
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMerging polyacenes and cationic helicenes: from weak to intense chiroptical properties in the far red region†
Romain
Duwald
a,
Johann
Bosson
 a,
Simon
Pascal
a,
Stéphane
Grass
a,
Francesco
Zinna
ab,
Céline
Besnard
c,
Lorenzo
Di Bari
b,
Denis
Jacquemin
*d and
Jérôme
Lacour
*a
a,
Simon
Pascal
a,
Stéphane
Grass
a,
Francesco
Zinna
ab,
Céline
Besnard
c,
Lorenzo
Di Bari
b,
Denis
Jacquemin
*d and
Jérôme
Lacour
*a
aDepartment of Organic Chemistry, University of Geneva, Quai Ernest Ansermet 30, 1211 Geneva 4, Switzerland. E-mail: jerome.lacour@unige.ch
bDipartimento di Chimica e Chimica Industriale, Università di Pisa, Via Moruzzi 13, 56124 Pisa, Italy
cLaboratoire de Cristallographie, University of Geneva, Quai Ernest Ansermet 24, 1211 Geneva 4, Switzerland
dCEISAM, UMR CNRS 6230, Université de Nantes, 2 rue de la Houssinière, 44322 Nantes, France. E-mail: Denis.Jacquemin@univ-nantes.fr
First published on 5th December 2019
Abstract
A series of helical tetracenes and pentacenes was synthesized from cationic [6] and [4]helicene precursors. These colorful acenes fluoresce in the far red region. While [4]helicene-based pentacenes exhibit chiroptical properties mainly in the UV region, [6]helicene-derived tetracenes show enhanced ECD in the visible range, in addition to clear CPL responses. This difference is rationalized using first principles.
Polyacenes, linearly fused polycyclic aromatic hydrocarbons or PAHs, are strongly studied for their electronic properties.1 Thanks to their narrow HOMO–LUMO gap, these compounds are efficient charge transport materials,2 with potential applications in semi-conductors, thin-film and light emitting devices.1c,e,3 Their heterocyclic analogs, and more specifically aza heteroacenes,1e,f,4 show improved resistance to oxidative degradation5 while maintaining their exceptional electronic behavior. Alternatively, helicenes, that are ortho fused polyaromatic systems and inherently chiral molecules, can be used.6 These entities are involved in various applications spanning from physical chemistry to biochemistry and catalysis.7 Helicenes generally possess optical properties in the blue part of the visible light spectrum, although both push–pull systems8 and cationic helicenes9 see their optical properties shifted toward the red domain. In addition, the chiroptical properties of helicenes have been intensively scrutinized,10 including their ability to emit circularly polarized light.10,11 Herein, the privileged polyacene and helicene motifs are merged into single frameworks to provide polyaza helical acenes 1–6 (Fig. 1).12 These derivatives, prepared by late-stage introduction of N-phenylquinolone motifs at internal or terminal extremities of cationic [4] or [6]helicenes 7 and 8 (Scheme 1, 3–5 steps),13 are colorful dyes emitting in the far red region.14 Tetracenes 4–6 exhibit strong electronic circular dichroism (ECD) and circularly polarized luminescence (CPL) properties in the whole visible domain while pentacenes 1–3 mainly display ECD in the UV spectral range, an outcome rationalized using ab initio calculations.
 | ||
| Fig. 1 Helical pentacenes 1–3 and tetracenes 4–6. M enantiomers shown. | ||
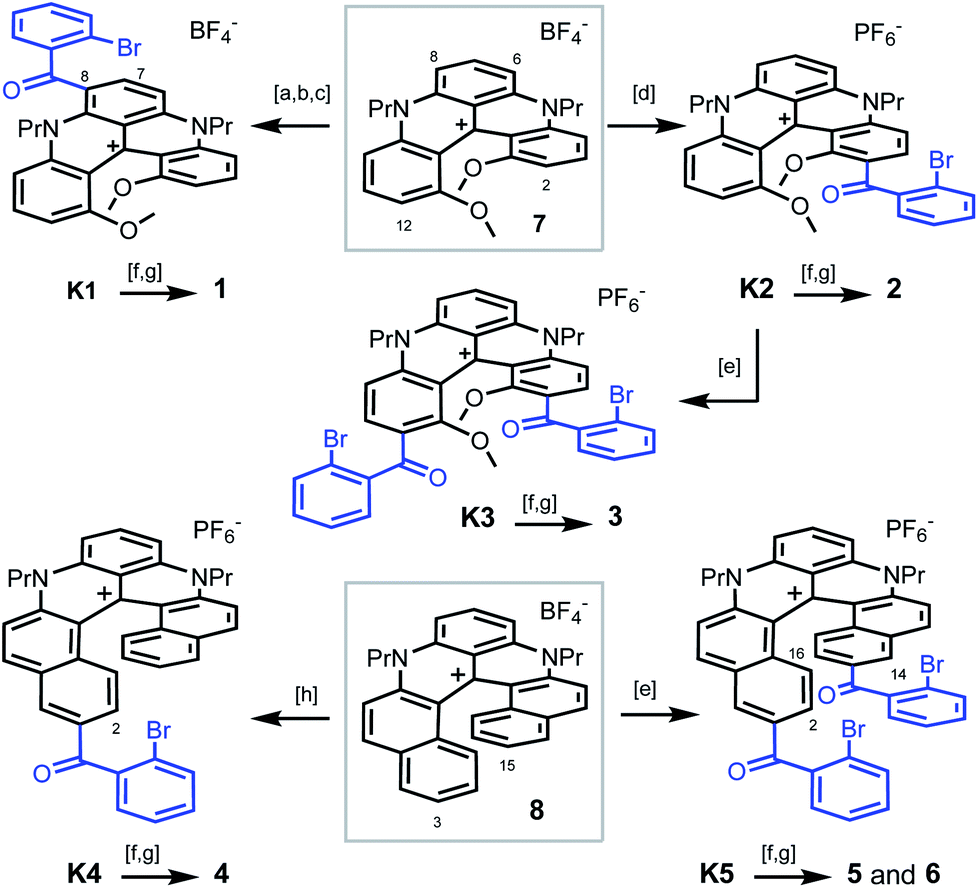 | ||
| Scheme 1 Synthesis of pentacenes 1–3 from [4]helicene 7 (top) and tetracenes 4–6 from [6]helicene 8 (bottom): [a] POCl3, DMF, 90 °C; [b] [(2-bromophenyl)magnesium bromide·LiCl],19 CH2Cl2, 0 °C; [c] PCC, CH2Cl2, 40 °C; [d] 2-bromobenzoic acid, Eaton's reagent, 60 °C; [e] 2-bromobenzoic acid, TfOH, P2O5, 50 or 80 °C; [f] PhNH2, Pd(OAc)2, rac-BINAP, Cs2CO3, DMF, 90 or 100 °C; [g] CuI, 2,2′-bipyridine, DMF or NMP, air, 90 or 130 °C; [h] 2-bromobenzoic acid, PPA, 100 °C. 1 is isolated as a BF4− salt, 2–6 as PF6− salts. | ||
To prepare polyaza acenes 1–6, recent developments on the late-stage functionalization of cationic [4] or [6]helicenes were used.9a,15 With 7 as substrate, three regioselective routes to pentacenes 1–3 were developed (Scheme 1, top). Under mild conditions,9a ketone K1 was obtained in three steps via a selective formylation of 7 at position 6 or 8 (46% overall yield). In strongly acidic conditions, benzoylations of 7 occurred at positions 2 and/or 12 instead.15 With an excess of 2-bromobenzoic acid in Eaton's reagent (MsOH, P2O5),16 ketone K2 was obtained in 93% yield. From K2, in even more acidic conditions (TfOH, P2O5), a second regioselective benzoylation was possible to afford K3 (40%). Then, with bromide K1–K3 in hand, final aza ring closures were achieved by sequences of Pd-catalyzed Buchwald–Hartwig reactions (introduction of NPh groups in place the bromine atoms),17 and Cu-catalyzed oxidative (air) C–N bond formations closing the acridone ring and thus completing the acene extension.18 Starting from 7, pentacenes 1, 2 and 3 were thus afforded in 24%, 41% and 13% yields in 5-, 3- and 4-step procedures respectively.
With [6]helicene 8 as substrate, tetracenes 4–6 were prepared using similar procedures and conditions to that above described. They are summarized at the bottom of Scheme 1.15 Depending on the acidic medium used,20 intermediates K4 and K5 were prepared in single steps. They were then engaged directly in the Buchwald–Hartwig/oxidative ring closure sequence. Using K4, after the Pd-catalyzed Br → NPh exchange, the aza ring closure occurred under oxidative coupling conditions at position 2 preferentially to yield 4 (combined yield 15% from 8). From K5, two regioisomers were isolated after the second oxidative acridone ring closure, C1-symmetric 5 (C-2 and C-14 reactivity) along with C2-symmetric regioisomer 6 (C-2 and C-16 reactivity). Products 5 and 6 were isolated in 10% and 9% combined yields from 8, respectively. In this series, and of importance to explain both the lower yields and some of the later properties, let us underline that the phenylaniline substituents are primarily introduced in the sterically encumbered interior and not on the outer rim of the helicene as in compounds 1 to 3.
Helical polyacenes 1–6 were bench and moisture stable and the BF4− or PF6− salts could be characterized as any other organic material (see the ESI†). Solid-state structures of acenes 2, 4, and 6 were also studied by X-ray diffraction analyses (Fig. 2). Key structural features like the dihedral angles and helical pitches are provided in Table 1 and in the ESI.† Unsurprisingly, the presence of aromatic phenyl groups on the inner rim of the helicenes (inside the fjord region) strongly increases the helicity from 2 to 4 and then 6. Nevertheless, there is enough space inside the groove to accommodate a quasi-perpendicular arrangement of the extra Ph ring(s). The angles between the mean planes of the pyridinone and phenyl motifs are 75° for 4, and 79° and 86° for Ph groups of 6 respectively. With this amplification and based on precedents,21 larger chiroptical responses were foreseen for the later derivatives. The helical pitch determined by DFT (Density Functional Theory) in solution are very similar (vide infra).
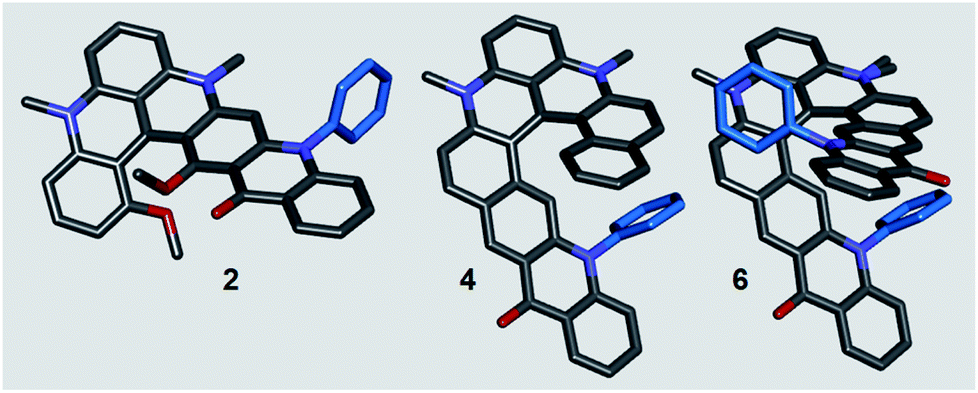 | ||
| Fig. 2 Stick view of the crystal structures of 2, 4 and 6. For clarity reasons, hydrogen atoms, propyl side chains and counterions are omitted or truncated. N-phenyl groups are drawn in blue. For 6, due to disorder, two side chain residues appear on one N-atom, see ESI.† | ||
| Helical acene | Dihedral angle (°) | Helical pitch (Å) |
|---|---|---|
| a Dihedral angles defining the fjord region are common descriptors for [4] and [6]helicene scaffolds (see ESI). The helical pitch is the distance between the first two overlapping atoms of the helicene. | ||
| 2 | 46.8 | 2.66 |
| 4 | 44.7 | 3.09 |
| 6 | 50.8 | 3.28 |
As mentioned, helicenes 1–6 are colorful dyes and efficient fluorophores. Their optical properties were investigated in acetonitrile (ca. 10−5 M, see the ESI†). In the [4]helicene series (1–3, Fig. 3 top), strong modifications of the absorption spectra were observed in the visible range as compared to parent 7 that presents a lowest energy transition at 616 nm with a shoulder at higher energy.9a Pentacene 1 exhibits a lowest energy transition peaking at 576 nm. This blue-shift, compared to 7, is inherent to the electron-withdrawing ketone functional group at the C-6/C-8 position. Conversely, pentacene 2 is red-shifted compared to 1 with a λmax of absorption at 620 nm, and three maxima of similar intensities appear in the 500–600 nm range. Further bathochromic shift is observed for the bis pentacene 3 that presents a broad absorption between 500 and 700 nm with a λmax at 572 nm. Overall, relative to parent 7, the lowest energy transitions of 2 and 3 are hardly shifted. The same trend is observed in fluorescence with blue and red-shifts of the emissions for 1, and 2–3 compared to parent [4]helicene 7. The fluorescence quantum yield of 1–3 is similar to that of 7 (ϕ = 0.15, 0.09 and 0.04 vs. 0.13 for 1, 2, 3 and 7 respectively). In contrast, the elongation of the naphthalene part of [6]helicene 8 has only a marginal influence on the optical properties (Fig. 3, bottom).9b In fact, for 4 and 6, the lowest energy transitions are centered around 614–616 nm with a shoulder at higher energies and present a moderate hypochromic shift compared to parent 8. The absorption of 5 is very slightly red-shifted to 625 nm. In terms of fluorescence, the emissions are centered at 653, and 656 nm for 4 and 6, and at 658 nm for parent 8. Again a small bathochromic shift is observed for 5 that fluoresces at 663 nm. The fluorescence quantum yields in acetonitrile (0.29, 0.25 and 0.21 for 4, 5 and 6) are similar to that of 8 (ϕ = 0.31).9b
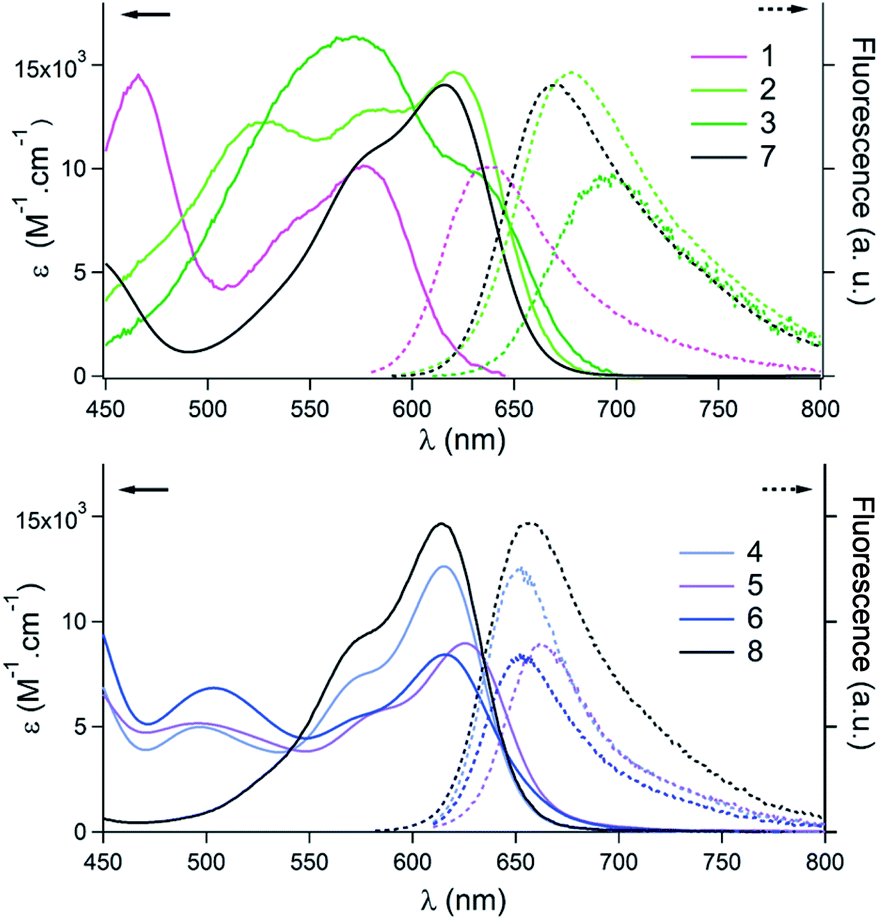 | ||
| Fig. 3 Absorption (plain lines) and emission (dashed lines) spectra in acetonitrile. Top: pentacenes 1–3 and comparison to [4]helicene 7. Bottom: tetracenes 4–6 and comparison to [6]helicene 8. | ||
Regarding the chiroptical properties,22 striking differences are noted, this time, in favor of [6]helicene-derived 4–6 over pentacenes 1–3. For all compounds, single enantiomers were obtained by mean of chiral stationary phase (CSP) HPLC (see Fig. S1–S12†). Noticeably, the ECD spectra of pentacenes 1 and 2 exhibit only very weak Cotton effects in the visible range (Fig. 4, top). Bis pentacene 3 presented a more intense response but only for the absorption band in the 520–600 nm region (Δε = + or −15 M−1 cm−1 at 574 nm) and not for the lowest-energy peak.23 For derivatives 4–6, Cotton effects were observed for the lowest energy transitions around 620 nm (Fig. 4, middle). Interestingly, an increase is noted from mono 4 to “distorted” 5 and finally to 6 (Δε = + or −7, 10 and 18 M−1 cm−1 for 4, 5 and 6, respectively).23 The circularly polarized luminescence (CPL) of helicenes 1–6 was also recorded in acetonitrile solutions.9b,c,11,24 For derivatives 1–3, the glum values were below the detection limit and are not displayed. In sharp contrast, helicenes 4–6 were effective CPL emitters (Fig. 4, bottom). While mono tetracene 4 presented at the fluorescence wavelength only an anisotropy factor glum of 1 × 10−4, non-symmetrical 5 exhibited a glum value of 6 × 10−4 at 650 nm. Finally, bis tetracene 6 was characterized by a glum value of 1.4 × 10−3 at 640 nm. This trend of the glum values of 4–6 is similar to that of the gabs values for the most red-shifted Cotton effects. This is an indication that major geometry variations do not occur between ground and emitting excited states; first principles calculations will confirm this observation (vide infra).25 With derivative 6, CPL was also measured in the solid state. For that purpose, samples were dispersed at a 0.5% weight level in methyl polymethylmethacrylate (PMMA) and the mixture was deposed on a quartz plate affording a glum value of 8 × 10−4 at 640 nm.26 In view of the marked contrast between the (chir)optical properties of pentacenes 1–3 on one side and 4–6 on the other, care was taken to gain a larger understanding of the dichotomic behavior through first principles analysis.
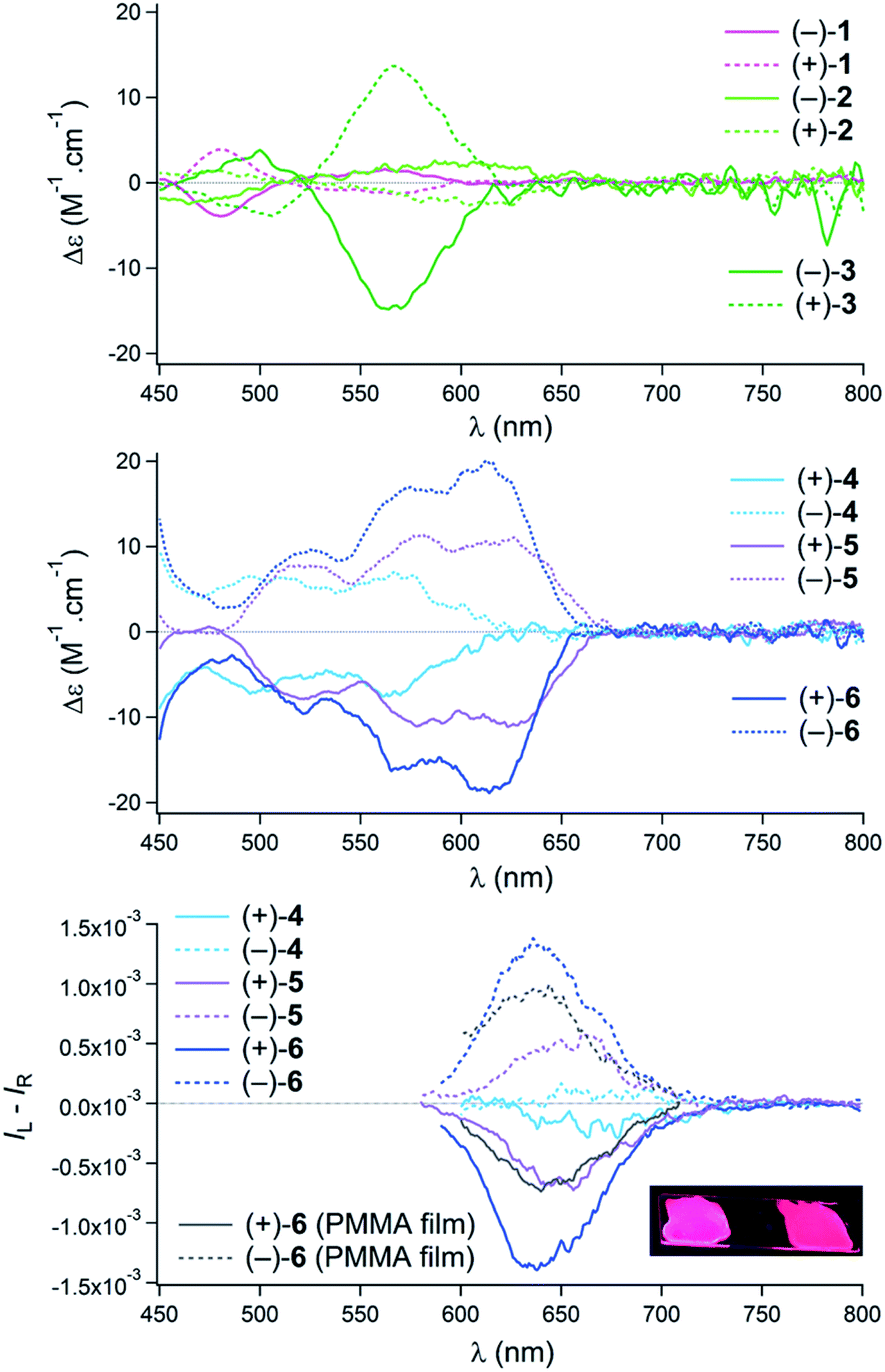 | ||
| Fig. 4 Top: ECD spectra in acetonitrile for 1 eluted (plain lines) and 2nd eluted (dashed lines) enantiomers of pentacenes 1–3. Middle: ECD spectra in acetonitrile for 1 eluted (plain lines) and 2nd eluted (dashed lines) enantiomers of tetracenes 4–6. Bottom: CPL of 4–6 in acetonitrile and 6 in PMMA films for 1 eluted (plain lines) and 2nd eluted (dashed lines) enantiomers; insert: picture of the films under UV light irradiation. | ||
The details of the applied theoretical protocol are given in the ESI.† The helical pitches determined by DFT in solution are 2.69, 2.61, 2.59, 3.18, 3.15, and 3.20 Å for 1, 2, 3, 4, 5, and 6, respectively. In the instance of compounds 2, 4 and 6, featuring experimental XRD values (Table 1), an excellent agreement is noticed, suggesting modest changes in solution. Clearly, helical pitches are significantly larger in the tetracene 4–6 series than in pentacenes 1–3. In the lowest excited-states, pitches remain very similar: 2.71, 2.63, 2.62, 3.27, 3.09, and 3.34 Å for 1, 2, 3, 4, 5, and 6, respectively, confirming the rigidity of the systems, which is consistent with the rather moderate Stokes shift noted experimentally (Fig. 3).
The vertical transition wavelengths determined with TD (Time-Dependent)-DFT are 459, 499, 503, 507, 512, 507, 491, and 504 nm for 1, 2, 3, 4, 5, 6, 7, and 8 respectively. These values are obviously blue-shifted compared to the measurements (Fig. 3), which is explainable by the nature of the considered systems and the neglect of vibronic couplings (see the ESI† for discussion, and calculations with higher levels of theory). Nevertheless, the ranking of the TD-DFT and experimental values perfectly agree, with almost no shift in the tetracene series but for a very small red-shift of 5, and more sizeable effects in the pentacene series with a large blue-shift of 1 as compared to the other members of the series. In Fig. 5, we show density difference plots corresponding to the lowest excited state (S1) for four selected dyes (see the ESI† for other compounds). One finds the typical pattern for such derivative,9b,c with alternating lobes of density gain and depletion, the central carbon atom (formally positively charged) acting has a strong acceptor (in red) and the “top” phenyl ring as an electron donor (mostly in blue). In 1, the pattern of the state is globally preserved (with limited delocalization on the added moiety), but the presence of the carbonyl group renders the donating character of the top phenyl weaker, hence explaining the blue-shift. Nevertheless, one notes overall that the delocalization of the excited state over the added phenyl rings is very limited in all cases, hence the nature of this lowest excited state is mostly preserved in the full series of compounds.
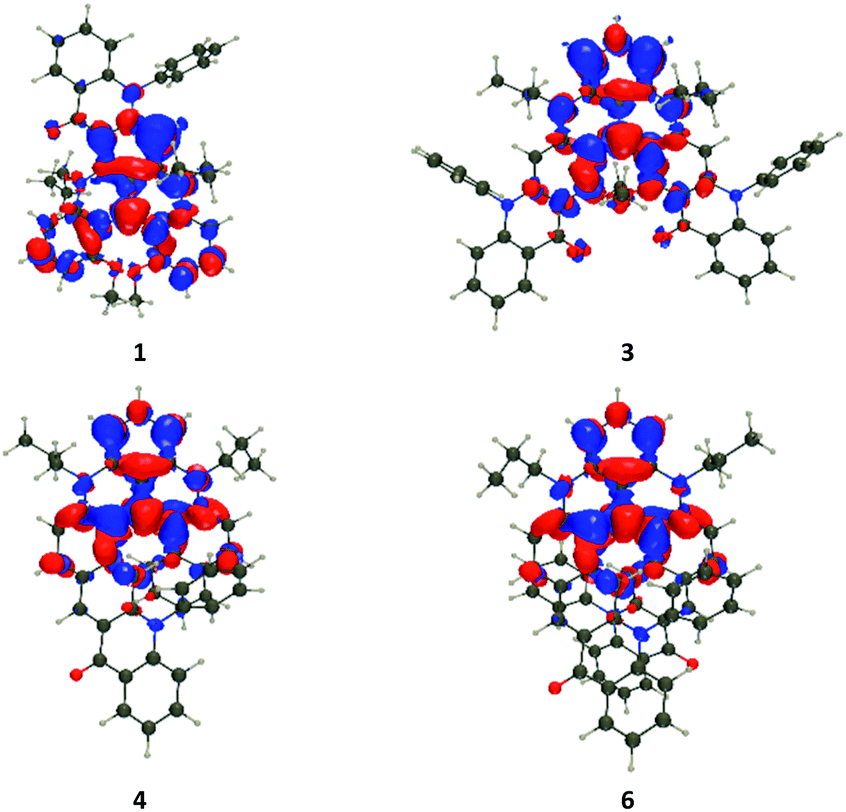 | ||
| Fig. 5 Density difference plots for the lowest transition in selected compounds. Blue and red regions indicate decrease and increase of electron density upon electronic excitation, respectively (contour threshold: 0.001 au). | ||
In Table S5 in the ESI,† we report the computed rotatory strengths (R) for the five lowest singlet states. For the lowest transition, we note rather small responses (<40 cgs, ca. <10 M−1 cm−1 in the Δε scale) in the pentacene series 1–3, but significantly higher responses (up to 218 cgs or ca. 38 M−1 cm−1 for 6) in the tetracene series 4–6, which is clearly in line with the experimental data (Fig. 4, top and middle). Theory also restores the outcome that the response is maximal for 5 and 6 and rather negligible for 1 and 2. As seen above, the electronic nature of the S1 state is globally preserved within all compounds, so that we qualitatively relate the larger ECD response in the tetracene series to their higher helicity, as illustrated by the pitch values (vide supra). For the higher-lying transitions, one notices in Table S5† that R values are very diverse in both series, e.g., for S2, TD-DFT predicts |R| of 97 cgs for 1, 176 cgs for 3, 48 cgs for 4, and 282 cgs for 6. This is consistent with the rather large responses of 3 and 6 at shorter wavelength as compared to 1 and 4 (Fig. 3). This variation as compared to S1 can be explained by the different natures of the S2 excited states that, as can be seen in Fig. S26 in the ESI,† now involve much more importantly the added acene rings as compared to 7 and 8. Thus, at higher energies, the helical pitch is not a sufficient parameter to understand the experimental responses.
Conclusions
Through late-stage functionalizations of cationic [6] or [4]helicenes, series of helical tetracenes and pentacenes were prepared. Due to a larger helical pitch (ca. 3.2 Å vs. 2.6–2.7 Å) and as demonstrated through first principles analyses, only the class of tetracene derivatives exhibit strong ECD and CPL properties in the far red domain, including in the solid state. Based on these enhanced properties, a wealth of developments can be considered.26b,d,fConflicts of interest
Authors declares no conflicts of interest.Acknowledgements
We thank the University of Geneva, the DIP-State of Geneva and the Swiss National Science for funding and support (SNF 200020-172497 and 200020-184843). We also acknowledge the contributions of the Sciences Mass Spectrometry (SMS) platform at the Faculty of Sciences, University of Geneva. This work used the computational resources of the CCIPL center installed in Nantes thanks to the support of Région des Pays de la Loire.Notes and references
- (a) J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed; (b) G. J. Richards, J. P. Hill, T. Mori and K. Ariga, Org. Biomol. Chem., 2011, 9, 5005–5017 RSC; (c) Q. Miao, Synlett, 2012, 2012, 326–336 CrossRef; (d) H. F. Bettinger and C. Toenshoff, Chem. Rec., 2015, 15, 364–369 CrossRef CAS PubMed; (e) J. Li and Q. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 28049–28062 CrossRef CAS PubMed; (f) U. H. F. Bunz, Acc. Chem. Res., 2015, 48, 1676–1686 CrossRef CAS PubMed.
- F. Paulus, J. U. Engelhart, P. E. Hopkinson, C. Schimpf, A. Leineweber, H. Sirringhaus, Y. Vaynzof and U. H. F. Bunz, J. Mater. Chem. C, 2016, 4, 1194–1200 RSC.
- D.-K. Kim and J.-H. Choi, Opt. Mater., 2018, 76, 359–367 CrossRef CAS.
- (a) U. H. F. Bunz, Pure Appl. Chem., 2010, 82, 953–968 CAS; (b) P.-Y. Gu, Z. Wang and Q. Zhang, J. Mater. Chem. B, 2016, 4, 7060–7074 RSC.
- S. S. Zade and M. Bendikov, J. Phys. Org. Chem., 2012, 25, 452–461 CrossRef CAS.
- (a) I. G. Stara and I. Stary, Sci. Synth., 2010, 45b, 885–953 Search PubMed; (b) Y. Shen and C.-F. Chen, Chem. Rev., 2011, 112, 1463–1535 CrossRef PubMed; (c) C.-F. Chen and Y. Shen, Helicene Chemistry From Synthesis to Applications, Springer, Berlin, Heidelberg, 2017 CrossRef.
- (a) N. Saleh, C. Shen and J. Crassous, Chem. Sci., 2014, 5, 3680–3694 RSC; (b) J. Bosson, J. Gouin and J. Lacour, Chem. Soc. Rev., 2014, 43, 2824–2840 RSC; (c) P. Aillard, A. Voituriez and A. Marinetti, Dalton Trans., 2014, 43, 15263–15278 RSC; (d) M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC.
- (a) M. Li, H.-Y. Lu, C. Zhang, L. Shi, Z. Tang and C.-F. Chen, Chem. Commun., 2016, 52, 9921–9924 RSC; (b) M. Li, Y. Niu, X. Zhu, Q. Peng, H.-Y. Lu, A. Xia and C.-F. Chen, Chem. Commun., 2014, 50, 2993–2995 RSC; (c) M. Li, W. Yao, J.-D. Chen, H.-Y. Lu, Y. Zhao and C.-F. Chen, J. Mater. Chem. C, 2014, 2, 8373–8380 RSC.
- (a) I. H. Delgado, S. Pascal, A. Wallabregue, R. Duwald, C. Besnard, L. Guenee, C. Nancoz, E. Vauthey, R. C. Tovar, J. L. Lunkley, G. Muller and J. Lacour, Chem. Sci., 2016, 7, 4685–4693 RSC; (b) J. Bosson, G. M. Labrador, S. Pascal, F.-A. Miannay, O. Yushchenko, H. Li, L. Bouffier, N. Sojic, R. C. Tovar, G. Muller, D. Jacquemin, A. D. Laurent, B. Le Guennic, E. Vauthey and J. Lacour, Chem.–Eur. J., 2016, 22, 18394–18403 CrossRef CAS PubMed; (c) S. Pascal, C. Besnard, F. Zinna, L. Di Bari, B. Le Guennic, D. Jacquemin and J. Lacour, Org. Biomol. Chem., 2016, 14, 4590–4594 RSC.
- H. Isla and J. Crassous, C. R. Chim., 2016, 19, 39–49 CrossRef CAS.
- E. M. Sánchez-Carnerero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem.–Eur. J., 2015, 21, 13488–13500 CrossRef PubMed.
- (a) K. Goto, R. Yamaguchi, S. Hiroto, H. Ueno, T. Kawai and H. Shinokubo, Angew. Chem., Int. Ed., 2012, 51, 10333–10336 CrossRef CAS PubMed; (b) D. Sakamaki, D. Kumano, E. Yashima and S. Seki, Angew. Chem., Int. Ed., 2015, 54, 5404–5407 CrossRef CAS PubMed; (c) X. Geng, J. P. Donahue, J. T. Mague and R. A. Pascal Jr, Angew. Chem., Int. Ed., 2015, 54, 13957–13960 CrossRef CAS PubMed.
- For a recent study describing terminal-end growth of helical skeletons up to [13]helicene frameworks, see S. K. Pedersen, K. Eriksen and M. Pittelkow, Angew. Chem., Int. Ed., 2019, 58(51), 18419–18423 CrossRef CAS PubMed.
- (a) B. W. Laursen and F. C. Krebs, Angew. Chem., Int. Ed., 2000, 39, 3432–3434 CrossRef CAS; (b) C. Herse, D. Bas, F. C. Krebs, T. Bürgi, J. Weber, T. Wesolowski, B. W. Laursen and J. Lacour, Angew. Chem., Int. Ed., 2003, 42, 3162–3166 CrossRef CAS PubMed; (c) F. Torricelli, J. Bosson, C. Besnard, M. Chekini, T. Bürgi and J. Lacour, Angew. Chem., Int. Ed., 2013, 52, 1796–1800 CrossRef CAS PubMed.
- R. Duwald, S. Pascal, J. Bosson, S. Grass, C. Besnard, T. Bürgi and J. Lacour, Chem.–Eur. J., 2017, 23, 13596–13601 CrossRef CAS PubMed.
- P. E. Eaton, G. R. Carlson and J. T. Lee, J. Org. Chem., 1973, 38, 4071–4073 CrossRef CAS.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- S. H. Cho, J. Yoon and S. Chang, J. Am. Chem. Soc., 2011, 133, 5996–6005 CrossRef CAS PubMed.
- N. Gommermann, C. Koradin and P. Knochel, Synthesis, 2002, 2002, 2143–2149 Search PubMed.
- For the formation of K4, the use of polyphosphoric acid (PPA) at 100 °C for 20 hours was preferred over Eaton's reagent.
- H. Tanaka, M. Ikenosako, Y. Kato, M. Fujiki, Y. Inoue and T. Mori, Commun. Chem., 2018, 1, 38 CrossRef.
- For a review on the influence of the twisting of polyacenes on their optical properties, see A. Bedi and O. Gidron, Acc. Chem. Res., 2019, 52, 2482–2490 CrossRef CAS PubMed.
- Strong ECD signals are observed in the UV domain for 1–3, with Δε up to 107 M−1 cm−1 for 3 at 298 nm. It is also the case for 4–6, with Δε up to 240 M−1 cm−1 for 6 at 286 nm.
- (a) Y. Yamamoto, H. Sakai, J. Yuasa, Y. Araki, T. Wada, T. Sakanoue, T. Takenobu, T. Kawai and T. Hasobe, Chem.–Eur. J., 2016, 22, 4263–4273 CrossRef CAS PubMed; (b) T. Biet, T. Cauchy, Q. Sun, J. Ding, A. Hauser, P. Oulevey, T. Burgi, D. Jacquemin, N. Vanthuyne, J. Crassous and N. Avarvari, Chem. Commun., 2017, 53, 9210–9213 RSC; (c) T. Otani, A. Tsuyuki, T. Iwachi, S. Someya, K. Tateno, H. Kawai, T. Saito, K. S. Kanyiva and T. Shibata, Angew. Chem., Int. Ed., 2017, 56, 3906–3910 CrossRef CAS PubMed; (d) Z. Dominguez, R. Lopez-Rodriguez, E. Alvarez, S. Abbate, G. Longhi, U. Pischel and A. Ros, Chem.–Eur. J., 2018, 24, 12660–12668 CrossRef CAS PubMed; (e) M. Satoh, Y. Shibata and K. Tanaka, Chem.–Eur. J., 2018, 24, 5434–5438 CrossRef CAS PubMed; (f) H. Tanaka, Y. Kato, M. Fujiki, Y. Inoue and T. Mori, J. Phys. Chem. A, 2018, 122, 7378–7384 CrossRef CAS PubMed.
- H. Tanaka, Y. Inoue and T. Mori, ChemPhotoChem, 2018, 2, 386–402 CrossRef CAS.
- (a) T. Kaseyama, S. Furumi, X. Zhang, K. Tanaka and M. Takeuchi, Angew. Chem., Int. Ed., 2011, 50, 3684–3687 CrossRef CAS PubMed; (b) T. Amako, K. Nakabayashi, A. Sudo, M. Fujiki and Y. Imai, Org. Biomol. Chem., 2015, 13, 2913–2917 RSC; (c) J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS PubMed; (d) M. Gon, R. Sawada, Y. Morisaki and Y. Chujo, Macromolecules, 2017, 50, 1790–1802 CrossRef CAS; (e) H. Nishimura, K. Tanaka, Y. Morisaki, Y. Chujo, A. Wakamiya and Y. Murata, J. Org. Chem., 2017, 82, 5242–5249 CrossRef CAS PubMed; (f) K. Takaishi, T. Yamamoto, S. Hinoide and T. Ema, Chem.–Eur. J., 2017, 23, 9249–9252 CrossRef CAS PubMed; (g) C. Maeda, K. Nagahata, K. Takaishi and T. Ema, Chem. Commun., 2019, 55, 3136–3139 RSC; (h) A. Taniguchi, D. Kaji, N. Hara, R. Murata, S. Akiyama, T. Harada, A. Sudo, H. Nishikawa and Y. Imai, RSC Adv., 2019, 9, 1976–1981 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental conditions, full characterizations, 1H NMR and 13C NMR spectra of all new compounds (PDF); CSP-HPLC traces; UV-vis, ECD, fluorescence and CPL spectra; computational details. In addition, the dataset for this article can be found at the following DOI: 10.26037/yareta:zsmeadwn3vbgpag2jqno3lj554. It will be preserved for 10 years. CCDC 1960498–1960500. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05407c |
| This journal is © The Royal Society of Chemistry 2020 |