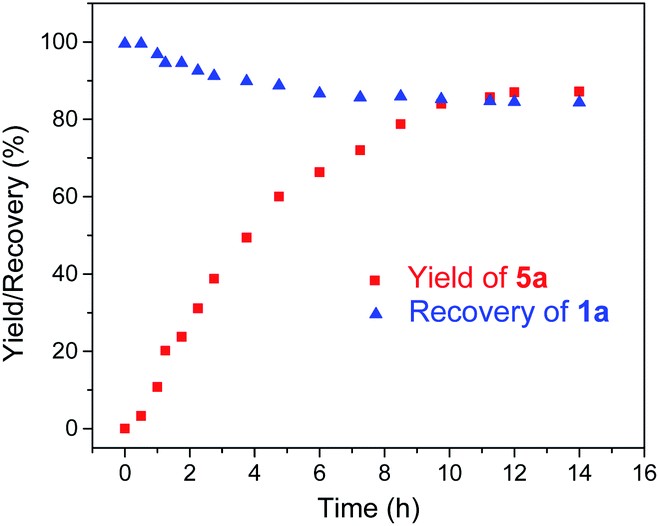
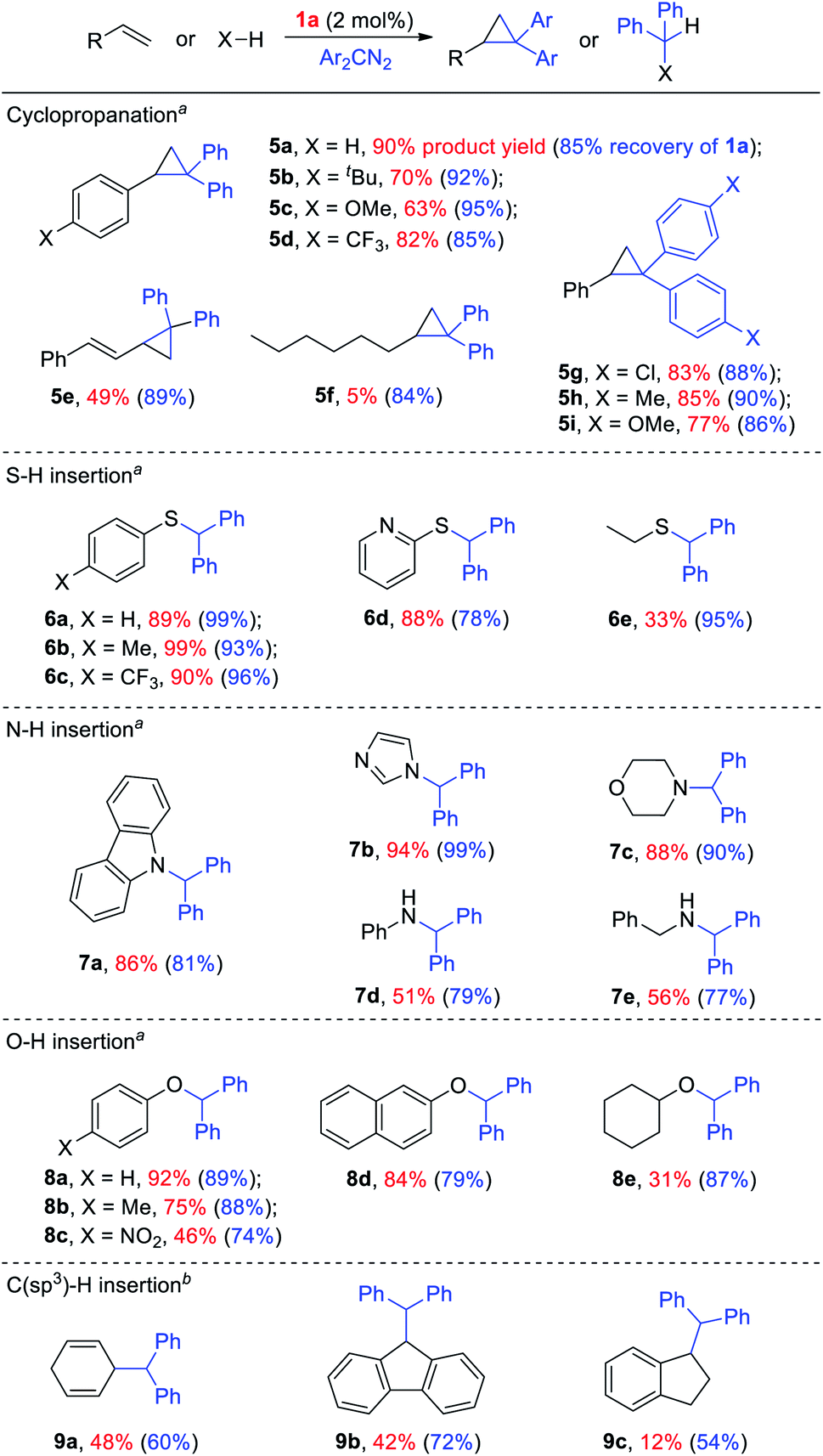
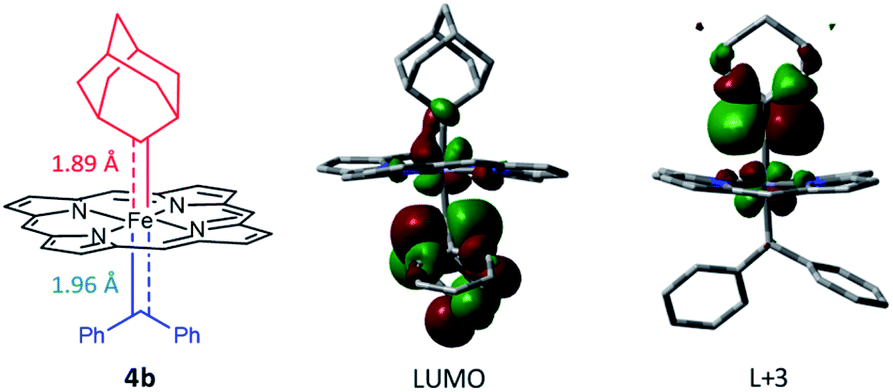
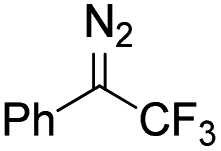
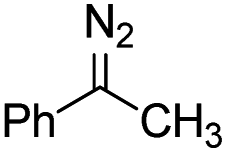
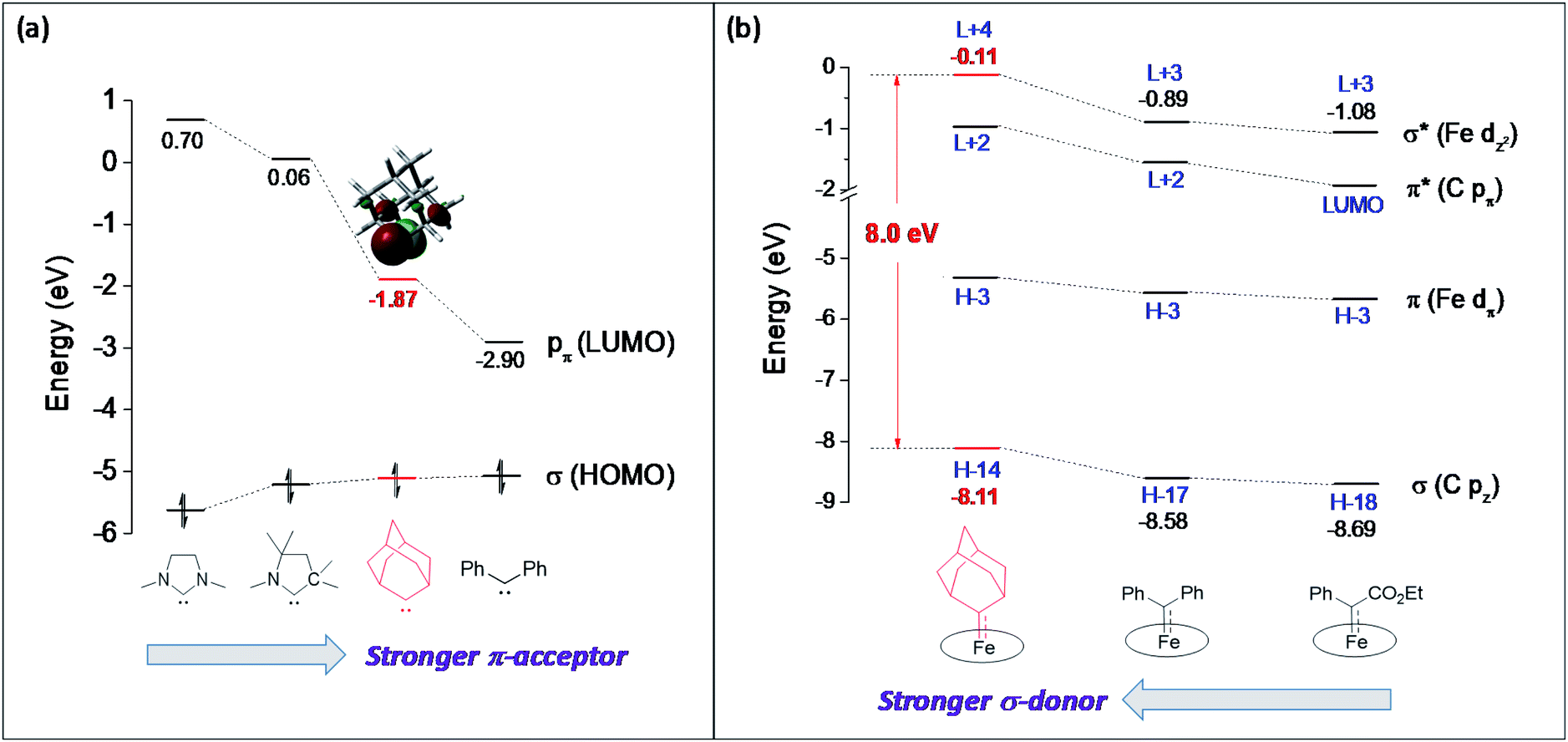
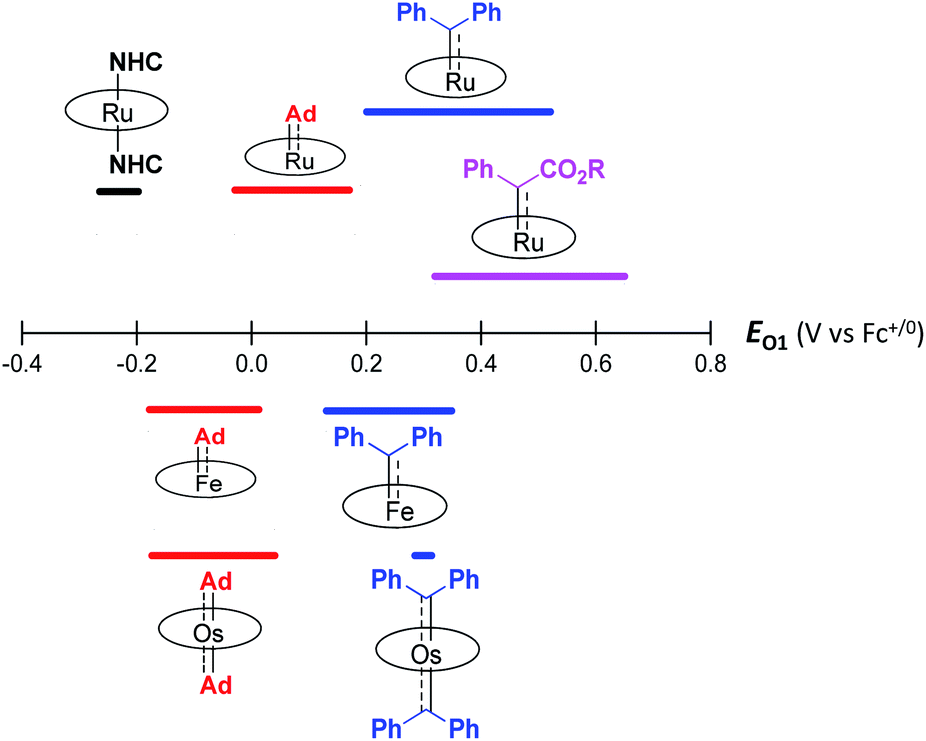
 complexes (especially the Fe and Ru complexes) are uniquely stable as compared to other examples of Fe/Ru-dialkylcarbene complexes which could undergo a 1,2-hydride/alkyl shift and/or carbene transfer reaction.7e,h,10,19b,20b,50 Such a striking difference is attributable to the rigid structure of Ad. On the one hand, its diamondoid skeleton suppresses the inner-sphere 1,2-hydride shift both thermodynamically and kinetically (Fig. 15a).22c On the other hand, the angle at carbene carbon is relatively fixed in Ad complexes while it is more flexible in other dialkylcarbene such as CMe2, as revealed by DFT calculations (Fig. 15b). Such an angle is closely related to the reactivity of carbene species47 as the energy level of the CMe2 pπ orbital decreases significantly upon increasing this angle (Fig. S9†); in contrast, the Ad complex stays preferentially around the local minimum of its potential energy curve which is apparently steeper than that of the CMe2 complex, thus making it less susceptible to outer-sphere nucleophilic attack.
complexes (especially the Fe and Ru complexes) are uniquely stable as compared to other examples of Fe/Ru-dialkylcarbene complexes which could undergo a 1,2-hydride/alkyl shift and/or carbene transfer reaction.7e,h,10,19b,20b,50 Such a striking difference is attributable to the rigid structure of Ad. On the one hand, its diamondoid skeleton suppresses the inner-sphere 1,2-hydride shift both thermodynamically and kinetically (Fig. 15a).22c On the other hand, the angle at carbene carbon is relatively fixed in Ad complexes while it is more flexible in other dialkylcarbene such as CMe2, as revealed by DFT calculations (Fig. 15b). Such an angle is closely related to the reactivity of carbene species47 as the energy level of the CMe2 pπ orbital decreases significantly upon increasing this angle (Fig. S9†); in contrast, the Ad complex stays preferentially around the local minimum of its potential energy curve which is apparently steeper than that of the CMe2 complex, thus making it less susceptible to outer-sphere nucleophilic attack.