
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChain propagation determines the chemo- and regioselectivity of alkyl radical additions to C–O vs. C–C double bonds†
Tiffany O.
Paulisch‡
a,
Felix
Strieth-Kalthoff‡
a,
Christian
Henkel
b,
Lena
Pitzer
a,
Dirk M.
Guldi
 b and
Frank
Glorius
*a
b and
Frank
Glorius
*a
aWestfälische Wilhelms-Universität Münster, Organisch-Chemisches Institut, Corrensstraße 40, 48149 Münster, Germany. E-mail: glorius@uni-muenster.de
bFriedrich-Alexander-Universität Erlangen-Nürnberg, Department of Chemistry and Pharmacy, Egerlandstraße 3, 91058 Erlangen, Germany
First published on 26th November 2019
Abstract
Investigations into the selectivity of intermolecular alkyl radical additions to C–O- vs. C–C-double bonds in α,β-unsaturated carbonyl compounds are described. Therefore, a photoredox-initiated radical chain reaction is explored, where the activation of the carbonyl-group through an in situ generated Lewis acid – originating from the substrate – enables the formation of either C–O or the C–C-addition products. α,β-Unsaturated aldehydes form selectively 1,2-, while esters and ketones form the corresponding 1,4-addition products exclusively. Computational studies lead to reason that this chemo- and regioselectivity is determined by the consecutive step, i.e. an electron transfer, after reversible radical addition, which eventually propagates the radical chain.
Introduction
Radical chemistry has been investigated for decades and, owing to the high reactivity of open-shell species, represents a complementary approach to classical closed-shell chemistry towards the synthesis of organic molecules.1 Considering that the selectivity of radical reactions is essential for targeted synthesis,2 numerous attempts to control these highly reactive intermediates have been made.3 Especially recent advances in controlled radical generation by means of photoredox catalysis have further expanded the field of selective radical reactions.4Owing to their high reactivity, radicals usually undergo only low activation barrier pathways. Selective radical reactions are therefore often enabled by rapid, innate chains (usually requiring fast and exothermic steps) or efficient transformations to long-lived (non-radical) intermediates (i.e. radical-polar crossover). To enable efficient chains, a polarity match of radical and reaction partner has to be given, otherwise non-productive side reactions with lower activation barriers will be the preferred pathways for these “impatient” intermediates.5
Amongst the elementary steps within radical chain reactions, radical additions to different π-bonds,6 which represent an efficient means to C–C bond formation, are quite well understood.7 In principle, selectivity in radical additions for two competitive reaction steps is determined by the difference in activation barriers: this kinetic control is usually governed by a polarity match between an electrophilic radical and a nucleophilic double bond, or vice versa. It is, however, rather challenging to control such a selectivity, when multiple addition sites with matching polarity and, in turn, similarly low barriers are available. As an example, the classical Minisci-type addition of nucleophilic radicals to electron-deficient heterocycles such as pyridine often yields a mixture of 2- and 4-substitution. In such systems, as well as e.g. for benzene rings or asymmetrical dienes, it remains challenging to predict the site selectivity of the radical addition of a given radical (see Fig. 1).8
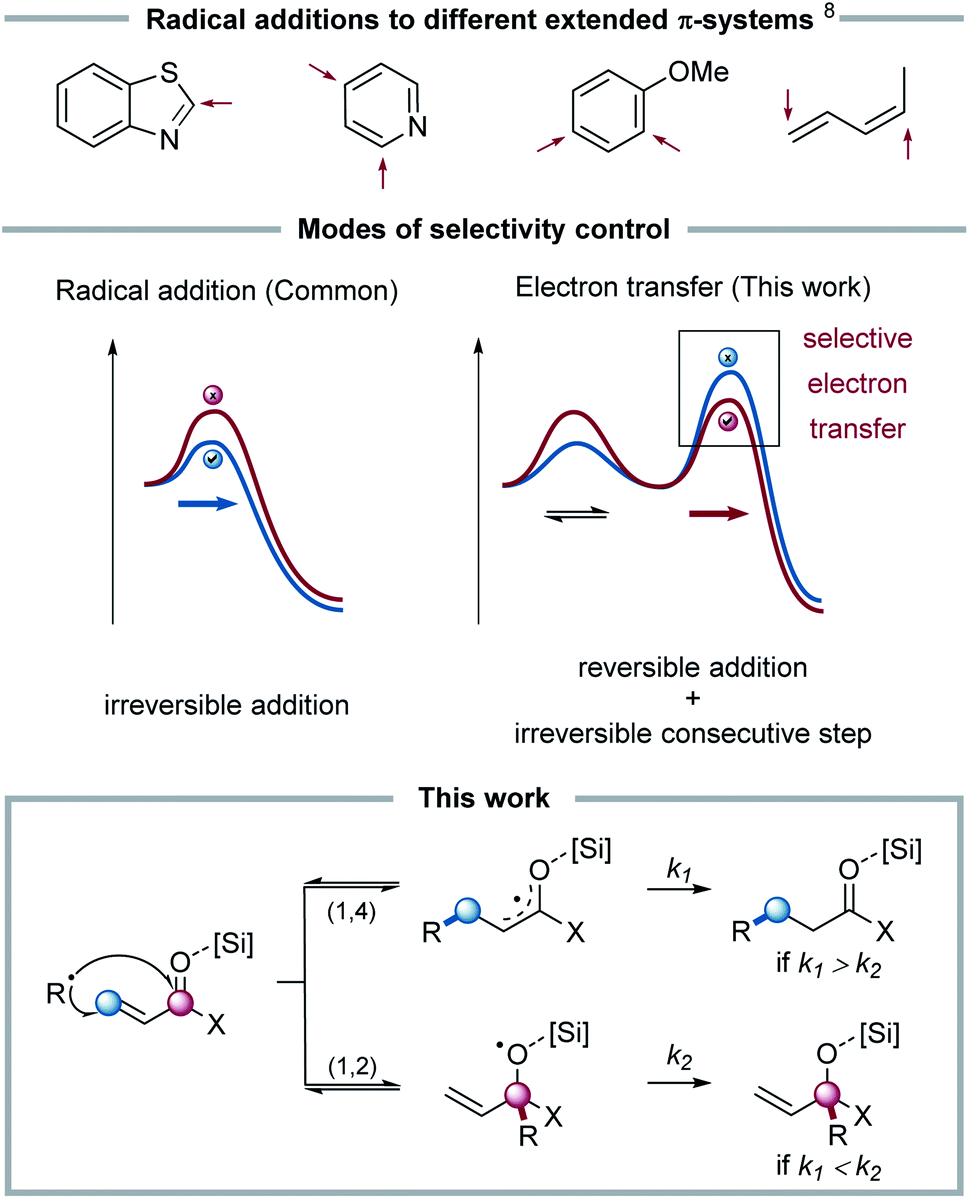 | ||
| Fig. 1 Radical additions to π-systems with indication where the addition can take place. Selectivity of product formation governed in the radical addition or a consecutive electron transfer step. This work: radical addition to α,β-unsaturated carbonyls shows 1,4- (blue) for R = OMe, Me and 1,2- (red) addition product for R = H. k: rate constant. | ||
Building on our recent study on the radical addition to carbonyls, in which aromatic aldehydes and ketones act as radical acceptors for alkyl radicals under Brønsted acid activation, we investigated the selectivity of the radical addition to the C–O vs. C–C double bond in Michael acceptors.9
In these systems (see Fig. 1), which exhibit two electrophilic addition sites, perfect regio- and chemoselectivity of alkyl radical additions was observed. We hypothesized that this selectivity is not determined by the addition itself, but by the consecutive electron transfer. Interestingly, this concept of radical selectivity control through consecutive steps has only been rarely applied, e.g. for controlling a radical metalation through a consecutive favoured/disfavoured cyclization,10 or to direct a reversible radical cyclization by selective radical-polar crossover.11 To the best of our knowledge, using electron transfer steps for controlling the selectivity of radical chain reactions has remained, to this date, elusive.
Results and discussion
To investigate the selectivity of the radical addition to C–O vs. C–C double bonds, Michael acceptors were chosen as radical acceptors. To enable an overall redox-neutral transformation, silyl ketene acetals were applied as radical precursors. In combination with an oxidizing photoredox catalyst, these substrates form amphiphilic alkyl radicals12 after single electron oxidation, which subsequently can add to π-bonds.Irradiating silyl ketene acetal 1 in the presence of the photocatalyst [Ir(dF-CF3-ppy)2(dtbpy)]PF6 (PC-1) and different Michael acceptors 2, an unexpected chemoselectivity switch was observed: whereas the addition to α,β-unsaturated esters and ketones afforded the Giese-type 1,4-addition product, the reaction of α,β-unsaturated aldehydes selectively formed allylic alcohols as products of a formal 1,2-addition. To rationalize this switch in reactivity, experimental and computational mechanistic analyses were conducted.
Mechanistic studies
As a first model system the reaction between trimethyl silyl ketene acetal (1a) and methyl acrylate (2a) was investigated in detail. In the absence of either light or photocatalyst, no product was detected, which corroborates a photochemical reaction pathway. In steady-state absorption measurements, the photocatalyst was confirmed to be the only visible-light-absorbing component in the reaction mixture. Stern–Volmer analysis and nanosecond-transient absorption spectroscopy (ns-TAS) suggest an effective quenching of the electronically exited state of PC-1 by silyl ketene acetal 1. Upon irradiation PC-1 gives rise to a MLCT triplet state featuring a 2.49 ± 0.06 μs lifetime. In ns-TAS experiments, the latter is effectively quenched when silyl ketene acetal 1 is added and gives rise to newly developing transient absorption features. A comparison with the spectro-electrochemical measurements (see ESI for further details†) infers the reduction of PC-1 to afford PC-1•−.13 The underlying electron transfer from the silyl ketene acetal 1 to photoexcited catalyst PC-1 occurs with diffusion-controlled dynamics: kET = (1.25 ± 0.02) 109 L mol−1 s−1. A high quantum yield (φ = 15.6) suggests that this quenching step initiates an efficient chain reaction.14 Considering that the addition of trimethylsilyl trifluoromethanesulfonate (TMSOTf, without irradiation) did not lead to product formation, a radical pathway rather than a Lewis-acid-mediated mechanism is likely to be operative.With these results in hand, we propose the following general mechanism (see Fig. 2): silyl ketene acetal 1 is oxidized by photocatalyst PC-1. The resulting radical cation A then fragments to the respective α-carbonyl radical B,15 which adds to the Lewis-acid-activated α,β-unsaturated carbonyl C to yield radical cation D.16 Intermediate D is readily reduced by the excess of silyl ketene acetal 1,17 which closes the radical chain cycle and yields intermediate E. Removal of the trimethylsilyl group by aqueous or fluoride workup leads to the final product.
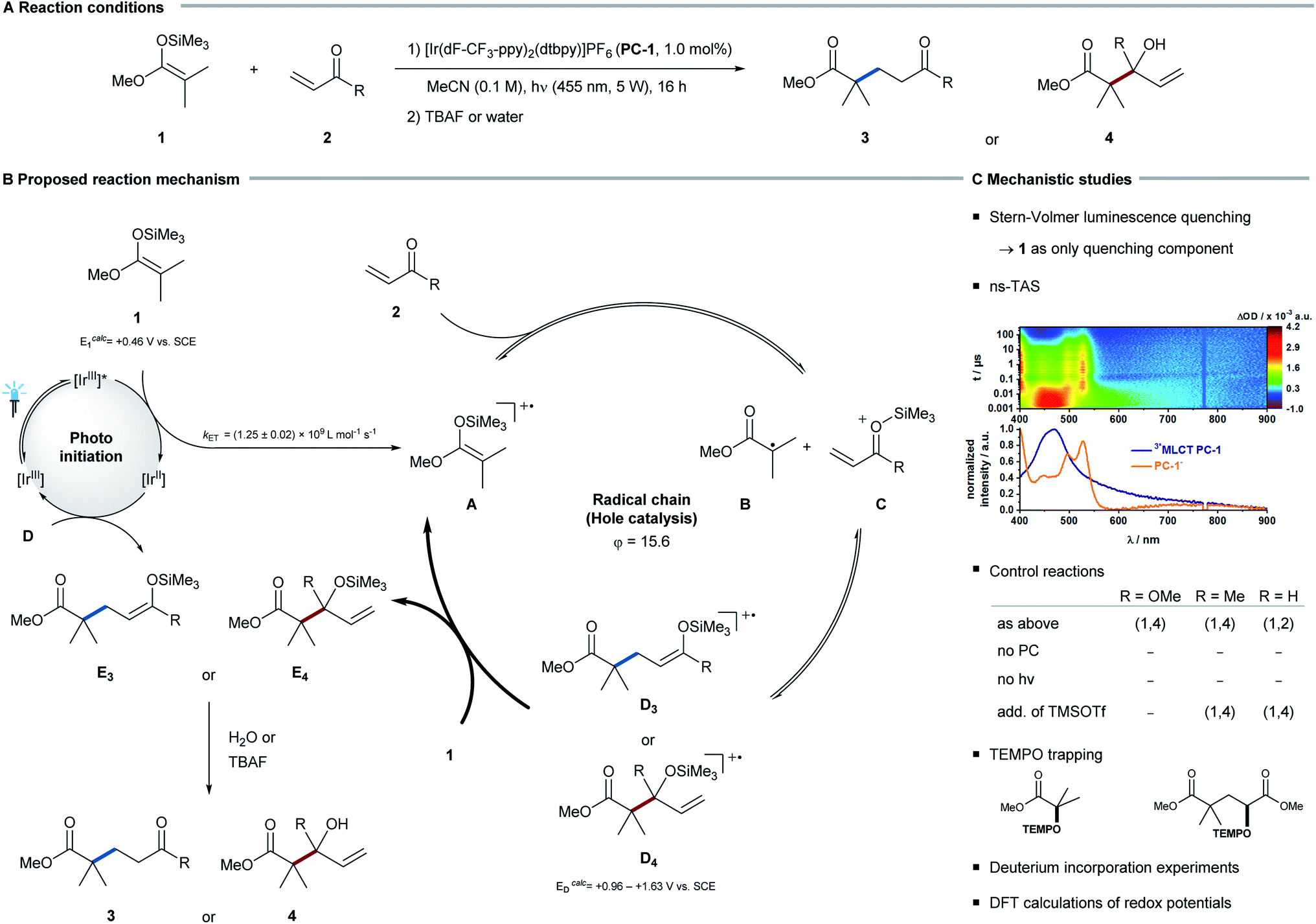 | ||
| Fig. 2 (A) Reaction conditions: silyl ketene acetal 1, Michael acceptor 2, PC-1, MeCN, 455 nm LEDs, workup with H2O or TBAF. (B) Proposed reaction mechanism for the reaction of silyl ketene acetals 1 with Michael acceptors 2 in a 1,2- or 1,4-fashion. (C) Stern–Volmer luminescence quenching studies and ns-transient absorption spectroscopy (ns-TAS) suggest a reductive quenching by silyl ketene acetal 1. Control reactions investigating a competing Lewis-acid-catalysed pathway (observed products are indicated as 1,2- or 1,4-addition respectively). TEMPO trapping experiments hint towards the presence of intermediary radical species. Deuterium incorporation experiments exclude HAT from solvent molecules. DFT-computed redox potentials substantiate the proposed chain propagation steps. | ||
Additional support for such a reaction mechanism came from further mechanistic experiments (see ESI for further details†): upon addition of TEMPO to the reaction mixture, TEMPO adducts of intermediates B and D could be detected, while intermediate E was identified by means of ESI-MS. Furthermore, experiments using deuterated reaction solvent excluded the possibility of a hydrogen atom transfer (HAT) event from a solvent molecule. Overall, the thermodynamic feasibility of the chain propagation step was supported by the calculated redox potentials, which underline that oxidation of silyl ketene acetal 1 by intermediate D is feasible (E1 = +0.46 V vs. SCE, ED = +0.96 V vs. SCE).18
For α,β-unsaturated aldehydes the observed 1,2-addition can be rationalized by an analogous hole catalysis mechanism (ED = +1.63 V vs. SCE). Here, photocatalyst and light are required for the reaction to proceed as well. Notably, the addition of TMSOTf (without irradiation) leads to exclusive formation of the 1,4-addition product. Since no 1,2-addition product was observed under these Lewis acid conditions, a radical pathway rather than a Lewis-acid-mediated mechanism is likely to be operative under the standard reaction conditions. For α,β-unsaturated ketones, however, a competing Lewis-acid-mediated chain reaction could not be fully excluded.19
To understand the difference in behaviour of α,β-unsaturated aldehydes and esters, a computational analysis of the proposed radical chain processes was conducted. Density functional theory calculations were performed at the B3LYP-D3/def2-TZVPP/CPCM(MeCN) level. Activation barriers for the electron transfer steps were estimated using the four-point approximation to Marcus–Hush theory (see ESI for further details†).20 Although not widely established, this method was applied to different cases in organic chemistry to estimate electron transfer barriers.21 Therefore, we believe that this methodology should result in a reasonable estimation of the electron transfer activation barrier (see Fig. 3).
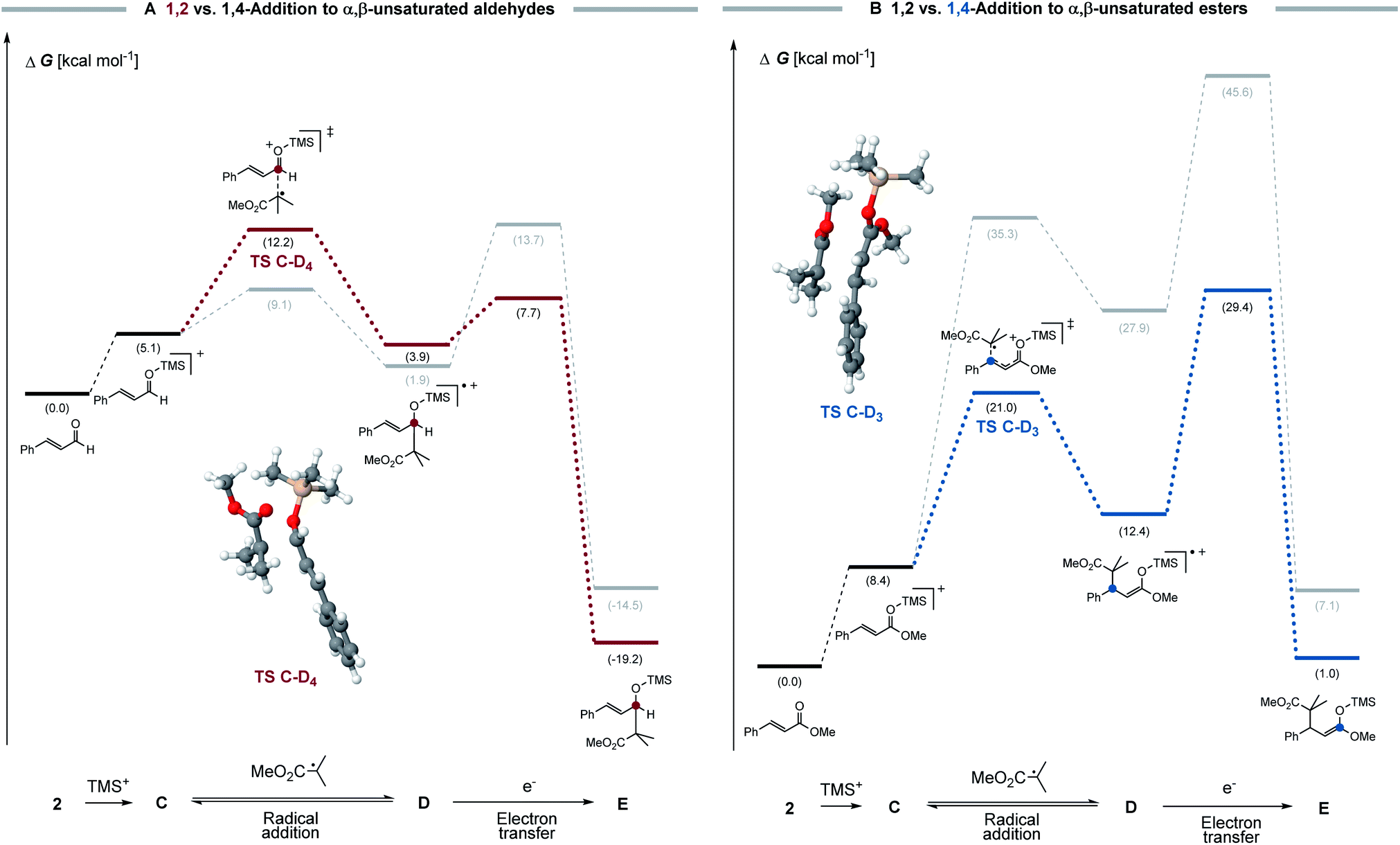 | ||
| Fig. 3 (A) 1,2- (red) vs. 1,4- (grey) radical addition to α,β-unsaturated aldehydes (B) 1,2- (grey) vs. 1,4- (blue) radical addition to α,β-unsaturated esters and the subsequent electron transfer step towards product E. Calculated for cinnamaldehyde and methyl cinnamate with trimethyl silyl ketene acetal (B3LYP-D3/def2-TZVPP/CPCM(MeCN)). | ||
For α,β-unsaturated aldehydes (see Fig. 3A), a highly oxidizing radical cation D4 can be formed via a reversible radical C–O addition, which can undergo a fast electron transfer as consecutive reaction. Crucial for the selectivity is the electron transfer barrier, which is seemingly lower for the 1,2-addition product (ΔG‡ = 3.8 kcal mol−1) than for the respective 1,4-addition product (ΔG‡ = 11.8 kcal mol−1). As such, we propose that, in a Curtin–Hammett-type scenario, the electron transfer is the selectivity determining step, which accounts for the 1,2- over 1,4-preference. This conclusion is in agreement with the computed redox potentials, since the 1,2-addition product (ED = +1.63 V vs. SCE) shows a higher oxidation potential than the 1,4-addition product (ED = +1.21 V vs. SCE) and therefore undergoes a faster electron transfer.
For α,β-unsaturated esters, a different selectivity is observed (see Fig. 3B). Here, a rather high activation barrier is calculated for the 1,2-radical addition (ΔG‡ = 26.9 kcal mol−1), which renders the reaction less favourable compared to the 1,4-addition product (ΔG‡ = 12.6 kcal mol−1). Furthermore, the lower activation barrier of the electron transfer for the 1,4-addition product compared to the 1,2-addition product, additionally accounts for the selectivity towards the 1,4-addition. We reason that also here the observed selectivity is determined by the final chain-propagating electron transfer step. Both computed reaction profiles are in good agreement with the experimental observation and support our hypothesis of selectivity control through the consecutive step.22
Reaction scope
With these mechanistic insights in hand, we sought to investigate the applicability of this protocol to functionalize aldehydes in 1,2- as well as esters and ketones in 1,4-fashion. The reaction was applied to several different silyl ketene acetals and Michael acceptors. As standard conditions, a solution of Michael acceptor 2 (1.0 eq.), silyl ketene acetal 1 (1.5 eq.) and PC-1 (1.0 mol%) in MeCN (0.1 M) was irradiated with 455 nm LEDs for 16 h (see Fig. 4).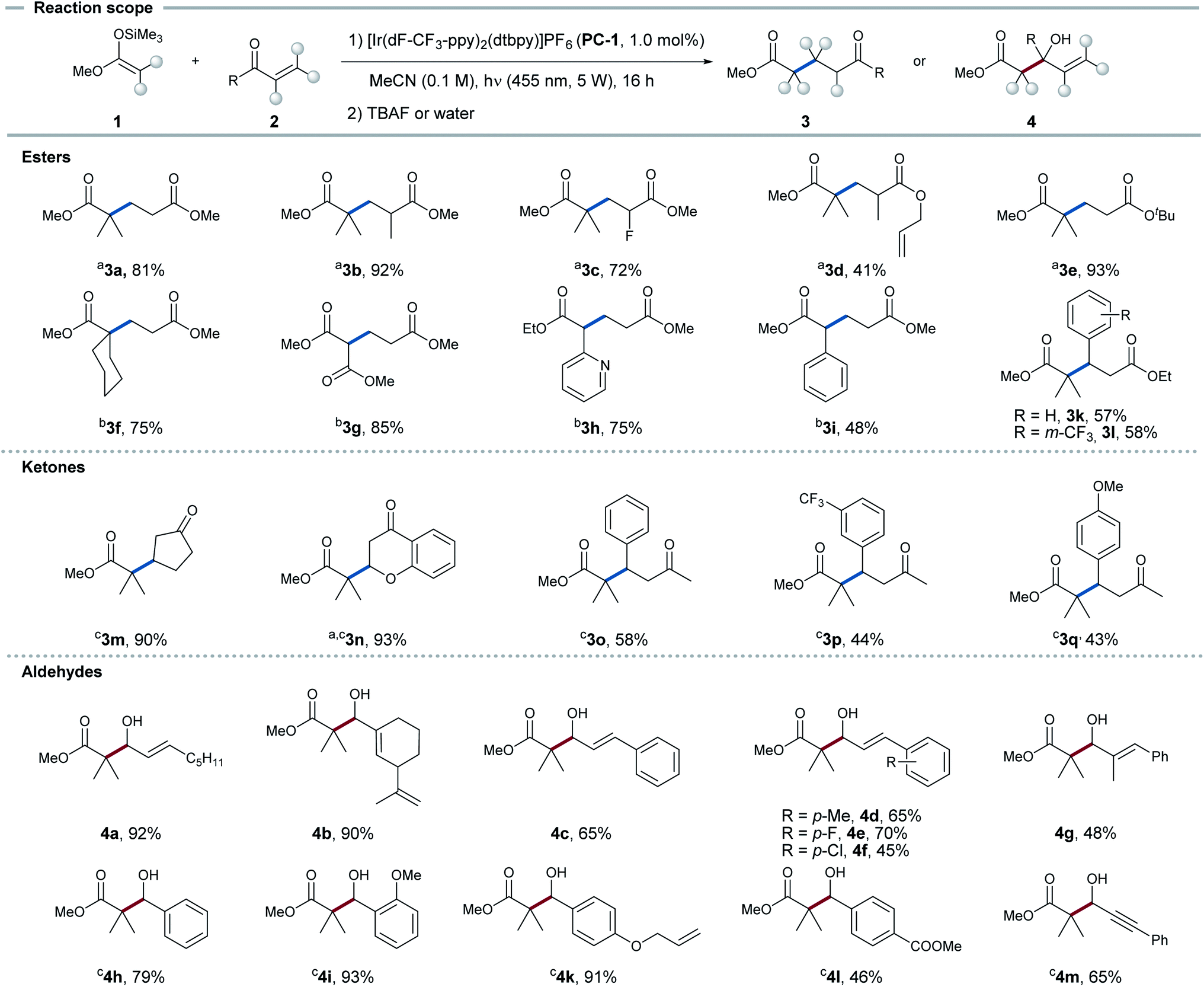 | ||
| Fig. 4 Scope of the 1,4-radical addition to α,β-unsaturated esters and ketones and the 1,2-radical addition to aldehydes. Standard conditions: Michael acceptor (2, 0.4 mmol), silyl ketene acetal (1, 0.6 mmol), PC-1 (0.004 mmol), MeCN (0.1 M), 16 h, 455 nm LEDs, workup with TBAF. (a) Workup with aq. HCl, 0.5 mmol scale, 1 (3.0 eq.). (b) Preformation of 1 without purification and subsequent coupling, 0.5 mmol scale. (c) Addition of TMSOTf (in the absence of light) showed product formation. | ||
Applying these conditions, esters and ketones showed high regio- and chemoselectivity towards the 1,4-addition product. Substituents at the α-position (3b, 3c) and alkene functionalities (3d) in the acceptor were well tolerated. Furthermore, diverse secondary and tertiary silyl ketene acetals could be applied (3g–i). For α,β-unsaturated ketones, 1,4-addition could be observed exclusively, although full conversion could not be achieved in all cases. This reaction was shown to be applicable for the functionalization of cyclopentenone (3m) and chromone (3n) in high yields. 4-Phenylbutenones (3o–q) could also be applied leading to decent yields. Employed α,β-unsaturated aldehydes could be functionalized with exclusive selectivity for the 1,2-addition product. Aliphatic α,β-unsaturated aldehydes underwent the transformation in good yields (4a–b). Moreover, cinnamaldehydes with different substituents in the aromatic ring (4c–f) and in α-position (4g) could be transformed in decent yields. The same reaction conditions could be applied to benzaldehydes (4h–l) in synthetically useful yields, while tolerating double bonds or esters in the side chains.
Conclusions
With this method, a radical alternative to the well-studied ionic Mukaiyama–Michael/Aldol-reaction was developed.23 This variant does not require any external Lewis acid, allowing for good functional group tolerance and for the generation of quaternary carbon centers under mild reaction conditions. α,β-Unsaturated esters and ketones show a high selectivity towards the 1,4-addition products, while α,β-unsaturated aldehydes form the 1,2-addition product exclusively. Computational studies suggest that the high regio- and chemoselectivity of the reaction is not determined by the radical addition itself, but by the subsequent electron transfer step, which propagates the radical chain. The control of regioselectivity of radical additions through a consecutive step represents a previously underestimated strategy which shows potential for further reaction design towards switchable selective radical additions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Frederik Sandfort, Dr Eloisa Serrano and Dr Michael James for helpful discussions. Financial support by Deutsche Forschungsgemeinschaft (SPP2102; FSK, CH) is gratefully acknowledged.Notes and references
- (a) D. P. Curran, Synthesis, 1988, 417 CrossRef CAS; (b) D. P. Curran, Synthesis, 1988, 489 CrossRef CAS; (c) B. Giese, Angew. Chem., Int. Ed. Engl., 1985, 24, 553 CrossRef.
- (a) B. M. Trost, Science, 1983, 219, 245 CrossRef CAS PubMed; (b) B. Giese, Angew. Chem., Int. Ed. Engl., 1983, 22, 753 CrossRef.
- (a) S. Zhu, A. Das, L. Bui, H. Zhou, D. P. Curran and M. Rueping, J. Am. Chem. Soc., 2013, 135, 1823 CrossRef CAS PubMed; (b) X. Liu, X. Ye, F. Bureš, H. Liu and Z. Jiang, Angew. Chem., Int. Ed., 2015, 54, 11443 CrossRef CAS PubMed; (c) C. Zhang, S. Li, F. Bureš, R. Lee, X. Ye and Z. Jiang, ACS Catal., 2016, 6, 6853 CrossRef CAS; (d) C. Yu, N. Iqbal, S. Park and E. J. Cho, Chem. Commun., 2014, 50, 12884 RSC.
- (a) A. Albini and S. Protti, Photochemistry, Royal Society of Chemistry, Cambridge, 2019 RSC; (b) M. Silvi, C. Verrier, Y. P. Rey, L. Buzzetti and P. Melchiorre, Nat. Chem., 2017, 9, 868 CrossRef CAS PubMed; (c) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed.
- (a) S. Wilsey, P. Dowd and K. N. Houk, J. Org. Chem., 1999, 64, 8801 CrossRef CAS PubMed; (b) L. Hernández-García, L. Quintero, M. Sánchez and F. Sartillo-Piscil, J. Org. Chem., 2007, 72, 8196 CrossRef PubMed; (c) J. Hartung, T. Gottwald and K. Špehar, Synthesis, 2002, 1469 CrossRef CAS; (d) G. Fuller and F. F. Rust, J. Am. Chem. Soc., 1958, 80, 6148 CrossRef CAS; (e) H. Miyabe, M. Ueda and T. Naito, Synlett, 2004, 1140 CAS; (f) G. K. Friestad, Tetrahedron, 2001, 57, 5461 CrossRef CAS; (g) A. G. Fallis and I. M. Brinza, Tetrahedron, 1997, 53, 17543 CrossRef CAS; (h) P. Devin, L. Fensterbank and M. Malacria, Tetrahedron Lett., 1999, 40, 5511 CrossRef CAS.
- (a) J. M. Tedder and J. C. Walton, Acc. Chem. Res., 1976, 9, 183 CrossRef CAS; (b) F. De Vleeschouwer, P. Jaque, P. Geerlings, A. Toro-Labbé and F. De Proft, J. Org. Chem., 2010, 75, 4964 CrossRef CAS PubMed; (c) H. Fischer and L. Radom, Angew. Chem., Int. Ed., 2001, 40, 1340 CrossRef CAS; (d) B. Giese, Angew. Chem., Int. Ed. Engl., 1983, 22, 753 CrossRef.
- (a) F. Minisci, R. Galli, M. Cecere, V. Malatesta and T. Caronna, Tetrahedron Lett., 1968, 54, 5609 CrossRef; (b) G. P. Gardini and F. Minisci, Ann. Chim., 1970, 60, 746 CAS; (c) F. Minisci, R. Galli, V. Malatesta and T. Caronna, Tetrahedron, 1970, 26, 4083 CrossRef CAS; (d) F. Minisci, Synthesis, 1973, 1 CAS; (e) F. Minisci, Chim. Ind., 1988, 70, 82 CAS; (f) F. Minisci, E. Vismara and F. Fontana, Heterocycles, 1989, 28, 489 CrossRef CAS; (g) R. S. J. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419 CrossRef CAS PubMed; (h) R. A. Garza-Sanchez, A. Tlahuext-Aca, G. Tavakoli and F. Glorius, ACS Catal., 2017, 7, 4057 CrossRef CAS; (i) A. Tlahuext-Aca, R. Aleyda Garza-Sanchez and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 3708 CrossRef CAS PubMed; (j) W. A. Thaler, A. A. Oswald and B. E. Hudson Jr, J. Am. Chem. Soc., 1965, 87, 311 CrossRef CAS; (k) K.-I. Yamada, H. Umeki, M. Maekawa, Y. Yamamoto, T. Akindele, M. Nakano and K. Tomioka, Tetrahedron, 2008, 64, 7258 CrossRef CAS.
- (a) L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, J. Am. Chem. Soc., 2017, 139, 13652 CrossRef CAS PubMed; (b) L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 16219 CrossRef CAS PubMed.
- (a) S. Mondal, R. K. Mohamed, M. Manoharan, H. Phan and I. V. Alabugin, Org. Lett., 2013, 15, 5650 CrossRef CAS PubMed; (b) R. K. Mohamed, S. Mondal, B. Gold, C. J. Evoniuk, T. Banerjee, K. Hanson and I. V. Alabugin, J. Am. Chem. Soc., 2015, 137, 6335 CrossRef CAS PubMed.
- A. Gansäuer, T. Lauterbach and D. Geich-Gimbel, Chem.–Eur. J., 2004, 10, 4983 CrossRef PubMed.
- B. Giese, J. He and W. Mehl, Chem. Ber., 1988, 121, 2063 CrossRef CAS.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803 RSC.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
- The fragmentation of A can facilitate oxidation of the silyl ketene acetal through equilibrium shift, see: M. A. Syroeshkin, I. B. Krylov, A. M. Hughes, I. V. Alabugin, D. V. Nasybullina, M. Y. Sharipov, V. P. Gultyai and A. O. Terent’ev, J. Phys. Org. Chem., 2017, 30, e3744 CrossRef.
- DFT studies show that a radical addition to the Lewis-acid-activated α,β-unsaturated carbonyl C is kinetically and thermodynamically more favored compared to the direct addition to 1 (see ESI for more detail†).
- Within the course of the chain propagation, radical cation A is converted to radical cation D, which is a stronger oxidant then A. In the context of Alabugin's recent work, this could be described as upconversion of an oxidant, see: M. A. Syroeshkin, F. Kuriakose, E. A. Saverina, V. A. Timofeeva, M. P. Egorov and I. V. Alabugin, Angew. Chem., Int. Ed., 2019, 58, 5532 CrossRef CAS PubMed.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714 CAS.
- Analogous to α,β-unsaturated aldehydes and esters, the hole catalysis cycle is thermodynamically feasible for α,β-unsaturated ketones (ED= +1.08 V vs. SCE). However, the addition of TMSOTf in the absence of light led to significant 1,4-addition product formation, so that two competing mechanisms are likely to be operative.
- (a) R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599 CrossRef CAS; (b) O. López-Estrada, H. G. Laguna, C. Barrueta-Flores and C. Amador-Bedolla, ACS Omega, 2018, 3, 2130 CrossRef PubMed.
- (a) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429 CrossRef CAS PubMed; (b) S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2007, 129, 3683 CrossRef CAS PubMed; (c) H.-S. Ren, M.-J. Ming, J.-Y. Ma and X.-Y. Li, J. Phys. Chem. A, 2013, 117, 8017 CrossRef CAS PubMed; (d) V. Lemaur, M. Steel, D. Beljonne, J.-L. Brédas and J. Comil, J. Am. Chem. Soc., 2005, 127, 6077 CrossRef CAS PubMed.
- It is important to consider that a certain amount of product can be formed through the re-oxidation of the photocatalyst. However, since a quantum yield of 15.6 suggests an efficient chain, the major amount of product should be formed through the described hole catalytic cycle.
- (a) T. Mukaiyama, K. Narasaka and K. Banno, Chem. Lett., 1973, 2, 1011 CrossRef; (b) K. Narasaka, K. Soai and T. Mukaiyama, Chem. Lett., 1974, 3, 1223 CrossRef; (c) J.-I. Matsuo and M. Murakami, Angew. Chem., Int. Ed., 2013, 52, 9109 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04846d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |