
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights into copper-catalyzed aerobic oxidative coupling of N–N bonds†
Michael C.
Ryan
 a,
Yeon Jung
Kim
a,
James B.
Gerken
a,
Fei
Wang
a,
Michael M.
Aristov
a,
Joseph R.
Martinelli
b and
Shannon S.
Stahl
*a
a,
Yeon Jung
Kim
a,
James B.
Gerken
a,
Fei
Wang
a,
Michael M.
Aristov
a,
Joseph R.
Martinelli
b and
Shannon S.
Stahl
*a
aDepartment of Chemistry, University of Wisconsin–Madison, 1101 University Avenue, Madison, Wisconsin 53706, USA. E-mail: stahl@chem.wisc.edu
bSmall Molecule Design and Development, Lilly Research Laboratories, Eli Lilly and Company, Indianapolis, Indiana 46285, USA
First published on 10th December 2019
Abstract
Catalytic N–N coupling is a valuable transformation for chemical synthesis and energy conversion. Here, mechanistic studies are presented for two related copper-catalyzed oxidative aerobic N–N coupling reactions, one involving the synthesis of a pharmaceutically relevant triazole and the other relevant to the oxidative conversion of ammonia to hydrazine. Analysis of catalytic and stoichiometric N–N coupling reactions support an “oxidase”-type catalytic mechanism with two redox half-reactions: (1) aerobic oxidation of a CuI catalyst and (2) CuII-promoted N–N coupling. Both reactions feature turnover-limiting oxidation of CuI by O2, and this step is inhibited by the N–H substrate(s). The results highlight the unexpected facility of the N–N coupling step and establish a foundation for development of improved catalysts for these transformations.
Introduction
Catalytic methods for oxidative N–N bond formation are the focus of increasing attention and have applications ranging from chemical synthesis1 to energy conversion (e.g., ammonia fuel cells).2 N–N coupling methods provide a valuable strategy to prepare azole-type heterocycles that are featured prominently in pharmaceuticals and agrochemicals,3,4 and provide a means to prepare bulk chemicals, such as azo dyes5 and hydrazine.6 Methods for N–N bond formation commonly involve oxidized nitrogen intermediates, such as those bearing N–OR or N–X (X = halide) bonds, that react with an amine or other nitrogen-based nucleophiles to form the N–N bond.1 Recent advances have led to alternative strategies that bypass the need for undesirable stoichiometric oxidants, independent synthesis of reactive intermediates, or the formation of wasteful byproducts. These strategies include electrochemical7–10 and aerobic oxidative coupling methods.11–18 Homogeneous Cu catalysts are featured prominently in the latter methods (Scheme 1A), but the mechanism of these reactions has received little attention. Here, we report kinetic and mechanistic studies of two important oxidative N–N coupling reactions (Scheme 1B). The first, reported by Nagasawa and coworkers,13 features base-promoted addition of amidines to nitriles to afford substituted imidoylamidines, followed by in situ oxidative N–N coupling to generate the 1,2,4-triazole. A number of related reactions have been reported subsequently by others,16,17,19,20 including process scale applications in the pharmaceutical industry.21 The second, reported by Hayashi and coworkers,22 features the oxidative homocoupling of benzophenone imine to afford the corresponding azine. This reaction is the key step in a multi-step process that has been considered for commercial production of hydrazine.6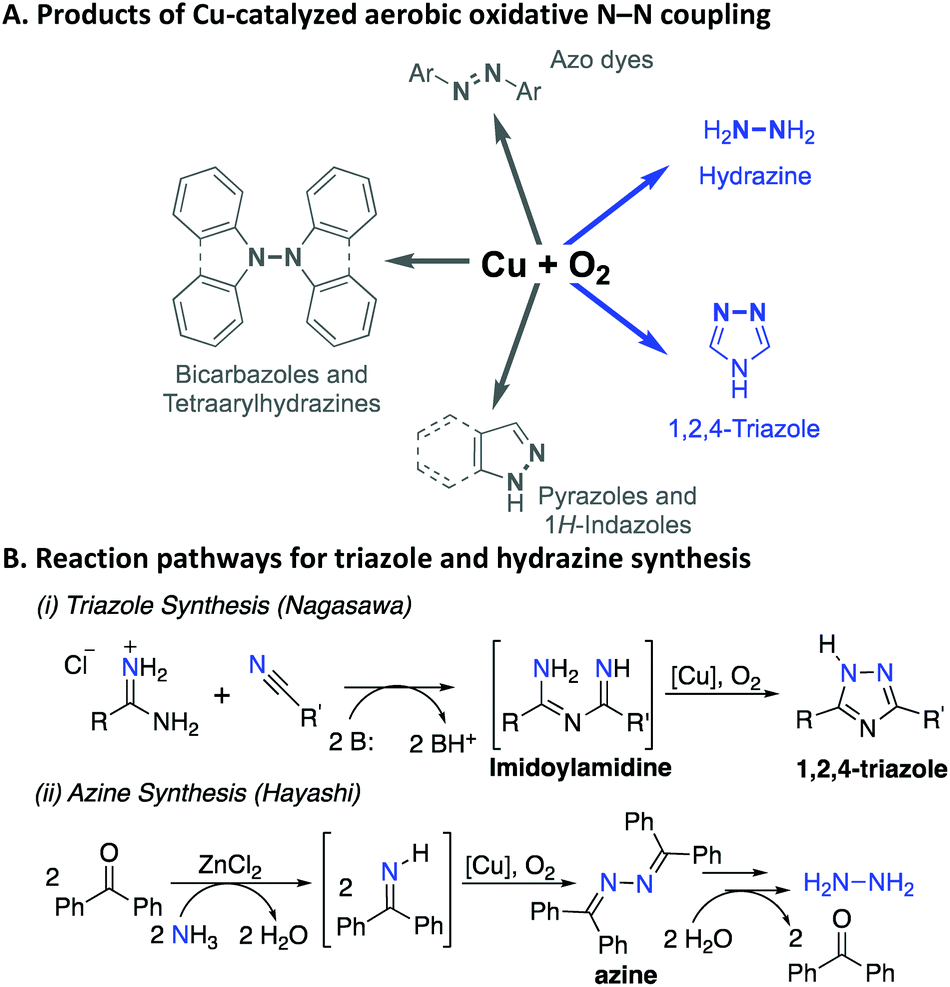 | ||
| Scheme 1 Cu-catalyzed aerobic N–N coupling reactions (A), and further details of the two applications that are the focus of the present mechanistic study (B). | ||
The results elaborated below demonstrate that N–N bond formation is remarkably facile, and that the turnover-limiting step during catalysis in both cases is aerobic oxidation of the CuI catalyst species. Analyses of catalytic and stoichiometric reactions support an “oxidase”-type aerobic oxidation mechanism consisting of two redox half-reactions, similar to other classes of Cu-catalyzed aerobic oxidative coupling reactions.23,24
Results and discussion
Kinetic studies of Cu-catalyzed N–N coupling reactions
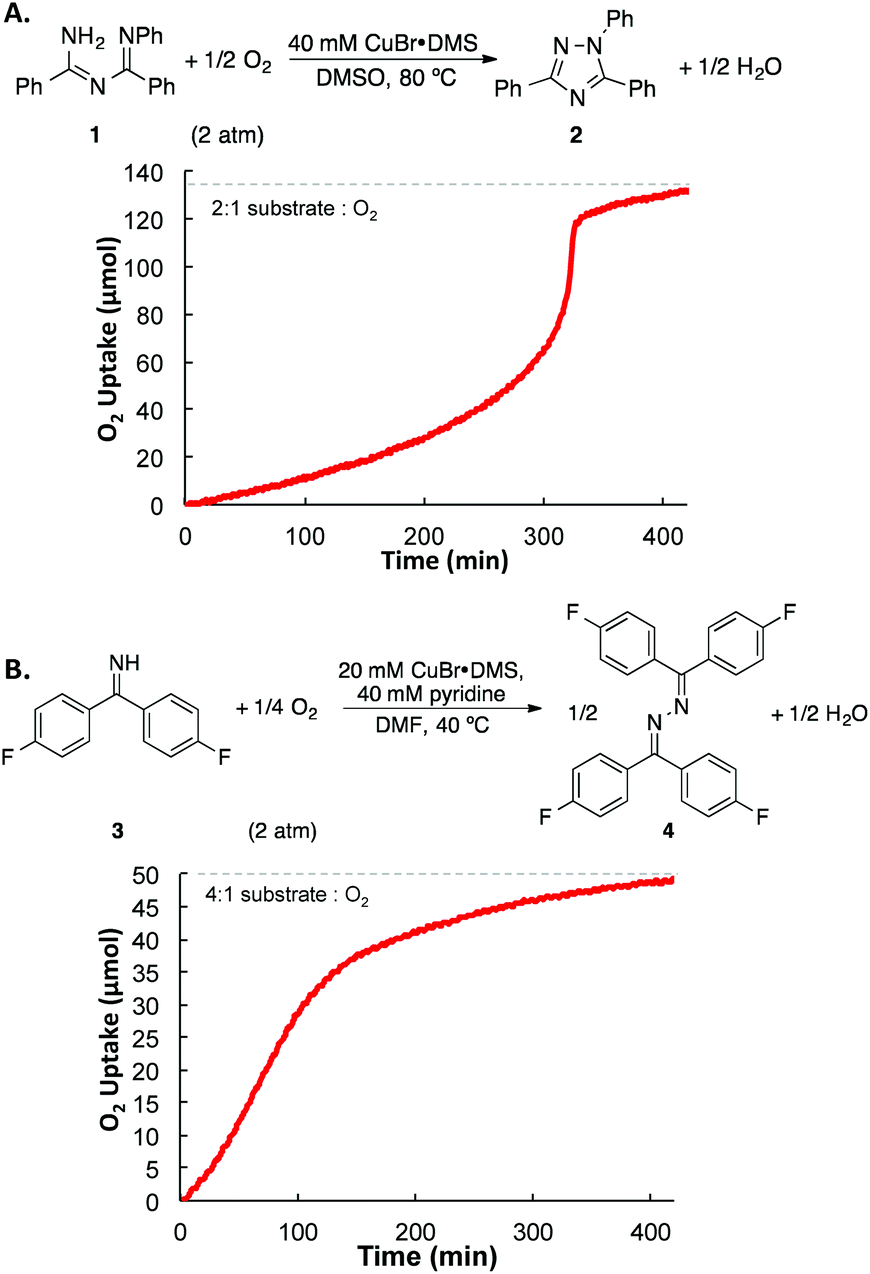
Both of the N–N coupling reactions of interest here were originally featured in multistep, one-pot processes (cf.Scheme 1B). In order to facilitate the acquisition of relevant mechanistic insights, we elected to analyze the Cu-catalyzed N–N coupling reactions as isolated steps, starting from the imidoylamidine and benzophenone imine precursors, respectively.Imidoylamidine 1 was selected as a representative precursor for the investigation of the triazole synthesis, and use of conditions similar to those originally reported by Nagasawa13 led to a 90% yield of triazole 2 (Fig. 1A). Initial-rate kinetic studies were carried out to gain insight into the catalytic rate law (Fig. 1B). A stock solution of CuBr·DMS in DMSO was prepared, and the reaction was initiated by adding the Cu solution to a DMSO solution of 1 at 80 °C under an atmosphere of O2. These data reveal a second-order dependence of the rate on the Cu concentration and a first order dependence on pO2.25 Increasing the imidoylamidine concentration, however, inhibited the reaction.
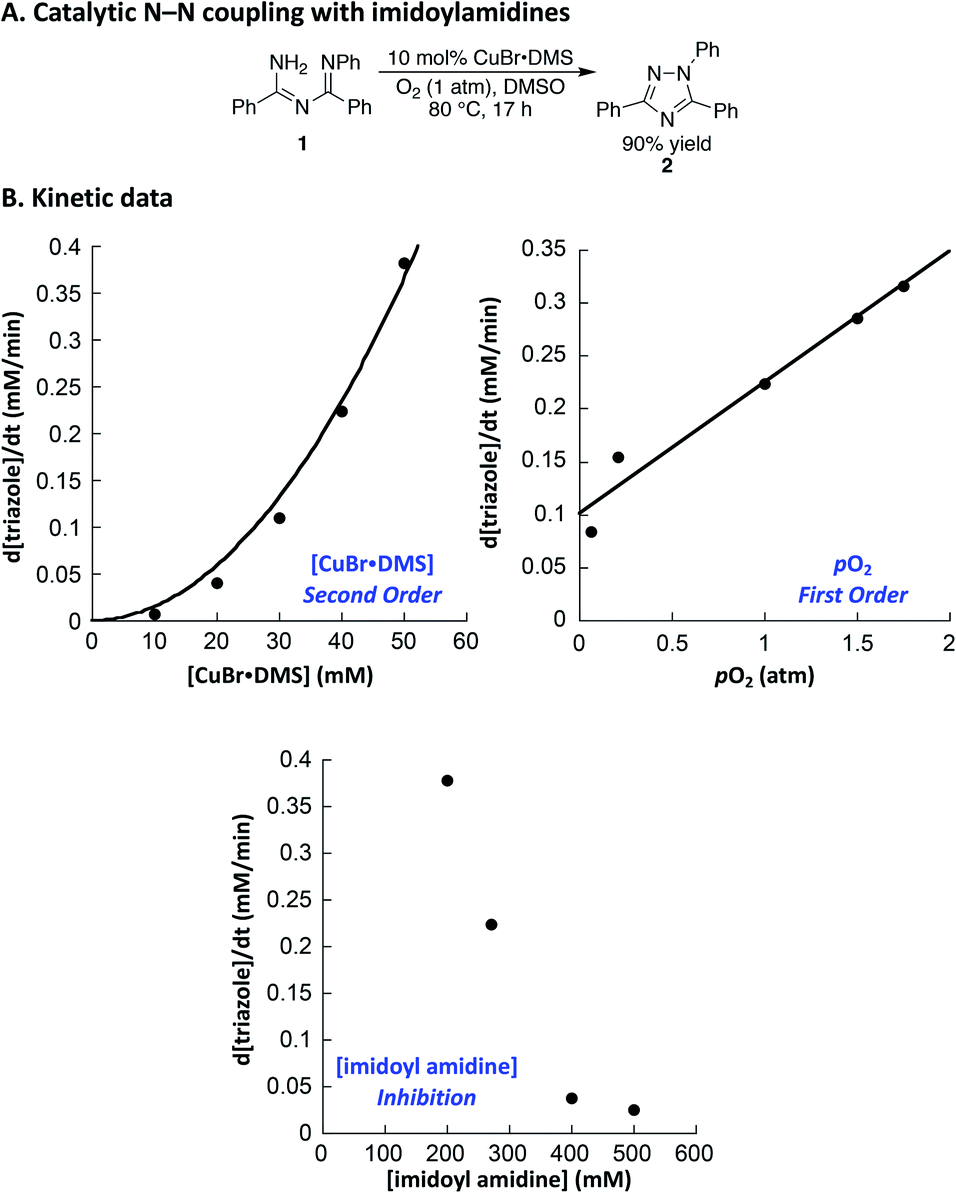 | ||
| Fig. 1 (A) Cu-catalyzed 1,2,4-triazole synthesis. (B) Kinetic data. Standard conditions: 270 mM 1, 40 mM CuBr·DMS, 1 atm O2, DMSO, 80 °C. The curve for the [CuBr·DMS] dependence reflects a second order fit, i.e. rate = c1x2. | ||
Drawing on the original studies of Hayashi,22,26 in addition to a recent report by Evano and coworkers, we optimized reaction conditions for the oxidative homocoupling of benzophenone imine.27 Using 4,4′-difluorobenzophenone imine 3 as the substrate to enable product analysis by 19F NMR spectroscopy, we evaluated a number of different copper sources, ancillary ligands, and solvents (see the ESI† for additional details). These efforts led to the identification of very mild conditions for the reaction. Use of CuBr·DMS/pyridine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) as the catalyst in DMF led to azine 4 in high yield at room temperature within 5 h (Fig. 2A). These conditions provided the basis for initial rate kinetic studies of the catalytic reaction. Data similar to those obtained in the imidoylamidine N–N coupling reaction were obtained (cf.Fig. 1B and 2B). The catalytic rate exhibits a second order dependence on [(pyr)2CuBr], and a first order dependence on [O2]. Increasing the imine substrate concentration inhibits the reaction; however, the rate can be increased by adding more pyridine. The linear dependence on [pyridine] exhibits a non-zero intercept, indicating that azine formation does not require pyridine, but it proceeds with a slower rate in the absence of pyridine. The kinetic data in Fig. 1 and 2 support a mechanism in which oxidation of CuI by O2 is the turnover limiting step of the catalytic reaction. The second-order dependence on [Cu] in both cases implicates a bimetallic pathway for reaction with O2.28,29 The first-order dependence on pO2 does not reflect gas–liquid mass transfer, as revealed by the increase in rate at higher [Cu]. If the rate were limited by gas–liquid mass transfer, the rate would plateau at the mass-transfer rate and not increase at higher catalyst loading.
:2) as the catalyst in DMF led to azine 4 in high yield at room temperature within 5 h (Fig. 2A). These conditions provided the basis for initial rate kinetic studies of the catalytic reaction. Data similar to those obtained in the imidoylamidine N–N coupling reaction were obtained (cf.Fig. 1B and 2B). The catalytic rate exhibits a second order dependence on [(pyr)2CuBr], and a first order dependence on [O2]. Increasing the imine substrate concentration inhibits the reaction; however, the rate can be increased by adding more pyridine. The linear dependence on [pyridine] exhibits a non-zero intercept, indicating that azine formation does not require pyridine, but it proceeds with a slower rate in the absence of pyridine. The kinetic data in Fig. 1 and 2 support a mechanism in which oxidation of CuI by O2 is the turnover limiting step of the catalytic reaction. The second-order dependence on [Cu] in both cases implicates a bimetallic pathway for reaction with O2.28,29 The first-order dependence on pO2 does not reflect gas–liquid mass transfer, as revealed by the increase in rate at higher [Cu]. If the rate were limited by gas–liquid mass transfer, the rate would plateau at the mass-transfer rate and not increase at higher catalyst loading.
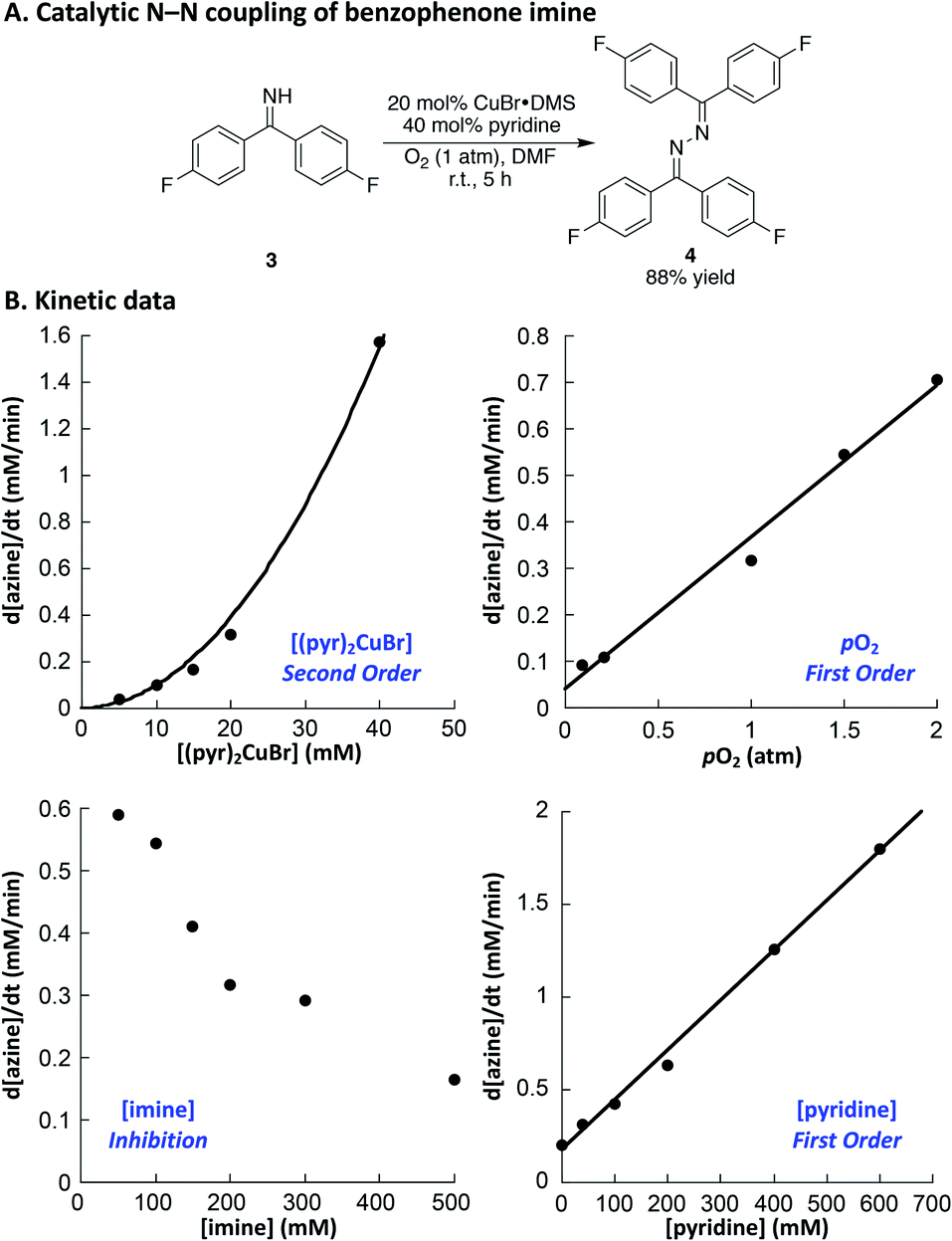 | ||
| Fig. 2 (A) Cu-catalyzed azine synthesis. (B) Kinetic data. Standard conditions: 200 mM imine, 20 mM CuBr·DMS, 40 mM pyridine, 1 atm O2, DMF, 40 °C. The curve for the [(pyr)2CuBr] dependence reflects a second order fit, i.e. rate = c1x2. | ||
Further insights into the imine homocoupling reaction were obtained from cyclic voltammetry studies (Fig. 3). Cu(OTf)2 displays a redox feature with an approximate mid-point potential of −0.46 V vs. Fc+/0 in DMF. Addition of one equivalent of bromide leads to a significant increase in this potential (−0.03 V), and the CuII/I redox feature is more reversible than that observed with Cu(OTf)2. Addition of 10 equiv. of the imine substrate shifts the CuII/I redox potential to a somewhat lower potential (−0.10 V), and addition of pyridine further lowers the CuII/I potential.
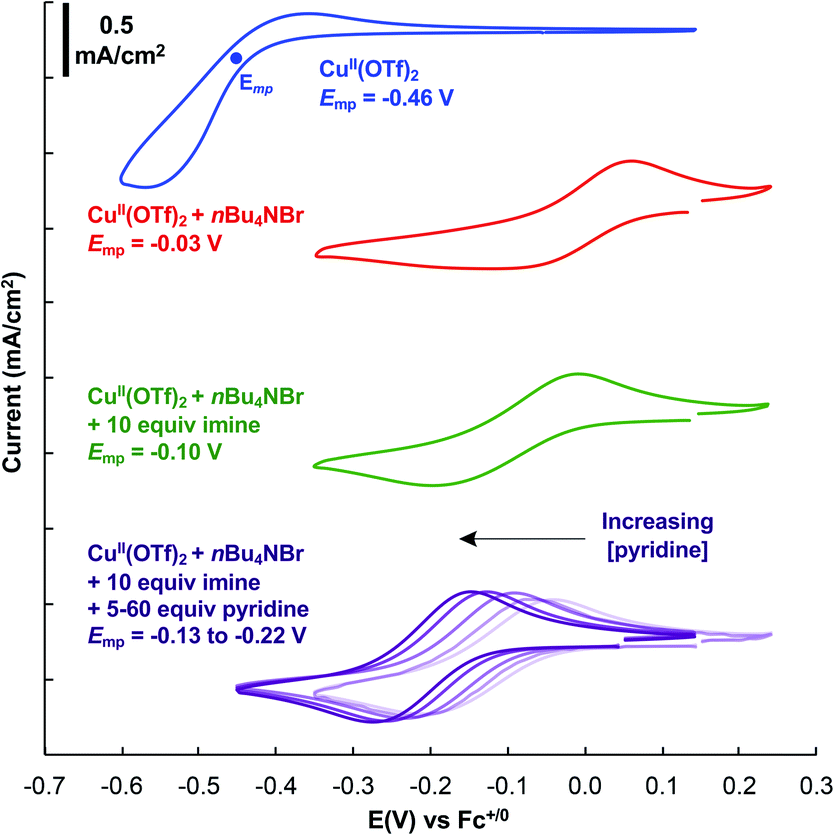 | ||
Fig. 3 Cyclic voltammetry of CuII in DMF. Conditions: 5 mM Cu(OTf)2, 5 mM nBu4NBr, 50 mM Ph2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH, 25–300 mM pyridine, 1 atm N2, 100 mM nBu4NPF6, DMF (10 mL), scan rate = 100 mV s−1. See ESI† for details. NH, 25–300 mM pyridine, 1 atm N2, 100 mM nBu4NPF6, DMF (10 mL), scan rate = 100 mV s−1. See ESI† for details. | ||
These electrochemical data provide complementary insights. Identification of a CuBr source as the optimal catalyst precursor undoubtedly reflects the significant influence of bromide on the CuII/I redox potential, as a CuII species with higher potential will more effectively promote the N–N bond forming step. This hypothesis is supported by stoichiometric reactivity studies described below. Meanwhile, the beneficial effect of pyridine on the reaction may be attributed its ability to lower the CuII/I potential and thereby support more facile oxidation of CuI by O2. The role of pyridine could also be attributed to a steric effect, as the smaller size of pyridine relative to the imine substrate probably enables more effective reaction of two equivalents of CuI with O2.
O2 stoichiometry in the catalytic reactions
The O2 stoichiometry for each of the catalytic reactions was determined by measuring the gas consumed within a sealed reaction vessel equipped with a pressure transducer (Fig. 4). The time-course traces reveal a lag-phase at the beginning of the reaction in both cases, with a longer induction period observed for the intramolecular reaction of the imidoylamidine. This effect may be rationalized by the inhibitory effect of the substrates, which results in acceleration of the reaction rates as the substrate is consumed. Both reactions, however, consume O2 in a stoichiometry corresponding to full reduction of O2 to H2O. N–N coupling of the imidoylamidine 1 approaches the theoretical stoichiometry of 2:1 1:O2 (Fig. 4A). Imine homocoupling exhibits a 4:1 3:O2 stoichiometry (2:1 4:O2), reflecting the one-electron oxidation of each imine to form azine 4 (Fig. 4B).
 | ||
| Fig. 4 Gas uptake time courses associated with Cu-catalyzed oxidative N–N coupling of (A) 1 and (B) 3, which reveal O2:product stoichiometry corresponding to four-electron reduction of O2 to H2O. | ||
Investigation of stoichiometric CuII-promoted N–N coupling
N–N bond formation cannot be probed directly under catalytic reaction conditions because the reaction of CuI and O2 is the turnover-limiting step. Nonetheless, it was possible to probe stoichiometric reactions between the substrates with CuII under anaerobic conditions. We prepared the previously reported imidoylamidine–CuIICl2 complex (ImAm)CuCl2 (ref. 30) and independently prepared the bis-ligated CuII complex [(ImAm)2Cu](PF6)2.31 An X-ray crystal structure of [(ImAm)2Cu](PF6)2 was obtained by slow diffusion of Et2O into a solution of [(ImAm)2Cu](PF6)2 in MeCN (Fig. 5). The CuII center has an approximately square-planar coordination geometry with two PF6− counterions weakly associated with the Cu center in the axial positions (dCu–F ≈ 2.53 Å). N3 and N6 are positioned trans to one another, presumably reflecting the ability of this geometry to avoid unfavorable steric interactions between the N-aryl substituents. The imidoylamidine ligand is closely related to the betadiketimines that have been widely used as ancillary ligands in Cu-based complexes and catalysts.32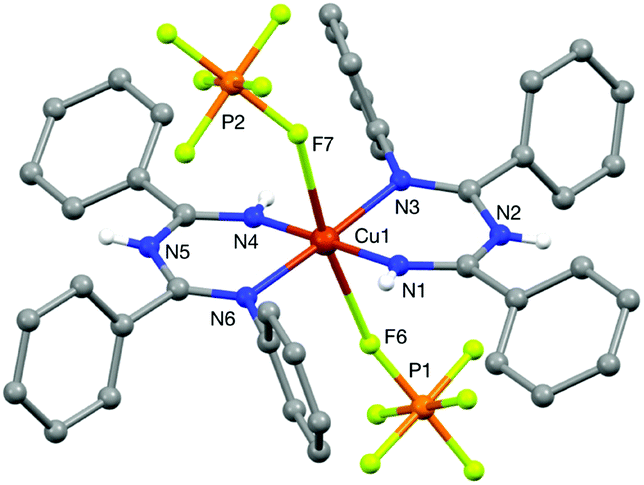 | ||
| Fig. 5 X-ray crystal structure of [(ImAm)2Cu](PF6)2. Two molecules of Et2O and the C–H hydrogen atoms are omitted for clarity. See ESI† for details. | ||
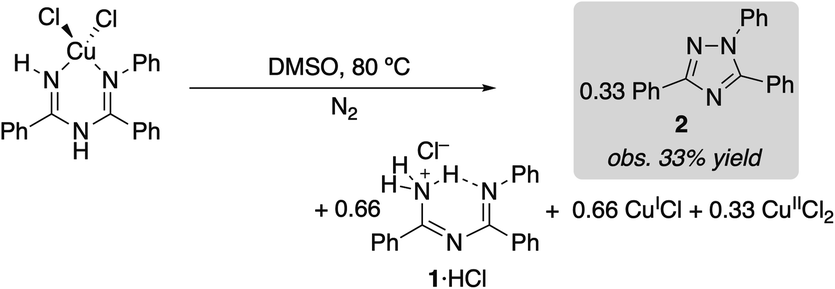
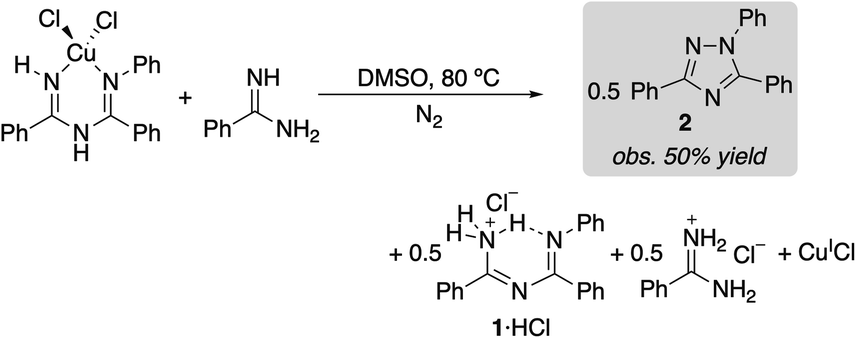
(ImAm)CuCl2 was dissolved in DMSO and heated at 80 °C under N2. The reaction afforded a 33% yield of triazole 2 within 5 min (eqn (1)), and the amount of 2 observed did not change with increased reaction time (>2 h). The same experiment was then repeated in the presence of 1 equiv. of benzamidine as a Brønsted base. In this case, a 50% yield of triazole 2 was observed (eqn (2)). These product yields are rationalized according to the balanced equations shown in eqn (1) and (2), which feature CuII as a one electron oxidant and require a Brønsted base to react with both equivalents of proton in the 2H+/2e− oxidative N–N coupling reaction. In the absence of added benzamidine as a base, the imidoylamidine serves as the base. (Under catalytic conditions, the protons are consumed in the reduction of O2 to H2O).
 | (1) |
 | (2) |
The bis-ligated [(ImAm)2Cu](PF6)2 was also heated in DMSO under anaerobic conditions. In this case, the reaction was slower and triazole 2 was obtained in 42% yield after 20 h (eqn (3)). This value approaches the 50% theoretical yield associated with the balanced equation in eqn (3), which features use of 2 equiv. of CuII and 2 equiv. of base in the reaction (eqn (3)).
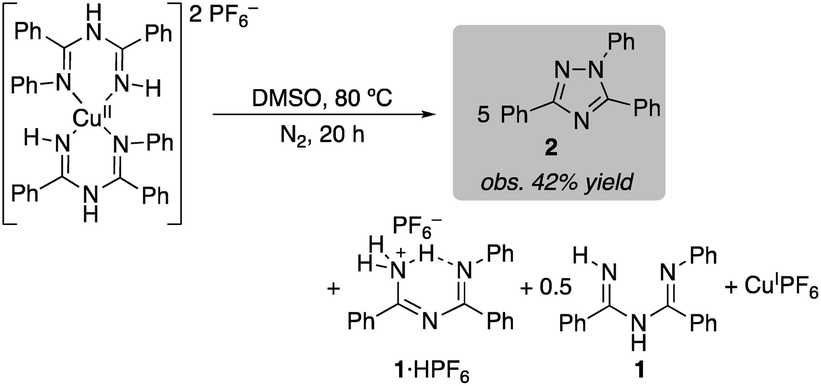 | (3) |
Stoichiometric CuII-mediated N–N coupling of benzophenone imine 3 was also investigated with Cu(OTf)2. The reaction was evaluated with a number of different Brønsted bases, such as pyridine, tetramethylpiperidine (TMP), NaHCO3 and Na2CO3 (see Section 9 of the ESI† for details). Low yields were observed in the absence of base (17% and 3%, with and without Br−, respectively). Pyridine was not effective (9% and 2%, with and without Br−), consistent with the proposal that pyridine contributes to catalyst reoxidation by O2 (cf.Fig. 3 and associated text). The best yield was obtained with TMP, affording a 77% yield of azine 4 in the presence of bromide (eqn (4); only 32% in the absence of Br−). The uniformly improved results observed in the presence of bromide is attributed to the increase the CuII/I redox potential (cf.Fig. 3), which should promote N–N coupling. Overall, these results are consistent with the data in eqn (1)–(3), which show that 2 equiv. of CuII and 2 equiv. of a Brønsted base are essential to promote the N–N coupling reaction.
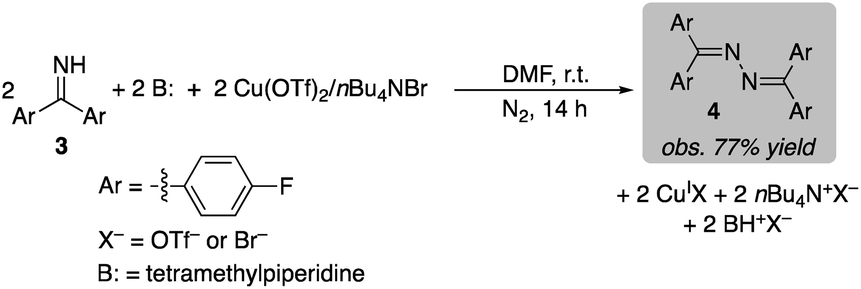 | (4) |
Catalytic mechanism
The data presented above support a catalytic mechanism composed of two redox half-reactions: (1) CuII/base-promoted oxidative N–N coupling and (2) oxidation of CuI by O2. The corresponding catalytic cycle in Scheme 2 resembles “ping-pong” mechanisms employed by oxidase enzymes.33 This cycle features the various reaction stoichiometries observed in the reactions described above: 2:1 N–N product:O2, 1:2 N–N product:CuII, and 4:1 CuI:O2. The kinetic data in Fig. 1 and 2 reveal that CuI re-oxidation by O2 is the turnover-limiting half-reaction in this sequence and that CuI is the catalyst resting state.
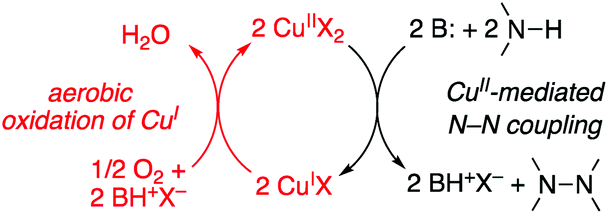 | ||
| Scheme 2 Simplified oxidase-type mechanism for Cu-catalyzed aerobic oxidative N–N coupling reactions. | ||
Inhibition of the reactions by the substrates suggests that one or more coordinated substrate molecules must dissociate from CuI prior to the reaction with O2.34 For imine homocoupling, the reaction of Cu with O2 is promoted by pyridine. This observation could arise from electronic effects, as pyridine is observed to lower the CuII/I potential, or a steric effect arising from the smaller steric influence of pyridine relative to the benzophenone imine. Steric effects will be amplified by the involvement of two Cu species in the turnover limiting reaction with O2, evident by the second order dependence of the reaction on [Cu] (Fig. 1B and 2B).
A rather unexpected outcome of this study is the facility of N–N bond formation, and efforts have been initiated to gain further insights into this step. Two observations that will be relevant to any mechanistic pathway for N–N coupling include (a) the 2:1 stoichiometry between CuII and the N–N-coupled product and (b) the role of a Brønsted base in promoting N–N bond formation.35 We postulate that N–N bond formation is triggered by deprotonation of a CuII-ligated substrate to form an anionic nitrogen ligand. Charge transfer to CuII can lead to delocalization of spin-density onto the nitrogen atom that could facilitate N–N coupling.36 As a complementary consideration, imine homocoupling could proceed via a binuclear Cu2(bis-μ-iminyl) intermediate structurally analogous to the Cu2(μ-O)2 species involved in reversible Cu-mediated O–O bond formation.29
Conclusions
The study described herein provides rare mechanistic insights into Cu-catalyzed N–N coupling reactions, relevant to pharmaceutical and chemical synthesis. The kinetic studies show that the most challenging step in the reactions investigated here is the oxidation of CuI by O2, not N–N bond formation. This insight has important implications for the development of new reactions, as it suggests that identification of ancillary ligands that control the coordination environment of the Cu center and facilitate aerobic oxidation of CuI could provide the basis for more effective catalysts. Alternatively, electrochemical methods for catalyst regeneration could bypass the kinetic limitations evident in the present reactions and enable more effective catalytic transformation. Efforts to explore both of these opportunities have been initiated.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Eli Lilly and Company (Lilly Research Award Program, LRAP) and the DOE (DE-FG02-05ER15690) for funding. Spectroscopic instrumentation was partially supported by a generous gift from Paul J. and Margaret M. Bender, the NIH (1S10 OD020022-1) and the NSF (CHE-1048642).References
- Q. Guo and Z. Lu, Synthesis, 2017, 49, 3835–3847 CrossRef CAS.
- D. J. Little, M. R. Smith and T. W. Hamann, Energy Environ. Sci., 2015, 8, 2775–2781 RSC.
- C.-H. Zhou and Y. Wang, Curr. Med. Chem., 2012, 19, 239–280 CrossRef CAS PubMed.
- L. M. Blair and J. Sperry, J. Nat. Prod., 2013, 76, 794–812 CrossRef CAS PubMed.
- E. Merino, Chem. Soc. Rev., 2011, 40, 3835–3853 RSC.
- H. Hayashi, Catal. Rev.: Sci. Eng., 1990, 32, 229–277 CrossRef CAS.
- B. R. Rosen, E. W. Werner, A. G. O'Brien and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5571–5574 CrossRef CAS PubMed.
- T. Gieshoff, D. Schollmeyer and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 9437–9440 CrossRef CAS PubMed.
- T. Gieshoff, A. Kehl, D. Schollmeyer, K. D. Moeller and S. R. Waldvogel, J. Am. Chem. Soc., 2017, 139, 12317–12324 CrossRef CAS PubMed.
- A. Kehl, T. Gieshoff, D. Schollmeyer and S. R. Waldvogel, Chem.–Eur. J., 2018, 24, 590–593 CrossRef CAS PubMed.
- A. Grirrane, A. Corma and H. García, Science, 2008, 322, 1661–1664 CrossRef CAS PubMed.
- C. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2010, 49, 6174–6177 CrossRef CAS PubMed.
- S. Ueda and H. Nagasawa, J. Am. Chem. Soc., 2009, 131, 15080–15081 CrossRef CAS PubMed.
- H. Xu, Y. Jiang and H. Fu, Synlett, 2013, 24, 125–129 CAS.
- D.-G. Yu, M. Suri and F. Glorius, J. Am. Chem. Soc., 2013, 135, 8802–8805 CrossRef CAS PubMed.
- F. Wang, Q. You, C. Wu, D. Min, T. Shi, Y. Kong and W. Zhang, RSC Adv., 2015, 5, 78422–78426 RSC.
- C.-Y. Chen, G. Tang, F. He, Z. Wang, H. Jing and R. Faessler, Org. Lett., 2016, 18, 1690–1693 CrossRef CAS PubMed.
- M. C. Ryan, J. R. Martinelli and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 9074–9077 CrossRef CAS PubMed.
- H. Wang, Y. Wang, C. Peng, J. Zhang and Q. Zhu, J. Am. Chem. Soc., 2010, 132, 13217–13219 CrossRef CAS PubMed.
- A. K. Bagdi, M. Rahman, S. Santra, A. Majee and A. Hajra, Adv. Synth. Catal., 2013, 355, 1741–1747 CrossRef CAS.
- (a) B. Bartels, C. G. Bolas, P. Cueni, S. Fantasia, N. Gaeng and A. S. Trita, J. Org. Chem., 2015, 80, 1249–1257 CrossRef CAS PubMed; (b) B. Bartels, S. M. Fantasia, A. Flohr, K. Puentener and S. Wang, WO patent 117610A1, 2013.
- H. Hayashi, A. Kainoh, M. Katayama, K. Kawasaki and T. Okazaki, Ind. Eng. Chem. Prod. Res. Dev., 1976, 15, 299–303 CrossRef CAS.
- For reviews, see: (a) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062–11087 CrossRef CAS PubMed; (b) B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824–8838 CrossRef CAS PubMed; (c) S. D. McCann and S. S. Stahl, Acc. Chem. Res., 2015, 48, 1756–1766 CrossRef CAS PubMed.
- A. E. King, T. C. Brunold and S. S. Stahl, J. Am. Chem. Soc., 2009, 131, 5044–5045 CrossRef CAS PubMed.
- The non-zero intercepts of the O2 dependences reflects the fact that stock solutions of the catalyst are pre-oxidized by air prior to their addition to reaction solutions..
- H. Hayashi, K. Kawasaki, M. Fujii, A. Kainoh and T. Okazaki, J. Catal., 1976, 41, 367–372 CrossRef CAS.
- A. Laouiti, M. M. Rammah, M. B. Rammah, J. Marrot, F. Couty and G. Evano, Org. Lett., 2012, 14, 6–9 CrossRef CAS PubMed.
- For similar observations in a different Cu-catalyzed aerobic oxidation reaction, see: J. M. Hoover, B. L. Ryland and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 2357–2367 CrossRef CAS PubMed.
- For reviews of fundamental Cu/O2 reactivity, which often shows second-order [Cu] dependence, see: (a) L. M. Mirica, X. Ottenwaelder and T. D. P. Stack, Chem. Rev., 2004, 104, 1013–1046 CrossRef CAS PubMed; (b) E. A. Lewis and W. B. Tolman, Chem. Rev., 2004, 104, 1047–1076 CrossRef CAS PubMed; (c) L. Q. Hatcher and K. D. Karlin, Adv. Inorg. Chem., 2006, 58, 131–184 CrossRef CAS.
- N. Heße, R. Fröhlich, I. Humelnicu and E.-U. Würthwein, Eur. J. Inorg. Chem., 2005, 2005, 2189–2197 CrossRef.
- A triflate salt of [(ImAm)2Cu]2+ has been reported previously (see ref. 30).
- See, for example: (a) N. W. Aboelella, S. V. Kryatov, B. F. Gherman, W. W. Brennessel, V. G. Young, R. Sarangi, E. V. Rybak-Akimova, K. O. Hodgson, B. Hedman, E. I. Solomon, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2004, 126, 16896–16911 CrossRef CAS PubMed; (b) E. S. Jang, C. L. McMullin, M. Käβ, K. Meyer, T. R. Cundari and T. H. Warren, J. Am. Chem. Soc., 2014, 136, 10930–10940 CrossRef CAS PubMed; (c) A. Bakhoda, Q. Jiang, J. A. Bertke, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2017, 56, 6426–6430 CrossRef CAS PubMed.
- For context, see: S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400–3420 CrossRef CAS PubMed.
- Benzophenone imine complexes of CuI have been characterized previously: W. Schneider, A. Bauer and H. Schmidbaur, Chem. Ber., 1997, 130, 947–950 CrossRef CAS.
- For a stoichiometric precedent involving reactions of anionic (i.e., deprotonated) nitrogen nucleophiles, see: T. Kauffmann, J. Albrecht, D. Berger and J. Legler, Angew. Chem., Int. Ed., 1967, 6, 633–634 CrossRef CAS.
- For leading references, see: (a) R. G. Hicks, Angew. Chem., Int. Ed., 2008, 47, 7393–7395 CrossRef CAS PubMed; (b) S. Hong, L. M. R. Hill, A. K. Gupta, B. D. Naab, J. B. Gilroy, R. G. Hicks, C. J. Cramer and W. B. Tolman, Inorg. Chem., 2009, 48, 4514–4523 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization data, kinetics time courses, and electrochemical and X-ray crystallographic data. CCDC 1949421. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04305e |
| This journal is © The Royal Society of Chemistry 2020 |