
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeparation of halogenated benzenes enabled by investigation of halogen–π interactions with carbon materials†
Eisuke
Kanao
a,
Takuya
Morinaga
a,
Takuya
Kubo
 *a,
Toyohiro
Naito
a,
Takatoshi
Matsumoto
b,
Tomoharu
Sano
c,
Hideshi
Maki
de,
Mingdi
Yan
f and
Koji
Otsuka
a
*a,
Toyohiro
Naito
a,
Takatoshi
Matsumoto
b,
Tomoharu
Sano
c,
Hideshi
Maki
de,
Mingdi
Yan
f and
Koji
Otsuka
a
aGraduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: kubo.takuya.6c@kyoto-u.ac.jp; Fax: +81-75-383-2450; Tel: +81-75-383-2448
bInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1, Katahira, Aoba-ku, Sendai 980-8577, Japan
cCenter for Environmental Measurement and Analysis, National Institute for Environmental Studies, Onogawa 16-2, Tsukuba, Ibaraki 305-8506, Japan
dCenter for Environmental Management, Kobe University, 1-1 Rokkodai-cho, Nada-ku, Kobe 657-8501, Japan
eDepartment of Chemical Science and Engineering, Graduate School of Engineering, Kobe University, 1-1 Rokkodai-cho, Nada-ku, Kobe 657-8501, Japan
fDepartment of Chemistry, University of Massachusetts Lowell, One University Ave., Lowell, MA 01854, USA
First published on 18th November 2019
Abstract
The halogen–π (X–π) interaction is an intermolecular interaction between the electron-poor region of bonded halogen atoms and aromatic rings. We report an experimental evaluation of the halogen–π (X–π) interaction using liquid chromatography with carbon-material coated columns providing strong π interactions in the normal phase mode. A C70-fullerene (C70)-coated column showed higher retentions for halogenated benzenes as the number of halogen substitutions increased as a result of X–π interactions. In addition, the strength of the X–π interaction increased in the order of F < Cl < Br < I. Changes to the UV absorption of C70 and the brominated benzenes suggested that the intermolecular interaction changed from the π–π interaction to X–π interaction as the number of bromo substitutions increased. Computer simulations also showed that the difference in dipole moments among structural isomers affected the strength of the π–π interaction. Furthermore, we concluded from small peak shifts in 1H NMR and from computer simulations that the orbital interaction contributes to the X–π interactions. Finally, we succeeded in the one-pot separation of all isomers of brominated benzenes using the C70-coated column by optimizing the mobile phase conditions.
Introduction
Advances in computational chemistry have enabled investigations of large charge deflections in halogen atoms. The low electron density region along the σ bonding axis of bonded halogen atoms, known as the “σ hole,” interacts propitiously with electron donor species.1–5 This halogen bonding interaction has attracted interest in many fields, such as molecular biology6,7 and crystal engineering,8,9 due to its high molecular selectivity and linear directionality.10–14 In particular, halogen bonding has been positively applied to develop new functional materials15–18 since Nguyen et al. first synthesized liquid crystals designed with halogen bonds in 2004.19 An example is the Rotaxane structure with iodo groups synthesized by Barendt et al. that selectively recognized nitrogen anions via halogen bonding, producing drastic color changes.20 Another example is the iodoperfluoroarene-substituted polystyrene synthesized by Vanderkooy et al. that controlled the self-assembly of nanostructures of polymer aggregates by halogen bonding.21 According to recent reports, halogen bonding greatly contributes to molecular recognition in vivo and has thus been applied in designing drugs,22–24 such as aldose reductase inhibitors25 and HIV-1 inhibitors.26 Therefore, fully understanding the characteristics of halogen bonding may drastically facilitate the advancement of materials chemistry.Notably, halogen bonding between halogen groups and π electrons of aromatic rings or unsaturated bonds acting as electron donors, i.e., “halogen–π (X–π) interactions,” plays important roles in biological systems.27–30 Voth et al. reported the first X–π interactions in biological systems.31 They found that the interactions between the aromatic rings of phenyl residues inhibited the protein kinases CDK2 and CK2. Data on X–π interactions in biological systems rapidly grew following Voth's report, and the X–π interaction now occupies about 30% of the current reports on halogen bonding in protein–ligand complexation.32 The importance of X–π interactions is now recognized, and many properties (magnitude directionality and origin of the X–π interaction) have been clarified with high level computational methods based on theoretical chemistry.33,34
Despite the progress, additional experimental data on X–π interactions are necessary because the halogen bond is in physical competition with many other stronger noncovalent interactions, including the hydrophobic interaction, hydrogen bond, and electrostatic interaction.35–37 As a few reports, Nazaré et al. indicated the experimental approach to reveal the strength of halogen–π interaction using bicyclic N-arylimide based molecular torsion balances system.38 Although the study showed the presence of halogen–π interaction in intramolecular system, few studies reported regarding halogen–π interaction for intermolecular system. On the other hand, high performance liquid chromatography (HPLC) is a powerful separation technique that utilizes the difference in partition coefficients between the solutes of the mobile and stationary phases, a factor that sensitively reflects the strength of the noncovalent interaction.39,40 In our previous studies, we developed novel separation media immobilized with carbon materials.41–43 These media exhibited strong π interactions because of their many π electrons. Characteristics of weak interactions from aromatic rings were clarified by evaluating their retention behaviors with HPLC measurements. This included the π–π interaction based on the spherical recognition of C60- or C70-fullerenes (C60/C70), the difference in intensity of the CH or CD–π interaction, and the strong CH–π interaction between corannulene and planar polycyclic aromatic hydrocarbons.
We also uncovered the possibility for the separation of halogenated aromatic compounds resulting from the efficient halogen–π interactions in HPLC.44 The aromatic halogens, such as polychlorinated biphenyls (PCBs)45,46 and polybrominated diphenyl ethers (PBDEs),47–49 are widely known to be highly toxic; therefore, we need monitor the concentration of these pollutions in our environment. Usually, these aromatic halogens are analyzed by gas-chromatography or reverse-phase liquid chromatography, which involve complicated preparation processes. Even with these existing methods, the separation of the full variety of isomers by the direct analysis of the sample in oil or after extraction with non-polar solvents is difficult because only factors driving the separation are the differences in vaporization or hydrophobicity between the compounds. In addition, the halogen–π interaction has never been utilized for the separation of these aromatic halogens. Thus, we hypothesized that the effective halogen–π interaction would enable the efficient separation of the isomers of aromatic halogens and that the one-pot separation system will contribute to the straightforward monitoring of environmental pollutions like PCBs and PBDEs.
In the present study, we experimentally evaluated the strength of the X–π interaction between carbon materials and a variety of halogenated benzenes using normal phase liquid chromatography (NPLC). The hydrophobic interaction was completely suppressed in this technique, and thus the π interactions could be simply examined. Furthermore, we accurately evaluated the π interactions between halogenated benzenes and C70 using ultraviolet-visible (UV-Vis) and 1H NMR spectroscopy. Finally, we demonstrate the one-pot separation of the entirety of the brominated benzenes (11 analogues) by optimizing the mobile phase conditions to control the X–π interactions.
Results and discussion
Method
The detailed experimental procedures including chemicals, instruments, the synthesis of the molecules, the preparation of the monoliths, and computational methods are summarized in the ESI.†The silica-monolithic capillary was treated with 1.0 M aqueous sodium hydroxide at 40 °C for 3 h. After washing with water and methanol, 3-aminopropyltrimethoxysilane (APTMS) in methanol (10%, v/v) was passed through the silica-monolithic capillary for 24 h and washed with methanol. C60/C70-4-azido-2,3,5,6-tetrafluorophenyl succinate (PFPA-NHS) in toluene (3.0 mg mL−1) was charged into the NH2-monolith for 24 h at room temperature and washed with toluene and methanol (Scheme S2†). A column that was modified with only the linkers or PFPA column (Scheme S3†) was also prepared and used as a control. NH2 groups on the silica-monolith surface and C70-PFPA-NHS were reacted under the conditions in Table S2.† For comparing column performances, we evaluated retention factors and separation factors of phenanthrene and corannulene, which showed specific intermolecular interaction with C70 due to its spherical recognition. Fig. S2a† shows the retention factors of phenanthrene and corannulene obtained and the separation factor for each column. The chromatograms of the phenanthrene and corannulene mixture obtained with column 1 and column 4 are shown in Fig. S2b.† Column 4 showed the largest retention factor and separation factor. This result clearly suggested that the separation performance of the column was successfully improved by increasing the reaction time and concentration of C70-PFPA-NHS in the reaction solution. We then used column 4 to evaluate the separation performance of the structural isomers of halogenated benzenes (column 1 is defined as Type-1 and column 4 is defined as Type-2).
Retention of halogenated benzenes on carbon-material coated columns in NPLC
We evaluated the retention of halogenated benzenes on four different carbon-material (πNAP, 5PYE, C60, and C70) coated columns (Fig. 1). Here, when we employed octadecylsilyl bonded silica (C18) column, which is typically used in HPLC based on hydrophobic interaction, all the halogenated benzenes were not retained at all. Herefrom, the hydrophobic interaction was not worked in our LC system and we could ignore the effect of hydrophobicity in later discussions.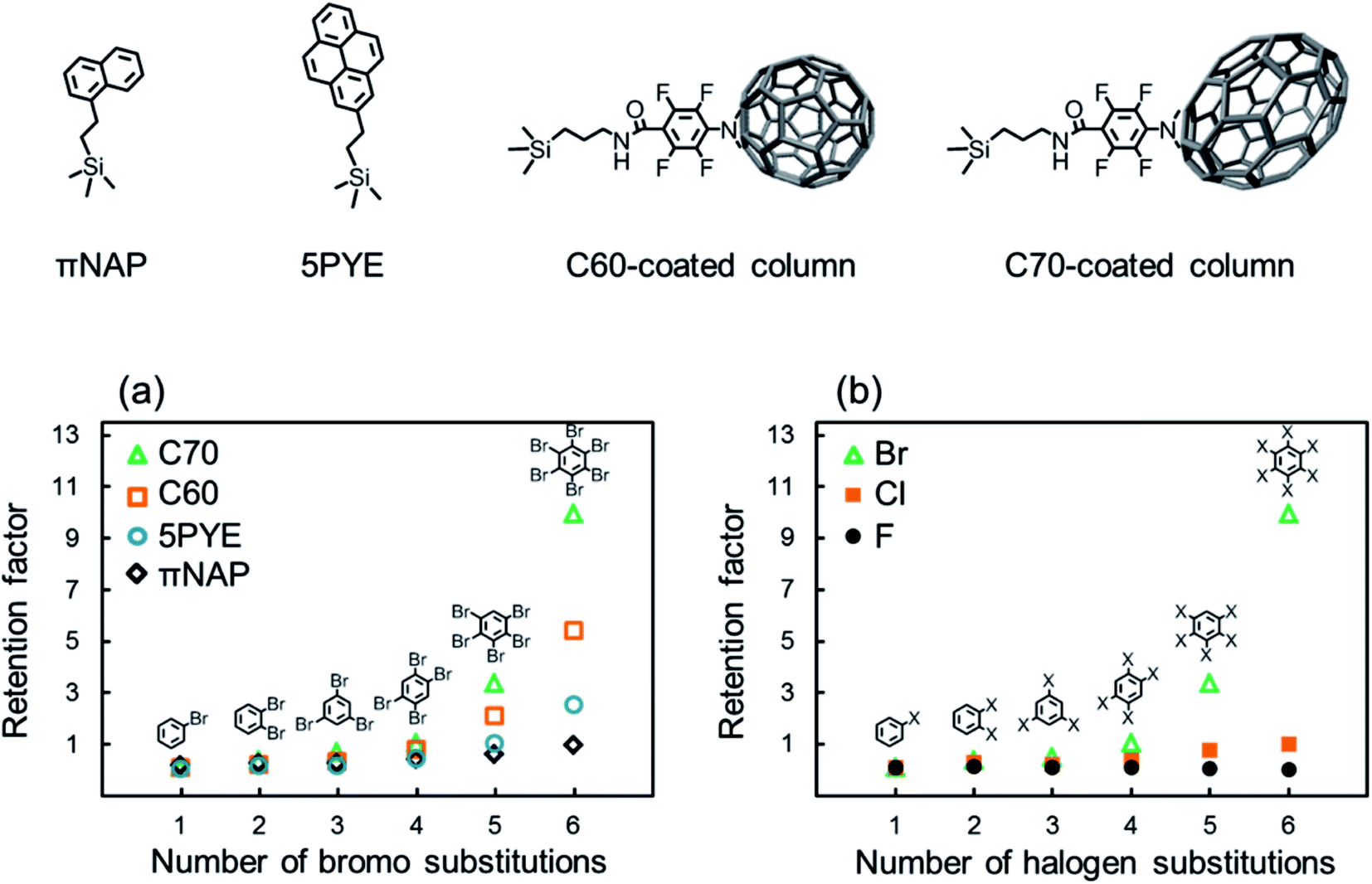 | ||
| Fig. 1 Structure of the stationary phases and retention behaviors of halogenated benzenes with carbon-material coated columns in NPLC. (a) Retention factors of brominated benzenes with each column and the number of brominated substitutions. (b) Retention factors of each halogenated benzene with the C70-coated column and the number of halogen substitutions. Conditions: column, C60-coated (28.8 cm × 100 μm i.d.), C70-coated (Type-1, 25.5 cm × 100 μm i.d.), 5PYE (150 mm × 4.6 mm i.d.), πNAP (50 mm × 2.0 mm i.d.); flow rate; 2.0 μL min−1 (C70, C60-coated), 1.0 mL min−1 (5PYE), 0.5 mL min−1 (πNAP); mobile phase, n-hexane; temperature, 25 °C; detection, UV 228 nm. | ||
Fig. 1a shows the retention factors k (k = (tR − t0)/t0 where tR is the retention time of a solute and t0 is the elution time of non-retained solute) of brominated benzenes on each carbon-material coated column against the number of bromo substitutions in the solutes. All the carbon-material coated columns showed higher retentions as the number of bromo substitutions increased. On contrary, fluorinated benzenes were not retained on the C70 column at all. As shown in a few previous studies, σ-hole was not identified because of small atom of fluorine, so that X–π interaction was not occurred in fluorinated molecules.50,51 Briefly, the presence of halogen–π interaction was confirmed except fluorinated molecules.2,52
The results indicated that the X–π interactions existed between the aromatic rings in the stationary phases and the bromo substitutions of the solutes. The C70-coated column showed significantly higher retentions for brominated benzenes. PFPA, the linker of the C70-coated column, did not provide such high retention, as shown in Table S1;† therefore, we concluded that the C70 moiety was responsible for the high retentions of the brominated benzenes.
Furthermore, in order to evaluate the impact of the halogen species in the solutes on retention behaviors, the retention factors of benzenes with various halogens on the C70-coated column were measured and plotted in Fig. 1b against the number of halogen substitutions in the solutes. The C70-coated column did not retain any fluorinated benzenes. This result suggested that fluorine substitutions were not capable of meaningful X–π interactions. Alternatively, the C70-coated column retained chlorinated and brominated benzenes and showed higher retentions as the number of halogen substitutions increased. However, the absolute retentions and the retention differences across the number of halogen substitutions of the chlorinated benzenes were much less than those of the brominated benzenes. Additionally, the iodinated benzenes were strongly retained on C70 column in the same condition, especially penta- and hexa-iodinated benzenes were not eluted, therefore, it is so hard to compare in this figure. The detailed discussions regarding stronger X–π interaction for iodinated benzenes are described later.
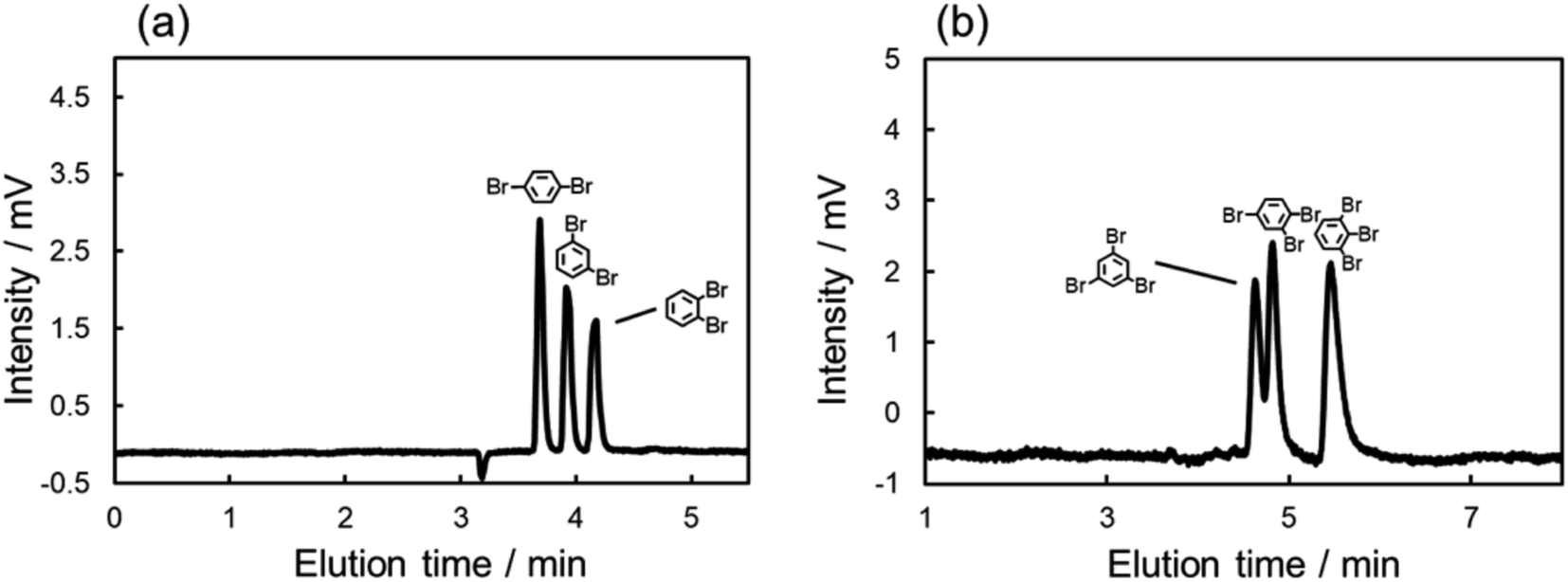
The chromatograms of chlorinated benzenes (Fig. 2a) and brominated benzenes (Fig. 2b) also clearly showed poorer resolution for the chlorinated benzenes. These results indicated that the X–π interactions of chlorine substitutions were weaker than those of their bromine counterparts.
 | ||
| Fig. 2 Chromatograms of the mixed sample of (a) chlorinated benzenes, (b) brominated benzenes, and (c) the mixture of hexa-halogenated benzenes with the C70-coated column. Conditions: (a) and (b) column, C70-coated (Type-1, 25.5 cm × 100 μm i.d.); flow rate; 2.0 μL min−1; mobile phase, n-hexane; temperature, 25 °C; detection, UV 228 nm; (c) column, C70-coated (Type-1, 75.0 cm × 100 μm i.d.); flow rate; 2.0 μL min−1; mobile phase, n-hexane/THF = 7/3; temperature, 25 °C; detection, UV 228 nm. | ||
Here, we hypothesized that the X–π interaction becomes stronger as the size of the substituting halogen increases. A few reports in the literature support our hypothesis; briefly, the size of the σ hole increases with the size of the halogen.53–55 In order to confirm our hypothesis, we evaluated the retention behaviors of the hexa-halogenated benzenes (C6F6, C6Cl6, C6Br6, C6I6) in NPLC with the C70-coated column. Fig. 2c shows the chromatograms of the mixed sample of hexa-halogenated benzenes. In consideration of the solubility of C6I6, tetrahydrofuran (THF) was added to the mobile phase in this separation. As shown in Fig. 2c, the elution order in the C70-coated column was C6F6 < C6Cl6 < C6Br6 < C6I6. As expected, larger halogen atoms enabled higher retentions; the differences in the X–π interactions should account for this trend.
Evaluation of π–π interactions between brominated benzenes and C70 by UV-Vis spectroscopy
In the interaction between halogenated benzenes and aromatic carbon-materials, typical π–π interactions could also contribute in addition to the X–π interactions. In order to evaluate the contributions of π–π interactions to the retention of brominated benzenes, we focused on the differences in UV-Vis absorption spectra of halogenated benzenes with/without C70. Several previous studies reported that the π–π interaction attenuated a certain absorbance region in UV-Vis.41,56 The absorption spectra of o-dibromobenzene (10 μM) with/without C70 (10 μM) in n-hexane are shown in Fig. 3a. Interestingly, the spectrum dramatically changed when C70 was added to the solution. Similar spectral alterations were observed for other brominated benzenes (Fig. S1†). These results suggested that the π–π interaction also contributes to the intermolecular interaction between brominated benzenes and C70. Alternatively, the absorption spectra with larger numbers of bromine substitutions (n > 3), especially hexabromobenzene, were almost unchanged. We assumed that the extent of contributions of the π–π interactions to the intermolecular interaction with C70 was different for these solutes. To evaluate the difference in detail, we calculated the difference in absorbance at the maximum wavelength (around 205 nm, typical specific absorption for the π–π* transition57) between columns with and without C70 (ΔA). Fig. 3b summarizes ΔA for each solute against the number of bromine substitutions. Based on this figure, we supposed that fewer bromine substitutions allowed stronger π–π interactions while higher bromine substitutions were mainly dominated by the X–π interaction. Briefly, the degree of the contribution by the π–π interaction between brominated benzenes and C70 followed a trend opposite to that of retention strength of brominated benzenes on the C70-coated column (Fig. 1). In higher bromine substitutions, the relatively stronger X–π interaction may dominantly contribute to the higher retentions on the C70-coated column. From these results, we conclude that both the X–π interaction and the π–π interaction are competitively involved in the intermolecular interaction between halogenated benzenes and C70. | ||
| Fig. 3 Absorption spectra of o-dibromobenzene in n-hexane with/without C70 (a) and difference of absorbance in maximum wavelength (205 nm) of brominated benzenes with/without C70 (b). | ||
Separation of the structural isomers enabled by differences in dipole moments
As described above, large differences of ΔA were also observed among the isomers of dibromo and tribromobenzenes. These absorbance changes were expected to indicate the strength of the π–π interaction between each structural isomer and C70. Thus, we anticipated that the C70-coated column could efficiently separate the isomers by optimizing the LC conditions to exploit the differences in the strength of the interactions. To achieve such separations, we improved the procedure of preparing a C70-coated column; in brief, we attempted to increase the concentration of C70-PFPA-NHS in the reaction solution to improve the immobilization amount of C70 (see ESI†). Then, we obtained another C70-coated column (Type-2), in which a high density of C70 was immobilized.We evaluated the separation behavior of the mixture of dibromobenzenes or tribromobenzenes with the Type-2 column. Fig. 4 shows chromatograms for the mixture of isomers. As we expected, the separation of these structural isomers was successfully achieved. Furthermore, the elution orders on the column, i.e., o-dibromobenzene > m-dibromobenzene > p-dibromobenzene, 1,2,3-tribromobenzene > 1,2,4-tribromobenzene > 1,3,5-tribromobenzene, followed the trend of π–π interaction contributions (Fig. 3b). Consequently, we anticipated that the retention differences were a result of the differences in the strength of the π–π interaction.
 | ||
| Fig. 4 Chromatograms of the mixture of (a) di- or (b) tri-brominated benzenes with the C70-coated column (the C70-coated, Type-2). Conditions: column, C70-coated (Type-2, 75.0 cm × 100 μm i.d.); flow rate; 2.0 μL min−1; mobile phase, n-hexane; temperature, 25 °C; detection, UV 228 nm. | ||
To investigate the differences in strength of the π–π interactions, we considered the dipole moments of these structural isomers. The strength of the π interaction is considered to be determined by the induced dipole–dipole interaction or the induced dipole–induced dipole interaction.33,58–62 The potential energy of the induced dipole–dipole interaction is given as follows:
| Ginduced dipole–dipole = −μ2α1/(4πε0εr)2r6 | (1) |
| Ginduced dipole–induced dipole = −Aα1α2/(4πε0εr)2r6 | (2) |
In a fashion similar to the above experiments, we evaluated the effect of the π–π interaction for chlorine and iodine substitutions. Fig. S3† shows the chromatograms of di-chlorinated and di-iodinated benzenes. The separation of these structural isomers was confirmed even though the separation efficiency was poorer than that of the bromine substitutions. Here, we again evaluated the retention factor and ΔA of each solute and calculated their dipole moments (Table S3†). The absorption spectra of structural isomers of di-chloro and di-iodo benzenes (10 μM) with/without C70 (10 μM) in n-hexane are shown in Fig. S4.†Fig. 5c and d show the retention factors and the ΔA of di-chlorinated and iodinated benzenes against μ2. Chlorinated benzenes showed larger retention factors and ΔA as their dipole moments increased in comparison to brominated benzenes (Fig. 5c). The results indicated that structural isomers of chlorinated benzenes showed the differences in strength of π–π interactions with the magnitudes of their dipole moments as we expected. Surprisingly, di-iodinated benzenes showed little change in ΔA, and no correlation between ΔA and their dipole moment was observed. We considered that the differences in the retentions of di-iodo benzenes were not a result of the π–π interaction. As mentioned above, the σ hole of iodine groups is the largest and shows the strongest halogen–π interactions amongst the halogen groups. Additionally, Präsang et al. reported that stronger X–π interaction is occurred by substitution of the electron withdrawing functional groups to next carbon atom on the aromatic rings.64 In our case, the X–π interaction strength could be shown in the order of o- > m- > p-di-iode benzenes. This is because iodine also has an electron withdrawing effect and the strength of the withdrawing effect follows the order of o- > m- > p-di-iode benzenes.
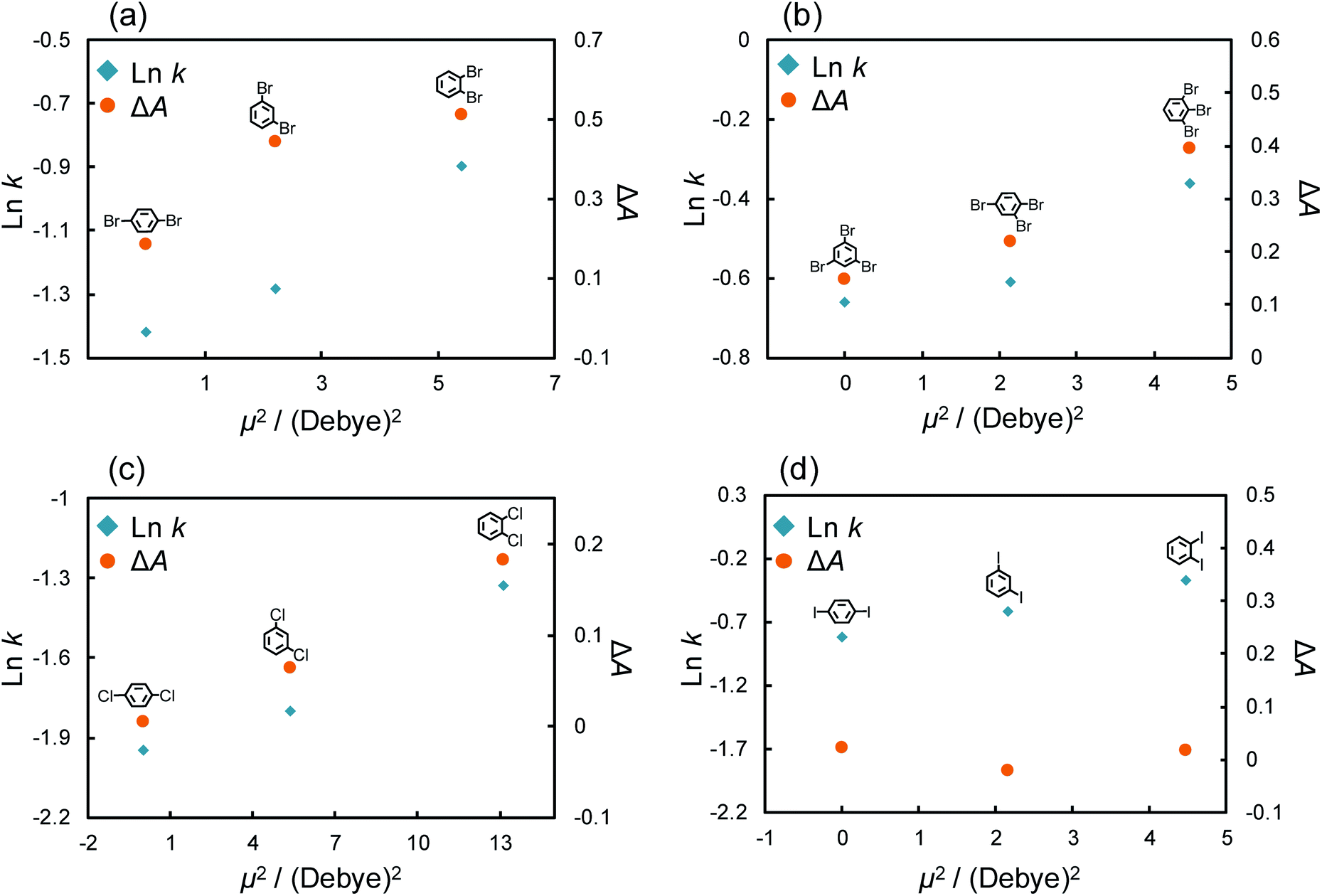 | ||
| Fig. 5 Logarithm of the retention factor or ΔA of (a) di- or (b) tri-brominated benzenes and (c) di-chlorinated or (d) di-iodinated benzenes with the C70-coated column against square of dipole moment in each solute. Conditions: column, C70-coated (Type-2, 75.0 cm × 100 μm i.d.); flow rate; 2.0 μL min−1; mobile phase, n-hexane; temperature, 25 °C; detection, UV 228 nm. | ||
Effect of dielectric constants in mobile phases
As shown in eqn (1) and (2), the induced-dipole interaction is strongly influenced by the dielectric constant (ε) of the solvents. Fig. 6a shows the chromatograms of the mixtures of halogenated benzenes in n-hexane or n-octane mobile phases with the C70-coated column. As expected, the retentions for halogenated benzenes with the C70-coated column were drastically lower with n-octane than with n-hexane. To consider the effect of ε, we evaluated the retention behaviors of halogenated benzenes in several normal alkanes (NAs), which have different ε. Fig. 6b shows a semilogarithmic plot of the retention factors for halogenated benzenes obtained with each NA against 1/ε2. The plot shows that ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k increased linearly with 1/ε2. This result was consistent with the expected trend given by eqn (1) and (2) and clearly indicated that the retentions of halogenated benzenes were significantly impacted by the induced-dipole interaction.
k increased linearly with 1/ε2. This result was consistent with the expected trend given by eqn (1) and (2) and clearly indicated that the retentions of halogenated benzenes were significantly impacted by the induced-dipole interaction.
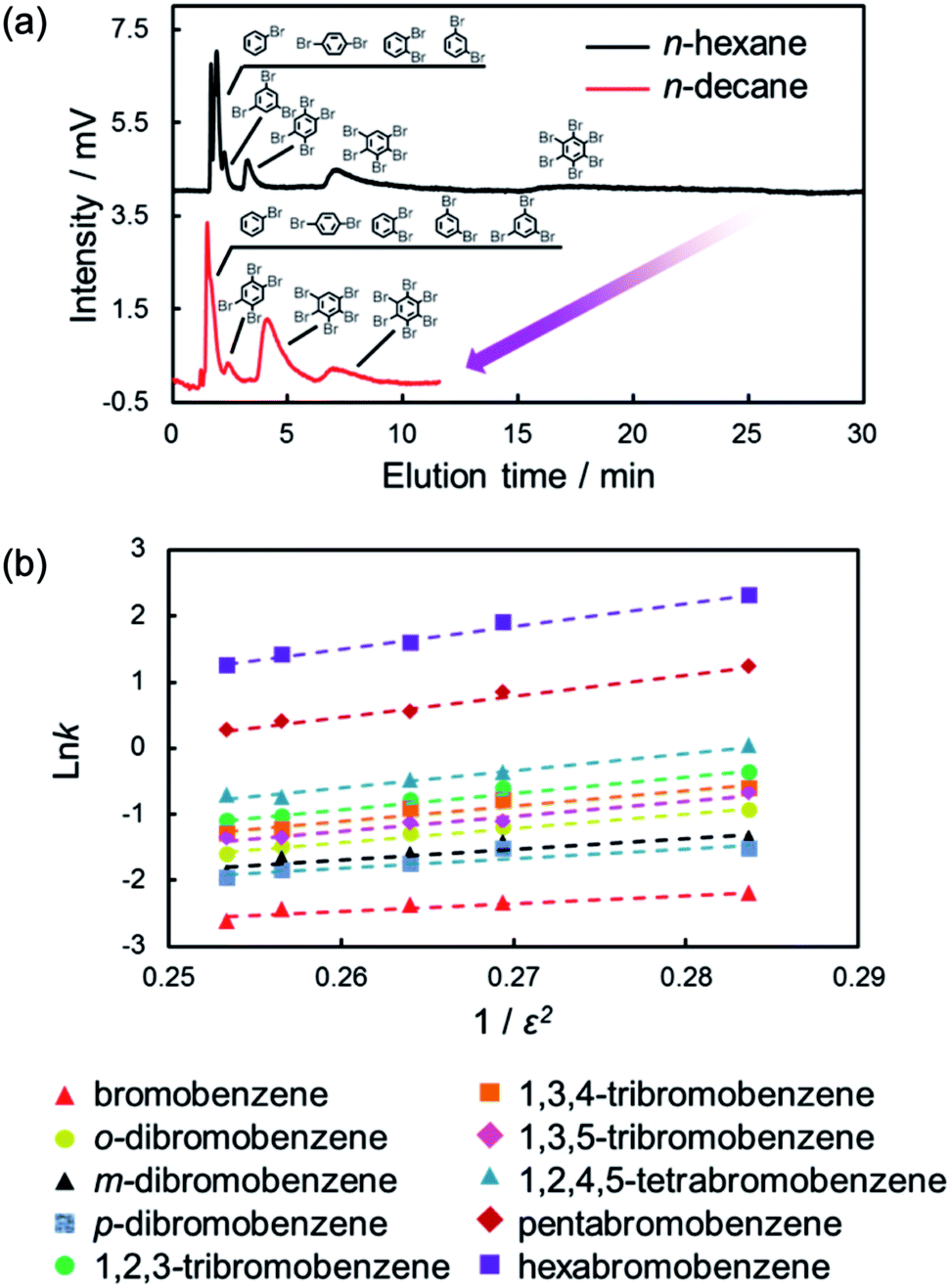 | ||
| Fig. 6 Chromatograms of the mixture of brominated benzenes with the C70-coated column (a) and lnk against 1/ε2 in each NA mobile phase (b). Conditions: column, C70-coated (Type-1, 25.5 cm × 100 μm i.d.); flow rate; 2.0 μL min−1; mobile phase, n-hexane, n-heptane, n-octane, n-nonane, n-decane; temperature, 25 °C; detection, UV 228 nm. | ||
Possibility of orbital interactions influencing X–π interactions
Because the σ hole behaves as an electron acceptor in halogen bonding, the contributions of the orbital interaction as well as electrostatic interaction have to be considered. Early in 1950, Mulliken showed experimental evidence of changes in the electronic structure occur upon formation of a halogen bond.65 Furthermore, many computational results have also supported the notion that orbital interactions affect halogen bonding.66–69 However, several computational studies suggested an opposing view that the orbital interaction could not largely contribute to halogen bonding.70–72 In order to promote such a complicated argument, we evaluated the contribution of orbital energy to the X–π interaction by 1H NMR spectroscopy, which sensitively reflects the electronic state of protons. In our study, No-D 1H NMR was employed because protic solvents can be used by subtracting the solvents peaks, briefly we utilized protic n-hexane as a solvent. The No-D 1H NMR spectra of o-dibromobenzene and pentabromobenzene in the presence of C70 in n-hexane were recorded and compared with each other. Peak shifts were not observed in o-dibromobenzene when o-dibromobenzene and C70 were mixed (Fig. 7a). However, when pentabromobenzene was mixed with C70, the peak in the pentabromobenzene spectrum shifted slightly upfield (Fig. 7b). These results suggested that a proton on the pentabromobenzene was placed into a more electron rich (more shielded) environment and that pentabromobenzene, as an electron acceptor of C70, might be subjected to the orbital interaction. Briefly, five bromine atoms on pentabromobenzene became electron rich from the molecular interaction with C70; this meant that the aromatic ring and its proton were also in electron rich conditions. In addition, we carried out computer simulations to determine the differences in orbital energy between the HOMO in C70-PFPA-NHS and the LUMO in each brominated benzene (Fig. 7c). The differences in orbital energies of poly-brominated benzenes (n > 2) were much lower than that for dibromobenzenes and were more likely to cause orbital interactions with C70-PFPA-NHS. Furthermore, the LUMOs of poly-brominated benzenes (n > 2) extended in the direction of the σ bond in the C–Br of the bromine atoms and matched the positions showing the X–π interactions (Fig. S5†). These results also suggested that orbital interactions might contribute to the X–π interaction.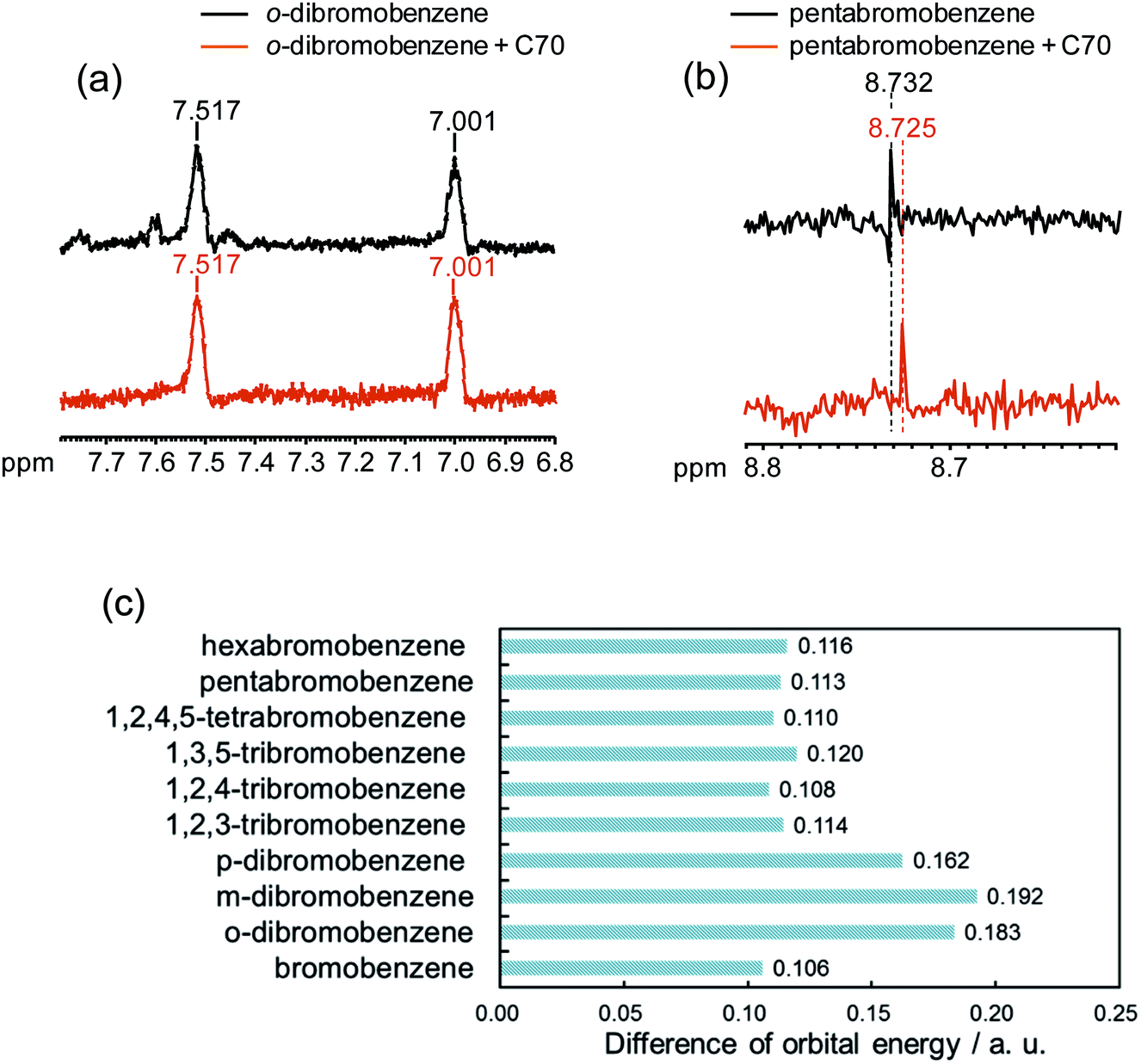 | ||
| Fig. 7 1H NMR spectra of o-dibromobenzene (a), pentabromobenzene (b) with/without C70 in n-hexane and the difference in orbital energy between HOMO in C70-PFPA-NHS and LUMO in each brominated benzene (c). | ||
One-pot separation of the entirety of brominated benzenes
In the above discussions, we clarified the presence of π–π and/or X–π interactions between halogenated benzenes and carbon-materials. In addition, we revealed the effect of the mobile phase as being against these π interactions. Finally, we were faced with the challenge of achieving the separation of halogenated benzenes in NPLC. This achievement will lead to efficient separations of aromatic halogens in oil. To realize this goal, we optimized the mobile phase conditions, in consideration of the dielectric effect, to achieve a one-pot separation of all isomers of brominated benzenes (11 analogues, Fig. 8). The separation of halogenated benzenes in oil samples is a major issue in the field of environmental chemistry, since many halogenated benzenes are in waste oil and are highly toxic.73–75 Therefore, the LC separation of halogenated benzenes has great importance in environmental chemistry and shows its potential for separating toxic aromatic halogens, such as polychlorinated biphenyls and polybrominated biphenyls. | ||
| Fig. 8 Chromatogram of all isomers of brominated benzenes with the C70-coated column in NPLC. Conditions: column, C70-coated (Type-2, 75.0 cm × 100 μm i.d.); flow rate; 2.0 μL min−1; mobile phase, n-hexane/n-decane = 8/2; temperature, 25 °C; detection, UV 228 nm. | ||
Conclusion
In this study, we experimentally evaluated the strength of the X–π interaction between carbon-materials and the variety of halogenated benzenes using NPLC, in which the hydrophobic interaction was completely suppressed. Higher retentions were observed as the number of Cl, Br, or I substitutions on the benzenes increased, especially for the C70-coated column, which showed higher retention efficiencies than other carbon materials. UV absorption spectra of mono-to tri-brominated benzenes were critically changed in the presence of C70, indicating that these compounds mainly interacted via the π–π interaction with C70. Furthermore, the number of bromine substitutions affected the strength of the X–π interaction with C70. The 1H NMR spectra of o-dibromobenzene and pentabromobenzene in the presence of C70 also showed the possibility for orbital interactions based on the X–π interaction. Thus, we supposed the existence of bimodal interactions, the π–π and X–π interactions, between the halogenated benzenes and aromatic materials. Using this new knowledge, we successfully demonstrated the effective separation of di- or tri-bromo benzene isomers and 11 brominated benzene analogues with a C70-coated column in NPLC by optimizing the mobile phase conditions. We believe that this report greatly contributes to elucidation of the X–π interaction and efficient separations of halogenated environmental pollutants.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Environment Research and Technology Development Fund of the Environmental Restoration and Conservation Agency of Japan (5-1953) National Science Foundation CHE-1808671, and Mukai Science and Technolgy Foundation.Notes and references
- M. H. Kolar and P. Hobza, Chem. Rev., 2016, 116, 5155–5187 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, J. Med. Chem., 2013, 56, 1363–1388 CrossRef CAS.
- S. Kozuch and J. M. L. Martin, J. Chem. Theory Comput., 2013, 9, 1918–1931 CrossRef CAS.
- M. Erdelyi, Nat. Chem., 2014, 6, 762 CrossRef CAS.
- E. Danelius, H. Andersson, P. Jarvoll, K. Lood, J. Gräfenstein and M. Erdélyi, Biochemistry, 2017, 56, 3265–3272 CrossRef CAS.
- M. Erdélyi, Biochemistry, 2017, 56, 2759–2761 CrossRef PubMed.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- H. D. Arman, E. R. Rafferty, C. A. Bayse and W. T. Pennington, Cryst. Growth Des., 2012, 12, 4315–4323 CrossRef CAS.
- C. Ouvrard, J.-Y. Le Questel, M. Berthelot and C. Laurence, Acta Crystallogr., Sect. B: Struct. Sci., 2003, 59, 512–526 CrossRef PubMed.
- H. S. El-Sheshtawy, B. S. Bassil, K. I. Assaf, U. Kortz and W. M. Nau, J. Am. Chem. Soc., 2012, 134, 19935–19941 CrossRef CAS PubMed.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed.
- M. J. Langton, S. W. Robinson, I. Marques, V. Félix and P. D. Beer, Nat. Chem., 2014, 6, 1039 CrossRef CAS PubMed.
- B. W. Tresca, A. C. Brueckner, M. M. Haley, P. H. Y. Cheong and D. W. Johnson, J. Am. Chem. Soc., 2017, 139, 3962–3965 CrossRef CAS PubMed.
- J. Noh, S. Jung, D. G. Koo, G. Kim, K. S. Choi, J. Park, T. J. Shin, C. Yang and J. Park, Sci. Rep., 2018, 8, 14448 CrossRef PubMed.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS.
- D. González-Rodríguez and A. P. H. J. Schenning, Chem. Mater., 2011, 23, 310–325 CrossRef.
- F. Fernandez-Palacio, M. Poutanen, M. Saccone, A. Siiskonen, G. Terraneo, G. Resnati, O. Ikkala, P. Metrangolo and A. Priimagi, Chem. Mater., 2016, 28, 8314–8321 CrossRef CAS PubMed.
- H. L. Nguyen, P. N. Horton, M. B. Hursthouse, A. C. Legon and D. W. Bruce, J. Am. Chem. Soc., 2004, 126, 16–17 CrossRef CAS.
- T. A. Barendt, A. Docker, I. Marques, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 11069–11076 CrossRef CAS PubMed.
- A. Vanderkooy, P. Pfefferkorn and M. S. Taylor, Macromolecules, 2017, 50, 3807–3817 CrossRef CAS.
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789 CrossRef CAS PubMed.
- Y. Lu, T. Shi, Y. Wang, H. Yang, X. Yan, X. Luo, H. Jiang and W. Zhu, J. Med. Chem., 2009, 52, 2854–2862 CrossRef CAS PubMed.
- L. A. Hardegger, B. Kuhn, B. Spinnler, L. Anselm, R. Ecabert, M. Stihle, B. Gsell, R. Thoma, J. Diez, J. Benz, J.-M. Plancher, G. Hartmann, D. W. Banner, W. Haap and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 314–318 CrossRef CAS PubMed.
- C. Yabe-Nishimura, Pharmacol. Rev., 1998, 50, 21 CAS.
- D. M. Himmel, K. Das, A. D. Clark, S. H. Hughes, A. Benjahad, S. Oumouch, J. Guillemont, S. Coupa, A. Poncelet, I. Csoka, C. Meyer, K. Andries, C. H. Nguyen, D. S. Grierson and E. Arnold, J. Med. Chem., 2005, 48, 7582–7591 CrossRef CAS PubMed.
- J. Cao, X. Yan, W. He, X. Li, Z. Li, Y. Mo, M. Liu and Y.-B. Jiang, J. Am. Chem. Soc., 2017, 139, 6605–6610 CrossRef CAS PubMed.
- C. Heroven, V. Georgi, G. K. Ganotra, P. Brennan, F. Wolfreys, R. C. Wade, A. E. Fernández-Montalván, A. Chaikuad and S. Knapp, Angew. Chem., Int. Ed., 2018, 57, 7220–7224 CrossRef CAS PubMed.
- H. Matter, M. Nazaré, S. Güssregen, D. W. Will, H. Schreuder, A. Bauer, M. Urmann, K. Ritter, M. Wagner and V. Wehner, Angew. Chem., Int. Ed., 2009, 48, 2911–2916 CrossRef CAS PubMed.
- M. B. Shah, J. Liu, Q. Zhang, C. D. Stout and J. R. Halpert, ACS Chem. Biol., 2017, 12, 1204–1210 CrossRef CAS PubMed.
- A. Voth Regier and P. Ho Shing, Curr. Top. Med. Chem., 2007, 7, 1336–1348 CrossRef PubMed.
- Y. Lu, Y. Wang and W. Zhu, Phys. Chem. Chem. Phys., 2010, 12, 4543–4551 RSC.
- S. Tsuzuki, T. Uchimaru, A. Wakisaka and T. Ono, J. Phys. Chem. A, 2016, 120, 7020–7029 CrossRef CAS PubMed.
- I. S. Youn, D. Y. Kim, W. J. Cho, J. M. L. Madridejos, H. M. Lee, M. Kołaski, J. Lee, C. Baig, S. K. Shin, M. Filatov and K. S. Kim, J. Phys. Chem. A, 2016, 120, 9305–9314 CrossRef CAS PubMed.
- F. F. Awwadi, D. Taher, S. F. Haddad and M. M. Turnbull, Cryst. Growth Des., 2014, 14, 1961–1971 CrossRef CAS.
- C. B. Aakeröy, M. Fasulo, N. Schultheiss, J. Desper and C. Moore, J. Am. Chem. Soc., 2007, 129, 13772–13773 CrossRef PubMed.
- B. Zha, M. Dong, X. Miao, S. Peng, Y. Wu, K. Miao, Y. Hu and W. Deng, Nanoscale, 2017, 9, 237–250 RSC.
- H. Sun, A. Horatscheck, V. Martos, M. Bartetzko, U. Uhrig, D. Lentz, P. Schmieder and M. Nazaré, Angew. Chem., Int. Ed., 2017, 56, 6454–6458 CrossRef CAS PubMed.
- K. Kimata, K. Hosoya, T. Araki and N. Tanaka, J. Org. Chem., 1993, 58, 282–283 CrossRef CAS.
- S. Chen and M. Meyerhoff, Anal. Chem., 1998, 70, 2523–2529 CrossRef CAS PubMed.
- T. Kubo, E. Kanao, T. Matsumoto, T. Naito, T. Sano, M. Yan and K. Otsuka, ChemistrySelect, 2016, 1, 5900–5904 CrossRef CAS.
- T. Kubo, Y. Murakami, M. Tsuzuki, H. Kobayashi, T. Naito, T. Sano, M. Yan and K. Otsuka, Chem.–Eur. J., 2015, 21, 18095–18098 CrossRef CAS PubMed.
- E. Kanao, T. Kubo, T. Naito, T. Matsumoto, T. Sano, M. Yan and K. Otsuka, Anal. Chem., 2019, 91, 2439–2446 CrossRef CAS PubMed.
- E. Kanao, T. Naito, T. Kubo and K. Otsuka, Chromatography, 2017, 38, 45–51 CrossRef CAS.
- P. Hadi, M. Xu, C. S. K. Lin, C. W. Hui and G. McKay, J. Hazard. Mater., 2015, 283, 234–243 CrossRef CAS.
- N. Quinete, A. Esser, T. Kraus and T. Schettgen, Environ. Int., 2016, 97, 171–179 CrossRef CAS PubMed.
- R. J. Law, A. Covaci, S. Harrad, D. Herzke, M. A. E. Abdallah, K. Femie, L. M. L. Toms and H. Takigami, Environ. Int., 2014, 65, 147–158 CrossRef CAS PubMed.
- E. Malliari and O. I. Kalantzi, Environ. Int., 2017, 108, 146–169 CrossRef CAS.
- T. J. McGrath, A. S. Ball and B. O. Clarke, Environ. Pollut., 2017, 230, 741–757 CrossRef CAS.
- Y.-Z. Zheng, N.-N. Wang, Y. Zhou and Z.-W. Yu, Phys. Chem. Chem. Phys., 2014, 16, 6946–6956 RSC.
- M. A. A. Ibrahim and A. A. M. Hasb, Theor. Chem. Acc., 2018, 138, 2 Search PubMed.
- M. Erdélyi, Chem. Soc. Rev., 2012, 41, 3547–3557 RSC.
- L. C. Roper, C. Präsang, A. C. Whitwood and D. W. Bruce, CrystEngComm, 2010, 12, 3382–3384 RSC.
- M. A. A. Ibrahim, J. Comput. Chem., 2011, 32, 2564–2574 CrossRef CAS.
- R. Vijay Solomon, S. Angeline Vedha and P. Venuvanalingam, Phys. Chem. Chem. Phys., 2014, 16, 7430–7440 RSC.
- V. A. Karachevtsev, A. M. Plokhotnichenko, M. V. Karachevtsev and V. S. Leontiev, Carbon, 2010, 48, 3682–3691 CrossRef CAS.
- T.-H. Wang, C.-S. Hsu, W.-L. Huang and Y.-H. Lo, Spectrochim. Acta, Part A, 2013, 115, 866–875 CrossRef CAS PubMed.
- J. Trnka, R. Sedlak, M. Kolář and P. Hobza, J. Phys. Chem. A, 2013, 117, 4331–4337 CrossRef CAS PubMed.
- A. Forni, S. Pieraccini, S. Rendine and M. Sironi, J. Comput. Chem., 2014, 35, 386–394 CrossRef CAS PubMed.
- S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc., 2002, 124, 104–112 CrossRef CAS PubMed.
- C. F. R. A. C. Lima, M. A. A. Rocha, L. R. Gomes, J. N. Low, A. M. S. Silva and L. M. N. B. F. Santos, Chem.–Eur. J., 2012, 18, 8934–8943 CrossRef CAS PubMed.
- H. G. Wallnoefer, T. Fox, K. R. Liedl and C. S. Tautermann, Phys. Chem. Chem. Phys., 2010, 12, 14941–14949 RSC.
- E. Kanao, T. Kubo, T. Naito, T. Matsumoto, T. Sano, M. Yan and K. Otsuka, J. Phys. Chem. C, 2018, 122, 15026–15032 CrossRef CAS.
- C. Präsang, A. C. Whitwood and D. W. Bruce, Cryst. Growth Des., 2009, 9, 5319–5326 CrossRef.
- R. S. Mulliken, J. Am. Chem. Soc., 1950, 72, 600–608 CrossRef CAS.
- M. Palusiak, J. Mol. Struct., 2010, 945, 89–92 CrossRef CAS.
- C. Wang, D. Danovich, Y. Mo and S. Shaik, J. Chem. Theory Comput., 2014, 10, 3726–3737 CrossRef CAS PubMed.
- L. P. Wolters and F. M. Bickelhaupt, ChemistryOpen, 2012, 1, 96–105 CrossRef CAS PubMed.
- R. Desiraju Gautam, P. S. Ho, L. Kloo, C. Legon Anthony, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 Search PubMed.
- K. E. Riley and P. Hobza, J. Chem. Theory Comput., 2008, 4, 232–242 CrossRef CAS PubMed.
- S. Tsuzuki, A. Wakisaka, T. Ono and T. Sonoda, Chem.–Eur. J., 2012, 18, 951–960 CrossRef CAS PubMed.
- J. Řezáč and A. de la Lande, Phys. Chem. Chem. Phys., 2017, 19, 791–803 RSC.
- F. M. Jaward, N. J. Farrar, T. Harner, A. J. Sweetman and K. C. Jones, Environ. Sci. Technol., 2004, 38, 34–41 CrossRef CAS PubMed.
- M. H. Jacobson, L. A. Darrow, D. B. Barr, P. P. Howards, R. H. Lyles, M. L. Terrell, A. K. Smith, K. N. Conneely, M. E. Marder and M. Marcus, Environ. Health Perspect., 2017, 125, 097020 CrossRef PubMed.
- S. Safe and O. Hutzinger, Crit. Rev. Toxicol., 1984, 13, 319–395 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04906a |
| This journal is © The Royal Society of Chemistry 2020 |