
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceα-Glucuronosyl and α-glucosyl diacylglycerides, natural killer T cell-activating lipids from bacteria and fungi†
Satvika
Burugupalli‡
a,
Catarina F.
Almeida‡
bc,
Dylan G. M.
Smith‡
a,
Sayali
Shah
a,
Onisha
Patel
d,
Jamie
Rossjohn
def,
Adam P.
Uldrich
bc,
Dale I.
Godfrey
 *bc and
Spencer J.
Williams
*ab
*bc and
Spencer J.
Williams
*ab
aSchool of Chemistry, Bio21 Molecular Science and Biotechnology Institute, University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: sjwill@unimelb.edu.au
bDepartment of Microbiology and Immunology, Peter Doherty Institute for Infection and Immunity, University of Melbourne, Parkville, Victoria 3010, Australia
cAustralian Research Council Centre of Excellence for Advanced Molecular Imaging, University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: godfrey@unimelb.edu.au
dInfection and Immunity Program, Department of Biochemistry and Molecular Biology, Biomedicine Discovery Institute, Monash University, Clayton, VIC 3800, Australia
eAustralian Research Council Centre of Excellence for Advanced Molecular Imaging, University of Monash, Monash, Victoria 3010, Australia
fInstitute of Infection and Immunity, Cardiff University School of Medicine, Cardiff CF14 4XN, UK
First published on 14th January 2020
Abstract
Natural killer T cells express T cell receptors (TCRs) that recognize glycolipid antigens in association with the antigen-presenting molecule CD1d. Here, we report the concise chemical synthesis of a range of saturated and unsaturated α-glucosyl and α-glucuronosyl diacylglycerides of bacterial and fungal origins from allyl α-glucoside with Jacobsen kinetic resolution as a key step. These glycolipids are recognized by a classical type I NKT TCR that uses an invariant Vα14-Jα18 TCR α-chain, but also by an atypical NKT TCR that uses a different TCR α-chain (Vα10-Jα50). In both cases, recognition is sensitive to the lipid fine structure, and includes recognition of glycosyl diacylglycerides bearing branched (R- and S-tuberculostearic acid) and unsaturated (oleic and vaccenic) acids. The TCR footprints on CD1d loaded with a mycobacterial α-glucuronosyl diacylglyceride were assessed using mutant CD1d molecules and, while similar to that for α-GalCer recognition by a type I NKT TCR, were more sensitive to mutations when α-glucuronosyl diacylglyceride was the antigen. In summary, we provide an efficient approach for synthesis of a broad class of bacterial and fungal α-glycosyl diacylglyceride antigens and demonstrate that they can be recognised by TCRs derived from type I and atypical NKT cells.
Introduction
An important branch of cellular immunity involves specialized T cell populations that can recognize assorted lipid-based antigens when displayed on antigen-presenting cells. Natural killer T (NKT) cells are unconventional T cells with the ability to recognize glycolipid antigens presented by the MHC-class I-like molecule CD1d.1 NKT cells are traditionally classified into two main groups: type I NKT cells and type II NKT cells.2 Type I NKT cells express an invariant TCR α-chain (Vα14-Jα18) in mice (Vα24-Jα18 in humans), and can recognize the prototypical glycolipid antigen α-galactosyl ceramide (α-GalCer). Conversely, CD1d-restricted type II NKT cells express a range of TCR α-and β-chains and do not recognize α-GalCer presented by CD1d. In an earlier study,3 we discovered a population of NKT cells in mice that responded to α-GalCer, yet utilised a different TCR α-chain (Vα10-Jα50) to that of type I NKT cells. These cells therefore share some characteristics of type I and type II NKT cells and were defined as ‘atypical’ NKT cells.4 Atypical NKT cells are present in most human individuals (11/19 human subjects), and in most mice.5 Atypical NKT cells appear to represent up to 10% of the number of CD1d-α-GalCer reactive NKT cells in human blood, and roughly 1% of CD1d-α-GalCer reactive NKT cells in mouse thymus (approx. 104 cells per thymus).5While atypical NKT cells can recognize α-GalCer when presented by CD1d, this synthetic antigen is of uncertain biological relevance. We demonstrated that atypical NKT cells exhibit unique reactivity to an α-glucuronosyl diacylglyceride (α-GlcADAG) from Mycobacterium smegmatis termed Gl-A (1), containing R-tuberculostearic acid (R-TBSA) at the sn-1 position and palmitic acid at the sn-2 position, hereafter referred to as α-GlcADAG-(R-C19:0/C16:0) 1 (Fig. 1).3 Recently, CD1d tetramers loaded with 1 were used to identify a polyclonal subset of NKT cells, including populations of α-GalCer-reactive type I NKT cells, atypical Vα10Jα50 NKT cells and α-GlcADAG reactive type II NKT T cells.6 A 3D structure of a type II-NKT TCR–CD1d-1 complex revealed how this microbial ligand is presented by CD1d to an NKT TCR.6
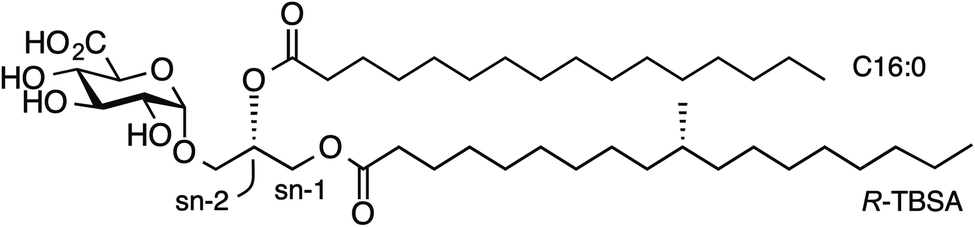 | ||
| Fig. 1 Structure of α-glucuronosyl diacylglyceride Gl-A (1, α-GlcADAG (R-C19:0/C16:0)) from Mycobacterium smegmatis. | ||
Brennan and coworkers reported the isolation of 1 from M. smegmatis MNC strain 13.7 In order to confirm its structural assignment, and to explore its biosynthesis and immunological activity, we completed its total synthesis.8 This synthetic route enabled the preparation of two analogues that exposed an intriguing structure–activity relationship for activation of NKT cells. Thus, while compound 1 stimulated the proliferation of Vα10Jα50 NKT cells from mouse thymus, the C16:0/R-C19:0 regioisomer did not, and a C18:0/C16:0 analogue lacking the distinguishing methyl branch of TBSA was also inactive.3 These results suggest that the methyl branch is critical for loading and/or correct presentation of 1 to the Vα10Jα50 NKT TCR. Brennan and coworkers also reported the isolation from M. smegmatis of the lipoform α-GlcADAG-(C18:1/C16:0), in which a double bond is present between C9-10 of the sn-1 fatty acid ester;7 an identical compound is present in C. glutamicum.8,9 Closely related compounds have been isolated from the pathogenic fungus Aspergillus fumigatus,10Pseudomonas diminuta11 and plants.12 Here we report the details of the approach used to obtain these compounds, a second generation synthesis of α-glucuronosyl diacylglycerides that dramatically shortens and simplifies the preparation of this class of compounds, and allows for the first time the synthesis of unsaturated members of this family. Using a range of analogues varying in the stereochemistry and nature of the lipid, and the carbohydrate head-group, we explore the molecular basis for NKT cell recognition of this class of molecule, and identify atypical TCRs with the ability to recognize a family of α-glucosyl and α-glucuronosyl diacylglycerides of bacterial and fungal origins.
Results
Concise synthesis of α-glucosyl and α-glucuronosyl diacylglycerides
We have reported that α-GlcADAG-(R-C19:0/C16:0) 1 is presented by mouse CD1d and recognised by populations of NKT cells expressing the atypical Vα10Jα50 TCR α-chain, the classical type I NKT TCR α-chain and also certain type II NKT TCRs.3 The original approach to synthesising 1 required 27 total steps involving preparation of three fragments with a longest linear route of 23 steps.8 This included an 8-step synthesis of R-TBSA, and involved 5 types of protecting groups, including the use of benzyl ethers as protecting groups which prevented the synthesis of unsaturated variants. We recently developed a concise approach to α-glucosyl diacylglycerides from allyl α-D-glucopyranoside involving epoxidation and hydrolytic kinetic resolution to build a diastereomerically pure glycidyl α-D-glucopyranoside that could be elaborated to α-GlcDAG derivatives.13 The use of methoxyacetyl groups enabled the preparation of the unsaturated variant Streptococcus pneumoniae α-GlcDAG-(C16:0/Δ11-C18:1; also known as SPN-GlcDAG).14This new approach provided the opportunity to develop a concise synthesis of compound 1 and analogues. We synthesized S-TBSA from (S)-citronellyl bromide, which was converted to alkene 9 with C6H13MgBr and Li2CuCl4,15 subjected to a tandem cross-metathesis/reduction to afford the ethyl ester 10, and then saponified to give S-TBSA 11 (Fig. 2A). R-TBSA was prepared analogously by tandem alkene cross-metathesis of (S)-2,6-dimethyltetradecene (prepared in one step from (R)-citronellyl bromide15) with ethyl 6-heptenoate using Hoveyda–Grubbs II catalyst and in situ reduction using hydrogen, followed by saponification.16
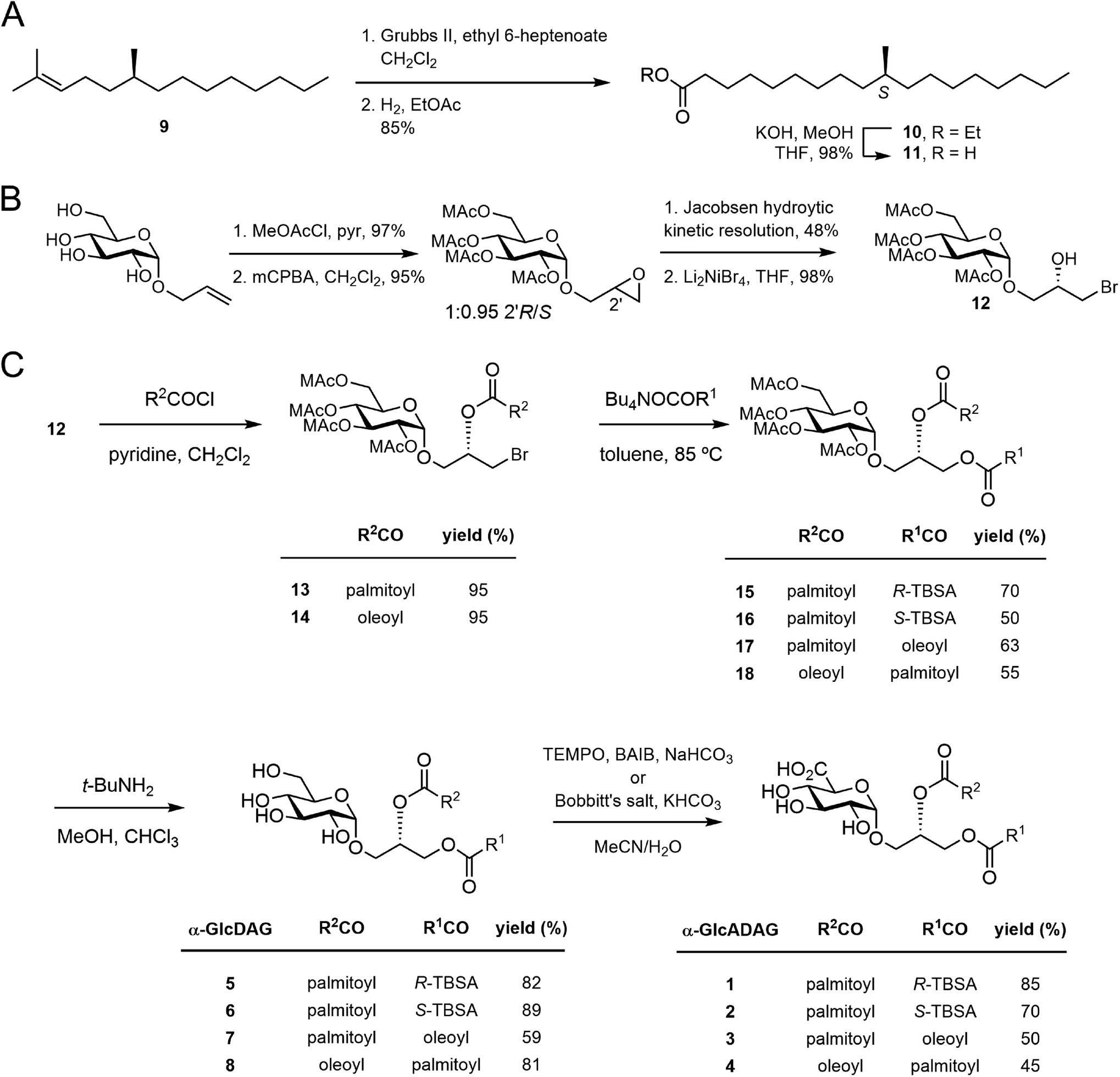 | ||
| Fig. 2 (A) Synthesis of S-TBSA. (B) Previously reported synthesis of the key bromohydrin (ref. 17). (C) Synthesis of α-glucosyl and α-glucuronosyl diacylglycerides. | ||
As reported by Shah et al.17 allyl α-D-glucopyranoside was converted to the methoxyacetate by treatment with MeOAcCl/pyr, treated with mCPBA to obtain a 1:0.95 mixture of 2′R/2′S epoxides, then subjected to hydrolytic kinetic resolution using S,S-Jacobsen's catalyst to provide the stereopure 2′R-epoxide (Fig. 2B). Regioselective brominolysis with Li2NiBr4 afforded bromohydrin 12, a key intermediate allowing introduction of an acyl group at the sn-2 position by direct acylation, followed by introduction of a second acyl group at the sn-1 position by nucleophilic substitution. We have previously used quantitative 13C NMR and 1-13C-labelled fatty acids to show that while stepwise acylation processes that proceed via hydroxyl-ester intermediates give imperfect regioselectivity (owing to intramolecular acyl transfer), this acylation/substitution strategy gives exquisite regioselectivity (R > 99%) at the limit of detection.14 While this analytical approach is limited to 1-13C-labelled fatty acids it is presumed that the regioselectivity observed in the published example extends to the present case. Thus, acylation of 12 with palmitoyl chloride gave monoester 13, and substitution with the tetrabutylammonium salt of R-TBSA afforded the diester 15 (Fig. 2C). The methoxyacetyl groups were removed by treatment with t-BuNH2 in MeOH to afford α-GlcDAG-(R-C19:0/C16:0) 12, and α-GlcADAG-(R-C19:0/C16:0) 1 was obtained by oxidation with TEMPO/BAIB in MeCN/H2O. Application of the same approach to monoester 13 but with S-TBSA afforded α-GlcDAG-(S-C19:0/C16:0) 6 and α-GlcADAG-(S-C19:0/C16:0) 2.
The compatibility of methoxyacetyl groups with alkene-containing fatty acids allowed the total synthesis of unsaturated variants. Thus by a similar approach involving substitution of monoester 13 with the tetrabutylammonium salt of oleic acid (→17), and cleavage of methoxyacetyl groups we obtained α-GlcDAG-(C18:1/C16:0) 7 (Fig. 2C). While catalytic TEMPO/BAIB could be applied to oxidize the primary alcohol of 7 to the acid, better results were obtained using stoichiometric Bobbitt's salt (4-(acetylamino)-2,2,6,6-tetramethyl-1-oxo-piperidinium tetrafluoroborate)18 affording α-GlcADAG-(C18:1/C16:0) 3. By a similar approach (12→14→18) the regioisomers α-GlcDAG-(C16:0/C18:1) 8 and α-GlcADAG-(C16:0/C18:1) 4 were also prepared. Collision-induced dissociation, multistage mass spectrometry was used to show that the synthetic materials had the indicated arrangement of fatty acyl groups. In particular, consistent with previously established rules,8,19 loss of the fatty acyl group at the sn-1 glycerol position produces a more intense peak than that derived from the loss of the sn-2-linked acyl chain.
Glycolipid recognition by type I and atypical NKT TCRs
We explored the ability of the various α-GlcADAGs and α-GlcDAGs produced in this study to be presented in the context of CD1d molecules to both type I and atypical NKT cell-derived TCRs. We used BW58 cell lines transduced with either a type I Vα14Jα18+ Vβ8.2+ NKT TCR (clone VB8-STD20), or two atypical Vα10Jα50+ NKT TCRs (clones A10B8.13 and A10B8.36) with different TCR-β chains. These assays directly measured binding of CD1d–antigen complex with the TCR-transduced cell lines using CD1d-tetramer staining and flow cytometry (Fig. 3A), and their ability to activate these cells as measured by interleukin (IL)-2 production (Fig. 3B).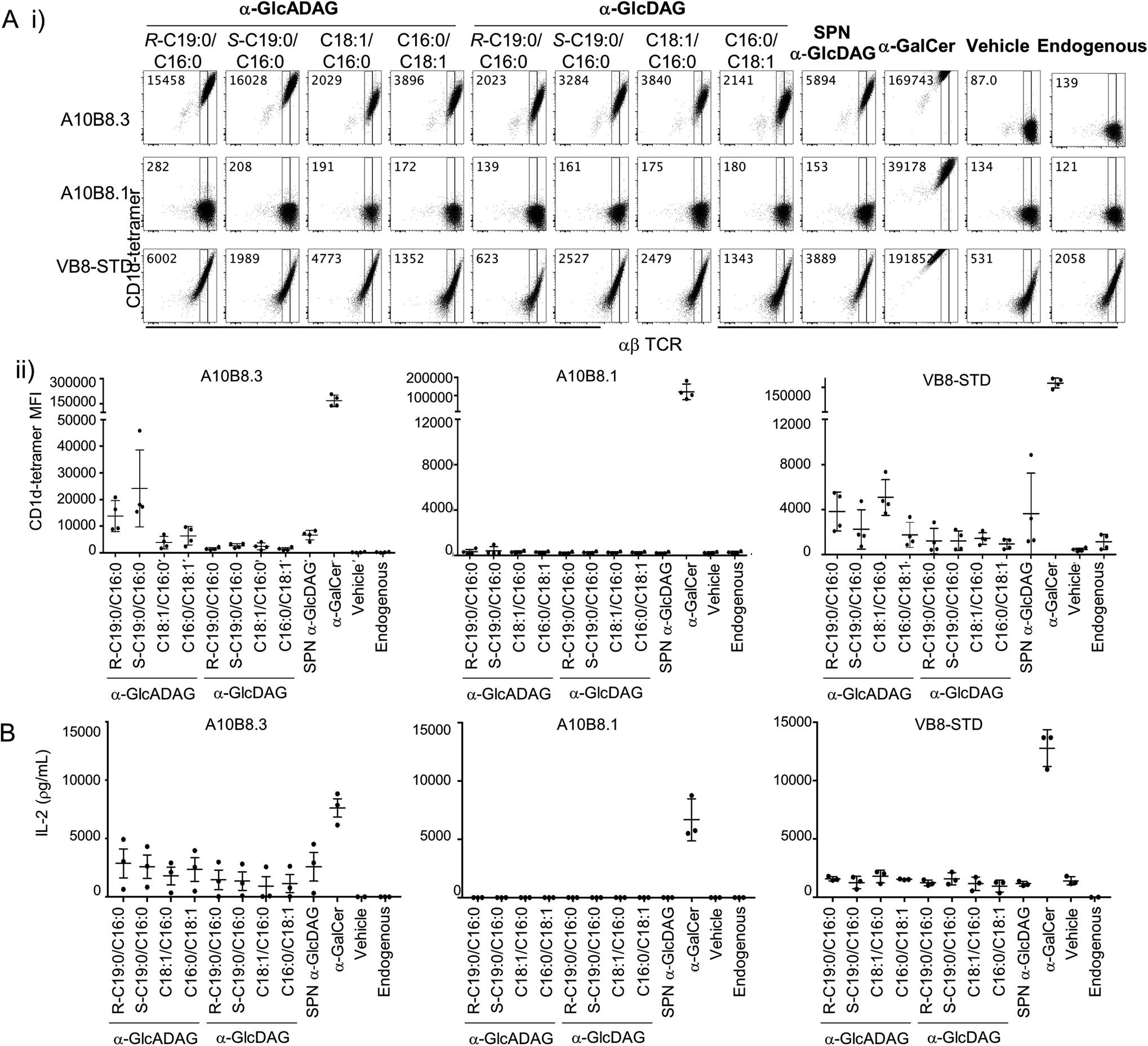 | ||
| Fig. 3 Reactivity of CD1d-restricted TCR+ cell clones to glycolipids. (A) (i) Representative plots of flow cytometry of type I (VB8-STD) and atypical (A10B8.1 and A10B8.3) TCR-transduced BW58 cells labelled with CD1d tetramers loaded with lipids. Numbers in the top left corners of representative plots indicate mean fluorescence intensity (MFI) for each tetramer. (ii) Graphs display MFI for CD1d-tetramers loaded with the indicated lipids (x-axes) for each cell line. Data are representative of 4 independent experiments. One of the 4 datapoints for the CD1d-α-GalCer, -Endo and -α-GlcADAG controls for A10B8.1, 8.3 and VB8-STD TCRs were part of a larger experiment with additional TCRs, previously published in ref. 6 (B) TCR+ cell lines were co-cultured overnight in the presence of plate-bound CD1d–glycolipid–Ag complex (pre-loaded at 10 μg mL−1). After 16 hours, the supernatants were collected and the presence of IL-2 measured by cytometric bead array. Graphs show IL-2 detected in the supernatant. Data is derived from 3 independent experiments. | ||
α-GalCer-loaded CD1d tetramers strongly stained all three NKT TCR+ BW58 cells (Fig. 3A) and plate-bound α-GalCer-loaded CD1d strongly activated these cells (Fig. 3B). Notably, while the VB8-STD type I NKT TCR+ cell line was also mildly reactive to vehicle-loaded CD1d tetramers (which contain endogenous (Endo) lipids incorporated during CD1d synthesis), the two atypical NKT TCR+ cell lines were not, indicating that they are more dependent on the lipid antigen presented by the CD1d–lipid complex. Similarly, plate-bound vehicle-loaded or unloaded CD1d elicited measurable responses (IL-2 secretion) by the VB8-STD type I NKT TCR+ cell line but not by either of the atypical NKT TCR+ cell lines (Fig. 3B). Importantly, the A10B8.3 TCR+ cells were clearly stained by CD1d tetramers loaded with all of the α-GlcDAG and α-GlcADAG variants examined, at levels higher than Endo- or vehicle-loaded CD1d tetramers, indicating that all of the glycolipid lipoforms synthesized in this study can be loaded and presented by CD1d (Fig. 3A). Furthermore, IL-2 was detected in the supernatants of A10B8.3 cells stimulated with plate-bound CD1d in the presence of each synthetic variant at levels above that of Endo- or vehicle-loaded CD1d (Fig. 3B). This indicates that all the synthetic glycolipids are NKT cell agonists. Conversely A10B8.1 TCR+ cells were not stained by CD1d tetramers loaded with any of the synthetic α-GlcDAGs and α-GlcADAGs, indicating that this TCR is highly selective for α-GalCer (Fig. 3A).3 Whilst both the A10B8.3 and VB8-STD TCR+ cell lines were stained by α-GlcADAG-loaded CD1d tetramers, and stimulated by α-GlcADAG-loaded CD1d, both cell lines gave lower mean fluorescent intensities (MFIs) and IL-2 levels than with CD1d-α-GalCer (Fig. 3A and B) indicating that they are weaker agonists than α-GalCer. CD1d tetramers loaded with α-GlcADAG-(S-C19:0/C16:0), -(R-C19:0/C16:0) or -(C16:0/C18:1) displayed the greatest abilities to stain and activate A10B8.3 TCR+ cells (Fig. 3A). Of note, the VB8-STD TCR+ cell line showed a different tetramer staining pattern across the glycolipids, with α-GlcADAG-(C18:1/C16:0) showing the highest (14-fold higher than CD1d–Endo) followed by α-GlcADAG-(C18:1/C16:0), -(R-C19:0/C16:0), -(S-C19:0/C16:0) and -(C16:0/C18:1) (Fig. 3A).
Our studies also included a range of α-glucosyl diacylglycerides (Fig. 3). In addition to those synthesized herein, we also studied SPN-GlcDAG, a cis-vaccenic acid-containing α-GlcDAG from Streptococcus pneumoniae (Fig. S1†).14 CD1d–SPN-GlcDAG tetramers provided moderate staining of the VB8STD TCR+ cells, which reflects moderate staining of type I NKT cells in a previous study,21 and a similar level of staining was seen with α-GlcDAG-(C18:1/C16:0); all other GlcDAGs exhibited little to no staining enhancement over Endo. The A10B8.1 TCR+ cells did not exhibit measurable staining or activation by any of the α-GlcDAGs when presented by CD1d. Conversely, A10B8.3 TCR+ cells were strongly stained (35-fold MFI, relative to Endo) by SPN-GlcDAG-loaded CD1d tetramers, with intermediate levels of staining by α-GlcDAG-(S-C19:0/C16:0), -(R-C19:0/C16:0), -(C18:1/C16:0) and -(C16:0/C18:1) loaded CD1d tetramers. IL-2 was detected in the supernatants of A10B8.3 TCR+ cells in response to plate-bound SPN-GlcDAG- and other GlcDAG-loaded CD1d molecules, demonstrating that they act as NKT cell agonists.
Collectively, these data show that the α-glucuronosyl and α-glucosyl diacylglycerides we have synthesised are capable of binding to CD1d and can stimulate certain NKT cells, including type I and atypical NKT cells. Furthermore, the reactivity of these cells can be influenced by the nature of the lipids, and their arrangement at the sn-1 and sn-2 positions on the glycerol.
CD1d binding footprints for glycolipid recognition by type I and atypical NKT TCRs
Given the divergent TCR usage and different patterns of Ag reactivity by type I and atypical TCRs, the binding footprints of A10B8.3, A10B8.1 or VB8-STD TCRs on CD1d–lipid complexed with α-GalCer, α-GlcADAG-(C19:0/C16:0), and endogenous lipid (Endo) were investigated. This was achieved using a panel of lipid-loaded mutant CD1d molecules in a plate-bound assay similar to that described above (Fig. 4). The CD1d molecules carry point mutations where individual solvent exposed amino acids have been converted to alanine at sites across the docking face of the protein. These amino acids were selected based on previous studies that demonstrated their involvement in contacts with type I20 or type II NKT TCRs.22,23 The impact of these mutations on the activation of the A10B8.3, A10B8.1 or VB8-STD TCR cell lines allowed an assessment of which residues were important for TCR binding. The results were used to generate surface-impact maps displayed on the structure of CD1d, which provide insight into the TCR binding footprint on CD1d.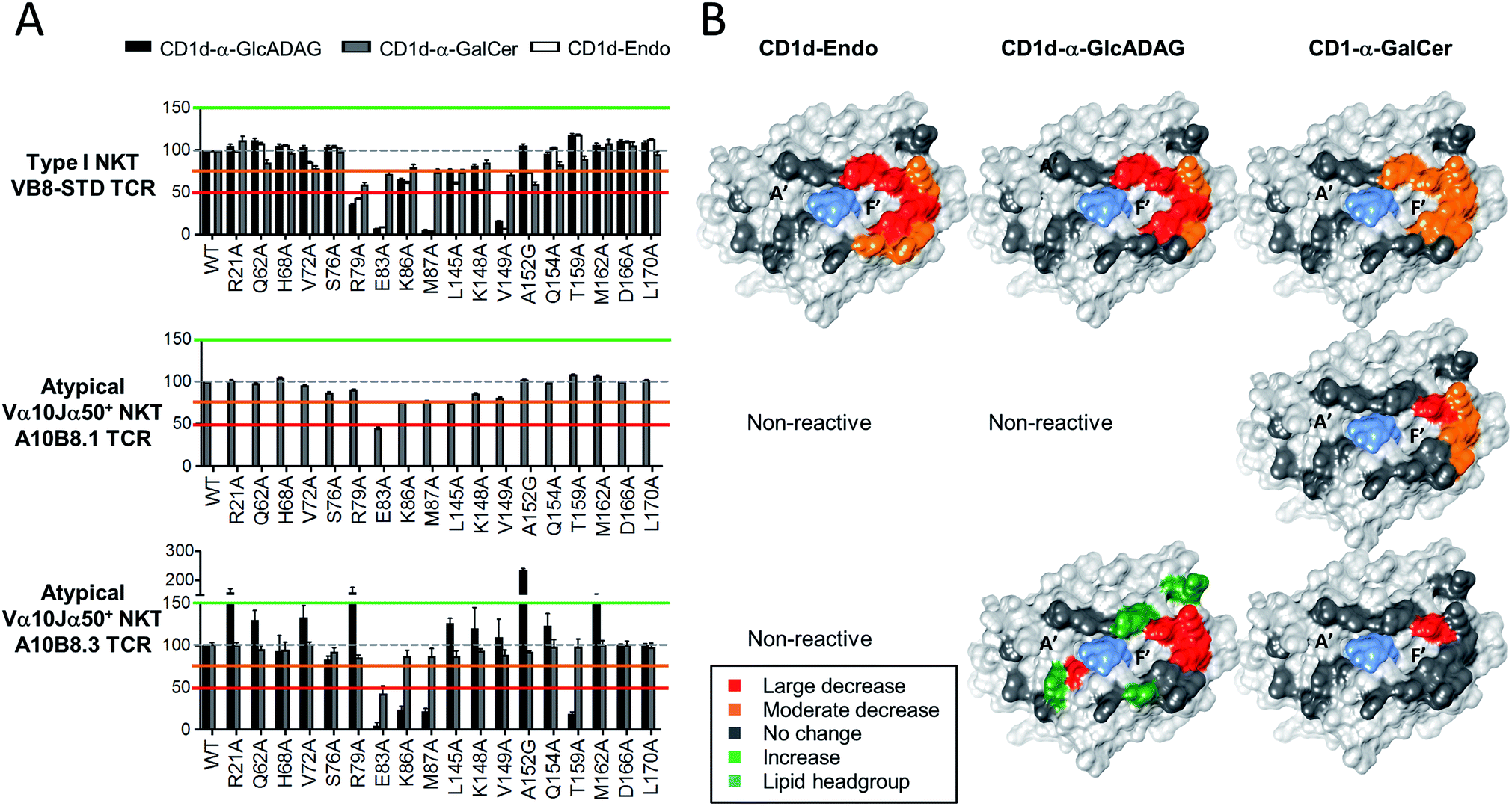 | ||
| Fig. 4 Determination of CD1d-binding patterns by different NKT cell TCRs. Soluble alanine mutants of mouse CD1d loaded with α-GalCer, α-GlcADAG-(C19:0/C16:0) or unloaded (Endo), were immobilized in culture plates and investigated for their ability to stimulate NKT cell lines expressing the A10B8.1 and A10B8.3 atypical NKT TCRs. The VB8-STD type I NKT TCR data referring to CD1d-α-GalCer and CD1d-Endo was published in ref. 6 and is included here as a control. After 16 hours of stimulation, cells were harvested and analysed by flow cytometry for CD69 expression. The level of activation elicited by each mutant was assessed by CD69 upregulation, in comparison to non-activated samples. Data is shown as percentage normalized to the response elicited by the control CD1d mutant (Asp226Ala); Asp226 is located distal to the antigen/TCR binding interface within the α3 domain of CD1d. (A) Graphs depict mean ± SEM derived from n = 2 independent experiments for A10B8.1 or n = 3 for VB8-STD and A10B8.3 TCRs. The data from each experiment was averaged from duplicate cultures. Responses to CD1d-α-GlcADAG (black), CD1d-α-GalCer (grey), and CD1d–Endo (white), are shown for relevant TCRs. (B) Corresponding surfaces of CD1d (PDB code 1Z5L), with impact maps, colour coded as per key. | ||
For the type I TCR (VB8-STD), the TCR footprint data shows that the CD1d residues required for optimal activation are skewed towards the F′ pocket of CD1d (Fig. 4), which is consistent with the type I NKT TCR-CD1d-α-GalCer crystal structure.20 Moreover, the footprints are broadly similar, irrespective of whether CD1d was loaded with α-GlcADAG-(R-C19:0/C16:0), α-GalCer or endogenous ligands (Endo). The footprint for CD1d-α-GalCer differed from the other two, through involvement of L145 and no involvement of K86. The binding footprint for α-GalCer-loaded CD1d mutants undergoes at most only minor to moderate changes in TCR binding, whereas those for the other two ligands are perturbed to a greater degree. This suggests that contacts provided by a high affinity antigen such as α-GalCer can compensate for the loss of certain interactions between CD1d and the type I NKT TCR.
As the atypical A10B8.1 TCR+ cell line could not be activated by CD1d–Endo or CD1d-α-GlcADAG-(C19:0/C16:0) 1, only the effect of CD1d mutants loaded with α-GalCer could be assessed. Again, mutants with variations around the F′ pocket had the most significant loss of activation, although fewer of the mutated residues had an impact relative to the type I TCR with the same ligand (Fig. 4). Among all of the amino acid mutations analysed, Glu83Ala resulted in a greater decrease in A10B8.1 TCR response than for the type I TCR, suggesting that this is a pivotal residue for A10B8.1 TCR recognition of CD1d loaded with this antigen. These results are consistent with structural studies with the Vα10Jα50 Vβ8.1 TCR, in which all these residues were involved in TCR contacts.3
For the atypical A10B8.3 TCR+ cell line, the footprints were investigated for both α-GlcADAG-(C19:0/C16:0) and α-GalCer (Fig. 4). Several mutation positions impacted on A10B8.3 TCR reactivity with the CD1d-α-GlcADAG complex, indicating engagement with CD1d residues Glu83, Lys86, Met87, Thr159. This suggests that the TCR footprint is positioned towards the F′ pocket of CD1d. Interestingly, the responses to Arg21Ala, Arg79Ala, Ser76Ala, Met162Ala and Ala152Gly mutants were greater (>150%) than those elicited by the control, suggesting that these CD1d amino acid side chains might normally impede interactions of the A10B8.3 TCR with CD1d. When α-GalCer was loaded into the mutant CD1d molecules, the A10B8.3 TCR was less affected by mutations in the side chains of CD1d during α-GalCer recognition, with the Glu83Ala mutation being the sole variant for which attenutated activation was observed. This result may reflect the tighter binding of the A10B8.3 TCR to CD1d-α-GalCer, allowing for cellular activation even when binding is partially perturbed at other sites.
Taken together, these data suggest that the α-GlcADAG-(R-C19:0/C16:0) antigen can be recognised in the context of a CD1d footprint skewed towards the F′ pocket of CD1d that is broadly similar for type I, type II,6 and atypical NKT cells, but that the individual amino acid TCR–CD1d contacts vary in their importance for cellular activation. Furthermore, in the case of the atypical TCR, several of the TCR–CD1d contacts appear to normally interfere with recognition of α-GlcADAG, but not of α-GalCer, possibly providing an explanation for the higher potency of the latter antigen.
Discussion
Here we present the total synthesis of important microbial glycolipid antigens that can be targeted by CD1d-restricted NKT cells. These antigens include α-GlcADAG 3 (from M. smegmatis and C. glutamicum), and a second generation total synthesis of α-GlcADAG 1 (from M. smegmatis), as well as a panel of analogues that include molecules representative of α-GlcADAGs from the fungus A. fumigatus. The present approach affords α-GlcADAG 1 in 8 linear steps from allyl α-D-glucopyranoside, and 10 steps in total including the preparation of R-TBSA, compared to the previously reported approach which required 23 linear steps and 27 steps overall. The step-change in efficiency arises from the use of a single protecting group, the avoidance of technically demanding α-glucosylation, the use of an acylation/substitution process to introduce two distinct fatty acyl chains, and the late-stage regioselective oxidation with TEMPO or, better still, Bobbitt's reagent. This approach is compatible with unsaturated fatty acids, and in addition provides the α-glucosyl analogues, which are formed as the penultimate intermediates.Most studies of NKT cells have focussed on the classical Vα14Jα18+ type I NKT cells, which is partly because of the use of the prototypic α-GalCer glycolipid antigen that uniformly detects, and stimulates, these cells. The existence of other types of CD1d-restricted NKT cells (broadly defined as type II NKT cells) has been known for many years,24 but a lack of knowledge of the specific lipid antigens to which they respond has limited investigations into the broader NKT cell family. In some cases lipid antigens such as sulfatide and phosphatidyl glycerol have been helpful for studying discrete subsets of these cells and more such studies are needed. Other studies have shown that some lipid antigens, such as α-GlcDAG or α-GlcADAG, appear to stimulate a subset of type-I NKT cells21 or a distinct population of ‘atypical’ Vα10Jα50+ canonical NKT cells.3,6 Atypical NKT cells do not neatly fit the mould of type I (α-GalCer specific, Vα14Jα18+) or type II (α-GalCer non-reactive, TCR-diverse) because they fall partway between, using TCR-α chains other than Vα14Jα18 (Vα24Jα18 in humans) but are still able to respond to α-GalCer presented by CD1d.3,6 While atypical NKT cells may seem like type I NKT cells by their common reactivity to α-GalCer, their specificity for other glycolipid antigens can differ substantially from type I NKT cells.3,5
We have demonstrated a role for TCR-β chain of atypical Vα10Jα50+ NKT cell TCRs in modulating antigen reactivity of α-GlcADAG and the variants synthesized in this study. Thus, the Vα10Jα50+ VB8.3+ (A10B8.3) TCR recognized a range of α-glucosyl and α-glucuronosyl diacylglycerides whereas the Vα10Jα50 Vβ8.1+ (A10B1.8) TCR did not recognise these α-glucosyl and α-glucuronosyl diacylglycerol lipids but still strongly reacted to the prototypic ligand α-GalCer, despite a conserved CD1d footprint over the F′ pocket of CD1d. This is reminiscent of how TCR-β chain can regulate type I NKT cell recognition of different glycolipid antigens.25 These findings highlight the value in investigating different types of glycolipid antigens in the context of subsets of NKT cells that express distinct TCR-α and TCR-β chains. Through such studies, we are likely to see different hierarchies of antigen recognition depending on the cell types and TCR chains involved, some of which may be overlooked if studies continue to focus exclusively on Vα14Jα18+ type I NKT cells.
An interesting feature of the A10B8.3 TCR is its reactivity to α-GlcADAG-(R-C19:0/C16:0) 1 and α-GlcADAG-(S-C19:0/C16:0) 2, which contain branched lipids. CD1d can tolerate some variation in lipid structure, including unsaturation and lipid chain length.4 However, more complex lipids such SMC124 (a cyclopropane-modified α-galactosylceramide related to plakoside A)26 and the terminal branch present within the agalasphins (the progenitors of α-GalCer)27 suggest that CD1d can tolerate more complex lipid structures within their lipid binding pockets. Indeed, another CD1 protein, CD1b, can load and present methyl-branched and cyclopropanated mycolic acids,28,29 and CD1c can present multiply methyl-branched mannose polyprenolphosphate.30 While some differences are seen between the R- and S-diastereoisomers of the TBSA-containing α-GlcADAGs they are not significant (see Fig. 3Aii), suggesting that CD1d can accommodate these differences in lipid structure without affecting TCR targeting of the CD1d–glycolipid complex. This data complements a recent study that showed a selection of the synthetic lipids described herein could activate type II NKT cells.6
Three-dimensional structures of glycolipid-loaded CD1d molecules complexed with various TCRs reveals that complex formation is driven by co-recognition of the CD1d molecule and the glycolipid head group by the complementarity-determining regions on the α- and β-chains of the TCR.1 Complexes of CD1d–glycolipid with type I TCRs typically display orientation of the TCR towards the F′-pocket of the CD1d molecule where the TCR-α chain is responsible for all antigen contact as well as CD1d, while the TCR-β chain contacts are limited to CD1d. Collectively, the CD1d footprint binding maps determined here for type I and atypical TCRs binding to CD1d-α-GalCer demonstrate similar footprints over the F′ pocket. The footprints are similar for α-GlcADAG-reactive type I, type II,6 and atypical TCRs, while the atypical A10B8.3 TCR requires the involvement of a more central CD1d residue. For all these Vβ8+ NKT TCRs, Glu83 acts as a ‘hot spot’ residue required for binding, regardless of their antigen reactivity.
In summary, we report the total synthesis of a range of α-glucosyl and α-glucuronosyl diacylglycerides from various Gram-positive bacteria, and fungi. These glycolipids bind to CD1d and stimulate type I and atypical Vα10Jα50 NKT cells, with the fine structures of the fatty acid tails of the antigens, and the TCR-β chains of the cells, acting as key determinants of antigen recognition. These findings extend the range of microbial glycolipids that can be presented by CD1d and emphasize that there is scope within the more diverse and less extensively studied atypical NKT cell repertoire for recognition of these antigens.
Conflicts of interest
D. I. G. is a shareholder and member of the scientific advisory board of Avalia Immunotherapies, a company working on NKT cell-based vaccines. The other authors declare no conflicts of interest.Acknowledgements
This work was supported by grants from the Australian Research Council (ARC) (DP160100597; CE140100011) and the National Health and Medical Research Council of Australia (NHMRC) ( 1113293 & 1016629). APU was supported by an ARC Future Fellowship (FT140100278); JR is supported by an Australian ARC Laureate Fellowship; DIG is supported by an NHMRC Senior Principal Research Fellowships (1117766); and SJW was supported by an ARC Future Fellowship (FT130100103).Notes and references
- J. Rossjohn, S. Gras, J. J. Miles, S. J. Turner, D. I. Godfrey and J. McCluskey, Annu. Rev. Immunol., 2015, 33, 169–200 CrossRef CAS PubMed.
- M. V. Dhodapkar and V. Kumar, J. Immunol., 2017, 198, 1015–1021 CrossRef CAS PubMed.
- A. P. Uldrich, O. Patel, G. Cameron, D. G. Pellicci, E. B. Day, L. C. Sullivan, K. Kyparissoudis, L. Kjer-Nielsen, J. P. Vivian, B. Cao, A. G. Brooks, S. J. Williams, P. Illarionov, G. S. Besra, S. J. Turner, S. A. Porcelli, J. McCluskey, M. J. Smyth, J. Rossjohn and D. I. Godfrey, Nat. Immunol., 2011, 12, 616–623 CrossRef CAS PubMed.
- J. Rossjohn, D. G. Pellicci, O. Patel, L. Gapin and D. I. Godfrey, Nat. Rev. Immunol., 2012, 12, 845–857 CrossRef CAS PubMed.
- J. Le Nours, T. Praveena, D. G. Pellicci, N. A. Gherardin, F. J. Ross, R. T. Lim, G. S. Besra, S. Keshipeddy, S. K. Richardson, A. R. Howell, S. Gras, D. I. Godfrey, J. Rossjohn and A. P. Uldrich, Nat. Commun., 2016, 7, 10570 CrossRef CAS.
- C. Almeida, S. Sundararaj, J. Le Nours, T. Praveena, B. Cao, S. Burugupalli, D. G. M. Smith, O. Patel, M. Brigl, D. G. Pellicci, S. J. Williams, A. P. Uldrich, D. I. Godfrey and J. Rossjohn, Nat. Commun., 2019, 10, 5242 CrossRef PubMed.
- B. A. Wolucka, M. R. McNeil, L. Kalbe, C. Cocito and P. J. Brennan, Biochim. Biophys. Acta, 1993, 1170, 131–136 CrossRef CAS.
- B. Cao, X. Chen, Y. Yamaryo-Botte, M. B. Richardson, K. L. Martin, G. N. Khairallah, T. W. Rupasinghe, R. M. O'Flaherty, R. A. O'Hair, J. E. Ralton, P. K. Crellin, R. L. Coppel, M. J. McConville and S. J. Williams, J. Org. Chem., 2013, 78, 2175–2190 CrossRef CAS PubMed.
- R. V. Tatituri, P. A. Illarionov, L. G. Dover, J. Nigou, M. Gilleron, P. G. Hitchen, K. Krumbach, H. R. Morris, N. Spencer, A. Dell, L. Eggeling and G. S. Besra, J. Biol. Chem., 2007, 282, 4561–4572 CrossRef CAS PubMed.
- T. Fontaine, C. Lamarre, C. Simenel, K. Lambou, B. Coddeville, M. Delepierre and J. P. Latge, Carbohydr. Res., 2009, 344, 1960–1967 CrossRef CAS.
- S. G. Wilkinson, Biochim. Biophys. Acta, 1969, 187, 492–500 CrossRef CAS.
- Y. Okazaki, H. Otsuki, T. Narisawa, M. Kobayashi, S. Sawai, Y. Kamide, M. Kusano, T. Aoki, M. Y. Hirai and K. Saito, Nat. Commun., 2013, 4, 1510 CrossRef PubMed.
- S. Shah, J. M. White and S. J. Williams, Org. Biomol. Chem., 2014, 12, 9427–9438 RSC.
- M. B. Richardson, D. G. Smith and S. J. Williams, Chem. Commun., 2017, 53, 1100–1103 RSC.
- I. O. Roberts and M. S. Baird, Chem. Phys. Lipids, 2006, 142, 111–117 CrossRef CAS PubMed.
- S. Burugupalli, M. B. Richardson and S. J. Williams, Org. Biomol. Chem., 2017, 15, 7422–7429 RSC.
- S. Shah, M. Nagata, S. Yamasaki and S. J. Williams, Chem. Commun., 2016, 52, 10902–10905 RSC.
- M. A. Mercadante, C. B. Kelly, J. M. Bobbitt, L. J. Tilley and N. E. Leadbeater, Nat. Protoc., 2013, 8, 666–676 CrossRef CAS PubMed.
- G. Guella, R. Frassanito and I. Mancini, Rapid Commun. Mass Spectrom., 2003, 17, 1982–1994 CrossRef CAS PubMed.
- D. G. Pellicci, O. Patel, L. Kjer-Nielsen, S. S. Pang, L. C. Sullivan, K. Kyparissoudis, A. G. Brooks, H. H. Reid, S. Gras, I. S. Lucet, R. Koh, M. J. Smyth, T. Mallevaey, J. L. Matsuda, L. Gapin, J. McCluskey, D. I. Godfrey and J. Rossjohn, Immunity, 2009, 31, 47–59 CrossRef CAS PubMed.
- Y. Kinjo, E. Tupin, D. Wu, M. Fujio, R. Garcia-Navarro, M. R. Benhnia, D. M. Zajonc, G. Ben-Menachem, G. D. Ainge, G. F. Painter, A. Khurana, K. Hoebe, S. M. Behar, B. Beutler, I. A. Wilson, M. Tsuji, T. J. Sellati, C. H. Wong and M. Kronenberg, Nat. Immunol., 2006, 7, 978–986 CrossRef CAS PubMed.
- E. Girardi, I. Maricic, J. Wang, T. T. Mac, P. Iyer, V. Kumar and D. M. Zajonc, Nat. Immunol., 2012, 13, 851–856 CrossRef CAS PubMed.
- O. Patel, D. G. Pellicci, S. Gras, M. L. Sandoval-Romero, A. P. Uldrich, T. Mallevaey, A. J. Clarke, J. Le Nours, A. Theodossis, S. L. Cardell, L. Gapin, D. I. Godfrey and J. Rossjohn, Nat. Immunol., 2012, 13, 857–863 CrossRef CAS PubMed.
- A. K. Singh, P. Tripathi and S. L. Cardell, Front. Immunol., 2018, 9, 1969 CrossRef PubMed.
- G. Cameron, D. G. Pellicci, A. P. Uldrich, G. S. Besra, P. Illarionov, S. J. Williams, N. L. La Gruta, J. Rossjohn and D. I. Godfrey, J. Immunol., 2015, 195, 4604–4614 CrossRef CAS PubMed.
- A. J. Tyznik, E. Farber, E. Girardi, A. Birkholz, Y. Li, S. Chitale, R. So, P. Arora, A. Khurana, J. Wang, S. A. Porcelli, D. M. Zajonc, M. Kronenberg and A. R. Howell, Chem. Biol., 2011, 18, 1620–1630 CrossRef CAS PubMed.
- M. Morita, K. Motoki, K. Akimoto, T. Natori, T. Sakai, E. Sawa, K. Yamaji, Y. Koezuka, E. Kobayashi and H. Fukushima, J. Med. Chem., 1995, 38, 2176–2187 CrossRef CAS.
- I. Van Rhijn, A. Kasmar, A. de Jong, S. Gras, M. Bhati, M. E. Doorenspleet, N. de Vries, D. I. Godfrey, J. D. Altman, W. de Jager, J. Rossjohn and D. B. Moody, Nat. Immunol., 2013, 14, 706–713 CrossRef CAS.
- A. Chancellor, A. S. Tocheva, C. Cave-Ayland, L. Tezera, A. White, J. R. Al Dulayymi, J. S. Bridgeman, I. Tews, S. Wilson, N. M. Lissin, M. Tebruegge, B. Marshall, S. Sharpe, T. Elliott, C. K. Skylaris, J. W. Essex, M. S. Baird, S. Gadola, P. Elkington and S. Mansour, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E10956–e10964 CrossRef CAS.
- D. B. Moody, T. Ulrichs, W. Muhlecker, D. C. Young, S. S. Gurcha, E. Grant, J. P. Rosat, M. B. Brenner, C. E. Costello, G. S. Besra and S. A. Porcelli, Nature, 2000, 404, 884–888 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05248h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |