
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal synthesis of (−)-penicimutanin a and related congeners†‡
Haiyong
Yu§
a,
Yan
Zong§
a and
Tao
Xu
 *ab
*ab
aKey Laboratory of Marine Drugs, Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, 5 Yushan Road, Qingdao 266003, China. E-mail: xutao@ouc.edu.cn
bLaboratory for Marine Drugs and Bioproducts, Open Studio for Druggability Research of Marine Natural Products, Pilot National Laboratory for Marine Science and Technology, 1 Wenhai Road, Qingdao 266237, China
First published on 20th November 2019
Abstract
The first total synthesis of penicimutanin A (1) was achieved within 10 steps (LLS). Key innovations in this synthesis consist of (1) a highly efficient electro-oxidative dearomatization; (2) an unprecedented bisoxirane-directed intermolecular aldol reaction from the sterically hindered face of the ketone and (3) the diastereoselective one-step Meerwein–Eschenmoser–Claisen rearrangement enabling the construction of vicinal quaternary stereocenters. Related family members e.g. penicimutanolone (3) and penicimutatin (5) have also been synthesized alongside, elucidating their absolute configurations, hence the absolute configuration of 1.
Introduction
Fungi have been exploited as rich sources to generate structurally appealing molecules featuring synthetically challenging motifs and consecutive stereocenters, which often display promising bioactivities towards life-threatening diseases.1 In particular, for deep sea-derived fungal strains, chemical mutagenesis is commonly used to activate “muted genes” for the discovery of new natural products.2 Penicimutanin A (1) and B (2) are two representative secondary metabolites reported by Cui and coworkers in 2014 (Fig. 1), isolated from a marine-derived fungal strain identified as Penicillium purpurogenum G59 through chemical mutagenesis.3 An interesting observation is that, in addition to 1, penicimutanolone (3) and penicimutatin (5) together with vicinal quaternary center-bearing pyrroloindoline-containing4c–g fructigenine A (4)4a,b were also isolated alongside, indicating their biosynthetic interrelationship. More importantly, it implied that chemical mutagenesis may have activated a “linchpin gene” of the marine-derived fungus which endogenously joined penicimutanolone (3) and fructigenine A (4) together, in contrast to the terrestrial-derived fungal strain where only analogues of 3 were isolated.5,6 These secondary metabolites and their congeners were found to possess a range of biological activities from anti-fungal as a histidine kinase inhibitor to anti-inflammatory as well as anti-tumor effects.3–5 However, the scarcity of these compounds from natural sources greatly hampered their further biological profiling. What's more, the absolute configuration of penicimutanin A (1) and penicimutanolone (3) remained undetermined.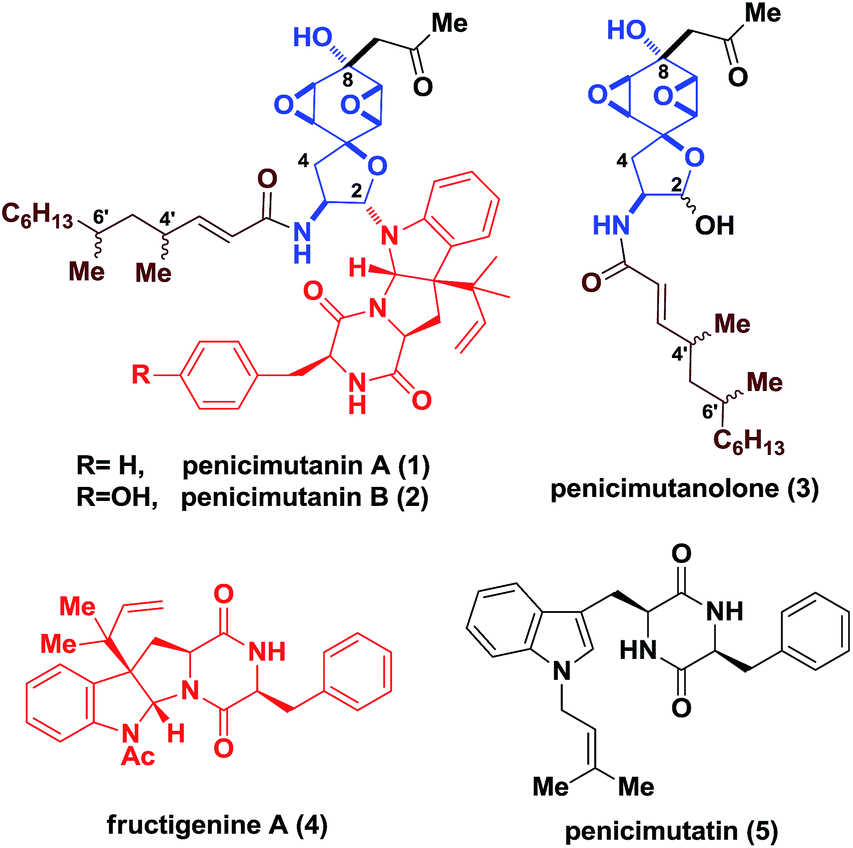 | ||
| Fig. 1 Penicimutanin A and the related congeners. | ||
It is clear that to address the aforementioned structural uncertainty issues, practical and reliable chemical synthesis of these natural products would be necessary. However, to the best of our knowledge, total synthesis of penicimutanin A and its congeners (e.g.2, 3 and 5) has not been reported.
The challenge for their synthesis could be three-fold. First, these natural products are characterized by a fully oxygenated cyclohexane ring which contains an acid/base-sensitive cis-bisoxirane moiety, and is part of a spirocyclic ring skeleton (Fig. 1). Second, the trans relationship between the C8 hydroxyl group and the bisoxirane represents a unique feature which indicated a non-directed expodiation. Third, controlling the stereochemistry for forming the labile C2 hemi-aminal ether is not trivial. On the other hand, the south fragment 4′ contains a diketopiperazine fused hexapyrroloindoline, which possessed a C3 1,1-dimethylpropenyl (reverse prenyl) group generating a vicinal all-carbon quaternary stereocenter.7 The highly oxidized hydrophilic core skeletons, together with the lipophilic aliphatic side chain of penicimutanin, were postulated to account for their observed biological activities.3
In this article, we describe our detailed development of an efficient and scalable approach for the asymmetric total synthesis of penicimutanin A (1), penicimutanolone (3) and penicimutatin (5).
Results and discussion
Biosynthetically, it was hypothesized that penicimutanin A (1) could be disconnected into deAc-fructigenine A (4′) and penicimutanolone (3) through the hemi-aminal ether formation (“linchpin” step). The south fragment 4′ could be accessed through a biomimetic aza-Claisen rearrangement followed by indoline formation from the natural congener 5, which itself was an N-prenylated diketopiperazine compound and could be reached from commercially available (S)-tryptophan ester 6. On the other hand, the northern fragment 3 could be easily accessed through a side chain attaching after a diastereoselective aldol reaction from bisoxirane 7, which might be obtained through an oxidative dearomatization from the commercially available (S)-Cbz-tyrosine 8. This design features a biomimetic, convergent strategy, in the hope of obtaining four natural products from common starting materials (Scheme 1).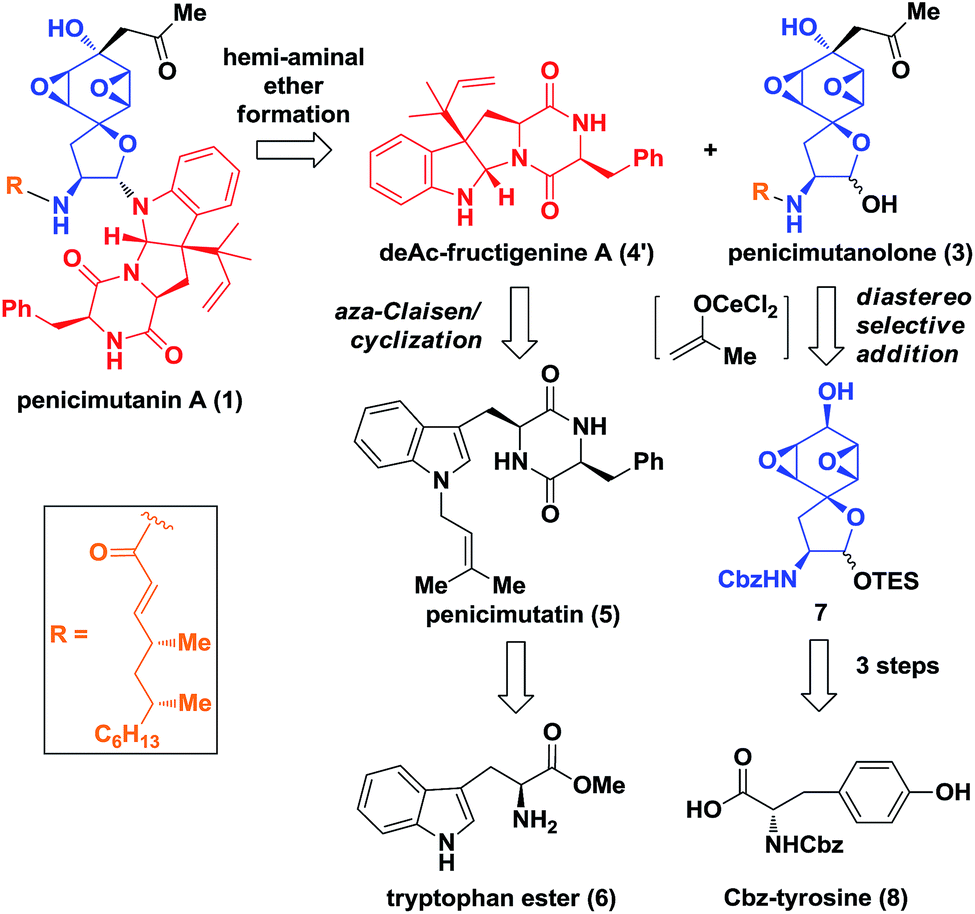 | ||
| Scheme 1 Retro-synthesis. | ||
Our synthesis of deacetyl fructigenine A (4′) started with selective prenylation of commercially available tryptophan ester 6 in 86% yield with isoprenyl bromide 9. The desired prenylation product 10 was condensed with Fmoc-L-tyrosine after Boc removal, affording compound 11 in 76% overall yield. Upon Fmoc deprotection under standard conditions, the diketopiperazine-containing penicimutatin 5 was achieved with 65% yield (Scheme 2A). The intended biomimetic aza-Claisen was proved unviable after extensive condition screening (see the ESI,‡ for details). Our rationale is that the intrinsic sp2 hybridized nitrogen atom of the indole framework inhibited the formation of the chair transition state TS-5, in which the N-atom was twisted into a transient sp3 hybridized form (TS-5 in Scheme 2A).
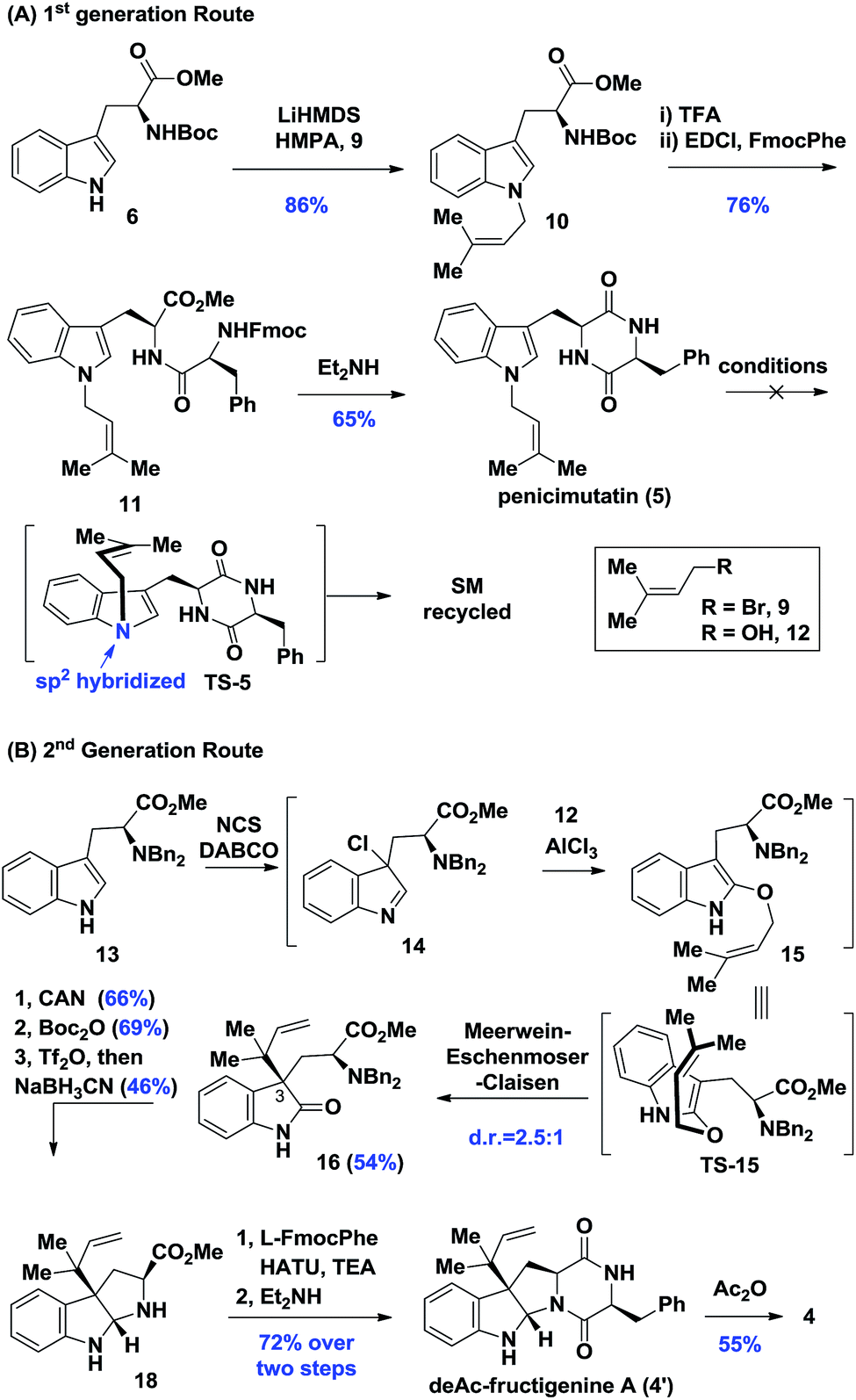 | ||
| Scheme 2 Synthesis of fructigenine A (4) and penicimutatin (5). | ||
Given the above results, a one-pot diastereoselective Meerwein–Eschenmoser–Claisen (MEC) rearrangement8 was developed to construct C3 reverse prenylated9 oxindoles from non-protected C3-ester indole and isoprenyl alcohol through an AlCl3 catalytic protocol. The L-tryptophan derivative 13 was subjected to our AlCl3-mediated conditions. To our delight, the oxindole 16 was achieved in a satisfactory 54% yield with synthetically acceptable stereocontrol (d.r. = 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Compound 16 possessed the key reversely prenylated vicinal quaternary oxindole motif, which was cyclized through reductive amination conditions after Bn-deprotection (66% yield) and Boc protection (69% yield) to achieve hexahydropyrroloindoline 18 in 46% yield. Compound 18 was coupled with L-Fmoc-phenylalanine and cyclized upon Fmoc removal, achieving the deAc-fructigenine 4′ in 72% overall yield. The known natural product fructigenine 4 (reported3a as [α]20D −129.3° (c 0.30, MeOH)) was also successfully obtained through acylation in 55% yield and spectroscopic data of 4 ([α]20D −128.0° (c 0.35, MeOH)) were in good agreement with those previously reported (see the ESI‡ for other spectroscopic data).
:1). Compound 16 possessed the key reversely prenylated vicinal quaternary oxindole motif, which was cyclized through reductive amination conditions after Bn-deprotection (66% yield) and Boc protection (69% yield) to achieve hexahydropyrroloindoline 18 in 46% yield. Compound 18 was coupled with L-Fmoc-phenylalanine and cyclized upon Fmoc removal, achieving the deAc-fructigenine 4′ in 72% overall yield. The known natural product fructigenine 4 (reported3a as [α]20D −129.3° (c 0.30, MeOH)) was also successfully obtained through acylation in 55% yield and spectroscopic data of 4 ([α]20D −128.0° (c 0.35, MeOH)) were in good agreement with those previously reported (see the ESI‡ for other spectroscopic data).
Our synthesis of the penicimutanolone (3) started with oxidative dearomatization of the commercially available (S)-Cbz-tyrosine (8).10 The recent development and innate sustainability of electrochemistry11 prompted us to develop the electro-oxidative dearomatization of 8 (Table 1) in a synthetically useful fashion. The idea is to use an in situ formed milder hypervalent iodine oxidant ii from PhI (i) (Table 1).12 We first tried the electro-oxidation with 1 equivalent of LiClO4 at a constant electrical current of 2.1 mA in TFE. Gratifyingly, the desired spirocycle 19 was isolated in 38% yield. We further investigated the electrical current (entries 2 and 3, Table 1), electrolyte (entries 2–5) and loading of i (entry 2–5) and concluded that the reaction can be optimized to 65% isolated yield when using optimal amounts of the aforementioned factors. Serendipitously, an electrical power cut happened during another trial, which left the reaction standing still overnight after only several hours of electrical oxidation. However the reaction yield was improved to 82% (entry 6, Table 1). A careful adjustment of other co-factors and stirring time enabled us to obtain the oxidative dearomatization reaction in a satisfactory 85% isolated yield.
|
|
|||
|---|---|---|---|
| Entrya | Conditions | Time | Yieldb (%) |
| a All reactions were run with LiClO4 as the electrolyte, PhI as the additive and a C(+)/Pt(−) anode in an undivided cell on a 0.31 mmol scale using 2,2,2-trifluoromethylethanol (TFE) as the solvent at a concentration of 0.04 M at ambient temperature for indicated hours with a constant electrical current unless otherwise noted. b Isolated yields. c The electrical current was stopped after 4 h but stirring was continued for 6 h. d The electrical current was stopped after 5 h but stirring was continued for 5 h. | |||
| 1 | LiClO4 (1 eq.), I = 2.1 mA, PhI (1 eq.) | 8 h | 38 |
| 2 | LiClO4 (10 eq.), I = 6.1 mA, PhI (4 eq.) | 4 h | 64 |
| 3 | LiClO4 (10 eq.), I = 6.1 mA, PhI (4 eq.) | 6 h | 65 |
| 4 | LiClO4 (13 eq.), I = 10 mA, PhI (5 eq.) | 6 h | 65 |
| 5 | LiClO4 (15.6 eq.), I = 10 mA, PhI (6 eq.) | 3 h | 65 |
| 6c | LiClO4 (13 eq.), I = 10 mA, PhI (5 eq.) | 4 + 6 h | 82 |
| 7 | LiClO 4 (15.6 eq.), I = 10 mA, PhI (6 eq.) | 5 + 5 h | 85 |
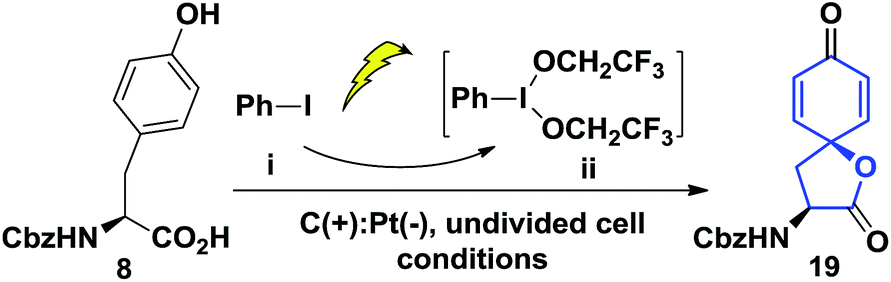
With enough amounts of 19 in hand, we set off to install the key bisoxirane moiety (Scheme 3). Due to 19's instability, a selective NaBH4 reduction followed by TES protection were carried out to release the strain of spirocyclic lactone, yielding dienone 20 in 45% yield over two steps. A diastereoselective Luche-reduction was performed, providing β-OH alcohol (d.r. = 3:1), which allowed the mCPBA mediated epoxidation to proceed smoothly affording 22 as a single diastereoisomer in 51% overall yield. The key reaction precursor 23 was obtained in 80% yield upon DMP oxidation. Crystallographic analysis showed unambiguous structural evidence of compound 23.13
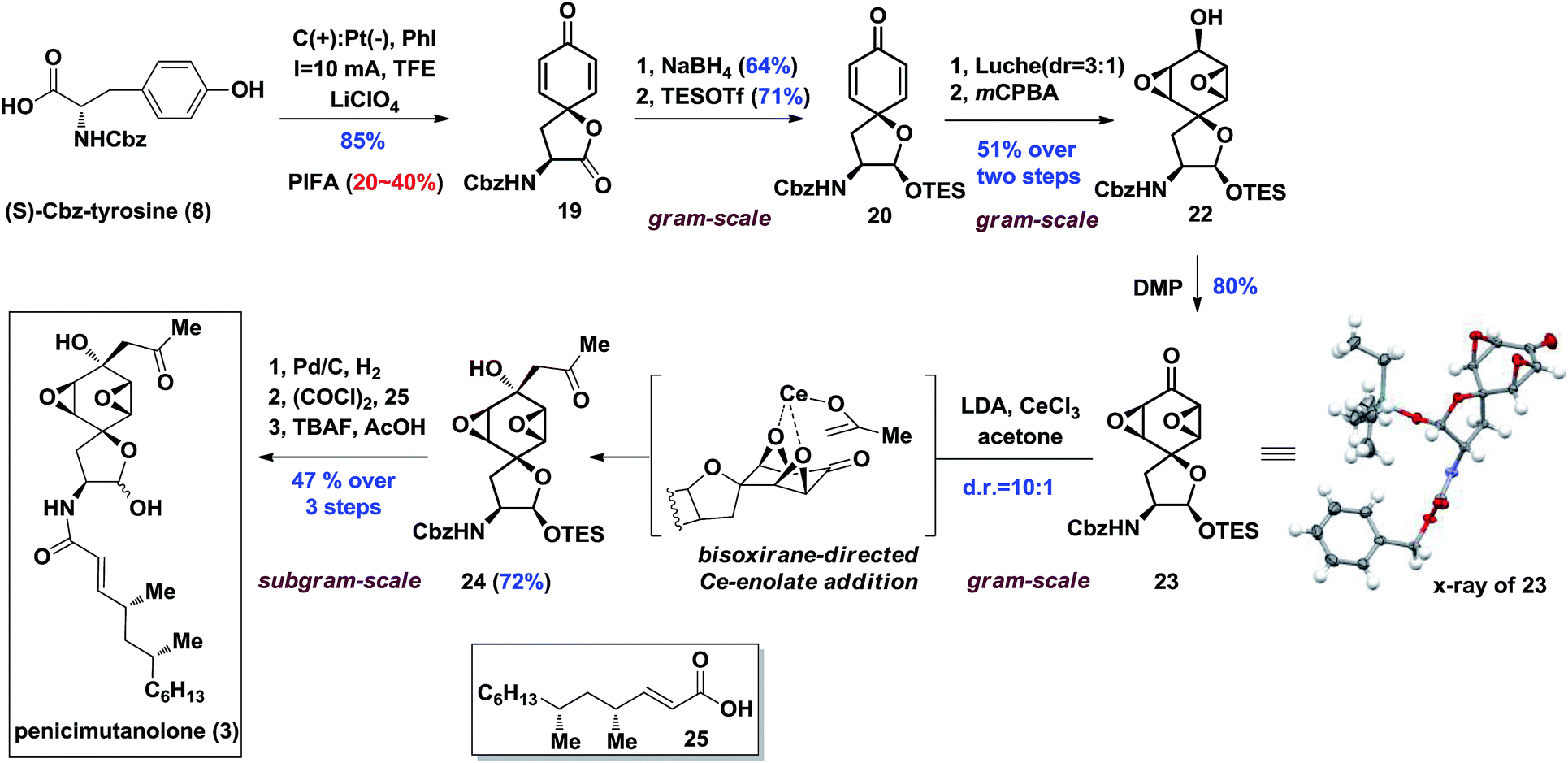 | ||
| Scheme 3 Synthesis of penicimutanolone 3. | ||
It was hypothesized that introduction of an acetonyl group at the C8 position could be delivered from the more sterically hindered β-face of 23, thus affording the desired stereoisomer by utilizing the bisoxirane moiety as a directing group (Scheme 3). With this assumption, the proposed bisoxirane-directed 1,2-addition was attempted by in situ generation of the oxophilic cerium-enolate instead of its traditional lithium counterpart. Gratifyingly, the transformation was realized in a 72% yield, with a resultant diastereoselective (d.r. = 10:1) formation of the desired α-OH isomer 24. The efficacy of this challenging diastereoselective 1,2-addition step was attributed to a better chelation effect of the cerium-enolate as well as its reduced basicity. With the core skeleton in hand, side chain 25 (see the ESI‡ for preparation and characterization of 25) was coupled with compound 24 over a three-step sequence, affording penicimutanolone 3 in an overall 47% yield. The spectroscopic data of synthetic C4′,C6′-(R,R)-penicimutanolone ([α]25D −1.0° (c 0.88, MeOH)) correspond to penicimutanolone (3) ([α]25D −3.5° (c 1.0, MeOH)) very well thus confirming the absolute configuration of C4′ and C6′ of the natural product.
At this point, the linchpin14 step conditions were investigated. Different substrates and conditions were screened (Table 2). Simply mixing the desilylation fragment 26 and south fragment 4′ didn't provide the product 29 (entry 1, Table 2). A Lewis acid promoted in situ formation of an electrophilic oxonium intermediate didn't yield any product either, while slight decomposition of 26 was observed (entries 2 and 3). It was hypothesized that the benzyl group of Cbz might block one side of the oxonium thus inhibiting its reactivity. Different substrates 27 and 28 were prepared and tested, but no fruitful results were obtained (entries 3 and 4). To our delight, 8% of desired 29 could be isolated when TMSCl and DMAP were used and the efficiency was further improved to 35% (55% brsm) yield when, unexpectedly, acidic conditions were employed (entry 6). In addition, an X-ray crystallographic analysis of 28 undisputedly verified all the stereochemistry as well as absolute configurations corresponding to natural product 1 (Table 2).
|
|
|||
|---|---|---|---|
| Entrya | Substrate | Conditions | Yieldb (%) |
| a All reactions were run with 1 equivalent of a Lewis acid and 2 equivalents of a base on a 0.1 mmol scale with DCM (1 mL) at 0 °C and stirring overnight unless otherwise noted. b Isolated yields. c PTSA was used in 1 equivalent and 4 Å MS was added, MeCN/AcOH = 10/1 mixture (1 mL) was used instead of DCM, and the reaction was stirred at 60 °C for 72 hours. | |||
| 1 | 26 | THF, rt, 4 h | 0 |
| 2 | 26 | TMSI, 0 °C, TEA, 4′ DCM | 0 |
| 3 | 26 | TMSCl, KBr, 0 °C, TEA, 4′ DCM | 0 |
| 4 | 27 | TMSBr, 0 °C, TEA, 4′ DCM | 0 |
| 5 | 28 | TMSBr, 0 °C, TEA, 4′ DCM | 0 |
| 6 | 26 | TMSCl, NaI, −20 °C, DMAP, 4′ DCM | 8 |
| 7 | 26 | PTSA, MS, 4′ MeCN/AcOH = 10/1 | 35 (55) |
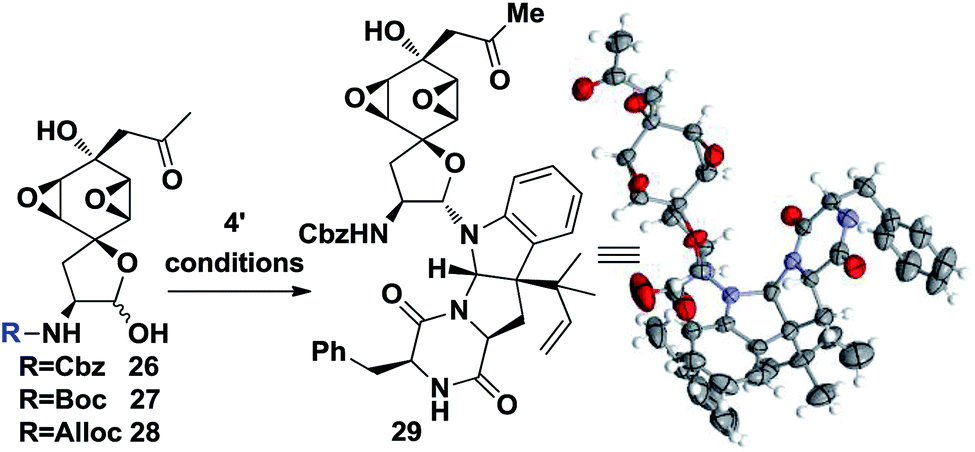
Although the coupling efficiency is still low, we nevertheless tried it on the real substrates of penicimutanolone 3 and deAc-fructigenine 4′. As shown in eqn (1), natural product penicimutanin A (1) was successfully achieved under modified conditions in 26% (77% brsm) yield demonstrating our biomimetic hypothesis. The spectroscopic data of synthetic C4′,C6′-(R,R)-penicimutanin A ([α]25D −91.1° (c 0.25, MeOH)) correspond to the natural sample of penicimutanin A ([α]25D −99.7° (c 0.5, MeOH)) very well thus confirming its absolute configuration (see Table S1 in the ESI‡ for a full comparison of NMR data).
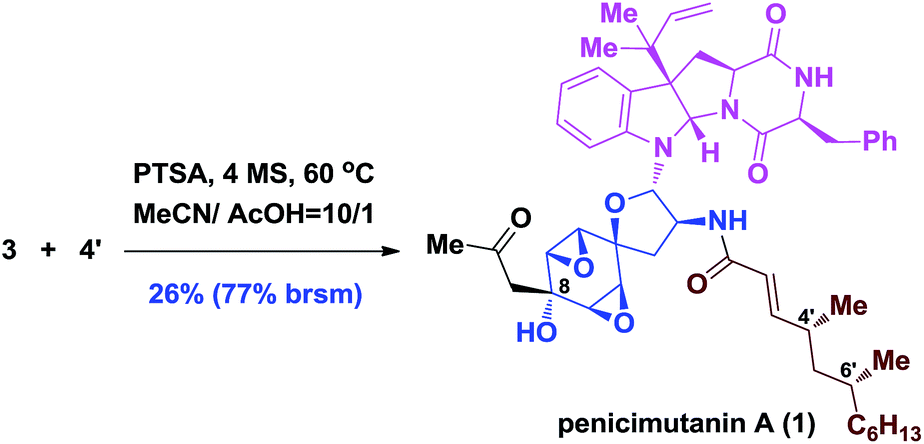 | (1) |
Conclusion
In summary, we have completed the first total synthesis and determined the absolute configurations of penicimutanin A (1) and its related congeners penicimutanolone (3), fructigenine A (4) and penicimutatin (5) in a highly convergent and biomimetic fashion from the commercially available (S)-tryptophan and (S)-tyrosine derivatives for the first time. The highly concise total synthesis of 3 (10 step LLS) was enabled by an improved and highly efficient electro-oxidative dearomatization as well as an unprecedented diastereoselective 1,2-addition directed by the bioxirane moiety. A modified one-pot diastereoselective MEC rearrangement was developed to reach fructigenine A (4) in only 6 steps. Further investigation of this methodology in natural product synthesis and biological profiling of penicimutans is currently on-going in our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank “1000 Talents Plan for Young Professionals” and OUC for a start-up fund. The project was partially funded by the NSFC (No. U1706213, U1606403 and 81973232, 81991522), National Science and Technology Major Project of China (No. 2018ZX09735-004) and SOA (MBRCU201802). T. X. is a Taishan Youth Scholar (tsqn20161012). Qingdao government is acknowledged for a grant of “Leading Innovative Scholars” Program. We thank professor Guangbin Dong of UChicago for helpful discussion and proofreading the manuscript. We thank Mr Jian-Yu Zhang of OUC for help with X-ray crystallography.Notes and references
- For selective comprehensive reviews, see: (a) G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, 2013, 1830, 3670 CrossRef CAS; (b) R. X. Tan and W. X. Zhou, Nat. Prod. Rep., 2001, 18, 448 RSC; (c) M. E. Retab and R. Ebel, Nat. Prod. Rep., 2011, 28, 290 RSC.
- (a) C. Hertweck, Nat. Chem. Biol., 2009, 5, 450 CrossRef CAS; (b) A. A. Brakhage and V. Schroeckh, Fungal Genet. Biol., 2011, 48, 15 CrossRef CAS.
- For isolation of penicimutan congeners and initial bioactivity, see: (a) S.-M. Fang, C.-J. Wu, C.-W. Li and C.-B. Cui, Mar. Drugs, 2014, 12, 1788 CrossRef; (b) N. Wang, C. W. Li, C. B. Cui, B. Cai, L. L. Xu and H. J. Zhu, Eur. J. Med. Chem., 2018, 158, 548 CrossRef CAS.
- For isolation, see: (a) K. Arai, K. Kimura, T. Mushiroda and Y. Yamamoto, Chem. Pharm. Bull., 1989, 37, 2937 CrossRef CAS; for total synthesis, see: (b) S. Takiguchi, T. Iizuka, Y. Kumakura, K. Murasaki, N. Ban, K. Higuchi and T. Kawasaki, J. Org. Chem., 2010, 75, 1126 CrossRef CAS; for pioneering synthesis of pyrroloindoline core bearing vicinal quaternary centers, see: (c) M. Bruncko, D. Crich and R. Samy, J. Org. Chem., 1994, 59, 5543 CrossRef CAS; (d) K. M. Depew, S. P. Marsden, D. Zatorska, A. Zatorski, W. G. Bornmann and S. J. Danishefsky, J. Am. Chem. Soc., 1999, 121, 11953 CrossRef CAS; (e) J. F. Austin, S.-G. Kim, C. J. Sinz, W.-J. Xiao and D. W. C. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5482 CrossRef CAS; (f) L. M. Repka, J. Ni and S. E. Reisman, J. Am. Chem. Soc., 2010, 132, 14418 CrossRef CAS; (g) Y. Sun, R. Li, W. Zhang and A. Li, Angew. Chem., Int. Ed., 2013, 52, 9201 CrossRef CAS.
- For isolation, see: (a) K. Roy, E. K. S. Vijayakumar, T. Mukhopadhyay, S. Chatterjee, R. G. Bhat, J. Blumbach and B. N. Ganguli, J. Antibiot., 1992, 45, 1592 CrossRef CAS; (b) T. Wantanabe, Y. Hashimoto, K. Yamamoto, Y. Hirao, A. Ishihama, M. Hino and R. Utsumi, J. Antibiot., 2003, 56, 1045 CrossRef; (c) C. W. Phoon, B. Somanadhan, S. C. H. Heng, A. Ngo, S. B. Ng, M. S. Butler, A. D. Buss and M. M. Sim, Tetrahedron, 2004, 60, 11619 CrossRef CAS; (d) L. Hammerschmidt, A. H. Aly, M. Abdel-Aziz, W. E. Muller, W. Lin, G. Daletos and P. Proksch, Bioorg. Med. Chem., 2015, 23, 712 CrossRef CAS; (e) F. Koizumi, T. Agatsuma, K. Ando, H. Kondo, Y. Saitoh, Y. Matsuda and S. Nakanishi, J. Antibiot., 2003, 56, 891 CrossRef CAS PubMed.
- (a) A. Mckillop, M. Lee, J. W. Robert, J. K. T. Richard and L. Norman, Tetrahedron Lett., 1993, 34, 5519 CrossRef CAS; (b) P. Wipf, Y. Kim and P. C. Fritch, J. Org. Chem., 1993, 58, 7195 CrossRef CAS.
- For recent total synthesis on constructing quaternary stereocenters, see: (a) K. Li, Y.-F. Wang, X.-M. Li, W.-J. Wang, X.-Z. Ai, X. Li, S.-Q. Yang, J. B. Gloer, B.-G. Wang and T. Xu, Org. Lett., 2018, 20, 417 CrossRef CAS; (b) B. Qiu, X.-T. Li, J.-Y. Zhang, J.-L. Zhan, S.-P. Huang and T. Xu, Org. Lett., 2018, 20, 7689 CrossRef CAS; (c) T. Sun, Y. Zhang, B. Qiu, Y. Wang, Y. Qin, G. Dong and T. Xu, Angew. Chem., Int. Ed., 2018, 130, 2909 CrossRef; (d) X. Zhang, B. N. Nakde, R. Guo, S. Yadav, Y. Gu and A. Li, Angew. Chem., Int. Ed., 2019, 58, 6053 CrossRef CAS; (e) Z. Lu, X. Zhang, Z. Guo, Y. Chen, T. Mu and A. Li, J. Am. Chem. Soc., 2018, 140, 9211 CrossRef CAS PubMed; (f) S. Zhou, K. Xia, X. Leng and A. Li, J. Am. Chem. Soc., 2019, 141, 13718 CrossRef CAS PubMed; (g) D. Wang, Z.-F. Song, W. Wang and T. Xu, Org. Lett., 2019, 21, 3963 CrossRef CAS PubMed.
- For the only two non-selective examples of one-pot Meerwein–Eschenmoser–Claisen rearrangement, see: (a) K. Booker-Milburn, M. Fedouloff, S. Paknoham, J. Strachan, J. Melville and M. Voyle, Tetrahedron Lett., 2000, 41, 4657 CrossRef CAS; (b) V. A. Ignatenko, P. Zhang and R. Viswanathan, Tetrahedron Lett., 2011, 52, 1269 CrossRef CAS.
- Based on our knowledge, only two examples of the one step C3 reverse prenylation of 3-substituted indoles are known, see: (a) J. Ruchti and E. M. Carreira, J. Am. Chem. Soc., 2014, 136, 16756 CrossRef CAS; (b) J. M. Muller and C. B. W. Stark, Angew. Chem., Int. Ed., 2016, 55, 4798 CrossRef.
- A seminal work by Scott and coworkers achieved this reaction but only in 15% yield, see: A. I. Scott, P. A. Dodson, F. McCapra and M. B. Meyers, J. Am. Chem. Soc., 1963, 85, 3702 CrossRef CAS.
- For selected leading reviews of electrochemistry in organic synthesis, see: (a) N. L. Weinberg and H. R. Weinberg, Chem. Rev., 1968, 68, 4449 CrossRef; (b) J. Utley, Chem. Soc. Rev., 1997, 26, 157 RSC; (c) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS; (d) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706 CrossRef CAS PubMed; (e) E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302 CrossRef CAS PubMed.
- (a) Y. Amano and S. Nishiyama, Tetrahedron Lett., 2006, 47, 6505 CrossRef CAS; (b) Y. Amano and S. Nishiyama, Heterocycles, 2008, 75, 1997 CrossRef CAS.
- CCDC 1843076 (23) and 1950968 (29) contain the ESI‡ crystallographic data for this paper.
- For the recent use of the “linchpin” concept for cyclization, see: M. J. Dai, Curr. Org. Chem., 2016, 20, 1850 CrossRef CAS.
Footnotes |
| † This article is dedicated to Professor Zhen Yang on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1843076 and 1950968. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc05252f |
| § Haiyong and Yan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |