
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrisulfur radical anion-triggered stitching thienannulation: rapid access to largely π-extended thienoacenes†
Feng
Su
a,
Shuqi
Chen
a,
Xiaogang
Mo
a,
Kongchuan
Wu
a,
Jiajun
Wu
a,
Weidong
Lin
a,
Zhiwei
Lin
a,
Jianbin
Lin
 ab,
Hui-Jun
Zhang
*a and
Ting-Bin
Wen
a
ab,
Hui-Jun
Zhang
*a and
Ting-Bin
Wen
a
aDepartment of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, Fujian, P. R. China. E-mail: meghjzhang@xmu.edu.cn
bMOE Key Laboratory of Spectrochemical Analysis and Instrumentation, Xiamen University, Xiamen 361005, P. R. China
First published on 20th December 2019
Abstract
Largely π-extended rylene diimide-fused thienoacenes, a new family of fully fused electron donor–acceptor (D–A) molecules, have been readily synthesized by a novel trisulfur radical anion (S3˙−)-triggered stitching thienannulation strategy. The ladder-type fused thiophene cores are constructed in a stitching manner through multiple carbon–sulfur bond formation between acetylenic rylene dyes and S3˙−. A detailed mechanistic study of these stitching thienannulations unveiled the multiple reactivities of S3˙−. Physical properties of the newly formed D–A, A–D–A, and D–A–D type thienoacenes have also been investigated, which revealed their precisely controllable electronic properties.
Introduction
Largely π-extended thienoacenes, diacene-fused thienothiophenes such as [1]benzothieno[3,2-b][1] benzo-thiophene (BTBT) and related compounds constitute a useful class of molecules in the field of organic semiconducting materials due to their rigid planar conformation, well delocalized π-conjugation, unique intermolecular sulfur–sulfur interactions, and improved air stability compared with large acenes.1,2 To date, four reliable annulation approaches toward these skeletons have been reported (Scheme 1a).1e,f,3–8 However, each of these methods require the use of uncommon ortho-bifunctionalized aromatics as cyclization precursors, which means the options available to access such skeletons are rather limited. In addition, rylene diimides (RDIs) such as naphthalene diimides (NDIs) and perylene diimides (PDIs) represent an important class of n-channel organic semiconductors. Effective π-extensions of the NDI and PDI units are recognized as valuable approaches toward new molecular materials with precisely controlled electronic properties.9 In this context, fusion of electron-deficient RDI units (acceptors, A) and electron-rich thieno[3,2-b]thiophene or other ladder-type fused thiophenes (donors, D) may afford a variety of n-channel, p-channel, and ambipolar materials, depending on their specific electronic structures.7b,10 As a result, general and efficient synthetic pathways toward these appealing structures are highly desirable.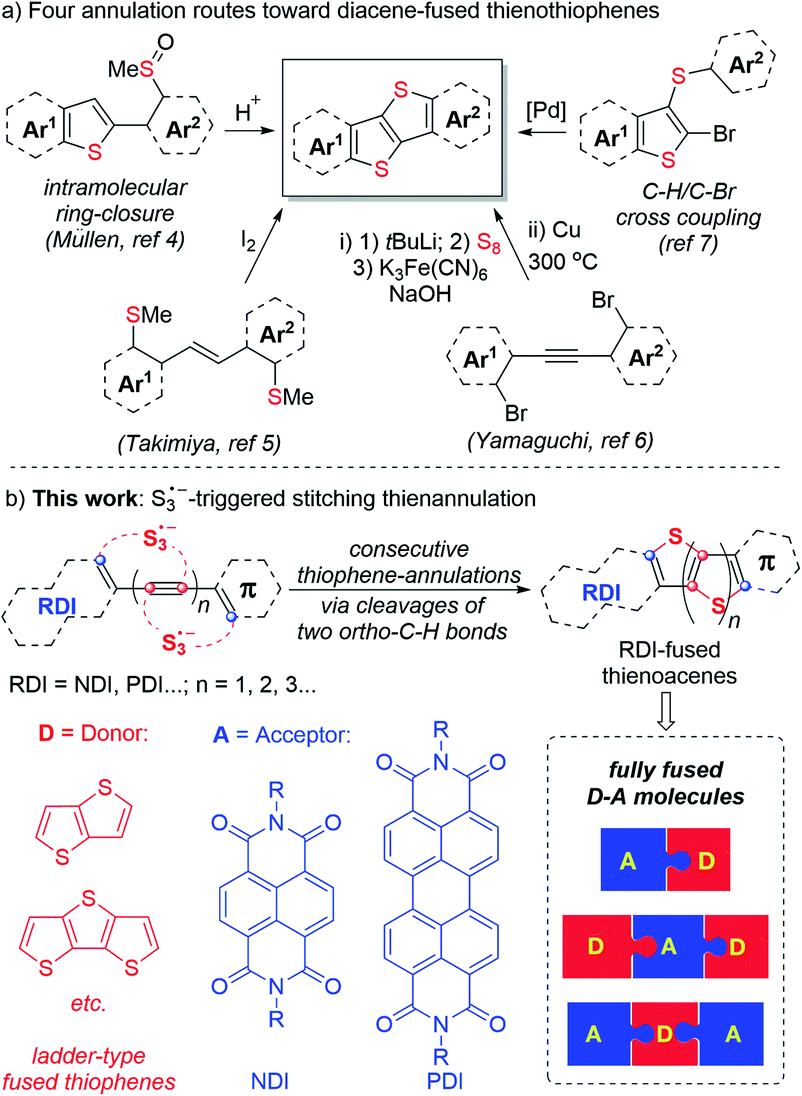 | ||
| Scheme 1 Four annulation routes toward diacene-fused thienothiophenes (previous work) and S3˙−-triggered stitching thienannulation (this work). | ||
The trisulfur radical anion (S3˙−), as a ubiquitous sulfur-centered radical, has only recently received increasing attention in synthetic chemistry.11,12 Until now, exploration of its unique and multiple reactivities and application in the synthesis of useful organosulfur compounds remain limited. Conventionally, intermolecular reactions of normal sulfur-centered radicals, such as thiyl radicals (RS˙) and sulfonyl radicals (RSO2˙), with alkynes predominantly led to the formation of S-functionalized alkene derivatives.13 In contrast, herein, we discovered that S3˙− could initiate consecutive thiophene-annulations (stitching thienannulation) of arylacetylenic RDIs via cleavage of two ortho-C–H bonds to afford a host of rylene diimide-fused largely π-extended soluble thienoacenes (Scheme 1b). With controllable electronic structures (D–A type, D–A–D type and A–D–A type), these RDI-fused thienoacenes are promising candidates for high-performance organic semiconductors.14
Results and discussion
Stitching thienannulation of phenylethynyl substituted RDIs
In recent years, Takimiya et al.,15 Wang et al.16 and our group17 have discovered that alkynyl-substituted rylene dyes could undergo direct thiophene–annulation reactions (thienannulations) with Na2S, S8 or K2S to form important thiophene-fused RDIs. Initially, to gain insight into the mechanisms of these thienannulation processes, the reaction of simple phenylethynyl substituted NDI 1a with different sulfur reagents at room temperature was studied (Scheme 2, eqn (1)). To our surprise, with addition of 6 equiv. of K2S in N,N-dimethylformamide (DMF) solution, 1a was entirely consumed within 10 min and afforded not only a normal thienannulation product 2a in 13% yield but also an abnormal disulfide product 3a′ in 60% yield (Scheme 2, eqn (1)).18 The reaction also took place in air and afforded similar results (2a, 9%; 3a′, 57%). Reducing the amount of K2S to 3 equiv. led to the formation of 3a′ in a lower yield (40%). Treatment of 1a with S8 in the presence of NaOH also produced 2a and 3a′ in 15% and 36% yields, respectively. Previously, Na2S·9H2O has been recognized as an efficient sulfur source for thienannulation of bis(trimethylsilyl)ethynyl substituted NDI to yield naphtho[2,3-b:6,7-b′]dithiophene diimide (NDTI).15,16a Nonetheless, the reaction of 1a with 6 equiv. of Na2S·9H2O at room temperature furnished only 2a in 22% yield. 38% of 1a was recovered, and disulfide product 3a′ was not formed at all. Realizing that S3˙− can be generated from the DMF solution of K2S and the DMF solution of a mixture of elemental sulfur and NaOH at room temperature,12a,c we explored whether S3˙− was responsible for this unique thienannulation process. UV-visible spectra of K2S, S8/NaOH and Na2S·9H2O in DMF were all recorded at room temperature (Fig. 1). A characteristic absorption peak of S3˙− at 617 nm (ref. 11) have been found for the DMF solutions of K2S and S8/NaOH. Clearly, the yields of 3a′ in the reactions are close related to the concentrations of S3˙− in the solutions, indicating that S3˙− was most likely involved in this reaction process.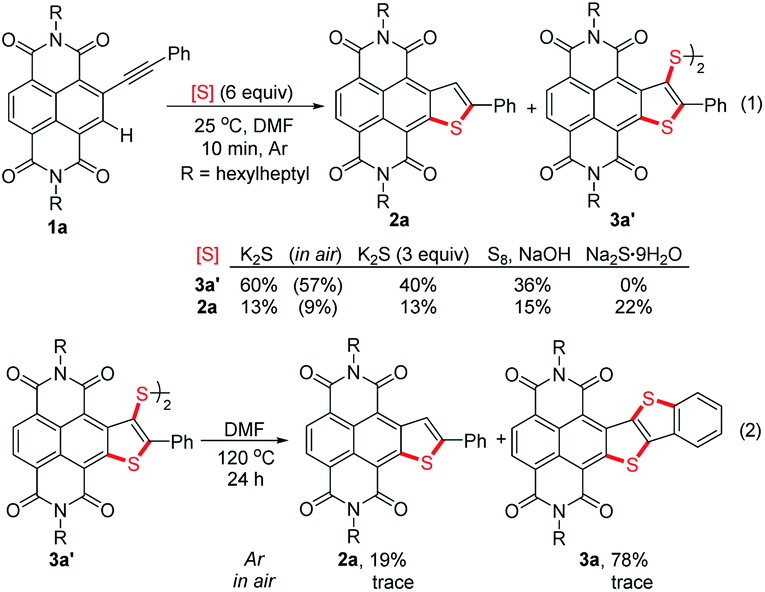 | ||
| Scheme 2 Thienannulations of 1a and 3a′. | ||
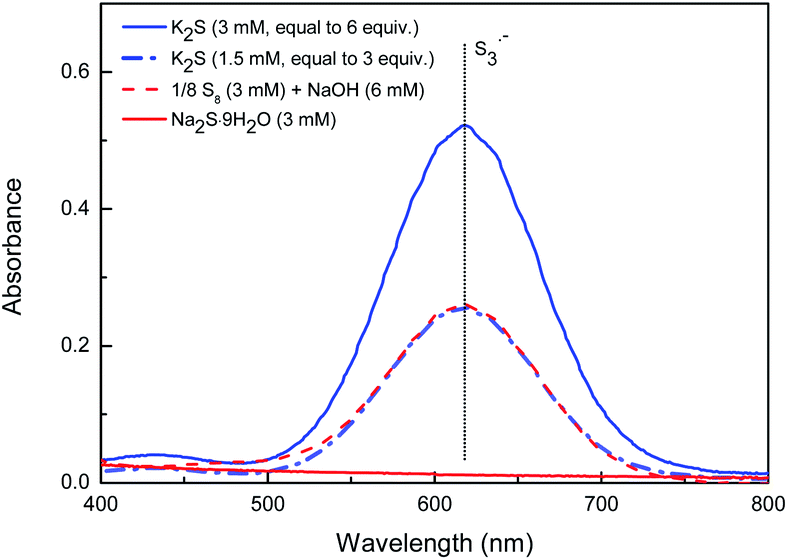 | ||
| Fig. 1 UV-visible spectra of K2S, S8/NaOH and Na2S·9H2O in DMF. | ||
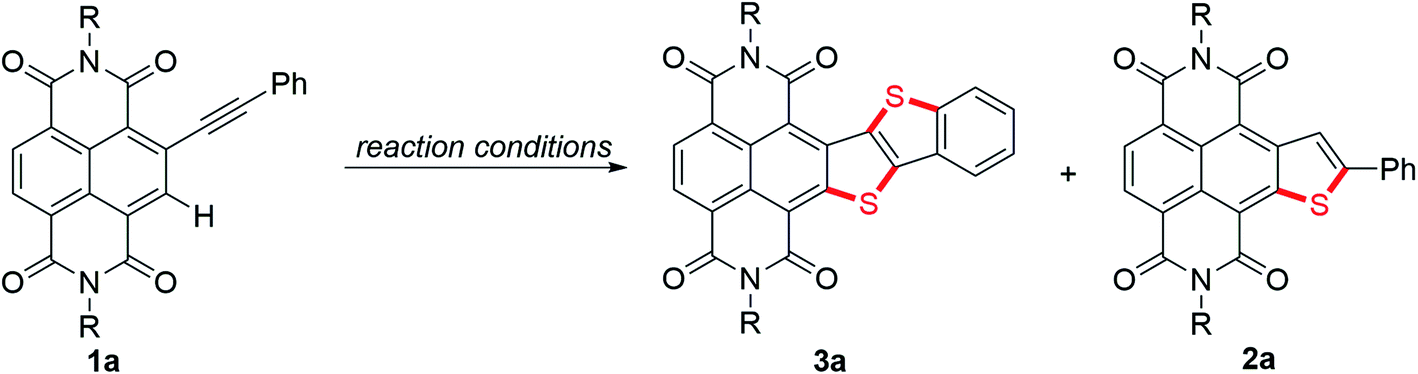
More interestingly, later we found that 3a′ could further undergo a direct thienannulation at 120 °C, without the addition of any transition-metal catalysts and additional oxidants,19 producing an NDI-fused thienoacene product 3a in 78% yield, accompanied by 2a in 19% yield (Scheme 2, eqn (2)). Notably, performing this reaction in air was detrimental and 70% of 3a′ was recovered after the reaction.
Motivated by these results, we expected to construct 3a directly from 1avia a one-pot consecutive thienannulation (stitching thienannulation) process (Table 1). To our delight, direct treatment of 1a with 6 equiv. of K2S at 120 °C for 24 h indeed furnished 3a in 51% yield (entry 1). Higher temperature was found to increase the yield of 3a to 66% (entry 2). As the amount of K2S was reduced, the yield of 3a was decreased, while the yield of 2a was dramatically increased (entries 3 and 4). Addition of 30 equiv. of H2O in the reaction had almost no adverse effects on the stitching thienannulation process (entry 5). Other sulfur reagents, such as Na2S·9H2O and S8, were also employed as sulfur sources for the synthesis of 3a albeit with much lower yields (entries 6 and 7). When the reaction between 1a and S8 was performed in the presence of 10 equiv. of Et3N, 2a was selectively formed in 98% yield (entry 8). Note that the yields of 3a in these reactions are also close related to the concentrations of S3˙− in the DMF solutions (see Fig. S1†).20 Gratifyingly, performing the reaction of 1a with 6 equiv. of K2S at 25 °C for 10 min and then at 140 °C for 24 h led to the formation of 3a in 83% yield (entry 9). This is obviously the fastest way to produce diarene-fused thienothiophenes (Scheme 1a).
|
|
|||
|---|---|---|---|
| Entry | Reaction conditions (reagent (equiv)) | % Yield of 3ab | % Yield of 2ab |
| a Reactions were run in 0.03 mmol scale for 24 h in Ar (R = hexylheptyl). b Isolated yields of 2a and 3a. c 40 h. d Before heating at 140 °C, the reaction mixture was stirred at 25 °C for 10 min. | |||
| 1 | K2S (6), 120 °C | 51 | 19 |
| 2 | K2S (6), 140 °C | 66 | 16 |
| 3 | K2S (4), 140 °C | 42 | 44 |
| 4 | K2S (2), 140 °C | 10 | 64 |
| 5c | K2S (6), H2O (30), 140 °C | 60 | 19 |
| 6 | Na2S·9H2O (6), 140 °C | 20 | 15 |
| 7 | S8 (6), 140 °C | 10 | 71 |
| 8 | S8 (6), Et3N (10), 140 °C | Trace | 98 |
| 9d | K2S (6), 140 °C | 83 | 16 |
Substrate scope
The scope of the double thienannulation was first examined with respect to four arylethynyl substituted NDIs 1b–e, three ortho-arylethynyl substituted PDIs 1f–h21 and one bay-phenylethynyl substituted PDI 1i (Table 2). The stitching thienannulations of (1-naphthyl)ethynyl-NDI 1b, (2-naphthyl)ethynyl-NDI 1c and (4-pyrenyl)ethynyl-NDI 1d, all polycyclic aromatic hydrocarbon (PAH) derivatives, proceeded smoothly even at room temperature and afforded the corresponding NDI-fused thienoacenes 3b–d in good yields (65–87%). The reaction between (2-benzothienyl)ethynyl substituted NDI 1e and K2S was performed at 140 °C and furnished 3e in 31% yield. Besides these acetylenic NDIs, acetylenic PDIs were also susceptible to this transformation. The reactions of (2-naphthyl)ethynyl-PDI 1f and (9-phenanthryl)ethynyl-PDI 1g with K2S were both carried out at 140 °C and afforded the corresponding PDI-fused thienoacenes 3f and 3g in 62% and 72% yields, respectively. Compared with the PAH derivatives 1f and 1g, less reactive ortho-phenylethynyl substituted PDI 1h underwent double thienannulations at a higher temperature (170 °C), affording the desired product 3h in 60% yield. In contrast, the reaction of bay-phenylethynyl substituted PDI 1i with K2S proceeded smoothly at 40 °C and furnished a thienobenzothiopyran product 4 in 63% yield. In this case, the desired PDI-fused thienoacene 3i was not formed perhaps due to the much higher reactivity of PDI unit compared with phenyl ring during the S3˙−-triggered thienannulation.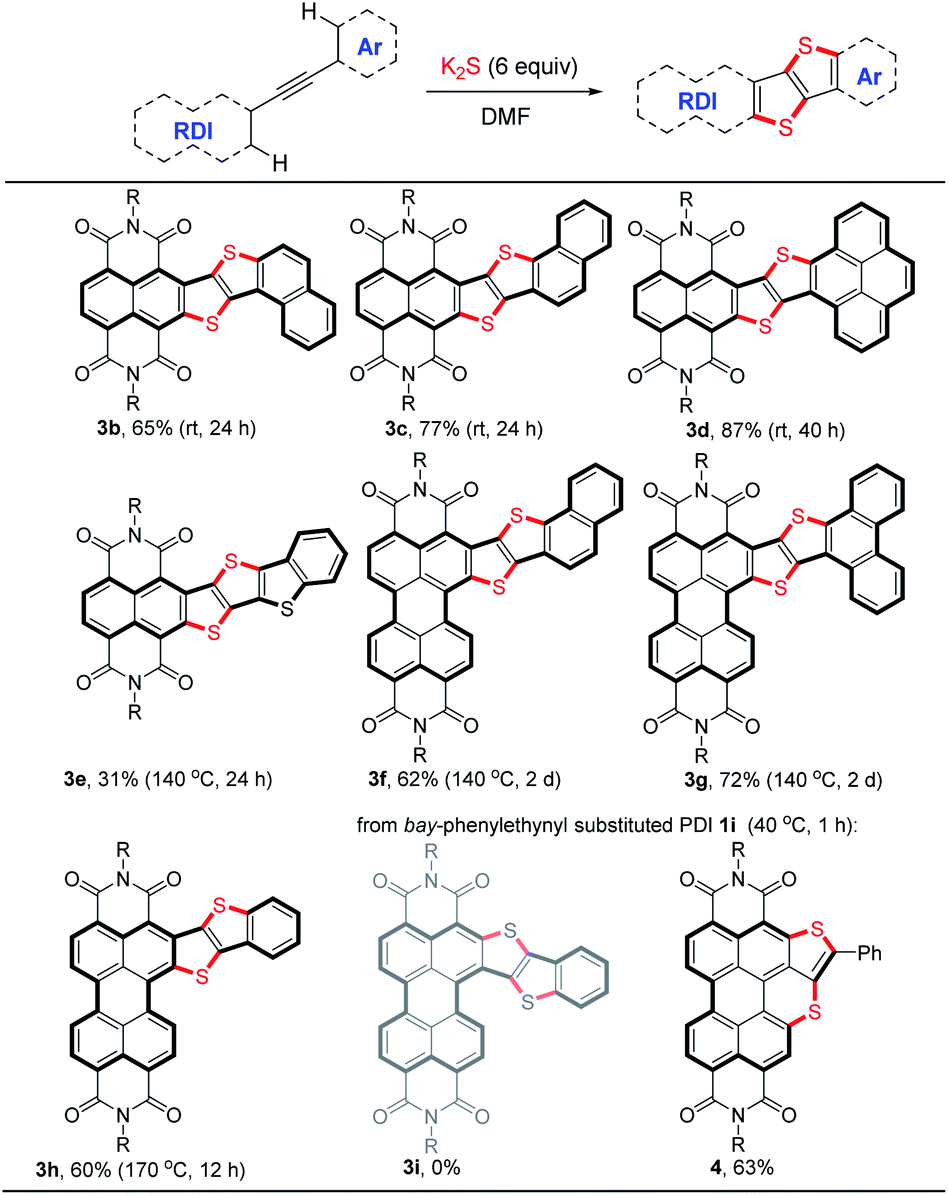
Ladder-type heteroacenes with planar molecular backbones and zero conformational disorder are potentially promising candidates for many optoelectronic applications.1f Using the S3˙−-triggered stitching thienannulation approach, two large NDI-fused ladder-type thienoacenes 3j and 3k could be readily synthesized from the corresponding bisarylethynyl substituted NDI 1j and p-phenylenediyne-linked NDI dimer 1k in good yields (Scheme 3). Notably, during both of these two reactions, four ortho-C–H bonds in the starting compound were cleaved and functionalized in one-pot.
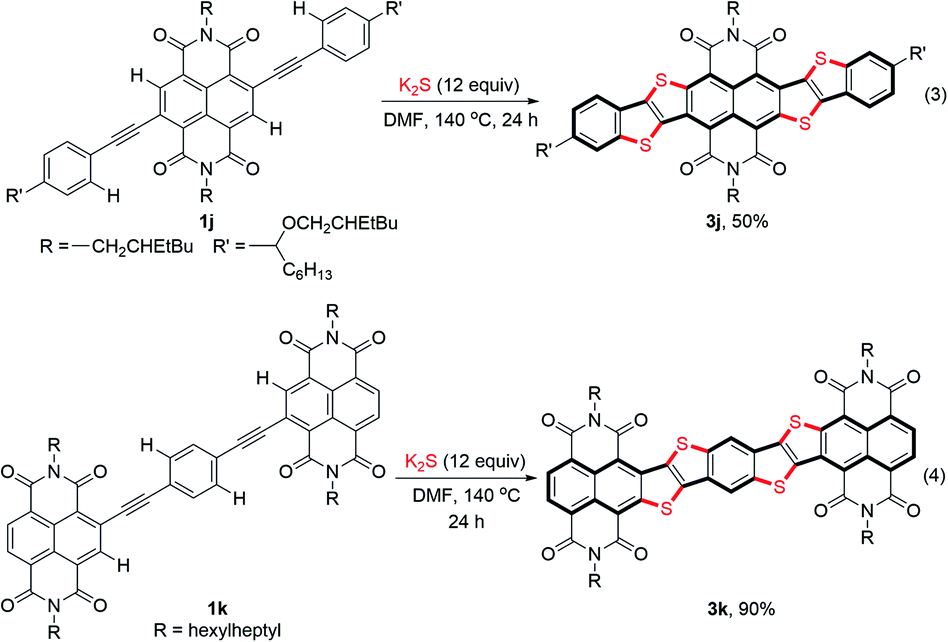 | ||
| Scheme 3 Stitching thienannulations of 1j and 1k. | ||
To further explore the potential applications of this stitching thienannulation strategy, ethyne-, butadiyne-, and hexatriyne-bridged RDI dimers 1l–q were examined (Table 3). With appropriate amounts of K2S, these oligoyne-tethered RDI dimers could directly undergo double, triple, or even quadruple thienannulations to furnish the corresponding π-extended thienoacenes 3l–q.22 The corresponding fused 1,2-dithiin products were not observed.6a
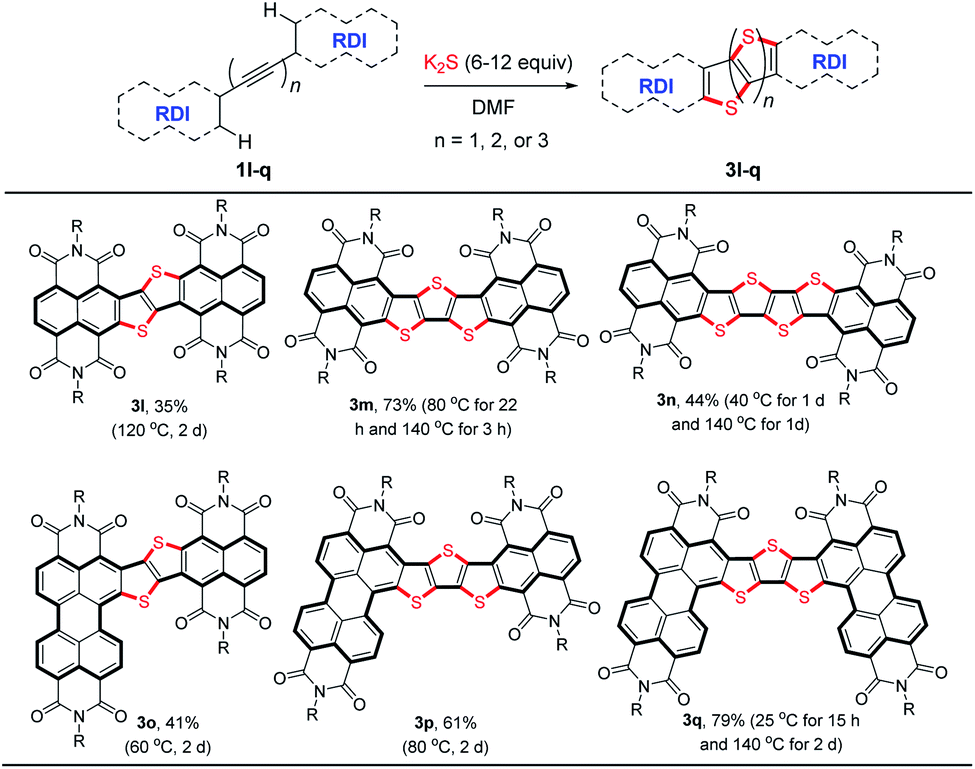
To figure out the detailed thienannulation process of oligoyne-bridged RDI dimers, both the reactions of butadiyne-bridged RDI dimer 1m and 1q with K2S were carefully investigated at lower temperatures (Scheme 4 and Table S4†). Treatment of 1m with 9 equiv. of K2S in DMF at room temperature afforded not only the triple thienannulation product 3m in 15% yield, but also a double thienannulation product 3m′ in 40% yield (Scheme 4).22 Directly heating the DMF solution of 3m′ at 140 °C for 2 h produced 3m in 82% yield. Similar reactions were also observed for 1q (Table S4†), which provided insight into the selective formation of π-extended thienoacenes rather than fused 1,2-dithiins.
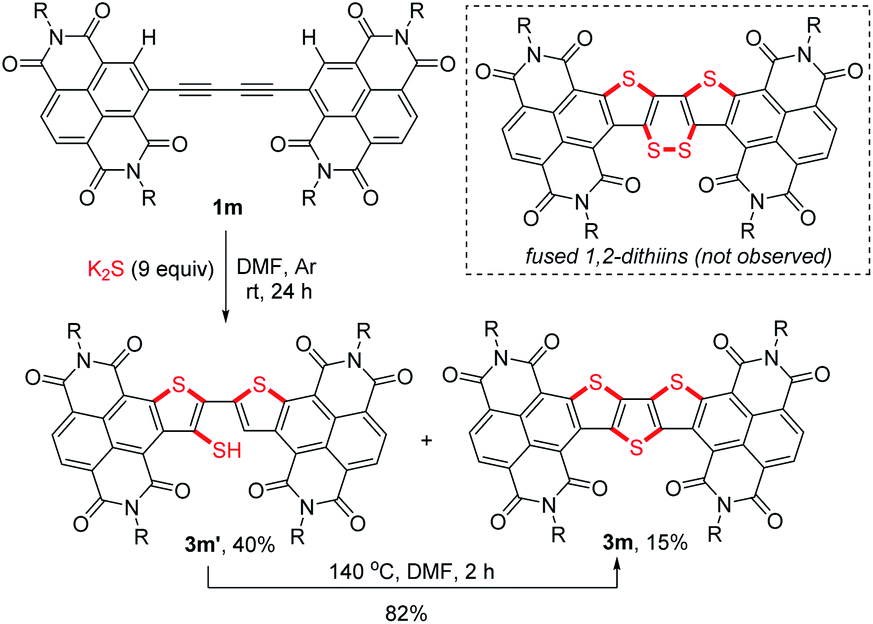 | ||
| Scheme 4 Consecutive thienannulations of 1m. | ||
Further mechanistic studies
To gain a deeper insight into the mechanism of the stitching thienannulation process, we then conducted additional experiments. First, the key step involving the formation of 2a and 3a′ was investigated in detail (Scheme 5). For the reaction of 1a with 6 equiv. of K2S at room temperature for 10 min, the presence of 30 equiv. of H2O did not significantly affect the yields of 2a and 3a′ (Scheme 5a). When 10 equiv. of TEMPO as radical scavenger was added, a TEMPO-adduct was formed instead of the disulfide 3a′ (Scheme 5b, S2 and Fig. S7†). These results confirmed a radical mechanism. Later, directly heating the isolated TEMPO-adduct in DMF at 140 °C afforded the double thienannulation product 3a. Based on this result, we speculate that a labile sulfurated thiophene intermediate that could be easily oxidized during work-up in air to produce 3a′ was formed in the reaction. During optimization of the reaction conditions (see Table S1†), we noticed that reducing the amount of K2S led to the formation of 3a′ in lower yields. Other than that, as the reaction time increased, the yield of 3a′ decreased, while the yield of 2a increased (for the reaction progresses monitored by UV-vis spectroscopy, see Fig. S5†). This changing trend of the yields is more evident for the reaction performed at a slightly higher temperature. For instance, although treatment of 1a with 3 equiv. of K2S at 35 °C for 10 min predominantly produced 3a′ in 36% yield, prolonging the reaction time to 24 h resulted in the selective formation of 2a in 73% yield (Scheme 5c). Thereafter, the latter transformation was further investigated with a deuteration experiment (Scheme 5c). When adding 10 equiv. of D2O in the reaction, 2a-D was isolated in 67% yield with 53% deuterium incorporation at the β-position of the thiophene ring. These results indicate that the labile sulfurated thiophene intermediate can be slowly decomposed in Ar to afford a thiophene anion intermediate which is protonated to afford 2a.23 Therefore, based on the substrate scope and the above results, we proposed that the stitching thienannulation should be initiated with the attack of S3˙− on either alkyne or RDI unit and a sulfurated thiophene intermediate is formed as a key intermediate (see Scheme S3† for a plausible mechanism).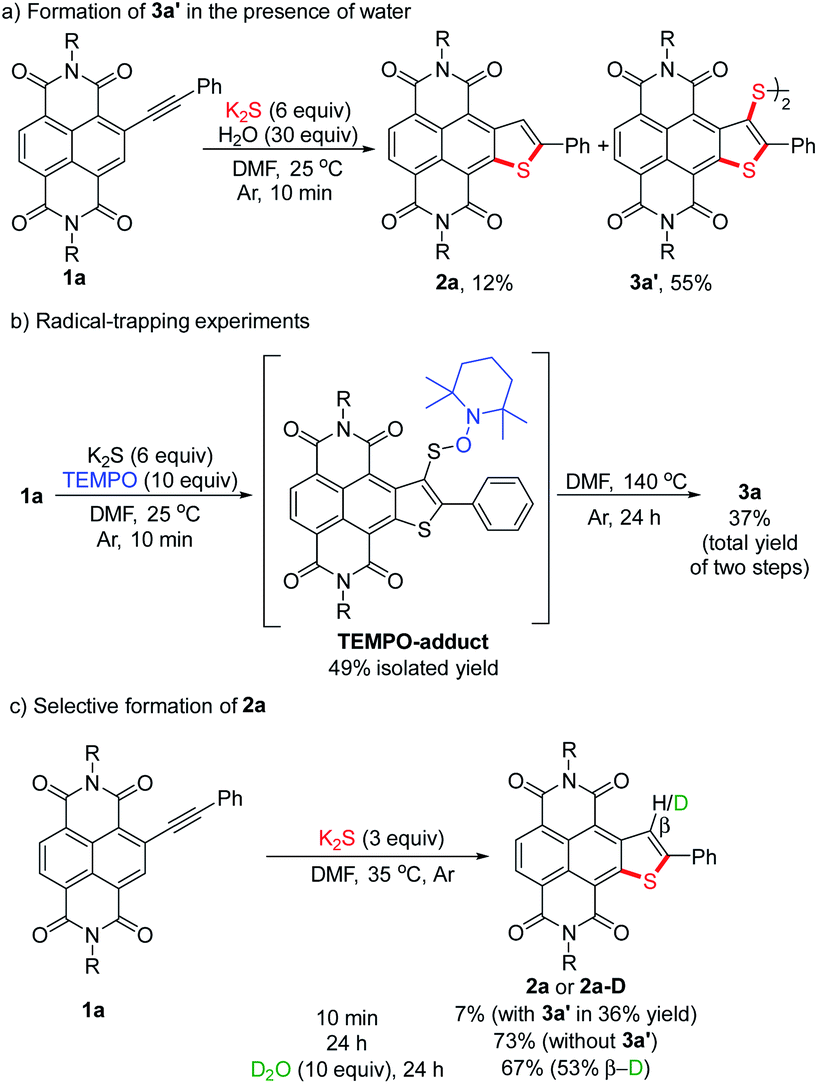 | ||
| Scheme 5 Mechanistic studies on the thienannulation of 1a: (a) formation of 3a′ in the presence of water, (b) radical-trapping experiments and (c) selective formation of 2a. | ||
Structural and physical properties of RDI-fused thienoacenes
These newly formed D–A (3a–h), D–A–D (3j) and A–D–A (3k–q) type thienoacenes possess three major structural advantages in exploration of excellent organic semiconductors: (1) the conformational rigidity that allows them to have reduced reorganization energies and favorable intermolecular π–π stackings; (2) extended π-conjugation lengths and increased electron delocalization, leading to reduced bandgaps and broadened absorptions; and (3) strong dipole–dipole and sulfur–sulfur interactions which lead to ordered intermolecular arrangements. In addition, these molecules all have good solubilities in common organic solvents, such as CH2Cl2, CHCl3, toluene and tetrahydrofuran, which is beneficial for solution processing. To gain a deeper insight into their photophysical properties, UV-vis absorption measurements of 3a–h and 3j–q were performed (Fig. S10–S12†). Compared with simple core-unsubstituted NDI, PDI and mono-thiophene-fused NDI 2a, these RDI-fused thienoacenes exhibited large bathochromic shifts in their UV-vis absorption spectra due to the extension of π-conjugation. Moreover, higher intensities of absorption peaks in the long-wavelength region suggested a strong intramolecular charge transfer in RDI-fused thienoacenes. Thereafter, the electrochemical properties of 3a–h and 3j–q were compared using cyclic voltammetry (Fig. 2, S13–S15 and Table S5†). Compared to 2a, eight D–A type thienoacenes 3a–h have similar reduction potentials, whereas their oxidation potentials are finely tuned by the variation of fused-thiophene-ring and conjugation length, indicating that the LUMOs tend to localize on the RDI skeleton and the HOMOs tend to delocalize in the lateral direction.15 Compared to these asymmetric fused D–A systems, symmetric D–A–D type thienoacene 3j has an obviously lower LUMO level, indicating its higher electron affinity.7b For A–D–A type thienoacenes 3k–q, the HOMO and LUMO energy levels increase as the donor length increase because the large conjugation enhances the electron donating abilities, resulting in narrowed bandgaps. Moreover, compared to NDI fused A–D–A type thienoacenes, PDI fused counterparts have higher LUMO levels and exhibit more red-shifted UV-vis spectra. All these results suggested that the variation in the relative strengths of donor (ladder-type fused thiophene rings) and acceptor (RDI units), fused-ring number, and ring-fusion sequence in these D–A, D–A–D and A–D–A systems allowed finetuning of their absorption profiles, energy levels and charge–transport properties, which may eventually enable practical applications as organic semiconducting materials. | ||
| Fig. 2 Energy level diagram of RDI-fused thienoacenes 3a–h and 3j–q. | ||
Conclusions
In conclusion, we have developed a novel S3˙−-triggered stitching thienannulation approach for the rapid construction of RDI-fused, largely π-extended thienoacenes from ethyne-, butadiyne-, and hexatriyne-linked π-systems. With this facile and straightforward strategy, thieno[3,2-b]thiophene-, dithieno[3,2-b:2′,3′-d]thiophene and tetrathienoacene substructures can be readily constructed in a stitching manner. Detailed mechanistic studies revealed the unique reactivity of S3˙− and displayed the high reactivities of π-deficient RDI rings toward the radical thienannulation process. Furthermore, interesting properties including planar conformation, extended conjugation and precisely controllable electronic properties are integrated into these novel fully fused electron donor–acceptor molecules, which are promising candidates for high performance organic semiconductors. These findings are anticipated to provide new opportunities to explore more general methods for the synthesis of useful organosulfur compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for the financial support from the National Natural Science Foundation of China (21572188, 21772162, 21772165), Natural Science Foundation of Fujian Province of China (2018J01014) and Fundamental Research Funds for the Central Universities (20720180031). H.-J. Zhang thanks Prof. Pierre H. Dixneuf and Dr Daniel Priebbenow (Monash University, Australia) for valuable discussions and corrections.Notes and references
- For reviews, see: (a) J. E. Anthony, Chem. Rev., 2006, 106, 5028 CrossRef CAS PubMed; (b) K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347 CrossRef CAS PubMed; (c) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed; (d) W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113 RSC; (e) M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036 CrossRef CAS PubMed; (f) Z. Cai, M. A. Awais, N. Zhang and L. Yu, Chem, 2018, 4, 2538 CrossRef CAS.
- For selected examples, see: (a) K. Takimiya, H. Ebata, K. Sakamoto, T. Izawa, T. Otsubo and Y. Kunugi, J. Am. Chem. Soc., 2006, 128, 12604 CrossRef CAS PubMed; (b) H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732 CrossRef CAS PubMed; (c) K. Niimi, S. Shinamura, I. Osaka, E. Miyazaki and K. Takimiya, J. Am. Chem. Soc., 2011, 133, 8732 CrossRef CAS PubMed; (d) A. Y. Amin, A. Khassanov, K. Reuter, T. Meyer-Friedrichsen and M. Halik, J. Am. Chem. Soc., 2012, 134, 16548 CrossRef CAS PubMed; (e) Y. Miyata, E. Yoshikawa, T. Minari, K. Tsukagoshi and S. Yamaguchi, J. Mater. Chem., 2012, 22, 7715 RSC; (f) J. Huang, H. Luo, L. Wang, Y. Guo, W. Zhang, H. Chen, M. Zhu, Y. Liu and G. Yu, Org. Lett., 2012, 14, 3300 CrossRef CAS PubMed; (g) T. Zheng, Z. Cai, R. Ho-Wu, S. H. Yau, V. Shaparov, T. Goodson III and L. Yu, J. Am. Chem. Soc., 2016, 138, 868 CrossRef CAS PubMed.
- For intramolecular ring closure of aromatic methyl sulfoxides, see: (a) H. Sirringhaus, R. H. Friend, C. Wang, J. Leuninger and K. Müllen, J. Mater. Chem., 1999, 9, 2095 RSC; (b) P. Gao, D. Beckmann, H. N. Tsao, X. Feng, V. Enkelmann, M. Baumgarten, W. Pisula and K. Müllen, Adv. Mater., 2009, 21, 213 CrossRef CAS.
- D. Cho, X. Yang, V. Enkelmann, M. Baumagarten and K. Müllen, Chem.–Eur. J., 2010, 16, 5119 CrossRef PubMed.
- For iodine-promoted ring closure of stilbene derivatives, see: (a) T. Yamamoto and K. Takimiya, J. Am. Chem. Soc., 2007, 129, 2224 CrossRef CAS PubMed; (b) T. Yamamoto, T. Nishimura, T. Mori, E. Miyazaki, I. Osaka and K. Takimiya, Org. Lett., 2012, 14, 4914 CrossRef CAS PubMed.
- For intramolecular multiple cyclizations of bis(3-bromo-2-thienyl)-acetylenes and bis(o-haloaryl)diacetylenes, see: (a) T. Okamoto, K. Kudoh, A. Wakamiya and S. Yamaguchi, Org. Lett., 2005, 7, 5301 CrossRef CAS PubMed; (b) T. Okamoto, K. Kudoh, A. Wakamiya and S. Yamaguchi, Chem.–Eur. J., 2007, 13, 548 CrossRef CAS PubMed.
- For the construction of thieno[3,2-b]thiophene skeleton via Pd-catalyzed C–H/C–Br couplings, see: (a) T. Mori, T. Nishimura, T. Yamamoto, I. Doi, E. Miyazaki, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 13900 CrossRef CAS PubMed; (b) J. Wen, C. Xiao, A. Lv, H. Hayashi and L. Zhang, Chem. Commun., 2018, 54, 5542 RSC.
- For other methods toward linearly condensed thiophenes, see: (a) K. Xiao, Y. Liu, T. Qi, W. Zhang, F. Wang, J. Gao, W. Qiu, Y. Ma, G. Cui, S. Chen, X. Zhan, G. Yu, J. Qin, W. Hu and D. Zhu, J. Am. Chem. Soc., 2005, 127, 13281 CrossRef CAS PubMed; (b) X. Zhang, A. P. Côté and A. J. Matzger, J. Am. Chem. Soc., 2005, 127, 10502 CrossRef CAS PubMed; (c) L. Meng, T. Fujikawa, M. Kuwayama, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2016, 138, 10351 CrossRef CAS PubMed.
- For reviews, see: (a) L. Chen, C. Li and K. Müllen, J. Mater. Chem. C, 2014, 2, 1938 RSC; (b) F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962 CrossRef PubMed; (c) C. Li, Z. Lin, Y. Li and Z. Wang, Chem. Rec., 2016, 16, 873 CrossRef CAS PubMed; (d) H. Zhylitskaya and M. Stępień, Org. Chem. Front., 2018, 5, 2395 RSC; (e) A. Nowak-Król and F. Würthner, Org. Chem. Front., 2019, 6, 1272 RSC.
- (a) P. E. Hartnett, H. S. S. R. Matte, N. D. Eastham, N. E. Jackson, Y. Wu, L. X. Chen, M. A. Ratner, R. P. H. Chang, M. C. Hersam, M. R. Wasielewski and T. J. Marks, Chem. Sci., 2016, 7, 3543 RSC; (b) Z. Cai, R. J. Vázquez, D. Zhao, L. Li, W.-y. Lo, N. Zhang, Q. Wu, B. Keller, A. Eshun, N. Abeyasing, H. Banaszak-Holl, T. Goodson III and L. Yu, Chem. Mater., 2017, 29, 6726 CrossRef CAS.
- T. Chivers and P. J. W. Elder, Chem. Soc. Rev., 2013, 42, 5996 RSC.
- (a) G. Zhang, H. Yi, H. Chen, C. Bian, C. Liu and A. Lei, Org. Lett., 2014, 16, 6156 CrossRef CAS PubMed; (b) W. Liu, C. Chen and H. Liu, Adv. Synth. Catal., 2015, 357, 4050 CrossRef CAS; (c) Z.-Y. Gu, J.-J. Cao, S.-Y. Wang and S.-J. Ji, Chem. Sci., 2016, 7, 4067 RSC; (d) J.-H. Li, Q. Huang, S.-Y. Wang and S.-J. Ji, Org. Lett., 2018, 20, 4704 CrossRef CAS PubMed; (e) L. Chen, H. Min, W. Zeng, X. Zhu, Y. Liang, G. Deng and Y. Yang, Org. Lett., 2018, 20, 7392 CrossRef CAS PubMed.
- (a) U. Wille, Chem. Rev., 2013, 113, 813 CrossRef CAS PubMed; (b) W. Guo, K. Tao, W. Tan, M. Zhao, L. Zheng and X. Fan, Org. Chem. Front., 2019, 6, 2048 RSC.
- (a) H. Dong, X. Fu, J. Liu, Z. Wang and W. Hu, Adv. Mater., 2013, 25, 6158 CrossRef CAS PubMed; (b) X. Gao and Z. Zhao, Sci. China: Chem., 2015, 58, 947 CrossRef CAS.
- (a) Y. Fukutomi, M. Nakano, J.-Y. Hu, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 11445 CrossRef CAS PubMed; (b) W. Chen, M. Nakano, J.-H. Kim, K. Takimiya and Q. Zhang, J. Mater. Chem. C, 2016, 4, 8879 RSC; (c) M. Nakano and K. Takimiya, Chem. Mater., 2017, 29, 256 CrossRef CAS; (d) K. Takimiya and M. Nakano, Bull. Chem. Soc. Jpn., 2018, 91, 121 CrossRef CAS.
- (a) W. Fan, C. Liu, Y. Li and Z. Wang, Chem. Commun., 2017, 53, 188 RSC; (b) C. Zeng, C. Xiao, X. Feng, L. Zhang, W. Jiang and Z. Wang, Angew. Chem., Int. Ed., 2018, 57, 10933 CrossRef CAS; (c) C. Zeng, D. Meng, W. Jiang and Z. Wang, Org. Lett., 2018, 20, 6606 CrossRef CAS PubMed.
- J. Wu, D. He, Y. Wang, F. Su, Z. Guo, J. Lin and H.-J. Zhang, Org. Lett., 2018, 20, 6117 CrossRef CAS PubMed.
- Before product isolation, the reaction mixture should be stirred in air for 1 h. Otherwise, three thienannulation products (2a, 3a′ and thiol 3a′′) could be formed. For details, see the ESI.†.
- For classical synthesis of dibenzothiophenes from 2-biphenylthiols or 2-biphenylyl disulfides, see: (a) T. Zhang, G. Deng, H. Li, B. Liu, Q. Tan and B. Xu, Org. Lett., 2018, 20, 5439 CrossRef CAS PubMed; (b) K. Nishino, Y. Ogiwara and N. Sakai, Chem.–Eur. J., 2018, 24, 10971 CrossRef CAS PubMed; (c) K. Nishino, Y. Ogiwara and N. Sakai, Eur. J. Org. Chem., 2017, 5892 CrossRef CAS.
- For UV-visible spectra studies of several sulfur reagents, please see Fig. S1.†.
- (a) J. Wu, D. He, L. Zhang, Y. Liu, X. Mo, J. Lin and H.-J. Zhang, Org. Lett., 2017, 19, 5438 CrossRef CAS PubMed; (b) F. Su, G. Chen, P. A. Korevaar, F. Pan, H. Liu, Z. Guo, A. P. H. J. Schenning, H.-J. Zhang, J. Lin and Y.-B. Jiang, Angew. Chem., Int. Ed., 2019, 58, 15273 CrossRef CAS PubMed.
- The structures of 3l, 3m and 3m′ were all confirmed by single-crystal X-ray diffraction analysis (Fig. S3 and S9†)..
- Note that 2a can also be formed through a direct addition of sulfide anion to the ethyne moiety followed by a subsequent intramolecular cyclization (see entry 9 in Table S1† and Fig. 1)..
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1960794 and 1960809. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05332h |
| This journal is © The Royal Society of Chemistry 2020 |