DOI:
10.1039/C9SC05036A
(Edge Article)
Chem. Sci., 2020,
11, 2155-2160
Coordinating bioorthogonal reactions with two tumor-microenvironment-responsive nanovehicles for spatiotemporally controlled prodrug activation†
Received
6th October 2019
, Accepted 9th January 2020
First published on 13th January 2020
Abstract
Precise activation of prodrugs in tumor tissues is critical to ensuring specific antitumor efficacy, meanwhile reducing the serious adverse effects. Here, a spatiotemporally controlled prodrug activation strategy was provided by integrating the inverse electron demand Diels–Alder (IEDDA) reaction with two tumor-microenvironment-responsive nanovehicles. The prodrug (Dox-TCO) and [4-(6-methyl-1,2,4,5-tetrazin-3-yl)phenyl]methanamine (Tz) were separately camouflaged into low pH and matrix metalloproteinase 2 (MMP-2) sensitive micellar nanoparticles. After systemic administration, only in the tumor tissues could both the nanovehicles dissociate via responding to two special tumor microenvironments, with Dox-TCO and Tz released and then immediately triggering the prodrug activation through the IEDDA reaction. The hierarchically regulated and locally confined Dox liberation led to dramatically decreased side-effects that were much lower than those of the clinical Doxorubicin Hydrochloride Liposomal Injection (Doxil), while the antitumor therapeutic effect was potent.
Introduction
Prodrugs, carrying chemical moieties that block certain essential parts of the parent drugs, have long been used to reduce the serious adverse effects in cancer therapy.1–3 And the bioorthogonal cleavage reaction is enjoying widespread attention in prodrug activation because it can liberate potent cytotoxic drugs under physiological conditions.4–6 Especially, the inverse electron demand Diels–Alder (IEDDA) reaction between tetrazine and trans-cyclooctene (TCO) has shown sufficiently in vivo potential thanks to its satisfactory biocompatibility and fast elimination reaction kinetics.7–13 To specifically activate prodrugs in tumor sites through the IEDDA reaction, tetrazine groups, one of the reactive units, were introduced into tumors through local injection14 or in situ enzyme-directed supramolecular self-assembly,15 followed by the administration of prodrugs containing releasable TCO groups. Although effective, the former is limited to resectable tumors, and the latter is only suitable for tumors that over-express phosphatase. Moreover, despite the pre-accumulation of tetrazine groups, it is difficult for the intravenously (i.v.) injected small-molecule prodrugs to be entrapped into tumors due to their short half-life time. A more universal and efficient strategy that can augment the IEDDA reaction based local prodrug activation, thus enhancing the specific tumor therapeutic effect, is still urgently needed.
Nanovehicles are excellent candidates for anti-tumoral drug delivery.16–18 Moreover, the tumor microenvironment (TME) exhibits special physicochemical properties and structures.19 Therefore, smart nanovehicles responding to the unique TME have been developed to mimic Trojan Horse for drug delivery.20–22 Unfortunately, most single-response strategies potentially suffer from the issue of nonspecific activation since other tissues or sites often share certain features of TME.23–25 In contrast, few normal tissues or sites exhibit two or more similar tumoral characteristics.26 Hence a strategy that exploits more than one feature of TME27,28 should significantly improve the specificity of anticancer drug delivery, which would be further greatly enhanced if coordinated with the IEDDA reaction based local prodrug activation approach, but remains unexplored.
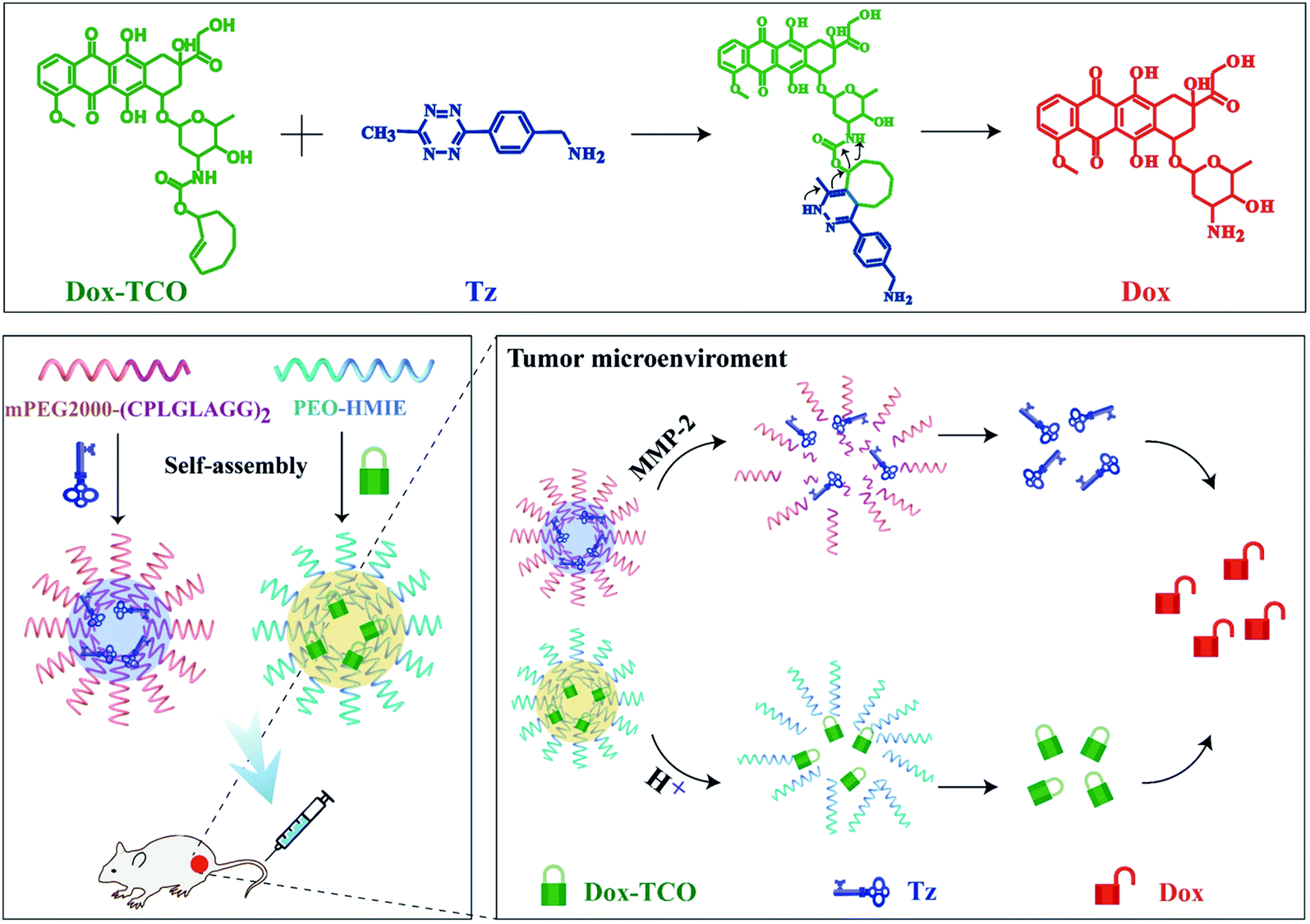
Here, we designed two micellar nanoparticles that individually respond to low pH29–32 and MMP-2,33–36 and prepared corresponding nanovehicles loaded with Dox-TCO or Tz. After systemic administration, they steadily accumulated in tumors and then dissociated via the special TME, wherein Dox-TCO and Tz were released and encountered each other. Immediately, the IEDDA reaction led to the local liberation of Dox, resulting in significantly decreased side-effects and highly efficient antitumor efficacy (Scheme 1). Such a strategy, which integrates key-to-lock bioorthogonal prodrug activation with two tumor-microenvironment-responsive nanovehicles,27,28 showed great promise for the development of a novel platform for highly specific cancer therapy.
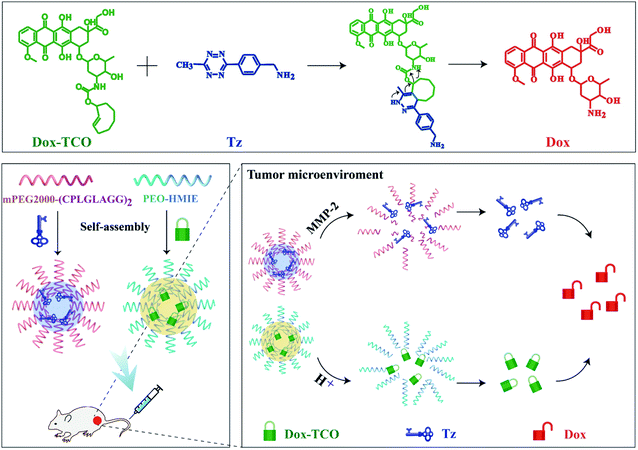 |
| | Scheme 1 Schematic illustration of the specific prodrug activation strategy integrating the IEDDA reaction with two tumor-microenvironment-responsive nanovehicles. | |
Results and discussion
Characterization of pH@Dox-TCO and MMP-2@Tz nanovehicles
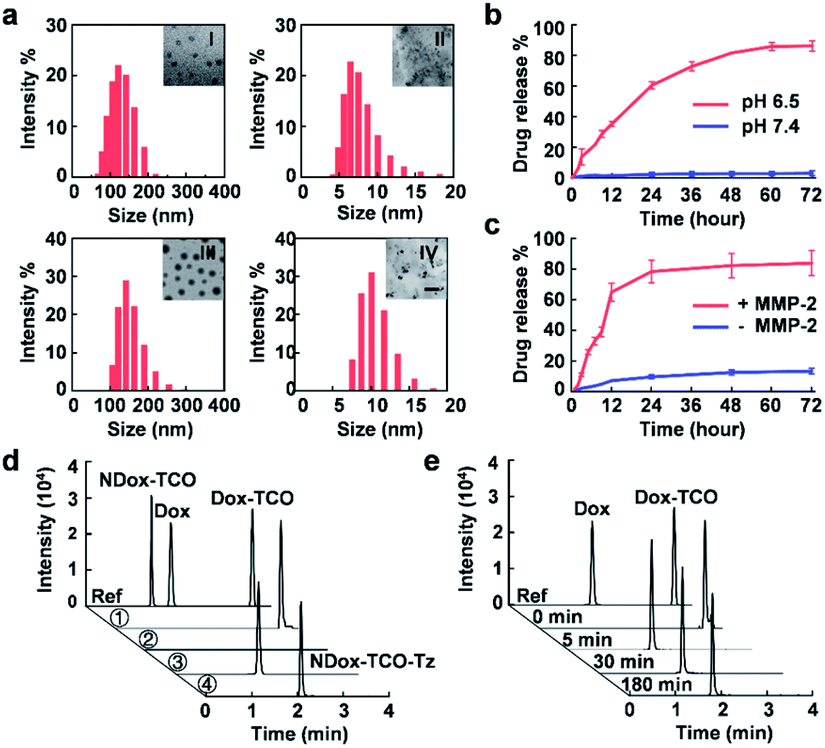
The bioorthogonal cleavable prodrug was prepared by modifying Dox with the axial tautomer of TCO.4 It exhibited a 10-fold higher EC50 (6.682 μM) than that of native Dox (0.614 μM) (Fig. S1†), indicating dramatically compromised cytotoxicity potency. Then low pH-responsive micellar nanoparticles loaded with Dox-TCO (pH@Dox-TCO) were fabricated via the supramolecular self-assembly of the tertiary amine containing block copolymer (PEO-HMIE) mixed with Dox-TCO.32 Similarly, MMP-2 sensitive micellar nanoparticles loaded with Tz (MMP-2@Tz) were assembled using the amphiphilic polyethylene glycol (PEG)-peptide (CPLGLAGG)2 diblock copolymer blended with Tz.35 Both the nanovehicles were monodisperse and uniform. When 5 mg of the low pH-responsive block copolymer and 1 mg of Dox-TCO were used, the average hydrodynamic size of pH@Dox-TCO was 127 ± 19 nm (Fig. 1a), while the corresponding micellar nanoparticles without Dox-TCO (pH NPs) were about 91.6 nm in size (Fig. S2a†). In this case, the encapsulation efficiency of Dox-TCO was 61.2%, and the corresponding drug loading was 5.62 × 105 molecules/particle. When 3.5 mg of MMP-2 responsive diblock copolymer and 0.5 mg of Tz molecules were used, the average hydrodynamic size of MMP-2@Tz was 150 ± 26 nm (Fig. 1a), increased by about 35 nm compared with that of MMP-2 responsive micellar nanoparticles (MMP-2 NPs) (Fig. S2b†), and the drug loading was 2.37 × 106 molecules/particle.
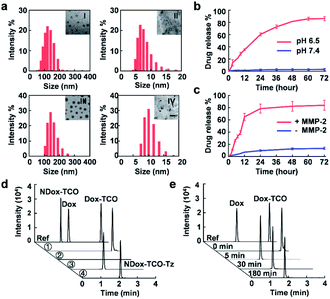 |
| | Fig. 1 Characterization of the two nanovehicles. (a) DLS and TEM imaging of pH@Dox-TCO ((I) pH 7.4, (II) pH 6.5) and MMP-2@Tz ((III) without MMP-2, and (IV) with MMP-2). The scale bar is 100 nm. (b and c) Time-dependent release of Dox-TCO or Tz under different conditions. (d) UHPLC-MS analysis of Dox-TCO activation in different media. Ref: references, ①: pH 6.5, ②: pH 7.4 with MMP-2, ③: pH 6.5 with MMP-2, and ④: NDox-TCO control. (e) Time-dependent activation of Dox-TCO in group ③ of (d) recorded by UHPLC-MS. All data represent the mean ± s.d. (n = 3). | |
The pH@Dox-TCO and MMP-2@Tz nanovehicles were biocompatible and stable. As a result, no obvious in vitro toxicity was observed (Fig. S3†), and the sizes hardly changed during one week of storage in water or FBS (Fig. S4†), but readily responded to low pH (pH 6.5) or MMP-2 (Fig. 1a), resulting in particle dissociation and payload release. The accumulated release percentages of Dox-TCO and Tz were up to 82.4% and 81.2% in 48 h, respectively (Fig. 1b and c). We also noted that the release of Tz was very faster in the first 12 h, while Dox-TCO was released in a much flatter manner, indicating different response rates of the two nanovehicles. To maximize the activation of Dox-TCO, we determined the optimal ratio of Dox-TCO and Tz. According to cell viability and ultra-high-performance liquid chromatography-mass spectrometry (UHPLC-MS) analysis results, when the molar ratio of Dox-TCO to Tz was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, Dox could be fully activated (Fig. S5†). Next, Dox-TCO activation was evaluated by the UHPLC-MS analysis. Both pH@Dox-TCO and MMP-2@Tz were dispersed in one of the following media: (1) pH 6.5, (2) pH 7.4 with MMP-2, and (3) pH 6.5 with MMP-2. To reveal the importance of cleavable tautomers in Dox-TCO, we also synthesized a prodrug containing an uncleavable equatorial tautomer of TCO (NDox-TCO) and prepared corresponding low pH sensitive nanovehicles (pH@NDox-TCO), which were mixed with MMP-2@Tz dispersed in the medium of pH 6.5 with MMP-2 (group 4). As can be seen in Fig. 1d, with Dox, Dox-TCO and NDox-TCO as references, the peak of Dox-TCO was unchanged and no Dox was detected in group 1. As expected, neither Dox-TCO nor Dox was observed in group 2 since it was difficult for pH@Dox-TCO to dissociate at pH 7.4. Only in group 3, the peak of Dox-TCO disappeared within 5 min, accompanied by the emergence of the Dox peak that remained the same between 5 and 180 min, illustrating the fast liberation of Dox in this case (Fig. 1e). Interestingly, the peak of NDox-TCO in group 4 was replaced by a new peak of NDox-TCO-Tz, illustrating that the prodrug could not be activated even after the IEDDA reaction if it was derived with an uncleavable TCO. These results together clearly demonstrated that the Dox-TCO camouflaged in nanovehicles could be specifically released under the synergistic control of low pH and MMP-2, immediately followed by IEDDA cleavage activation.
:2, Dox could be fully activated (Fig. S5†). Next, Dox-TCO activation was evaluated by the UHPLC-MS analysis. Both pH@Dox-TCO and MMP-2@Tz were dispersed in one of the following media: (1) pH 6.5, (2) pH 7.4 with MMP-2, and (3) pH 6.5 with MMP-2. To reveal the importance of cleavable tautomers in Dox-TCO, we also synthesized a prodrug containing an uncleavable equatorial tautomer of TCO (NDox-TCO) and prepared corresponding low pH sensitive nanovehicles (pH@NDox-TCO), which were mixed with MMP-2@Tz dispersed in the medium of pH 6.5 with MMP-2 (group 4). As can be seen in Fig. 1d, with Dox, Dox-TCO and NDox-TCO as references, the peak of Dox-TCO was unchanged and no Dox was detected in group 1. As expected, neither Dox-TCO nor Dox was observed in group 2 since it was difficult for pH@Dox-TCO to dissociate at pH 7.4. Only in group 3, the peak of Dox-TCO disappeared within 5 min, accompanied by the emergence of the Dox peak that remained the same between 5 and 180 min, illustrating the fast liberation of Dox in this case (Fig. 1e). Interestingly, the peak of NDox-TCO in group 4 was replaced by a new peak of NDox-TCO-Tz, illustrating that the prodrug could not be activated even after the IEDDA reaction if it was derived with an uncleavable TCO. These results together clearly demonstrated that the Dox-TCO camouflaged in nanovehicles could be specifically released under the synergistic control of low pH and MMP-2, immediately followed by IEDDA cleavage activation.
In vitro cytotoxicity evaluation of activated Dox-TCO
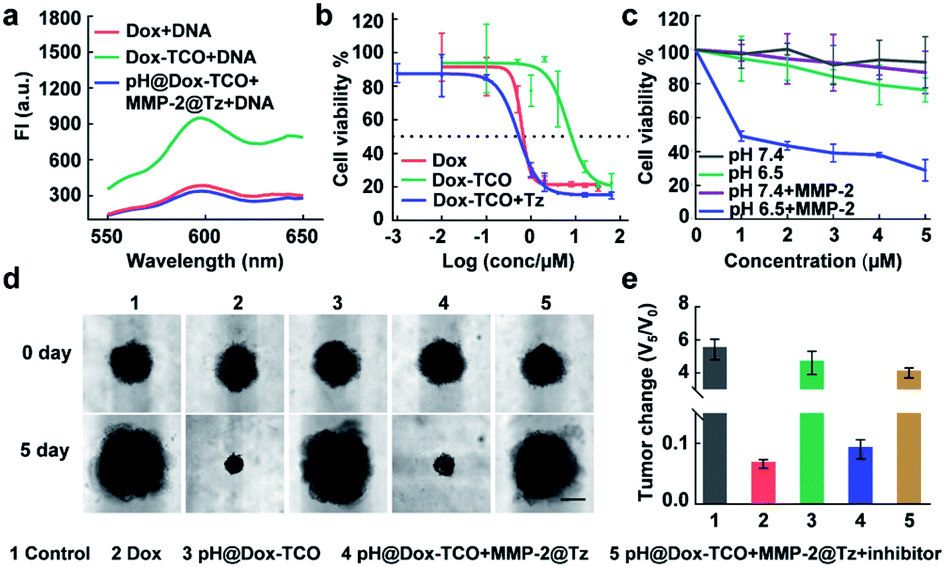
Upon addition of DNA to the solution of activated Dox-TCO, the fluorescence dramatically decreased to a comparable level to that of native Dox incubated with the same concentration of DNA, verifying the recovered DNA intercalation ability of activated Dox (Fig. 2a).37,38 Consequently, the cytotoxicity potency of activated Dox was noticeably recovered, with an EC50 value of 0.517 μM similar to that of native Dox (0.614 μM) (Fig. 2b). For further verification, the pH@Dox-TCO and MMP-2@Tz were divided into four groups and individually treated with different media for 48 h: (1) pH 7.4, (2) pH 6.5, (3) containing MMP-2 at pH 7.4, and (4) containing MMP-2 at pH 6.5. Then different amounts of the mixture were left to incubate with 4T1 cells for 48 h. As shown in Fig. 2c, only in group 4 the cell viability remarkably decreased along with increasing concentrations of Dox-TCO, while the decrease in the other groups was slight. To further closely mimic the TME and therefore provide a more accurate understanding, we also evaluated the performance of different formulations on a 3D multicellular tumor spheroid (MCTS) model. As expected, the MCTS volumes of the native Dox treated group decreased to a very small size, while those of only the pH@Dox-TCO treated group significantly increased. Indeed, the tumor growth of the group treated with both pH@Dox-TCO and MMP-2@Tz could be greatly inhibited, however this inhibition dramatically weakened upon addition of the MMP-2 inhibitor (Marimastat). All these results confirmed that Dox-TCO could be specifically activated by Tz at low pH together with MMP-2, with Dox liberated from Dox-TCO exhibiting potent anticancer efficacy (Fig. 2d and e).
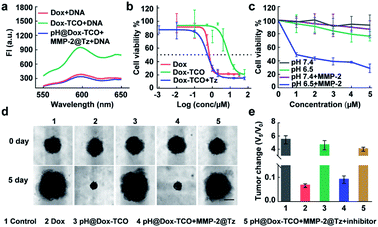 |
| | Fig. 2
In vitro cytotoxicity evaluation of activated Dox-TCO. (a) Fluorescence spectra of DNA solution incubated with different formulations of Dox. (b) Cytotoxicity of different Dox formulations against 4T1 cells. (c) Concentration-dependent cytotoxicity of the mixture of pH@Dox-TCO and MMP-2@Tz under different conditions. (d) Representative photos of the MCTS before and after different treatments for 5 days. The scale bar is 100 μm. (e) The corresponding volume change of (d). All data represent the mean ± s.d. (n = 6). | |
In vivo biodistribution of Dox after different treatments
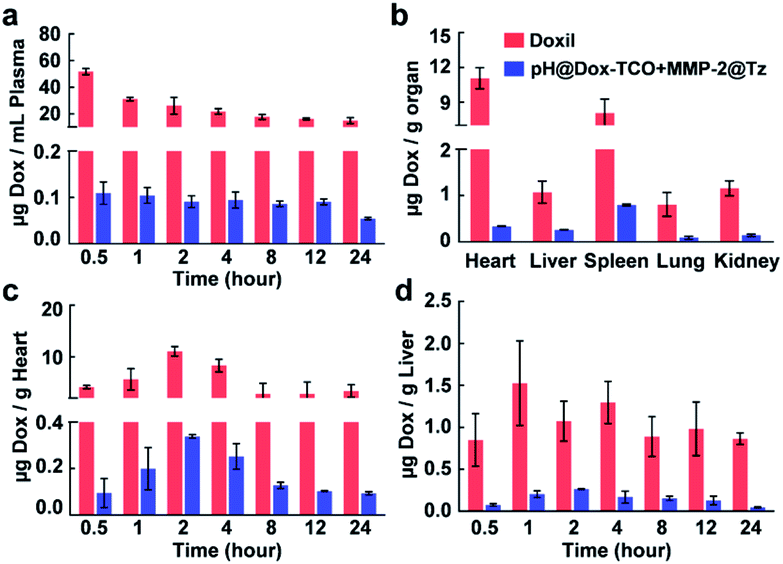
Since the activation of Dox-TCO could be precisely controlled, the in vivo safety of our strategy should be expected. For confirmation, the BALB/c mice were individually administered (i.v.) with a high dose of two nanovehicles or Doxorubicin Hydrochloride Liposomal Injection (Doxil) as a control (10 mg kg−1 Dox equiv.). Then the samples from blood and main organs were collected at different times and analyzed using UHPLC-MS. It was some surprise that the concentrations of free Dox in the group successively treated with pH@Dox-TCO and MMP-2@Tz were always much lower than that of the Doxil treated group in all samples (Fig. S7†). For example, it was only about 0.1 μg mL−1 in the plasma of the nanovehicle-treated group, while it was more than 20 μg mL−1 in the plasma of the Doxil treated group (Fig. 3a). The cardiotoxicity is a major limitation to the clinical application of Dox, and liver is the main accumulation site of nanoparticles after systemic administration. Fortunately, the contents of free Dox in both hearts and livers of the nanovehicle-treated group were scarce (Fig. 3c and d). In particular, it was individually 32.6 and 4.1 times lower than that of the Doxil treated group at 2 h post-injection (Fig. 3b). All these results together illustrated that Dox-TCO was hardly activated in normal tissues and sites by using our strategy. It was reasonable since Dox-TCO could only be activated when it ran across Tz, and this would exclusively happen at the site where it is of low pH as well as abundant MMP-2, which is very rare in vivo. As a result, Doxil treatment showed apparent systemic toxicity, with substantial decreases in the clinical chemistry parameters and body weight compared with those of the PBS control (Fig. S8†). Likewise, Dox enwrapped in low pH-sensitive nanoparticles (pH@Dox) also exhibited some adverse effects. In contrast, the side-effects of our nanovehicle-treated group were not evident. All the indexes were normal and the body weight of mice remained steady. These results clearly indicated that, compared with conventional liposomal and single-responsive strategies, our hierarchically triple-regulating strategy showed outstanding biosafety.
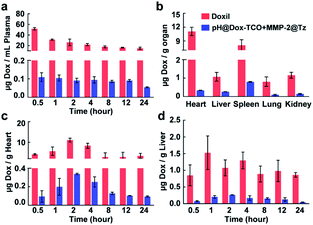 |
| | Fig. 3 Biodistribution of Dox in mice after different treatments analyzed by UHPLC-MS. (a) Concentrations of Dox in plasma after injection with MMP-2@Tz for different times. (b) Concentrations of Dox in main organs 2 h after injection with MMP-2@Tz. (c and d) Concentrations of Dox in hearts and livers after injection with MMP-2@Tz for different times. All data represent the mean ± s.d. (n = 6). | |
Biodistribution of pH@Dox-TCO and MMP@Tz nanovehicles
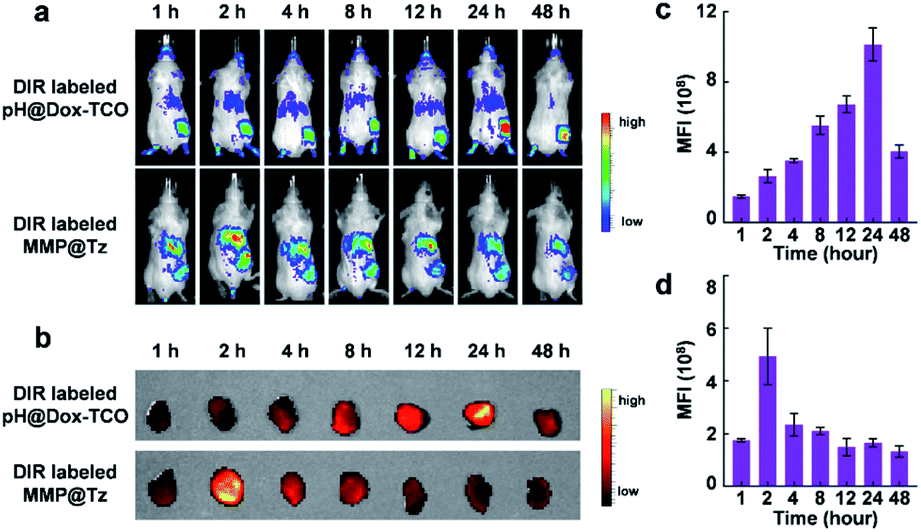
How about the in vivo anticancer performance of our strategy? At first, to evaluate the effective tumor accumulation of the two nanovehicles, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DIR) was loaded into the pH@Dox-TCO and MMP-2@Tz nanovehicles. Then, in vivo biodistribution of DiR-labeled pH@Dox-TCO or MMP-2@Tz nanovehicles was monitored in 4T1 tumor-bearing mice at different time points after i.v. injection. As shown in Fig. 4a, the fluorescence intensity of tumor tissues increased and respectively reached the maximum at 24 h post-injection of pH@Dox-TCO and 2 h post-injection of MMP-2@Tz, which was similar to reported results.35 The ex vivo fluorescence imaging of excised tumors further confirmed the in vivo imaging results (Fig. 4b–d). Finally, to ensure the encounter of the two nanovehicles in tumor tissues, MMP-2@Tz was administrated 22 h after the injection of pH@Dox-TCO in the following anticancer therapy study.
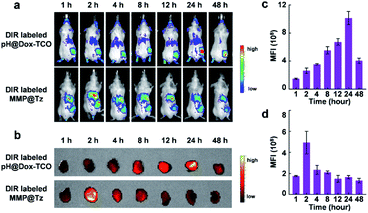 |
| | Fig. 4 Biodistribution of pH@Dox-TCO and MMP@Tz nanovehicles. (a) In vivo fluorescence images of DiR-labeled pH@Dox-TCO and MMP@Tz nanovehicles at different time points after i.v. administration. (b) Ex vivo fluorescence images of tumors excised from (a). (c and d) Quantitative statistics of the median fluorescence intensity (MFI) in (b). All data represent the mean ± s.d. (n = 6). | |
In vivo antitumor efficacy
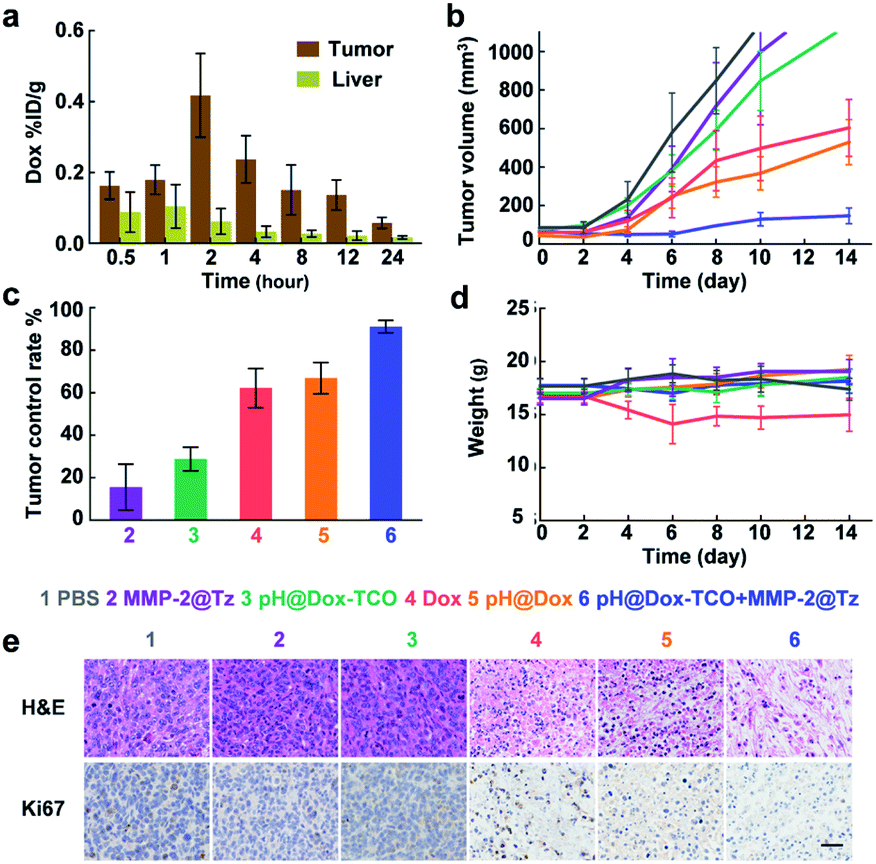
Besides the tumors, the accumulation of nanovehicles in the livers was also clear. But the UHPLC-MS analysis revealed a much higher content of free Dox in tumors than in livers (Fig. 5a) of mice sequentially treated with pH@Dox-TCO and MMP-2@Tz, again demonstrating the precise activation of Dox-TCO in tumors in vivo. Then 4T1 tumor-bearing mice were divided into 6 groups and individually treated with different formulations when the tumor volume was greater than 80 mm3. Compared with the PBS group, single MMP-2@Tz or pH@Dox-TCO treatment failed to yield a noticeable tumor inhibition. For the group that received Dox or pH@Dox, the tumor growth could be delayed to a certain extent, but the tumor control rate was only about 60% (Fig. 5b and c). Moreover, the body weight of the native Dox treated group decreased obviously due to the severe side-effects (Fig. 5d). Intriguingly, the tumor development could be effectively inhibited in the group treated with both pH@Dox-TCO and MMP-2@Tz, with a tumor control rate more than 90% (Fig. 5c). Correspondingly, the hematoxylin and eosin (H&E) staining results showed that the tumor cells were efficiently destroyed and the expression of cell proliferation indicator Ki67 was significantly inhibited in this group (Fig. 5e). In contrast, the major organs did not exhibit any distinct differences compared with the PBS control group based on the H&E staining results, further indicating the superior specificity and safety of our prodrug activation strategy for anticancer therapy (Fig. S10†).
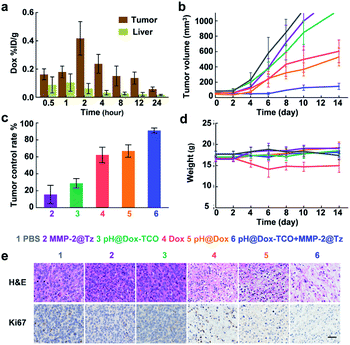 |
| | Fig. 5
In vivo antitumor efficacy after different treatments. (a) UHPLC-MS analysis of Dox in tumors and livers after i.v. injection of pH@Dox-TCO and MMP-2@Tz (5 mg Kg−1 Dox equiv.). (b) Tumor volumes of mice after different treatments. (c) Tumor control rate after different treatments. (d) Body weight of mice in each group. (e) H&E and Ki67 staining of tumor tissues. The scale bar is 30 μm. All data represent the mean ± s.d. (n = 8). | |
Conclusions
In summary, we have developed a hierarchically regulated and spatiotemporally controlled prodrug activation strategy for anticancer therapy. Dox-TCO and Tz were separately loaded in low pH and MMP-2 sensitive micellar nanoparticles. Only in TME could low pH and MMP-2 trigger simultaneous disaggregation of the two nanovehicles, and thus the release of Dox-TCO and Tz, followed by the IEDDA reaction of Dox-TCO with Tz to liberate free Dox. As a result, the adverse effects of Dox are significantly decreased and the tumor growth can be effectively inhibited, which is promising for highly specific and safe cancer treatment. Given that low pH and over-expression of MMP-2 are two typical features in TME, our strategy may be applied to most of the tumors, thus significantly expanding the scope of bioorthogonal cleavage reaction-based prodrug activation approaches. As we know, orthotopic models can provide the proper microenvironment for tumor growth and are more accurate for the treatment efficiency evaluation. In the future, we will continue to investigate the application feasibility of our strategy by using orthotopic cancer models.
Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals published in GB/T 35892-2018 and the experiments were approved by Institutional Animal Use and Care Committee of Peking University of China.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Biological & Medical Engineering Core Facilities (Beijing Institute of Technology) for providing advanced equipment. This work was supported by the National Natural Science Foundation of China (No. 81571813, 21874011 and 91859123) and National Science and Technology Major Project (No. 2018ZX10301405-001).
Notes and references
- J. Rautio, N. A. Meanwell, L. Di and M. J. Hageman, Nat. Rev. Drug Discovery, 2018, 17, 559–587 CrossRef CAS PubMed.
- S. Santra, C. Kaittanis, O. J. Santiesteban and J. M. Perez, J. Am. Chem. Soc., 2011, 133, 16680–16688 CrossRef CAS PubMed.
- K. Neumann, A. Gambardella, A. Lilienkampf and M. Bradley, Chem. Sci., 2018, 9, 7198–7203 RSC.
- R. M. Versteegen, R. Rossin, W. ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2013, 52, 14112–14116 CrossRef CAS PubMed.
- R. Rossin, S. M. van Duijnhoven, W. Ten Hoeve, H. M. Janssen, L. H. Kleijn, F. J. Hoeben, R. M. Versteegen and M. S. Robillard, Bioconjugate Chem., 2016, 27, 1697–1706 CrossRef CAS PubMed.
- S. S. Matikonda, D. L. Orsi, V. Staudacher, I. A. Jenkins, F. Fiedler, J. Y. Chen and A. B. Gamble, Chem. Sci., 2015, 6, 1212–1218 RSC.
- T. Caneque, S. Muller and R. Rodriguez, Nat. Rev. Chem., 2018, 2, 202–215 CrossRef.
- X. Fan, Y. Ge, F. Lin, Y. Yang, G. Zhang, W. S. C. Ngai, Z. Lin, S. Zheng, J. Wang, J. Zhao, J. Li and P. R. Chen, Angew. Chem., Int. Ed., 2016, 55, 14046–14050 CrossRef CAS PubMed.
- J. C. T. Carlson, H. Mikula and R. Weissleder, J. Am. Chem. Soc., 2018, 140, 3603–3612 CrossRef CAS PubMed.
- J. Li, S. Jia and P. R. Chen, Nat. Chem. Biol., 2014, 10, 1003–1005 CrossRef CAS.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- F. Lin, L. Chen, H. Zhang, W. S. C. Ngai, X. Zeng, J. Lin and P. R. Chen, CCS Chem., 2019, 226–236 Search PubMed.
- T. Peng and H. Hang, J. Am. Chem. Soc., 2016, 138, 14423–14433 CrossRef CAS PubMed.
- J. M. M. Oneto, I. Khan, L. Seebald and M. Royzen, ACS Cent. Sci., 2016, 2, 476–482 CrossRef PubMed.
- Q. Yao, F. Lin, X. Fan, Y. Wang, Y. Liu, Z. Liu, X. Jiang, P. R. Chen and Y. Gao, Nat. Commun., 2018, 9, 5032 CrossRef PubMed.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS PubMed.
- K. Maruyama, Adv. Drug Delivery Rev., 2011, 63, 161–169 CrossRef CAS PubMed.
- A. K. Iyer, G. Khaled, J. Fang and H. Maeda, Drug Discovery Today, 2006, 11, 812–818 CrossRef CAS PubMed.
- A. Jhaveri, P. Deshpande and V. Torchilin, J. Control. Release, 2014, 190, 352–370 CrossRef CAS PubMed.
- Z. Zhao, W. Wang, C. Li, Y. Zhang, T. Yu, R. Wu, J. Zhao, Z. Liu, J. Liu and H. Yu, Adv. Funct. Mater., 2019, 1905013 CrossRef CAS.
- Y. Chen, X. Liu, H. Yuan, Z. Yang, C. A. von Roemeling, Y. Qie, H. Zhao, Y. Wang, W. Jiang and B. Y. S. Kim, Adv. Sci., 2019, 6, 1802070 CrossRef PubMed.
- Y. Lee, S. Lee and S. Jon, Adv. Sci., 2018, 5, 1800017 CrossRef PubMed.
- Z. Cheng, A. A. Zaki, J. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed.
- M. Karimi, A. Ghasemi, P. S. Zangabad, R. Rahighi, S. M. M. Basri, H. Mirshekari, M. Amiri, Z. S. Pishabad, A. Aslani, M. Bozorgomid, D. Ghosh, A. Beyzavi, A. Vaseghi, A. R. Aref, L. Haghani, S. Bahrami and M. R. Hamblin, Chem. Soc. Rev., 2016, 45, 1457–1501 RSC.
- R. Cheng, F. Meng, C. Deng, H.-A. Klok and Z. Zhong, Biomaterials, 2013, 34, 3647–3657 CrossRef CAS.
- T. Ji, Y. Zhao, Y. Ding and G. Nie, Adv. Mater., 2013, 25, 3508–3525 CrossRef CAS.
- Z. Wang, X. Zhen, P. K. Upputuri, Y. Jiang, J. W. Lau, M. Pramanik, K. Pu and B. Xing, ACS Nano, 2019, 13, 5816–5825 CrossRef CAS.
- H. Ruan, Q. Hu, D. Wen, Q. Chen, G. Chen, Y. Lu, J. Wang, H. Cheng, W. Lu and Z. Gu, Adv. Mater., 2019, 31, 1806957 CrossRef PubMed.
- J. Liu, Y. Huang, A. Kumar, A. Tan, S. Jin, A. Mozhi and X. Liang, Biotechnol. Adv., 2014, 32, 693–710 CrossRef CAS PubMed.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., 2003, 115, 4788–4791 (
Angew. Chem., Int. Ed.
, 2003
, 42
, 4640–4643
) CrossRef.
- F. Meng, Y. Zhong, R. Cheng, C. Deng and Z. Zhong, Nanomedicine, 2014, 9, 487–499 CrossRef CAS PubMed.
- K. Zhou, Y. Wang, X. Huang, K. Luby-Phelps, B. D. Sumer and J. Gao, Angew. Chem., Int. Ed., 2011, 50, 6109–6114 CrossRef CAS PubMed.
- R. E. Vandenbroucke and C. Libert, Nat. Rev. Drug Discovery, 2014, 13, 904–927 CrossRef CAS PubMed.
- Q. Yao, L. Kou, Y. Tu and L. Zhu, Trends Pharmacol. Sci., 2018, 39, 766–781 CrossRef CAS.
- L. Shi, Y. Hu, A. Lin, C. Ma, C. Zhang, Y. Su, L. Zhou, Y. Niu and X. Zhu, Bioconjugate Chem., 2016, 27, 2943–2953 CrossRef CAS.
- Z. Peng and J. Kopecek, J. Am. Chem. Soc., 2015, 137, 6726–6729 CrossRef CAS.
- D. Agudelo, P. Bourassa, G. Bérubé and H.-A. Tajmir-Riahi, Int. J. Biol. Macromol., 2014, 66, 144–150 CrossRef CAS PubMed.
- Y. Zhou, F. Gan, Y. Zhang, X. He, C. Shen, H. Qiu and P. Liu, Adv. Sci., 2019, 1901341 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05036a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Peilin
Wang
a,
Guihong
Lu
a,
Jinfeng
Zhang
a,
Li-Li
Huang
c,
Peilin
Wang
a,
Guihong
Lu
a,
Jinfeng
Zhang
a,
Li-Li
Huang