
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDearomatization of aryl sulfoxides: a switch between mono- and dual-difluoroalkylation†
Xin
Huang‡
,
Yage
Zhang‡
,
Weijian
Liang
,
Qifeng
Zhang
,
Yaling
Zhan
,
Lichun
Kong
and
Bo
Peng
 *
*
Key Laboratory of the Ministry of Education for Advanced Catalysis Materials, Zhejiang Normal University, China. E-mail: pengbo@zjnu.cn
First published on 25th February 2020
Abstract
Herein we describe the dearomatization of aryl sulfoxides with difluoroenol silyl ether (DFESE) using a rearrangement/addition protocol. The selection of the sulfoxide activator determines whether one or two difluoroalkyl groups are incorporated into dearomatized products. Using TFAA can deliberately halt the reaction at the mono-difluoroalkylated dearomatized intermediate formed via a [3,3]-rearrangement, which can be further trapped by external nucleophiles to give mono-difluoroalkylated alicycles. In contrast, switching to Tf2O enhances the electrophilicity of dearomatized intermediates, thus allowing for the adoption of a second DFESE to produce dual-difluoroalkylated alicycles.
Introduction
Dearomatization is a powerful strategy that enables the direct conversion of readily available 2-D arenes to value-added 3-D alicyclic architectures in a step-economical manner.1 The dearomatization protocol has constantly received intensive research interest due to its great potential in the synthesis of natural products and bioactive compounds.2 Despite considerable advances in this context, the discovery of methods for dearomatizing a new class of arenes is still a useful endeavor.In the past few decades, ortho-C–H functionalization of aryl sulfoxides with certain nucleophiles via [3,3]-rearrangement has been validated and significantly progressed (Fig. 1a).3,4 Until now, an array of nucleophiles including allyl/propargyl silanes,5,6 carbonyl compounds,7 phenols,8 alkyl nitriles9 and cyclopropanols10 have been found to be suitable for this process. A noteworthy feature of these reactions is the unique [3,3]-rearrangement that allows for the transient formation of a dearomatization intermediate, which conventionally undergoes a rearomatization to yield rearomatized ortho-functionalized aryl sulfide. Based on this reaction pattern, we wondered if blocking the rearomatization of transiently formed dearomatized species could lead to dearomatization products. Interestingly, the dearomatization pathway we envisioned has been previously observed by Procter and co-workers who showcased an elegant dearomatization of benzothiophene S-oxides with phenols, in which a methyl group instead of hydrogen was introduced into the β-position for impeding the rearomatization process.11 As a result, the phenolic oxygen acting intramolecularly as a nucleophile trapped the dearomatized intermediate providing dearomatization products (Fig. 1b). In contrast, Yorimitsu and co-workers demonstrated the possibility of dearomatizing a nucleophilic counterpart such as 2,6-dimethyl- and 2,6-difluoro-phenols with the S(IV)-rearrangement protocol (Fig. 1b).8a,c However, to the best of our knowledge, there have been no examples of dearomatization of aryl sulfoxides, which is probably due to the relatively higher resonance energy of arenes than of heteroarenes.12 Herein we describe the dearomatization of aryl sulfoxides with difluoroenol silyl ether (DFESE) which has been identified as a highly effective rearrangement partner with aryl iodanes owing to the unique effect of fluorine in our recent studies.13 This protocol allows for the on-demand incorporation of one or two difluoroalkyl groups into dearomatized products by switching electrophilic activators (TFAA vs. Tf2O) (Fig. 1c). It is worth noting that the difluoroalkyl group plays critical roles in drug discovery since it can serve as a more-lipophilic isostere of carbinol, thiol, hydroxamic acid, or amide groups.14,15
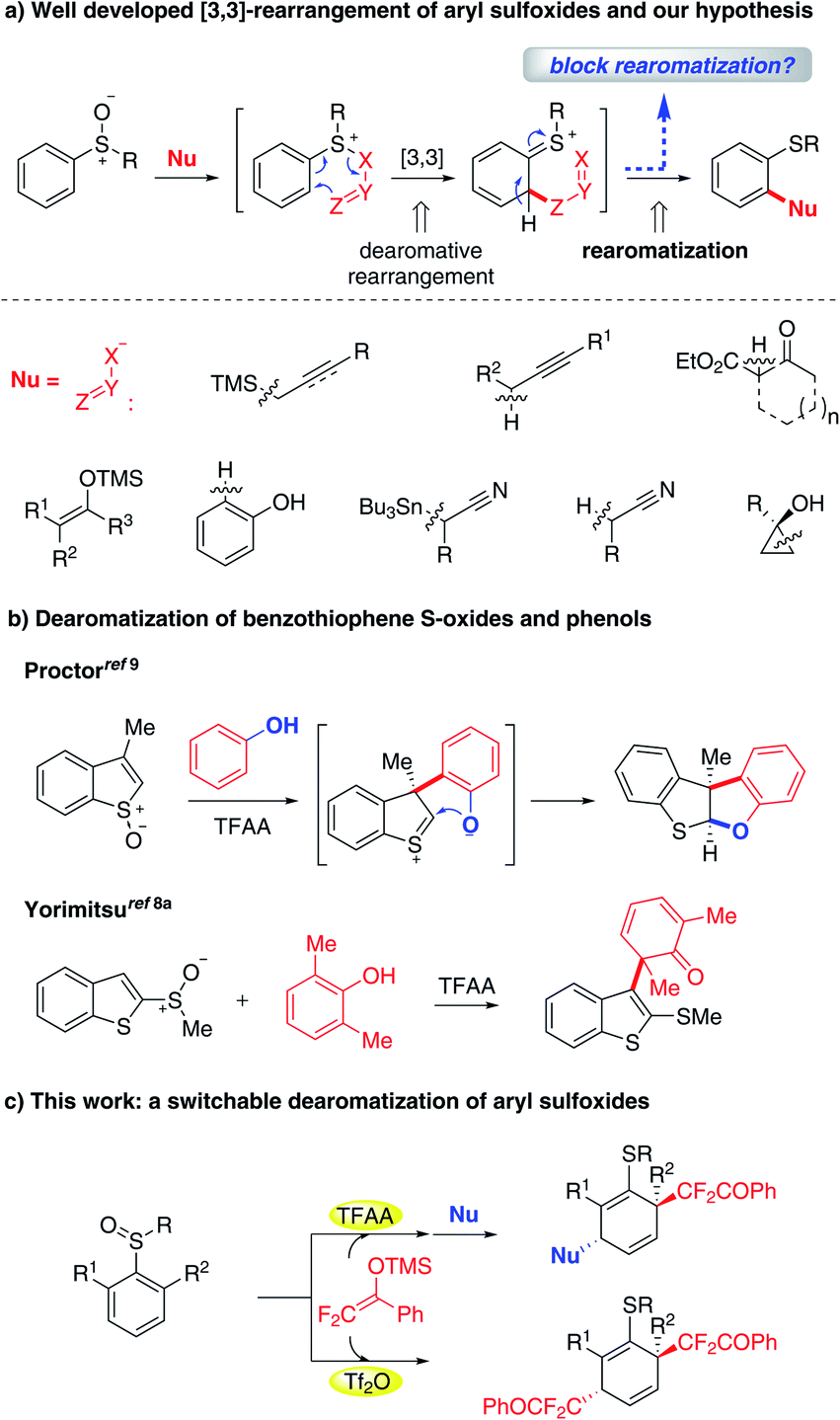 | ||
| Fig. 1 (a) Well developed [3,3]-rearrangement of aryl sulfoxides and our hypothesis, (b) dearomatization of benzothiophene S-oxides and phenols and (c) this work. | ||
We investigated the model reaction of ortho, ortho′-dimethyl phenyl sulfoxide 1a with DFESE 2a using Et3SiH as a reductive quencher (Table 1). Gratifyingly, the initial use of TFAA as the activator and MeCN as the solvent afforded mono-difluoroalkylated dearomatization product 3a in a modest yield (57%) (entry 1). It is worth mentioning that, in our recent studies, ortho, ortho′-dimethyl phenyl iodine(III) diacetate was also found to be suitable for a related dearomative rearrangement process.16 To our surprise, aside from the expected 3a, the reaction also produced a small amount of dual-difluoroalkylated product 4a (15%) which showed sufficient nucleophilicity of DFESE not only for the rearrangement with sulfoxide but also for the capture of the in situ-formed dearomatized intermediate. Notably, the reaction is unusually sensitive to the polarity of the solvent since slightly tuning the ratio of the combined solvents could exert a significant influence on the reaction outcome (entries 2–5). Switching to a nonpolar solvent (DCM) could completely inhibit the reaction (entry 2). The use of a 9/1 ratio of MeCN/DCM led to the best yield of 3a (65%) and showed the highest selectivity for 3a formation (entry 4). In contrast, slightly tuning the solvent ratio (10/1 and 8/1) dramatically deceased the efficiency of the reaction (entries 3 and 5). Further screening of different activators revealed that the use of Tf2O as an activator could exclusively afford the dual-difluoroalkylated product 4a albeit in rather poor yield (17%) (entry 7). To our delight, further optimization of the solvent and reaction temperature led to identification of the best conditions (Tf2O, DCM, −100 °C, 24 h) for dual-difluoroalkylation (entry 12). To the best of our knowledge, methods for dual-difluoroalkylation have not yet been reported.
|
|
||||
|---|---|---|---|---|
| Entry | Activator | Solvent | Temp. | Yieldb of 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4a :4a |
| a Unless otherwise noted, the reaction was performed with 1a (0.3 mmol), 2a (1.5 equiv.), activator (1.5 equiv.) and Et3SiH (3.0 equiv.). b The reaction of 1a and 2a (3.0 equiv.) was performed with Tf2O (1.5 equiv.) at the indicated temperature for 24 h and Et3SiH was not used herein. For the investigation of other reaction parameters, see the ESI. | ||||
| 1 | TFAA | MeCN | −50 °C | 57%:15% |
| 2 | TFAA | DCM | −50 °C | 0%:0% |
| 3 | TFAA | MeCN/DCM (10/1) | −50 °C | 50%:20% |
| 4 | TFAA | MeCN/DCM (9/1) | −50 °C | 65%:5% |
| 5 | TFAA | MeCN/DCM (8/1) | −50 °C | 27%:29% |
| 6 | Ts2O | MeCN/DCM (9/1) | −50 °C | 0%:trace |
| 7 | Tf2O | MeCN/DCM (9/1) | −50 °C | 0%:17% |
| 8 | (ClF2CO)2O | MeCN/DCM (9/1) | −50 °C | 61%:8% |
| 9 | Tf2O | DCM | −78 °C | 0%:48%b |
| 10 | Tf2O | MeCN | −50 °C | 0%:19%b |
| 11 | Tf2O | DCM | −50 °C | 0%:35%b |
| 12 | Tf2O | DCM | −100 °C | 0%:73%b |

With the best conditions in hand, we were curious about the intriguing dearomatized intermediate formed after rearrangement and prior to being trapped by the second nucleophile. Therefore, we attempted to identify the intermediate using in situ NMR at low temperature (−50 °C). To our surprise, the reaction of 1b with 2a using TFAA as the activator produced a bicyclic dearomatized intermediate IM1 with concurrent formation of C–C and C–O bonds (Fig. 2). Interestingly, IM1 could not only be reduced to the expected dearomatization product 3b, but could also be hydrolyzed to bicyclic hemiacetal 5, the structure of which was confirmed by single crystal X-ray structure analysis. It should be noted that we failed to attain the dearomatized intermediate using Tf2O as the activator. It is likely that the use of such a strong activator could facilitate the further trapping of the dearomatized intermediate, thus directly leading to the dual difluoroalkylated product.
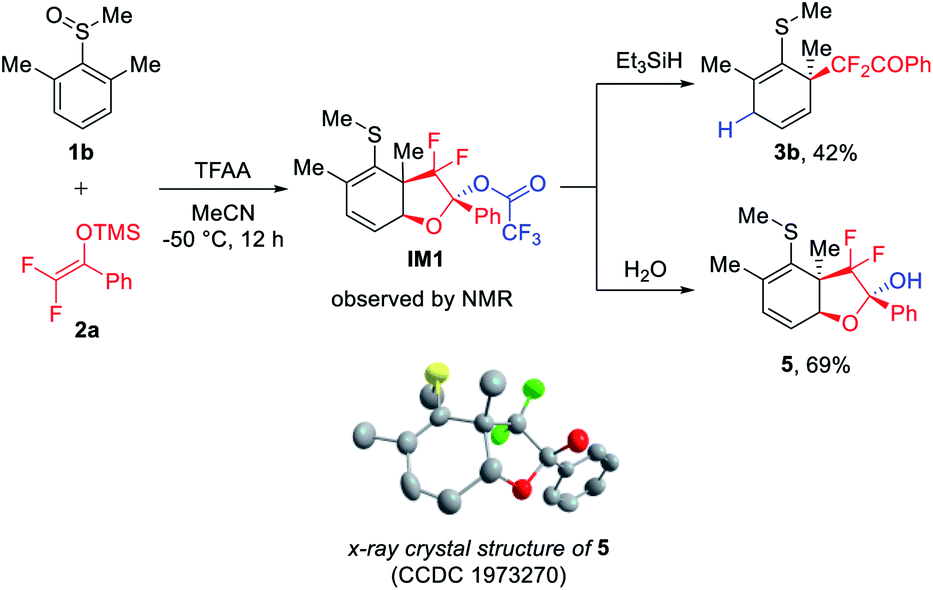 | ||
| Fig. 2 Identification of dearomatized key intermediate IM1. | ||
Next, we examined the generality of both methods under optimum conditions A and B (Table 2). Regardless of the length of alkyl chains on sulfur, both reactions proceeded smoothly to afford mono-difluoroalkylated products 3a–c and dual-difluoroalkylated products 4a–c in good yields (entry 1 to 3). Both electron-donating and -withdrawing groups (1d–l) at the para-position were generally well-tolerated in both reactions except the dual difluoroalkylation of para-alkynyl sulfoxide 1k (entry 7). This unsuccessful case was probably due to the instability of 1k with the alkynyl group which was found to deteriorate upon the addition of Tf2O. To our delight, both mono- and dual-difluoroalkylation reactions exhibited excellent functional group compatibility. Functional groups including alkyl/aromatic halides (1f, 1l, 1q, and 1s), ethers (1g and 1p), esters (1h, 1i, and 1q–s), and nitriles (1j) proved to be suitable for both reactions. Such functionalities provide a versatile platform for further asymmetric functionalization of the products. Remarkably, highly electrophilic benzylic chloride (1f) and α,β-unsaturated esters (1h and 1r) that can be problematic functional groups for conventional cross-coupling reactions were also tolerated herein (entries 3, 4 and 14). Except for 1n the reaction demonstrated an excellent regioselectivity when using aryl sulfoxides 1o–s (entries, 10–15). When using stereo-hindered substrate 1o, mono-difluoroalkylation proceeded smoothly to produce 3o in a good yield (74%) (entry 11). In contrast, the dual-difluoroalkylation of 1o merely furnished the expected 4o in a modest yield (43%). This is likely due to the relatively smaller size of Et3SiH than of DFESE, which allows the second nucleophile to easily approach the stereo-hindered dearomatization intermediate. In the case of sulfoxide 1t bearing chloro and methyl groups at the ortho positions, the use of different activators was found to be crucial to the regioselectivity of the rearrangement step (entry 16). Using TFAA as the activator exclusively afforded mono-difluoroalkylated product 3t, whereas the use of Tf2O resulted in poor regioselectivity yielding a mixture of regioisomers including dearomatized product 4t and dechlorinated, rearomatization product 4t′. In addition to aryl sulfoxides, furan sulfoxide 1u was also suitable for the dual-difluoroalkylation process. However, in the presence of TFAA, 1u could not afford any desired mono-difluloroalkylated product 3u. This was probably due to the less electrophilic nature of the relatively electron-rich furan moiety that inhibited the interaction of activated sulfoxide with nucleophiles.
| a Unless otherwise noted, the reaction was performed under the optimum conditions (condition A: Table 1, entry 5; condition B: Table 1, entry 13). The relative configurations of 4 were deduced from the X-ray structure of 4s. b After Et3SiH was added, the reaction mixture was slowly warmed to 0 °C for 1i, 1j, 1q, 1r, and 1s or 20 °C for 1k. c After warming to room temperature, 1k and 1u decomposed into complex mixtures. |
|---|

|
Encouraged by the obtained results in Table 2, we further studied the generality of second nucleophiles for trapping the dearomatized intermediate (Table 3). Gratifyingly, an array of carbon and heteroatom nucleophiles such as allyl silanes, ZnEt2, enol silyl ethers, thiophenes, indoles, naphthols, thiophenols, and Ts-NH2 were suitable for the reaction. As a result, a diverse set of densely functionalized alicyclic compounds 6a–h were obtained from simple aryl sulfoxide 1a which can be a challenging synthetic target using known methods. Interestingly, in contrast to aryl sulfoxides, naphthyl sulfoxide 1v and indole sulfoxide 1w could be converted to the desulfurated carbonyls 7a and 7b through electrophilic rearrangement and simple hydrolysis although they were not suitable for the aforementioned mono- and dual-difluoroalkylative dearomatization reactions (below the dashed line).17 It is likely that the “S(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C” bonds of these dearomatization intermediates formed via [3,3]-rearrangement were inert to the aforementioned nucleophiles (Et3SiH and DFESE) but could be quenched by H2O. This result also demonstrated the possibility of applying the current protocol for the synthesis of difluoroalkylated cyclohexenones. Unfortunately, the protocol of desulfurizing ketone formation could not be applicable to model sulfoxide 1a.
C” bonds of these dearomatization intermediates formed via [3,3]-rearrangement were inert to the aforementioned nucleophiles (Et3SiH and DFESE) but could be quenched by H2O. This result also demonstrated the possibility of applying the current protocol for the synthesis of difluoroalkylated cyclohexenones. Unfortunately, the protocol of desulfurizing ketone formation could not be applicable to model sulfoxide 1a.
| a Only one diastereoisomer was obtained in all cases. The relative configurations of 6 were deduced from the X-ray structure of 4s. |
|---|

|
Based on the obtained results, a plausible mechanism is shown in Fig. 3. Both TFAA and Tf2O can activate aryl sulfoxide (1a as a representative substrate) to yield highly electrophilic S(IV) species, which can be trapped by DFESE 2a to provide sulfonium enolates. Subsequent [3,3]-rearrangement of the “S(IV)–OTMS” linked enolates affords ortho-difluoroalkylated dearomatization S(IV) intermediates that readily undergo intramolecular addition to form bicyclic intermediates IM1 and IM2. The different leaving groups such as OCOCF3 and OTf anchored on IM1 and IM2 provide these intermediates with different electrophilicity. As a result, IM1 could be halted and characterized by NMR analysis and be trapped by different types of nucleophiles as demonstrated in Table 3. In contrast, due to its relatively high electrophilicity, IM2 is easily captured by the second DFESE 2a to furnish dual difluoroalkylated product 4b. As depicted in Fig. 3, the regioselectivity for introducing the second nucleophile into the 5-positions could be attributable to the stereo hindrance of the ortho-quaternary carbon of IM1 and IM2. The stereoselective formation of trans-cyclohexadiene products is probably due to the furan moieties of IM1 and IM2 that block one face of these intermediates. The outcome of this stereochemistry may also be understood via analysis of the X-ray crystal structure of 5 given in Fig. 2.
 | ||
| Fig. 3 Proposed mechanism and examination of difluoroenol silyl ethers 2b–2d. | ||
Encouraged by the success of DFESE 2a, we also examined other DFESEs by replacing the Ph group of 2a with n-Bu, OEt, and CF3 (Fig. 3, below the dashed line). Unfortunately, all these DFESEs (2b–2d) were found to be unsuitable for the two dearomatization processes.
To our delight, both dearomatization reactions on the gram-scale could produce the desired products in good yields which demonstrated the practicality of these protocols (Fig. 4). Inspired by the progress of sulfoxide mediated rearrangements,3 we attempted to apply the rearrangement protocol for further elaboration of the dearomatization product 3a. As illustrated in Fig. 4, after oxidation by m-CPBA, 3a could undergo a second rearrangement with nucleophiles such as DFESE and α-stannyl nitrile to produce highly substituted cyclohexenones 8a and 8b after desulfurization hydrolysis.17 Notably, the relative configuration of 8b was assigned according to NOE studies (for details, see the ESI†).
 | ||
| Fig. 4 Gram-scale reactions and conversion of 3a to highly substituted difluoroalkylated cyclohexenones. | ||
Conclusions
In summary, we have developed the dearomative difluoroalkylation of aryl sulfoxides. Through the choice of appropriate sulfoxide activators (Tf2O or TFAA), one or two difluoroalkyl groups according to the demand could be incorporated into the dearomatized products. The use of TFAA as the activator allowed determination of the structure of the key dearomatized intermediate IM1, which was elucidated to be a bicyclic dearomatized sulfide. Remarkably, both the mono- and dual-difluoroalkylation processes exhibited excellent functional group compatibility and excellent regioselectivity for asymmetric aryl sulfoxides. In addition to Et3SiH and DFESE, other carbon and heteroatom nucleophiles were also suitable for capturing the dearomatized difluoroalkylated intermediate. In brief, this study showcases the possibility of conducting the dearomative rearrangement in a very controlled manner which allows for the on-demand synthesis of a diverse array of valuable highly substituted alicycles from readily available aryl sulfoxides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. P. acknowledges Zhejiang Normal University and the support from the Zhejiang Provincial Natural Science Foundation of China (LR20B020001). X. H. thanks the Natural Science Foundation of Zhejiang Province (LQ19B020005).Notes and references
- (a) W. C. Wertjes, E. H. Southgate and D. Sarlah, Chem. Soc. Rev., 2018, 47, 7996 RSC; (b) C. Zheng and S.-L. You, Chem, 2016, 1, 830 CrossRef CAS; (c) F. C. Pigge, in Arene Chemistry, ed. J. Mortier, Wiley, Hoboken, 2015, pp. 399–423 Search PubMed; (d) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed; (e) F. López Ortiz, M. J. Iglesias, I. Fernández, C. M. Andújar Sánchez and G. R. Gómez, Chem. Rev., 2007, 107, 1580 CrossRef PubMed.
- (a) S. P. Roche and J. A. Porco Jr, Angew. Chem., Int. Ed., 2011, 50, 4068 CrossRef CAS PubMed; (b) C. Zheng and S.-L. You, Nat. Prod. Rep., 2019, 36, 1589 RSC; (c) Q. Ding, Y. Ye and R. Fan, Synthesis, 2013, 45, 1 CrossRef CAS; (d) L. Pouységu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66, 2235 CrossRef; (e) X. Huang, S. Klimczyk and N. Maulide, Synthesis, 2012, 2, 175 Search PubMed.
- (a) T. Yanagi, K. Nogi and H. Yorimitsu, Tetrahedron Lett., 2018, 59, 2951 CrossRef CAS; (b) H. Yorimitsu, Chem. Rec., 2017, 17, 1156 CrossRef CAS PubMed; (c) A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2016, 55, 9842 CrossRef CAS PubMed; (d) L. H. S. Smith, S. C. Coote, H. F. Sneddon and D. J. Procter, Angew. Chem., Int. Ed., 2010, 49, 5832 CrossRef CAS PubMed; (e) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701 CrossRef CAS PubMed; (f) L. Zhang, M. Hu and B. Peng, Synlett, 2019, 30, 2203 CrossRef CAS.
- For a comprehensive book on sulfur chemistry, see: (a) X. Jiang, Sulfur Chemistry, Springer, Berlin, 2019 CrossRef. For selected examples of mild synthesis of aryl sulfoxides, see: (b) Y. Li, S. A. Rizvi, D. Hu, D. Sun, A. Gao, Y. Zhou, J. Li and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 13499 CrossRef CAS PubMed; (c) Y. Li, M. Wang and X. Jiang, ACS Catal., 2017, 7, 7587 CrossRef CAS.
- (a) S. Akai, N. Kawashita, H. Satoh, Y. Wada, K. Kakiguchi, I. Kuriwaki and Y. Kita, Org. Lett., 2004, 6, 3793 CrossRef CAS PubMed; (b) A. Padwa, S. Nara and Q. Wang, Tetrahedron Lett., 2006, 47, 595 CrossRef CAS; (c) S. Akai, N. Kawashita, Y. Wada, H. Satoh, A. H. Alinejad, K. Kakiguchi, I. Kuriwaki and Y. Kita, Tetrahedron Lett., 2006, 47, 1881 CrossRef CAS; (d) A. J. Eberhart, J. E. Imbriglio and D. J. Procter, Org. Lett., 2011, 13, 5882 CrossRef CAS; (e) A. J. Eberhart, C. Cicoira and D. J. Procter, Org. Lett., 2013, 15, 3994 CrossRef CAS PubMed; (f) M. Šiaučiulis, S. Sapmaz, A. P. Pulis and D. J. Procter, Chem. Sci., 2018, 9, 754 RSC.
- (a) A. J. Eberhart and D. J. Procter, Angew. Chem., Int. Ed., 2013, 52, 4008 CrossRef CAS PubMed; (b) A. J. Eberhart, H. J. Shrives, E. Álvarez, A. Carrër, Y. Zhang and D. J. Procter, Chem.–Eur. J., 2015, 21, 7428 CrossRef CAS PubMed; (c) A. J. Eberhart, H. Shrives, Y. Zhang, A. Carrër, A. V. S. Parry, D. J. Tate, M. L. Turner and D. J. Procter, Chem. Sci., 2016, 7, 1281 RSC; (d) J. A. Fernández-Salas, A. J. Eberhart and D. J. Procter, J. Am. Chem. Soc., 2016, 138, 790 CrossRef PubMed.
- (a) X. Huang and N. Maulide, J. Am. Chem. Soc., 2011, 133, 8510 CrossRef CAS PubMed; (b) X. Huang, M. Patil, C. Farès, W. Thiel and N. Maulide, J. Am. Chem. Soc., 2013, 135, 7312 CrossRef CAS PubMed; (c) Ref 5a ; (d) Ref 5b ; (e) Ref 5c .
- (a) T. Yanagi, S. Otsuka, Y. Kasuga, K. Fujimoto, K. Murakami, K. Nogi, H. Yorimitsu and A. Osuka, J. Am. Chem. Soc., 2016, 138, 14582 CrossRef CAS PubMed; (b) H. J. Shrives, J. A. Fernández-Salas, C. Hedtke, A. P. Pulis and D. J. Procter, Nat. Commun., 2017, 8, 14801 CrossRef PubMed; (c) K. Okamoto, M. Hori, T. Yanagi, K. Murakami, K. Nogi and H. Yorimitsu, Angew. Chem., Int. Ed., 2018, 57, 14230 CrossRef CAS PubMed.
- (a) Y. Macé, C. Urban, C. Pradet, J. Blazejewski and E. Magnier, Eur. J. Org. Chem., 2009, 5313 CrossRef; (b) B. Pégot, C. Urban, P. Diter and E. Magnier, Eur. J. Org. Chem., 2013, 7800 CrossRef; (c) L. Shang, Y. Chang, F. Luo, J. He, X. Huang, L. Zhang, L. Kong, K. Li and B. Peng, J. Am. Chem. Soc., 2017, 139, 4211 CrossRef CAS PubMed; (d) F. Luo, Y. Lu, M. Hu, J. Tian, L. Zhang, W. Bao, C. Yan, X. Huang, Z.-X. Wang and B. Peng, Org. Chem. Front., 2018, 5, 1756 RSC; (e) L. Zhang, J.-N. He, Y. Liang, M. Hu, L. Shang, X. Huang, L. Kong, Z.-X. Wang and B. Peng, Angew. Chem., Int. Ed., 2019, 58, 5316 CrossRef CAS PubMed; (f) M. Hu, J.-N. He, Y. Liu, T. Dong, M. Chen, C. Yan, Y. Ye and B. Peng, Eur. J. Org. Chem., 2020, 193 DOI:10.1002/ejoc.201901577.
- D. Chen, Y. Fu, X. Cao, J. Luo, F. Wang and S. Huang, Org. Lett., 2019, 21, 5600 CrossRef CAS PubMed . In addition to [3,3]-sigmatropic rearrangement, the reaction of cyclopropanols with activated sulfoxides may also proceed via an intramolecular nucleophilic addition process..
- Z. He, H. J. Shrives, J. A. Fernández-Salas, A. Abenglzar, J. Neufeld, K. Yang, A. P. Pulis and D. J. Procter, Angew. Chem., Int. Ed., 2018, 57, 5759 CrossRef CAS PubMed.
- (a) N. Hu, H. Jung, Y. Zheng, J. Lee, L. Zhang, Z. Ullah, X. Xie, K. Harms, M. Baik and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 6242 CrossRef CAS PubMed; (b) F. Strieth-Kalthoff, C. Henkel, M. Teders, A. Kahnt, W. Knolle, A. Gómez-Suárez, K. Dirian, W. Alex, K. Bergander, C. G. Daniliuc, B. Abel, D. M. Guldi and F. Glorius, Chem, 2019, 5, 2183 CrossRef CAS; (c) K. Yang, A. P. Pulis, G. J. P. Perry and D. J. Procter, Org. Lett., 2018, 20, 7498 CrossRef CAS PubMed.
- For our recent work on the ortho-difluoroalkylation of aryliodanes with enol silyl ethers, see: (a) X. Huang, Y. Zhang, C. Zhang, L. Zhang, Y. Xu, L. Kong, Z.-X. Wang and B. Peng, Angew. Chem., Int. Ed., 2019, 58, 5956 CrossRef CAS PubMed. For related effect of flurine observed in other reactions, see: (b) J.-S. Yu, Y.-L. Liu, J. Tang, X. Wang and J. Zhou, Angew. Chem., Int. Ed., 2014, 53, 9512 CrossRef CAS PubMed; (c) Y.-L. Liu and J. Zhou, Chem. Commun., 2012, 48, 1919 RSC.
- For recent reviews on the biological aspects of difluoroalkyl groups, see: (a) J. Wang, M. Sánchez-Roselló, J. Luis Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (b) D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071 CrossRef; (c) J.-P. Begue and D. Bonnet-Delpon, Bioorganic and Medicinal Chemistry of Fluorine, Wiley-VCH, Weinheim, 2008 CrossRef; (d) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, John Wiley & Sons, Chichester, 2009 CrossRef.
- For selected examples on introduction of difluoroketone moiety to organic molecules, see: (a) S. Ge, W. Chaladaj and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4149 CrossRef CAS PubMed; (b) J. Yu, F. Liao, W. Gao, K. Liao, R. Zuo and J. Zhou, Angew. Chem., Int. Ed., 2015, 54, 7381 CrossRef CAS PubMed; (c) R. Doi, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2016, 55, 341 CrossRef CAS PubMed; (d) Ref. 13b . For recent reviews on difluoromethylation reactions, see: (e) D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem. Eur. J., 2017, 23, 14676 CrossRef CAS PubMed; (f) J. Rong, C. Ni and J. Hu, Asian J. Org. Chem., 2017, 6, 139 CrossRef CAS; (g) M.-C. Belhomme, T. Besset, T. Poisson and X. Pannecoucke, Chem. Eur. J., 2015, 21, 12836 CrossRef CAS PubMed; (h) B. Chen and D. A. Vicic, Top. Organomet. Chem., 2014, 52, 113 CrossRef.
- W. Zhao, X. Huang, Y. Zhan, Q. Zhang, D. Li, Y. Zhang, L. Kong and B. Peng, Angew. Chem., Int. Ed., 2019, 58, 17210 CrossRef CAS PubMed.
- For an elegant example of sulfur-traceless rearrangement of vinyl sulfoxides, see: D. Kaldre, I. Klose and N. Maulide, Science, 2018, 361, 664 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of all new compounds and details of DFT calculations. CCDC compound 5 (1973270) and compound 4s (1973271). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc00244e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |