
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling the structure and photophysics of fluorophore dimers using multiple cucurbit[8]uril clampings†
Guanglu
Wu
 a,
Youn Jue
Bae
b,
Magdalena
Olesińska
a,
Daniel
Antón-García
c,
István
Szabó
d,
Edina
Rosta
d,
Michael R.
Wasielewski
b and
Oren A.
Scherman
*a
a,
Youn Jue
Bae
b,
Magdalena
Olesińska
a,
Daniel
Antón-García
c,
István
Szabó
d,
Edina
Rosta
d,
Michael R.
Wasielewski
b and
Oren A.
Scherman
*a
aMelville Laboratory for Polymer Synthesis, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: oas23@cam.ac.uk
bDepartment of Chemistry, Institute for Sustainability and Energy at Northwestern, Northwestern University, Evanston, Illinois 60208-3113, USA
cDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
dDepartment of Chemistry, King's College London, 7 Trinity Street, London, SE1 1DB, UK
First published on 6th December 2019
Abstract
A modular strategy has been employed to develop a new class of fluorescent molecules, which generates discrete, dimeric stacked fluorophores upon complexation with multiple cucurbit[8]uril macrocycles. The multiple constraints result in a “static” complex (remaining as a single entity for more than 30 ms) and facilitate fluorophore coupling in the ground state, showing a significant bathochromic shift in absorption and emission. This modular design is surprisingly applicable and flexible and has been validated through an investigation of nine different fluorophore cores ranging in size, shape, and geometric variation of their clamping modules. All fluorescent dimers evaluated can be photo-excited to atypical excimer-like states with elongated excited lifetimes (up to 37 ns) and substantially high quantum yields (up to 1). This strategy offers a straightforward preparation of discrete fluorophore dimers, providing promising model systems with explicitly stable dimeric structures and tunable photophysical features, which can be utilized to study various intermolecular processes.
1 Introduction
Coupling two fluorophores within a sufficiently short distance for an extended period of time is crucial for both theoretical and experimental investigation of intermolecular processes such as charge transfer,1 excimer formation,2,3 long- or short-range exciton coupling,4,5 and singlet fission.6–8 Stacking together precisely two fluorophores in an aqueous solution, however, remains a substantial challenge as most aromatic hydrocarbons show a tendency to aggregate unpredictably (forming clusters of arbitrary numbers of molecules).9–11 To prevent fluorophores from aggregation in aqueous solution, a supramolecular approach has been established to “mechanically” separate fluorescent molecules through encapsulation by macrocycles.12–16 A popular class of macrocyclic hosts utilized for this purpose is cucurbit[n]uril (CB[n], n = 5–8, 10), which contains a cavity that enables the inclusion of various guest molecules and exhibits particularly high affinity towards positively-charged species.17,18 As an example, CB[7] is a promising host for the complexation of various fluorescent dyes,19 resulting in significant changes in photophysical properties such as anti-photobleaching20 and emission enhancement.21–23 This is attributed to the hydrophobic environment provided by the CB cavity as well as mechanical protection by the macrocycle against aggregation and quenching.12Dimeric fluorophore stacking, however, is unlikely to be realized by CB[7]-mediated complexation as its relatively small cavity only allows the complexation with one single guest molecule or, more strictly speaking, one binding moiety on a guest molecule. On the other hand, CB[8], a larger cucurbituril homologue, is capable of simultaneously encapsulating two guest moieties yielding either a heteroternary24 or homoternary complex.25 Although CB[8]-mediated ternary complexation may achieve stacking of two fluorophores, several limitations exist as the fluorophores are required to have the right shape, size and charge distribution to undergo complexation with CB[8].26 Moreover, they must align along the principal symmetry axis of the CB cavity limiting the way in which they stack.27 In case of a stepwise complexation of two guests with CB[8], formation of a dynamic ternary complex is evident by the significant signal broadening in NMR spectra.24,25 This dynamic complex results in a short-lived coupling between the two stacked fluorophores that is insufficient to allow for the investigation of specific intermolecular processes.
Recently, we have found that the dynamic exchange kinetics between the guests and CB[8] hosts are dramatically reduced through the formation of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 quaternary complexes,28 in which two elongated guests such as diarylviologen derivatives are “clamped” in place by two CB[8] hosts into a multicomponent complex. The simultaneous formation of two ternary motifs within a discrete complex decreases the likelihood of dissociation compared to a typical ternary complex.28 The formation of 2:2 complexes opposed to elongated supramolecular polymers requires a small change in the conformational entropy during complexation, i.e. a molecule with significant rigidity.28,29 For instance, various rigid molecular moieties such as benzidine,30 benzothiazole,31,32 arylpyridinium,28,33 arylterpyridyl,34 bipyridinium,35 and benzimidazole29 have been employed to produce CB[8]-mediated 2:2 complexes. Herein, we present a general and modular strategy towards the dimerization of arbitrary functional components (fluorophores in this work) by connecting them to multiple rigid modules that can be “clamped” together by CB[8] complexation. We use arylpyridinium moieties as the rigid “clamping” module (Fig. 1a) and exploit the modular strategy for designing fluorescent complexes in water comprised of two fluorophores that are stacked in a specific configuration with a constraint applied by CB[8] macrocycles at multiple points.
:2 quaternary complexes,28 in which two elongated guests such as diarylviologen derivatives are “clamped” in place by two CB[8] hosts into a multicomponent complex. The simultaneous formation of two ternary motifs within a discrete complex decreases the likelihood of dissociation compared to a typical ternary complex.28 The formation of 2:2 complexes opposed to elongated supramolecular polymers requires a small change in the conformational entropy during complexation, i.e. a molecule with significant rigidity.28,29 For instance, various rigid molecular moieties such as benzidine,30 benzothiazole,31,32 arylpyridinium,28,33 arylterpyridyl,34 bipyridinium,35 and benzimidazole29 have been employed to produce CB[8]-mediated 2:2 complexes. Herein, we present a general and modular strategy towards the dimerization of arbitrary functional components (fluorophores in this work) by connecting them to multiple rigid modules that can be “clamped” together by CB[8] complexation. We use arylpyridinium moieties as the rigid “clamping” module (Fig. 1a) and exploit the modular strategy for designing fluorescent complexes in water comprised of two fluorophores that are stacked in a specific configuration with a constraint applied by CB[8] macrocycles at multiple points.
 | ||
| Fig. 1 Modular strategy for designing fluorescent molecules (a) by plugging in a fluorophore module between two positively charged clamping modules, which herein are arylpyridinium moieties originated from diarylviologen derivatives. Following this strategy, (b) 2,7-naphthalene with non-parallel clamping modules, as an example, or (c) other fluorophores with various arrangement of clamping modules are expected to form a preorganized dimer constrained by CB[8]-mediated multiple clamping or a monomeric state of fluorophores protected by CB[7] from aggregation. The Cl− counterions are omitted for clarity. | ||
As illustrated in Fig. 1a, water-soluble fluorescent molecules are designed to incorporate a fluorescent core between two positively charged clamping modules, which in this work are arylpyridinium motifs originating from previously studied diarylviologen derivatives.28 When one equivalent (equiv.) of CB[8] is added to the system with one equiv. of guest molecule, two clamping modules are expected to bring together two guest molecules yielding a 2:2 quaternary complex. The fluorophore cores from each guest molecule are brought to close proximity to each other as a consequence of the assembly, resulting in preorganized dimeric fluorophore stacking. The preorganized dimer complex is stabilized by multiple CB[8] clamps, which ensures interaction between fluorophores for a sufficiently long period of time, endowing the complex with emergent photophysical properties. As the fluorophore modules are not encapsulated by CB[8] (Fig. 1c), a variety of fluorophores, including those with sizes substantially larger than the CB[8] cavity, can be employed as functional cores in this modular strategy (Scheme S1†). Moreover, the photophysical properties of the resultant complexes can be readily customized through altering fluorophores as well as the clamping modules. As exemplified in Fig. 1b, dimeric stacking still occurs even when the two clamping modules are non-parallel to each other (separated by an angle < 180°). The flexibility offered by this modular design provides a molecular toolbox and platform in which a wide range of fluorophores can be readily studied in their discrete monomeric or dimeric states facilitating future investigations of quantum optical phenomena.
2 Results and discussion
Fluorescent molecules are designed by bridging two arylpyridinium motifs with a central fluorophore core. Nine phenyl, naphthyl, or anthracenyl homologues are investigated as the fluorophore cores in this study (Fig. 1 and Scheme S1†). The general synthesis (Scheme S2†) of the molecules starts with Suzuki–Miyaura cross-coupling36 of two pyridin-4-yl groups onto the fluorophore core, followed by transformation of the pyridin-4-yl groups into arylpyridinium salts through a Zincke reaction.37–39 A complete study was carried out on Ant910Me, which contains a 9,10-anthracenyl (“Ant910”) as the central core and p-tolyl pyridiniums (“Me”) as clamping modules (Fig. 1a), which is presented here as a typical case prior to a further general discussion.Guest molecule (G) Ant910Me is found to form 1:2 complexes with CB[7], denoted G1–CB[7]2, and 2:2 complexes with CB[8], denoted G2–CB[8]2. The complex formations are verified by several NMR techniques including 1H NMR, 2D nuclear Overhauser spectroscopy (NOESY), and diffusion ordered spectroscopy (DOSY).
2.1 G1–CB[7]2: a discrete monomeric state
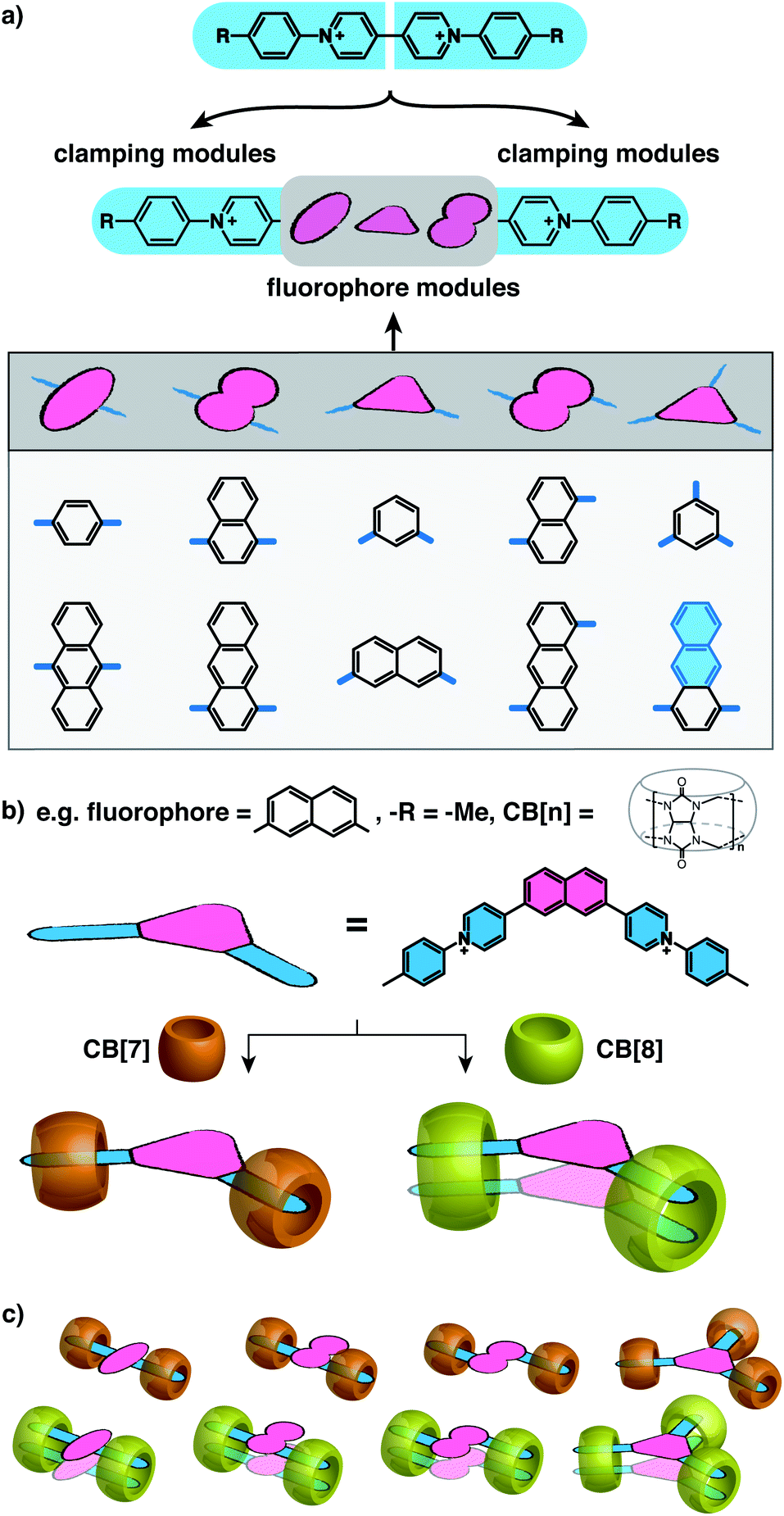
The way in which CB[n] binds to guest molecules can be precisely probed by 1H NMR. Protons residing inside the CB cavity typically exhibit upfield chemical shifts of ca. 1 ppm; while protons located outside and proximate to the CB portals will display downfield shifts.401H NMR spectra of Ant910Me (Fig. 2a) and its CB[7] complex (Fig. 2b) demonstrate significant upfield shift for the Hc,d,e,f upon complexation, which indicates that the entire tolyl moiety resides inside the CB[7] cavity along with a part of the pyridinium group. Meanwhile, a slight downfield shift of Hg,h confirms that the anthracenyl core is located outside the CB[7] portals.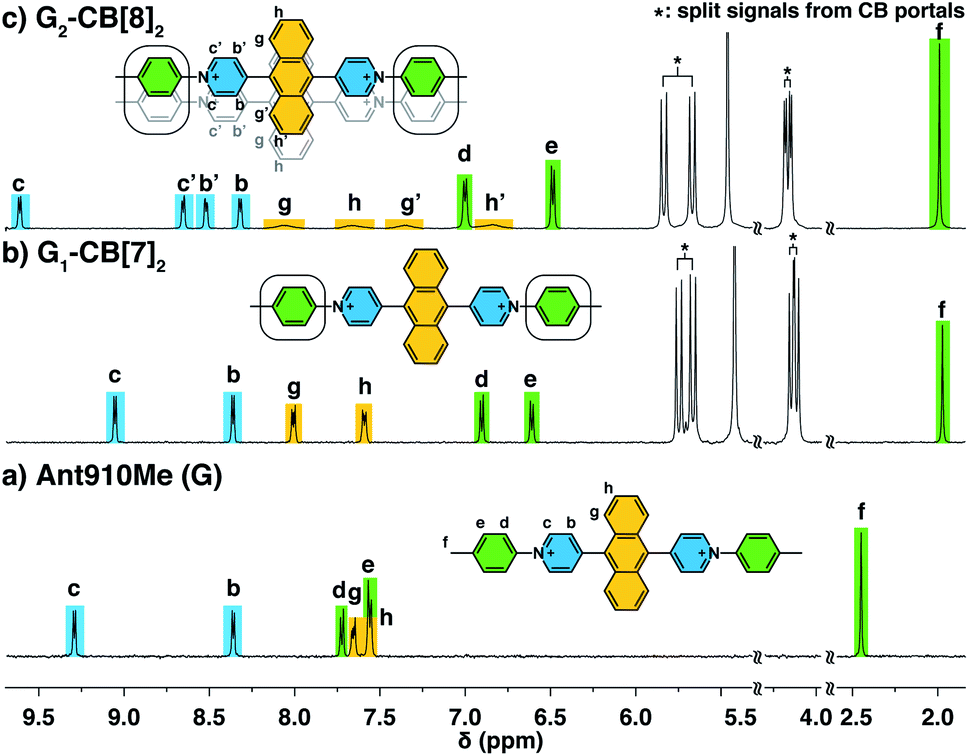 | ||
| Fig. 2 1H NMR spectra (500 MHz) of (a) Ant910Me (G), and (b) its complexation with 2 equiv. of CB[7] (G1–CB[7]2), or (c) with 1 equiv. of CB[8] (G2–CB[8]2) in D2O with a uniform guest concentration of 100 μM at 298 K. The Cl− counterions are omitted for clarity. | ||
CB proton signals ranging from 4 to 6 ppm split into two sets of equivalent doublets (Fig. 2b). The signal splitting suggests that the rate of CB[7] flipping around the tolyl moieties falls in the slow exchange limit with respect to the NMR time scale (500 MHz, 298 K). This slow flipping rate enables the direct observation of the two CB portals existing in distinctly different chemical environments.22,28,39,41 Signal splitting is observed throughout the titration of the guest into a CB[7] solution, leading to quantitative splitting at a ratio of 1:2, which confirms the stoichiometric formula of this CB[7] complex as G1–CB[7]2. Thus, each Ant910Me molecule is readily isolated in a monomeric state in aqueous solution when complexed by two CB[7] macrocycles.
2.2 G2–CB[8]2: preorganized π-stacked dimers
Upon titration of Ant910Me into a solution of CB[8] (Fig. 2c), splitting of the CB[8] protons are observed as well as the upfield shift of Hd,e,f. Both observations suggest that the CB[8] molecules remain at the tolyl moieties, with a slow flipping rate and asymmetric portal environment. Careful analysis of the signal splitting and proton integration confirms a binding stoichiometry of “1:1”, therefore, this CB[8]-mediated complex contains an equal number of hosts and guests. As elaborated in a previous work,28 this complex cannot be a 1:1 binary complex as CB[8]-mediated binary complexes exhibit much faster dynamics. An elongated polymeric Gn–CB[8]n complex (n = 1,2,3…), fabricated from the sequential stacking of tolyl groups,23 is also not possible as the head-to-tail alignment of two tolyl groups would result in a symmetric portal environment contrary to the observed splitting. Therefore, the most probable binding mode is a 2:2 complex (G2–CB[8]2), as illustrated in Fig. 2c, where two fluorescent molecules are constrained to overlap with each other. In this binding mode, the tolyl groups are head-to-head, thus resulting in an asymmetric portal environment for each CB[8]. The observed slow flipping rate of CB[8] and the signal splitting is explained by the tightly filled CB[8] cavities as well as the electrostatic interactions between multiple positive charges on one side of the CB portals.
We have learned from previous works28,30,39,41,42 that the diffusion coefficient (D) of a CB[n]-mediated complex is primarily determined by the number of CB macrocycles existing in the complex. Therefore, the formation of G2–CB[8]2 is further confirmed through a semi-quantitative analysis of D via DOSY experiments. As shown in Fig. 3a and Table S1,†D values of unbound guests (G) in aqueous solution range from 3.49 to 4.38, showing a standard deviation (SD) of 0.3. A much narrower distribution is observed for CB[8]-mediated complexes, ranging from 1.95 to 2.07 with a SD of 0.04 (Fig. 3a). These D values are much smaller than that of free CB[8] (D = 3.11) and typical binary complexes such as dzpy1–CB[8]1 (D = 3.04).41 However, the D values measured here are almost the same as for the 2:2 complexes produced by diarylviologen derivatives28,41 such as (VNMe2)2–CB[8]2 (D = 2.01). Therefore, the DOSY data fully supports the formation of a G2–CB[8]2 complex involving the stacking of two fluorescent molecules held together by two CB[8] hosts.
 | ||
| Fig. 3 Diffusion coefficients obtained from DOSY NMR for G (rectangles), G1–CB[7]2 (triangles), and G2–CB[8]2 (circles) with variation in (a) fluorophore cores or (b) substituted tails. (D2O solution with a uniform guest concentration of 100 μM at 298 K.) The Cl− counterions are omitted for clarity. | ||
The relative orientation of the two fluorophores with respect to each other is probed by proton and NOESY NMR in this dimeric system. As the proton spectrum recorded for G2–CB[8]2 (G = Ant910Me) exhibits more complicated signal splittings than that of G1–CB[7]2, COSY NMR (Fig. S2†) is used to identify each proton. Both the pyridinium and anthracenyl protons in this 2:2 complex split into two sets of equivalent peaks corresponding to Hb,b′,c,c′ and Hg,g′,h,h′ in Fig. 2c. The observation of two sets of signals suggests (i) a slow dynamic process and (ii) a certain asymmetry existing for the most probable configuration of the G2–CB[8]2 complex, which is consistent with a cofacial stacking and partial overlap of the two aromatic fluorophores as illustrated in Fig. 2c. Partial overlap of the two fluorophores with a slippage along their extended axis will result in one set of equivalent protons lying on top of or below an aromatic ring of the other molecule, while the other set of equivalent protons does not. The first set of equivalent protons are expected to display signals in a higher-field region on account of shielding by aromatic ring currents, compared to the latter set of equivalent protons, which is consistent with the observation of significantly lowered chemical shifts for Hg′,h′ compared to Hg,h. Similarly, the difference observed between Hc,b and Hc′,b′ is interpreted as two sets of protons that reside in different shielding and deshielding environments arising from the CB[8] portal.
The partial overlap of two aromatic fluorophores is also supported by the cross-correlation signals observed in NOESY NMR, which reveals the relative position of protons located in space. Proton Hb′,c′ (Fig. S3†), for instance, exhibits an intense cross-correlation with all anthracenyl signals (Hg,g′,h,h′), whereas Hb,c can only “feel” protons that are closer to the pyridinium protons, i.e. Hg,g′. This observation is consistent with the partial-overlap and stacking of the fluorophores where Hb′,c′ rather than Hb,c are closer to Hh,h′ in space (Fig. 2c and S3†).
2.3 Photophysics of the dimeric and monomeric Ant910Me
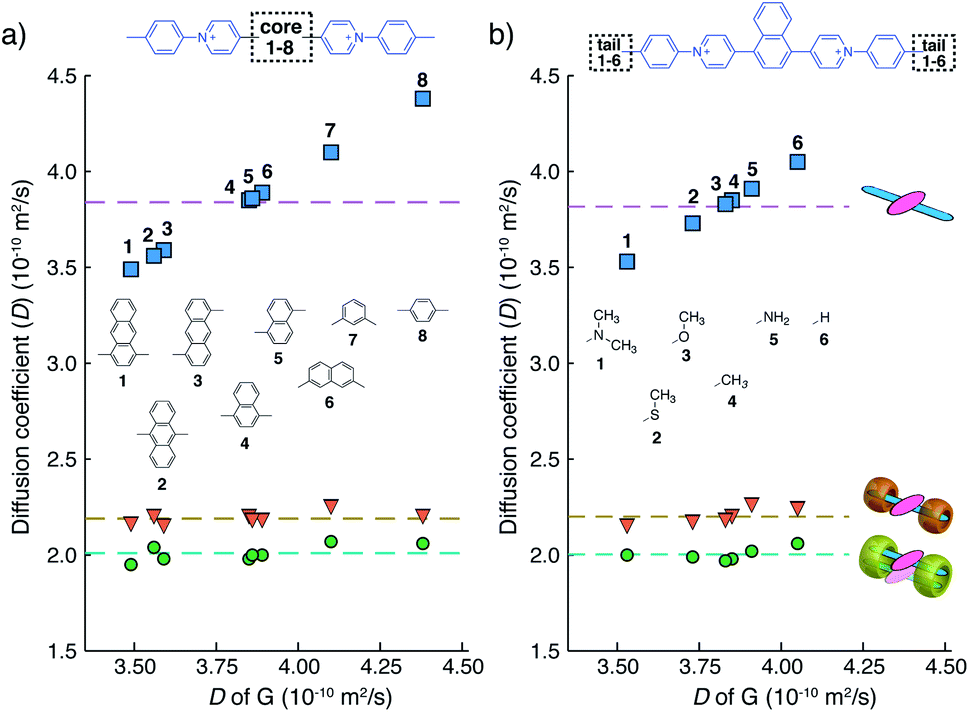
The well-resolved NMR spectrum of G2–CB[8]2 suggests that these complexes exist as discrete preorganized fluorescent dimers in aqueous solution without forming any larger aggregates. This is because the two CB[8] macrocycles mechanically block the interaction between multiple dimers. It also leads to a substantial change in the photophysical properties of Ant910Me upon complexation with CB[8] to G2–CB[8]2.As shown in Fig. 4a–c and Table 1, the anthracenyl moiety in G2–CB[8]2 exhibits a bathochromic shift of its absorption maximum (λabs = 469 nm) by over 50 nm compared to monomeric Ant910Me (λabs = 409 nm) in G1–CB[7]2 and unbound Ant910Me in pristine solution (λabs = 419 nm) (all compared at a concentration of 15 μM). The emission maximum of G2–CB[8]2 (λem = 578 nm) is also red-shifted relative to that of G1–CB[7]2 (λem = 537 nm), although Ant910Me in pristine solution exhibits the most bathochromic shift in emission (λem = 595 nm).
 | ||
| Fig. 4 Steady-state absorption (solid line) and emission (dash line with filling color) spectra of (a) Ant910Me (G), and (b) its G1–CB[7]2 complex, and (c) G2–CB[8]2 complex, whose time-dependent fluorescence decay is displayed in (d), (e), (f), respectively, along with corresponding lifetime (τs) and fluorescence quantum yield (ϕF) results. Aqueous solution of each species with a uniform guest concentration of 15 μM is tested at 298 K. The intensity is not normalized but scaled up by the same factor except the emission of G which is enlarged by an additional 4 times for a clear vision. Quantified data can be found in Table 1. Photographs of each species with a guest concentration of 20 μM before (g) and after (h) photoexcitation at 365 nm. | ||
| G | Species | λ abs/nm | λ em/nm | Δν/cm−1 (Δλ/nm) | τ s/ns | ϕ F | k nr/μs−1 | k r/μs−1 | ε(λabs)/103 M−1 cm−1 | ε(λabs) × ϕF/103 M−1 cm−1 |
|---|---|---|---|---|---|---|---|---|---|---|
| a Δν: wavenumber difference between λabs and λem (“Stokes shift”); ε(λabs): molar absorption coefficient; ϕF: fluorescence quantum yield; ε(λabs) × ϕF: emission brightness. | ||||||||||
|
G | 419 | 595 | 7060 (176) | 1.66 (96%) 9.50 (4%) | 0.04 | 486.4 | 20.3 | 9.1 | 0.4 |
| G1–CB[7]2 | 409 | 537 | 5828 (128) | 8.60 | 0.85 | 17.4 | 98.8 | 12.1 | 10.3 | |
| G2–CB[8]2 | 469 | 578 | 4021 (109) | 12.6 | 0.81 | 15.1 | 64.3 | 13.3 | 10.8 | |
|
G | 422 | 590 | 6748 (168) | 2.13 (97%) 8.35 (3%) | 0.12 | 379.9 | 51.8 | 14.8 | 1.8 |
| G1–CB[7]2 | 416 | 546 | 5723 (130) | 7.97 | 0.82 | 22.6 | 102.9 | 17.5 | 14.4 | |
| G2–CB[8]2 | 443 | 570 | 5030 (127) | 12.3 | 0.82 | 14.6 | 66.7 | 17.3 | 14.2 | |
|
G | 435 | 618 | 6807 (183) | 4.46 | 0.21 | 177.1 | 47.1 | 11.3 | 2.4 |
| G1–CB[7]2 | 426 | 585 | 6380 (159) | 8.74 | 0.60 | 45.8 | 68.6 | 13.8 | 8.3 | |
| G2–CB[8]2 | 478 | 650 | 5536 (172) | 1.60 (63%) 7.32 (37%) | 0.02 | 263.7 | 5.4 | 13.0 | 0.3 | |
| G2–CB[8]3 | 509 | 665 | 4609 (156) | 1.73 (41%) 7.20 (59%) | 0.01 | 199.7 | 2.0 | 11.1 | 0.1 | |
|
G | 335 | 477 | 8886 (142) | 8.85 | 0.96 | 4.5 | 108.5 | 52.1 | 50.0 |
| G1–CB[7]2 | 330 | 465 | 8798 (135) | 2.99 (36%) 10.3 (64%) | 0.94 | 7.8 | 122.6 | 57.5 | 54.0 | |
| G2–CB[8]2 | 351 | 531 | 9658 (180) | 36.8 | 0.55 | 12.2 | 14.9 | 62.0 | 34.1 | |
|
G | 371 | 484 | 6293 (113) | 4.34 | 1.00 | 0.0 | 230.4 | 20.0 | 20.0 |
| G1–CB[7]2 | 365 | 471 | 6166 (106) | 3.25 | 0.96 | 12.3 | 295.4 | 23.9 | 23.0 | |
| G2–CB[8]2 | 395 | 518 | 6011 (123) | 7.09 (39%) 17.3 (61%) | 0.95 | 3.8 | 71.3 | 27.0 | 25.6 | |
|
G | 358 | 482 | 7186 (124) | 5.66 | 0.94 | 10.6 | 166.1 | 23.6 | 22.2 |
| G1–CB[7]2 | 354 | 460 | 6509 (106) | 3.20 | 0.92 | 25.0 | 287.5 | 26.4 | 24.3 | |
| G2–CB[8]2 | 380 | 474 | 5219 (94) | 5.22 (57%) 9.76 (43%) | 0.92 | 11.2 | 128.3 | 30.4 | 28.0 | |
|
G | 341 | 454 | 7300 (113) | 1.97 | 0.77 | 116.8 | 390.9 | 54.4 | 41.9 |
| G1–CB[7]2 | 338 | 419 | 5719 (81) | 1.36 | 0.88 | 88.2 | 647.1 | 60.5 | 53.2 | |
| G2–CB[8]2 | 358 | 472 | 6747 (114) | 2.68 (28%) 10.7 (72%) | 0.82 | 21.3 | 97.0 | 59.0 | 48.4 | |
|
G | 314 | 431 | 8645 (117) | 2.36 | 0.63 | 156.8 | 266.9 | 35.9 | 22.6 |
| G1–CB[7]2 | 311 | 415 | 8058 (104) | 1.25 (21%) 2.66 (79%) | 1.00 | 0.0 | 423.0 | 37.2 | 37.2 | |
| G2–CB[8]2 | 330 | 446 | 7882 (116) | 13.3 | 0.84 | 12.0 | 63.2 | 37.7 | 31.6 | |
|
G | 314 | 441 | 9171 (127) | 2.20 | 0.71 | 131.8 | 322.7 | 40.6 | 28.8 |
| G1–CB[7]3 | 311 | 417 | 8174 (106) | 2.37 | 0.96 | 16.9 | 405.1 | 40.7 | 39.0 | |
| G2–CB[8]3 | 331 | 469 | 8890 (138) | 14.6 | 0.27 | 50.0 | 18.5 | 40.5 | 10.9 | |
After photoexcitation, fluorescence decay as well as the corresponding lifetime are recorded from time-correlated single photon counting (TCSPC) experiments, Fig. 4d–f. Excited anthracenyl dimers in the G2–CB[8]2 complexes display an excimer-like state that exhibits a lifetime (τs) of 12.6 ns, which is much longer than τs of 8.6 ns for its monomeric counterpart in G1–CB[7]2. A biexponential decay is observed for Ant910Me in pristine solution measured at the same concentration (15 μM) as that in G1–CB[7]2 and G2–CB[8]2, showing 96% of the intensity is due to a short-lived component of 1.66 ns and 4% arising from a long-lived component of 9.50 ns.
Fluorescence quantum yields (ϕF) for each species are measured by an absolute method using an integrating sphere. The emission of Ant910Me is significantly quenched in a pristine solution with a ϕF of 0.04, whereas its fluorescence intensity is dramatically enhanced upon complexation with either CB[7] or CB[8], showing a ϕF of 0.85 for G1–CB[7]2 and a ϕF of 0.81 for G2–CB[8]2 (Table 1).
The negligible quantum yield, bimodal decay, and red-shifted emission in a pristine solution of Ant910Me all suggest a certain extent of aggregation in aqueous solution, whose photophysical properties are highly concentration-dependent. On the other hand, complexation with CB[7] or CB[8] ensures a dispersion of discrete fluorophores in solution in either monomeric or dimeric fashion, respectively. It is known that the polarity of CB cavities is lower than that of water, which will also affect photophysical properties of dye molecules.19,43 Therefore, in the following discussion, a comparison is made between CB[7]- and CB[8]-mediated complexes on account of their similar cavity polarities. A comparison between G1–CB[7]2 and G2–CB[8]2 of Ant910Me shows that the stacking of anthracenyl moieties as a dimer, relative to monomer, exhibits (1) a significant bathochromic shift in absorption and emission, (2) an elongated excited-state lifetime, and (3) comparably high fluorescence efficiency.
2.4 Applicability and flexibility of the modular strategy
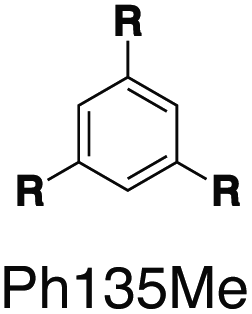
Although individual cases have demonstrated photophysical changes upon complexation with CB[8],31,35,39,44–47 the beauty and power of this work stems from the simple modular design. As illustrated in Fig. 1a, any selected fluorophore can be readily inserted between clamping modules, resulting in its monomeric or dimeric species through complexation with CB[7] or CB[8], respectively.Following this design strategy, a further eight fluorescent molecules were successfully synthesized, with similar topology to Ant910Me but with systematic variation in their structures. For example, the fluorophore cores are augmented between phenyl, naphthyl, and anthracenyl. Alternatively, the alignment between the two clamping modules is altered. While several derivatives exhibit both clamping modules in-line with one another (Ph14Me, Np14Me, Ant910Me, and Ant14Me) others have clamping modules that are not in-line but remain parallel to each other such as Np15Me and Ant15Me, or are no longer aligned in a parallel manner but with an angle < 180° (Ph13Me and Np27Me). Finally, one can readily add additional clamping motifs around the fluorophore core moiety, as demonstrated in the triply clamped systems Ph135Me and Ant14Me.
Results from 1H NMR and DOSY, as shown in Fig. 3a and in the ESI (Table S1 and Fig. S1–S17†), demonstrates that these fluorescent molecules all perform in a manner similar to Ant910Me. Despite their structural variation, they all generate a monomeric fluorophore in the presence of CB[7] and dimeric stacking of fluorophores with CB[8]. As CB[7] and CB[8] only bind the clamping modules (i.e. tolyl pyridinium moieties), choice of the fluorophores is no longer limited by the size and shape of the macrocycle cavities. Large fluorophores such as anthracenyl derivatives, which to date have only been shown to complex CB[7] or CB[8] along their principal symmetry axis, are easily incorporated using this strategy regardless of their substitution pattern. Moreover, small aromatic rings like phenyl moieties, whose binding is extremely dynamic inside a single CB[8] cavity, are now readily immobilized and constrained within a 2:2 complex.
2.5 Photophysical properties
In terms of photophysical properties, most fluorescent molecules also behave similarly to Ant910Me, with the exception of a few outliers that are discussed later in detail.Preorganized ground-state dimers are readily produced by the formation of G2–CB[8]2 in aqueous solution, corresponding to a considerable bathochromic shift in the absorption band (Fig. 5 and Table 1). An excimer-like emission with a broadened and structureless profile is observed for all fluorophores in their G2–CB[8]2 systems, exhibiting a red-shift in their emission maximum relative to their monomeric form in G1–CB[7]2 systems. Solutions of G2–CB[8]2 compared to their G1–CB[7]2 counterparts exhibit a smaller rate constant for non-radiative deactivation (knr), which corresponds to their observed elongated excited-state lifetime as well as comparably high quantum yields. Molar absorption coefficients for all fluorophores are slightly increased upon complexation with either CB[7] or CB[8] (Table 1) along with their high quantum yield, leading to reasonably high brightness (ε × ϕF) in aqueous solution.48 Considering their long fluorescence lifetimes, G2–CB[8]2 complexes in general should be promising candidates for time-gated imaging for biological systems.
 | ||
| Fig. 5 Steady-state absorption (solid line) and emission (dash line with filling color) spectra of non-associated fluorescent molecules (G in blue), and their CB[7]-mediated complexes (G1–CB[7]2 or G1–CB[7]3 in yellow) as well as CB[8] mediated complexes (G2–CB[8]2 or G2–CB[8]3 in red). The study covers the derivatives of three types of fluorophores including phenyl such as (a) Ph14Me, (b) Ph13Me, (c) Ph135Me, naphthyl such as (d) Np14Me, (e) Np27Me, (f) Np15Me, and anthracenyl such as (g) Ant910Me, (h) Ant14Me, (i) Ant15Me. Aqueous solutions of each species are tested under a uniform guest concentration at 298 K. The intensity is not normalized but scaled up by the same factor in most cases. Quantified data can be found in Table 1. | ||
Fluorescent molecules that contain anthracenyl cores all exhibit a low quantum yield in solution with a lifetime much shorter than that in their CB[7]- or CB[8]-mediated complexes. The discrete monomeric species of Ant910Me, Ant15Me, and Ant14Me in their corresponding G1–CB[7]2 complexes all display a lifetime around 8 ns (Table 1), similar to the lifetime (τs) of 9,10-diphenylanthracene (DPA), a typical anthracenyl standard.49 This recovery of lifetime to a value similar to DPA implies that the fluorescence of these CB[7] complexes is mainly contributed by their anthracenyl cores. In a solution of only free molecules (without the presence of CB), the excited anthracenyl cores are deactivated through certain pathways as evidence from the observed quenching of fluorescence. In particular, a typical deactivation pathway would be the photoinduced electron transfer (PET) from the anthracenyl core to π-deficient pyridinium moieties.50,51 However, the significant recovery of emission after complexation suggests that these deactivation pathways are forbidden or are at least largely restricted in both the G1–CB[7]2 and G2–CB[8]2 complexes. Quantum yields of the naphthyl and phenyl species are generally large (0.9–1.0 for Np, 0.6–1.0 for Ph) contrary to anthracenyl analogues, regardless of complexation, implying that PET from these two fluorophore cores to pyridinium moieties is not efficient.
Systematic variation in the alignment between the clamping modules also affects their photophysical properties. Np15Me and Ant15Me, with two parallel clamping modules that are not aligned, exhibit a red-shift in emission, which is not as large as for other species (Fig. 5) upon forming G2–CB[8]2 complexes. This non-aligned connectivity may force the two fluorophores to stack in a less J aggregate-like fashion.52 When the clamping modules are non-parallel, the G2–CB[8]2 of Np27Me displays a quantum yield of 0.55, which is almost half the value of the other naphthyl homologues (Table 1). However, this species exhibits a distinctively long-lived excited state with a τs up to 37 ns. Similar results are observed in CB[8]-mediated complexes of Ph13Me, which also possesses non-parallel clamping modules. The reduced fluorescence efficiency along with the elongated lifetime suggests that dimeric stacking in species with non-parallel clamping units may significantly suppress radiative pathways (i.e. see reduced kr values in Table 1).
2.6 Triple clamping
Ph135Me is a more complex version of non-parallel clamping, which forms dimeric stacks through triple clamping, denoted G2–CB[8]3 (Fig. S16†). Triple non-parallel clamping leads to a further decrease in kr (Table 1) compared to G2–CB[8]2 of Ph13Me, which results in a reduced quantum yield (0.27) of G2–CB[8]3 that is about one third of that of its CB[7]-mediated complex, G1–CB[7]3. Besides suppressing the radiative pathway, triple clamping exhibits a concerted feature of multivalency53 further stabilizing the dimeric stacking of two phenyl moieties. In a mixture consisting of 4 equiv. of Ph135Me and 3 equiv. of CB[8], excess guest molecule does not result in statistical complexes such as G2–CB[8]2 and G2–CB[8]1 (Fig. S17†). Instead, Ph135Me molecules exist either as G2–CB[8]3 complexes or as a free guest in aqueous solution.In addition to Ph135Me, which has three uniform clamping modules, Ant14Me with a protruding fluorophore core is also able to form a G2–CB[8]3 complex. Isothermal titration calorimetry and UV-Vis titration both confirm a binding stoichiometry of 2:3 (Fig. S12†). Its diffusion coefficient from DOSY NMR gives a D value similar to that of Ph135Me2–CB[8]3 (Table S1, Fig. S11 and S16†). Considering its T-shape topology, a third CB[8] in the Ant14Me G2–CB[8]3 complex binds with the two protruding, stacked anthracenyl cores. However, in contrast to Ph135Me, CB[8] complexation of Ant14Me (in excess) does not exhibit a self-sorting behavior. Addition of extra Ant14Me guest molecules gradually transforms the solution of G2–CB[8]3 into 2:2 complexes, in which two CB[8] macrocycles are bound with the two tolyl pyridinium moieties rather than the protruding anthracenyl cores (Fig. S10–S12†). This suggests that the affinity of CB[8] around the protruded binding site is substantially weaker than its binding with the clamping modules, which is confirmed by the ITC result in Fig. S12.†
2.7 Restricted intracomplex motion
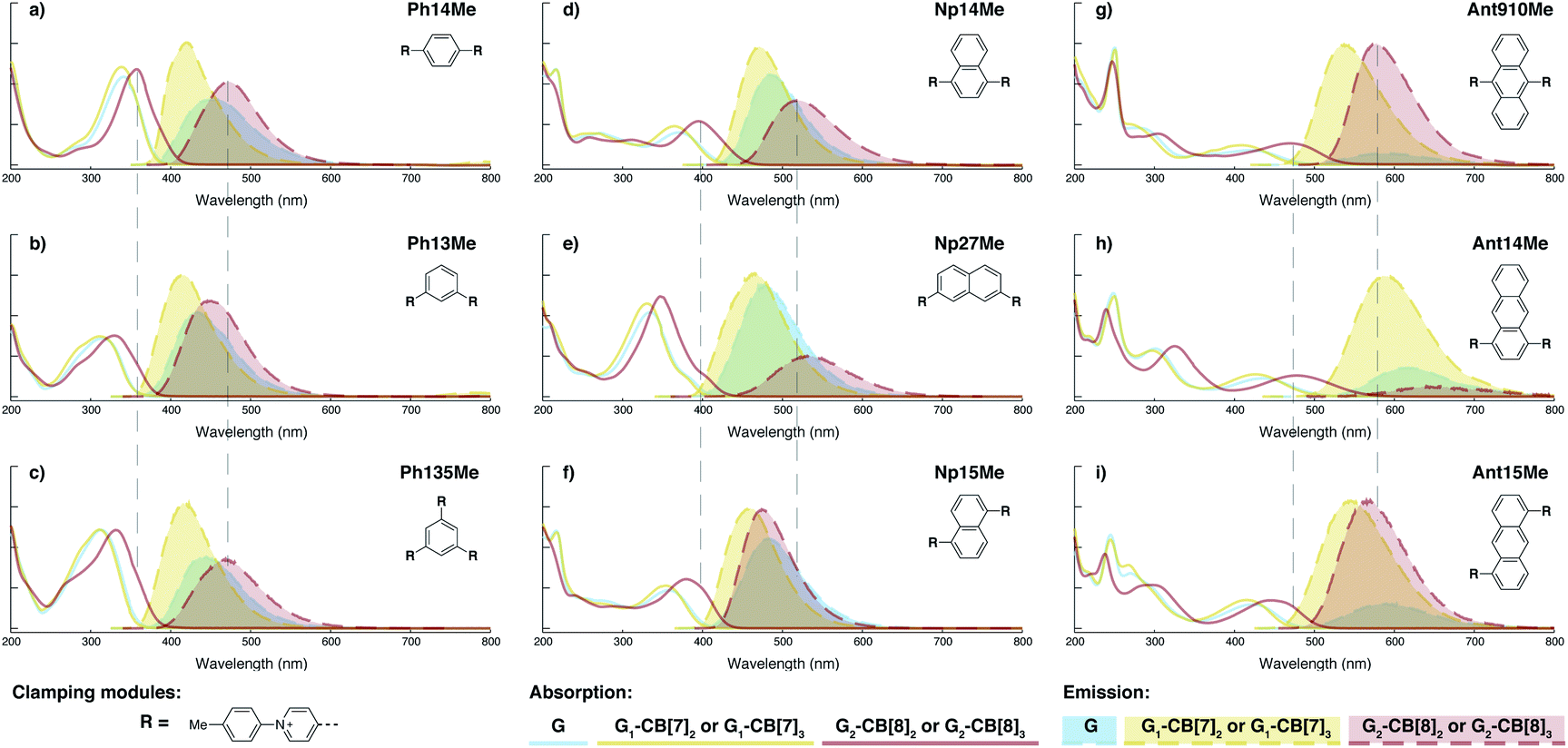
Dimeric fluorophore stacking in G2–CB[8]2 complexes generally exhibit an enhanced fluorescence efficiency, particularly in the case of employing anthracenyl motifs as cores. This observation implies that motion within the complex (intracomplex motion) of G2–CB[8]2 is extremely retarded and restricted, thus effectively suppressing deactivation pathways.Interconversion between the two states is further confirmed and quantified by variable-temperature nuclear magnetic resonance spectroscopy (VT-NMR). As shown in Fig. 6a, four signals of Hg,g′,h,h′ that correspond to two stacked anthracenyl cores broaden equally until coalescence is observed as the temperature rises from 278.6 K to 307.5 K on a high-field NMR spectrometer (500 MHz). A subsequent increase of temperature from 306.2 K to 362.6 K on a low-field NMR spectrometer (200 MHz) (Fig. 6b) leads to a gradual merging of the four signals into two broad peaks, which later become sharper as the temperature increases. The transition of the Hg,g′,h,h′ signals from the slow exchange limit to the fast exchange limit confirms the existence of a dynamic interconversion between two discrete states for the anthracenyl pair. By analysing the temperature-dependent line-broadening in the slow exchange limit54–56 (Fig. S19†), an activation energy of 43 kJ mol−1 is obtained for this interconversion.
 | ||
| Fig. 6 Variable-temperature 1H NMR spectra of the Ant910Me2–CB[8]2 complex in D2O solution (a) with a temperature increased from 278.6 K to 307.5 K (top to bottom) recorded by high-field spectrometer (500 MHz), and (b) with a temperature increased from 306.2 K to 362.6 K (bottom to top) recorded by low-field spectrometer (200 MHz), showing (c) restricted intracomplex rotations of tolyl, anthracenyl, and pyridinium pairs with non-uniform activation energies (Ea) of 22 kJ mol−1, 43 kJ mol−1, and 83 kJ mol−1, respectively, which are analysed from the temperature-dependent line-broadening (Fig. S19†). The temperature of 500 MHz and 200 MHz spectrometers is calibrated by MeOD (D, 99.8%) and ethylene glycol (80% in DMSO-d6), respectively. The Cl− counterions are omitted for clarity. | ||
As lowering the magnetic field is equivalent to severely heating the sample, the switch of VT-NMR from high-field to low-field enables us to witness and quantify a very slow exchange process such as that for the pyridinium pair in this study. In the spectra recorded by the high-field spectrometer (Fig. 6a), no significant line broadening is observed for pyridinium signals (Hb,b′,c,c′), whereas in the low-field VT-NMR, the signal broadening corresponding to an exchange in the slow limit is readily observed upon increase in temperature (Fig. 6b). The temperature-dependent signal broadening suggests an activation energy as large as 83 kJ mol−1 (Fig. S19†), implying a relatively slow interconversion of the pyridinium pair within the complex. The interconversion of protons in the stacked tolyl pair is already displayed in the fast exchange limit as demonstrated by the line sharpening of the Hd signal as the temperature is increased in the high-field spectrometer, exhibiting a relatively small exchange barrier of 22 kJ mol−1 (Fig. 6a and S19†).
Molecular dynamic (MD) simulations of the Ant910Me2–CB[8]2 complex in a cubic water box with 4000 H2O molecules was carried out in order to evaluate the stability of this complex under ambient conditions (298 K, 1 atm). The simulations indicate that the Ant910Me2–CB[8]2 complex (ESI† video media) remains as a single entity during the whole simulation period (>200 ns) without decomplexation or significantly altering its structure. This result is consistent with the analysis by NMR, which shows that the dimeric stack of fluorophores is constrained and stabilized by the CB[8]-mediated dual clamping. Moreover, the two stacked aromatic moieties, such as anthracenyl units, partially overlap one another and simultaneously rotate in a slow but coherent fashion during the MD simulation. For example, the two anthracenyl units (yellow) of the Ant910Me2–CB[8]2 complex in Fig. 6 must rotate or swing around the central axis of the complex in a coupled manner, which we refer to here as intracomplex motion. Thus, the activation energy obtained represents the energy barrier for each intracomplex rotation.
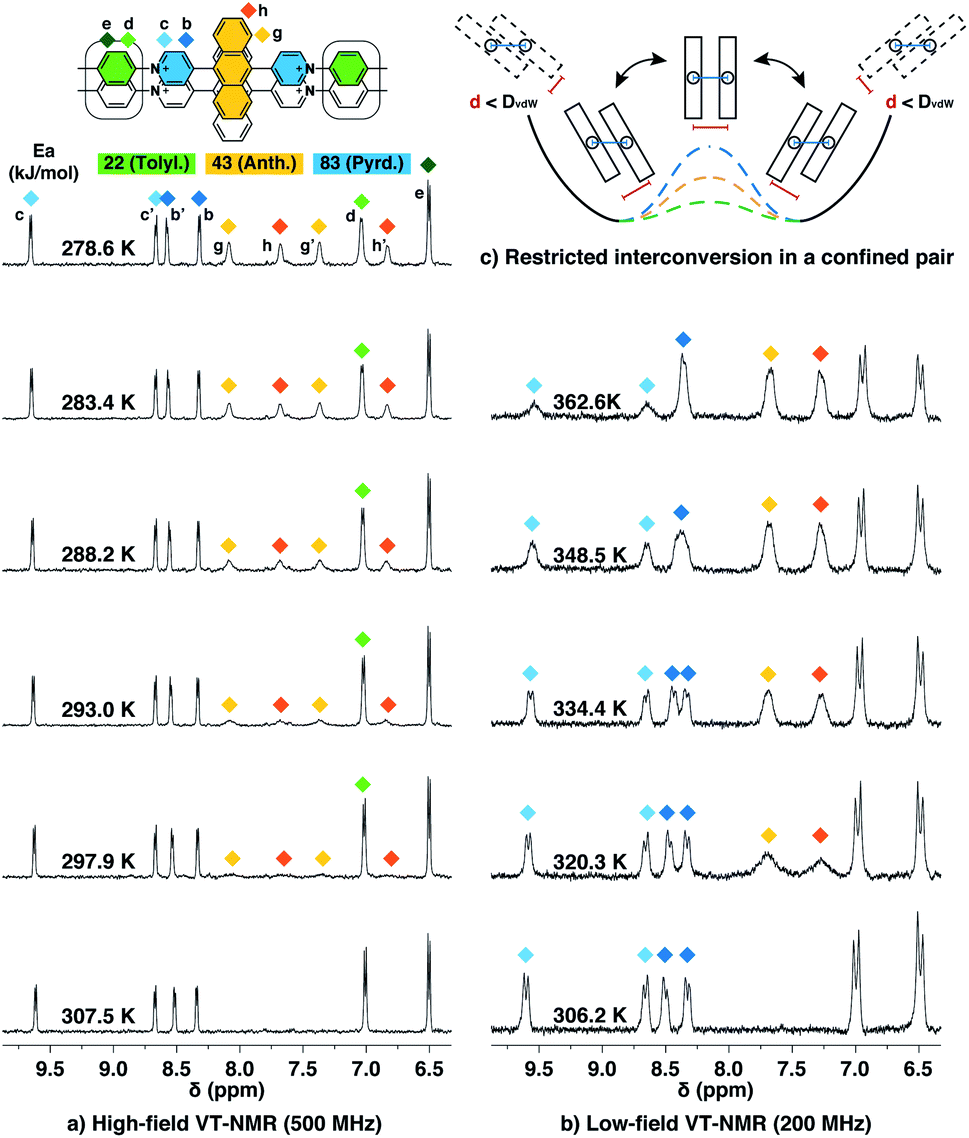
The height of the energy barrier reflects the steric hindrance present around the “rotor”. As exemplified by Ant910Me in Fig. 7, the rotation of the anthracenyl group is hindered by the presence of several pairs of adjacent protons between the anthracenyl core and pyridinium units. The rotation of the pyridinium moieties, in addition to steric hindrance from the anthracenyl core, are also impeded by the CB[8] portals, thus showing the highest rotational barrier. On the other hand, rotation of the tolyl groups is not significantly influenced by CB portals as they mainly reside within the CB[8] cavities. Their motion is also not significantly retarded by neighboring pyridinium protons, which present less steric clash than those between the anthracenyl and pyridinium units. Therefore, the tolyl groups exhibit the lowest rotational barrier and their proton signals always fall within the fast exchange limit (Fig. 6). It is worth noting that “rotation” does not necessarily refer to a full rotation. In the case of Ant910Me, it is more likely that the two anthracenyl groups swing coherently within a limited angle on account of van der Waals repulsion (Fig. 6c), where the activation energy represents the steric hindrance for swinging between two degenerate states.
 | ||
| Fig. 7 Linewidth comparison between G2–CB[8]2 and G1–CB[7]2 complexes for fluorescent molecules (Ant910Me, Np14Me, Ph13Me) with a variation in the extent of proton exclusion. The difference in steric hindrance of each fluorescent molecule is readily reflected by the signal broadening or splitting observed from both fluorophore and pyridinium moieties in G2–CB[8]2, but cannot be distinguished by those in G1–CB[7]2, indicating that the steric hindrance is largely amplified in a dimeric stacked complex. | ||
In contrast, the dimeric complexes of these molecules (G2–CB[8]2) display significant differences in both their fluorophore and pyridinium components in their 1H NMR spectra. As shown in Fig. 7a and c, proton signals of Ph13Me exhibit a narrow linewidth in the fast exchange limit for both the pyridinium and 1,3-phenyl groups, which is consistent with the fact that no severe steric clash exists in this molecule. However, signal broadening occurs with an increase of steric repulsion in the dimeric complex of Np14Me. Furthermore, proton signals from the anthracenyl and pyridinium groups in the dimeric complex of Ant910Me both fall into a slow exchange limit and split into two sets of peaks, which corresponds to much slower intracomplex motions. This observation stems from a further increase in steric hindrance and is amplified for the G2–CB[8]2 complexes as rotation of one moiety is not only retarded by covalently linked “neighbours” but also hindered by adjacent groups on the other stacked molecule. Careful comparison between the monomeric and dimeric systems verifies that formation of a constrained system largely restricts and slows down intracomplex motions in these dimers.
2.8 Ground and excited states of π-stacked dimers
In the case of G2–CB[8]2 (Fig. 5), however, a considerable bathochromic shift is generally observed in its steady-state absorption spectrum. Particularly, the vibronic progression is absent in the absorption of Ant910Me2–CB[8]2 (Fig. 4c) indicating a strong coupling and effective delocalization of π-electrons between the dimeric anthracenyl moieties at their ground states. This preorganized π-stacked ground-state dimer is excited as a precoupled entity to an excimer-like state, which is different from an excited monomer and, more importantly, does not require an additional diffusion-controlled process after photoexcitation. On the other hand, the excited G2–CB[8]2 complex will not exhibit an energy dissipation as significantly as during the formation of conventional excimers. This explains why Ant910Me in the G2–CB[8]2 complex exhibits a Stokes shift (wavenumber difference between λabs and λem) of 4012 cm−1 (109 nm) smaller than the value of 5828 cm−1 (128 nm) in its G1–CB[7]2 complex (Table 1). Due to the absence of diffusion-controlled steps in their excited state, one expects a mono-exponential fluorescence decay at pico- and nano-second timescale for G2–CB[8]2 complexes after photoexcitation, contrary to the bimodal decay of conventional excimers.58
Femtosecond (fsTA) and nanosecond (nsTA) transient absorption were employed to monitor the dynamic relaxation of both G2–CB[8]2 and G1–CB[7]2 complexes of Ant910Me after photoexcitation (Fig. S20–S25, Table S2†). As shown in Fig. 8a and b, two species are clearly detected in the excited state from fsTA for Ant910Me2–CB[8]2. Both exhibit a spectral feature of ground state bleaching (GSB) from 431 nm to 498 nm overlapping with an excited state absorption (ESA) from 431 nm to 800 nm and a stimulated emission (SE) from 573 nm to 654 nm. Upon photoexcitation, the first species A relaxes to species B with a fairly short lifetime of 3.6 ± 0.3 ps (Fig. 8c, d and S21†), and then back to its ground state with a lifetime of 12.9 ± 0.4 ns (Fig. 8e and f), consistent with the value of 12.6 ns measured from TCSPC (Table 1). Species B exhibits a similar ESA profile as species A except a slight red-shift in its absorption maximum (Fig. 8b), which suggests that the evolution from A to B with a picosecond time constant probably corresponds to excited state solvation.59 In addition to solvation, the excited complex relaxes back to its ground state in a mono-exponential manner without observing other competitive pathways. It is worth mentioning that the excited state absorption spectra of Ant910Me2–CB[8]2 (Fig. 8a) are quite broad suggesting a strong coupling also existing in the excited states.
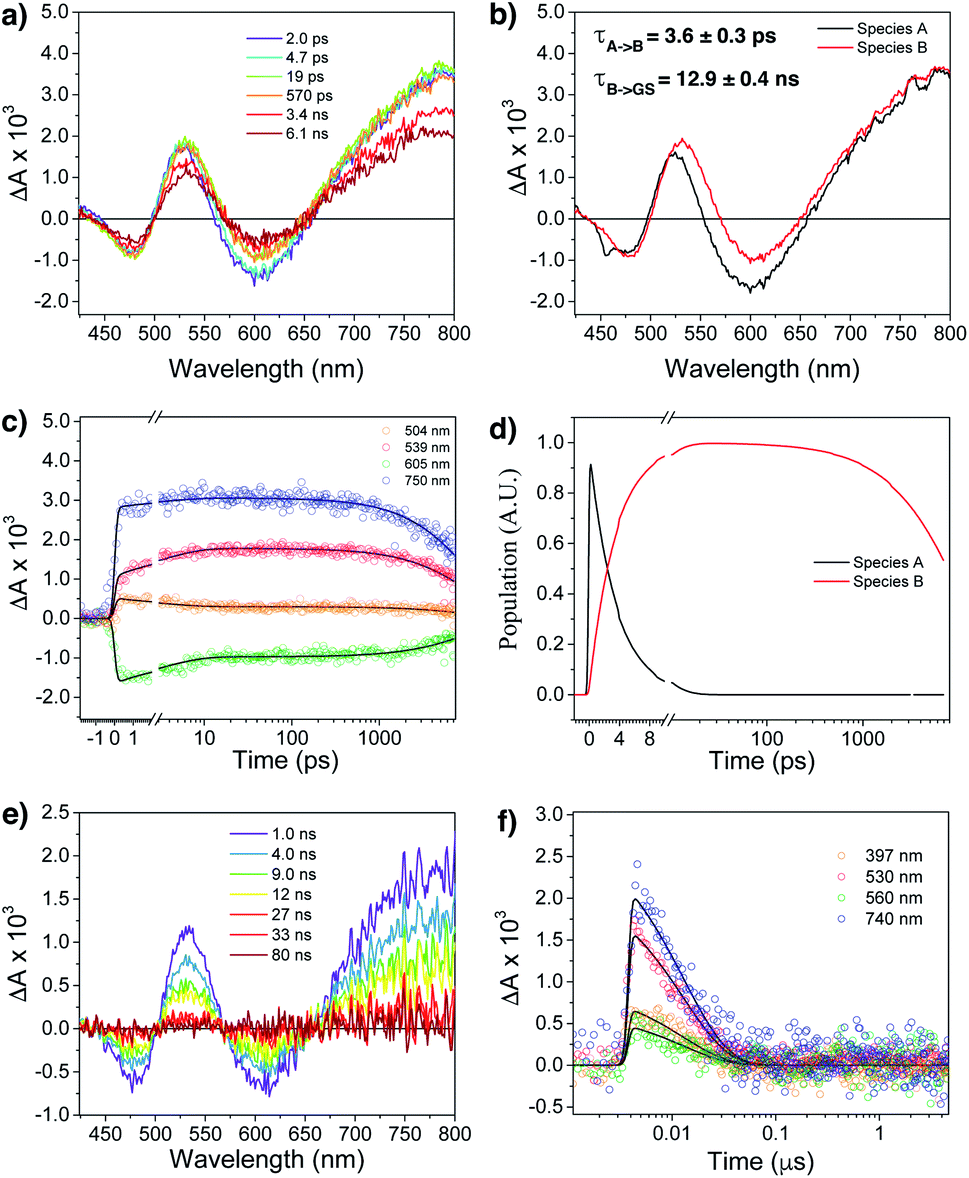 | ||
| Fig. 8 Femtosecond transient absorption spectra (fsTA) of (a) Ant910Me2–CB[8]2 complexes in D2O at 298 K following a laser pulse of 414 nm (1 μJ per pulse). Fitting from the raw data using a kinetic model A → B → GS gives (b) species associated spectra, (c) wavelength fitting, and (d) model population kinetics. (e) Nanosecond transient absorption spectra (nsTA) of species B and (f) its kinetic fitting at selected wavelength using mono-exponential model. | ||
Ant910Me1–CB[7]2 after photoexcitation (Fig. S23†) exhibits an absorption maximum at around 475 nm in its ESA profile, which is much smaller than that of Ant910Me2–CB[8]2 at around 530 nm (Fig. 8b). This observation confirms that G2–CB[8]2 complexes are directly pumped up to the excited state of precoupled dimers rather than the excited state of monomers. It is worth noticing that several species are detected (Fig. S23 and S24†) for Ant910Me1–CB[7]2 upon photoexcitation, which also includes a small amount of long-lived species with a time constant of 6 ± 1 μs. This long-lived species should come from a triplet state, as its lifetime increases significantly (>340 μs, Fig. S25†) after removal of oxygen from the solvent. The observed rich dynamic processes implies that after photoexcitation the fluorescent molecule within the G1–CB[7]2 complex has sufficient structural freedom to relax to various low-energy excited states. On the other hand, the singular excited state dynamic observed for G2–CB[8]2 complex suggests a restricted or retarded structural change even in its excited state, which may affect both the radiative and non-radiative pathways.
In addition to constrained complexation, the discrete nature of fluorophore dimers is also crucial to ensure high-efficiency fluorescence.60,61 The two CB[8] macrocycles that hold the fluorophore dimer together will mechanically block interactions from other dimers in aqueous solution, which effectively avoids the generation of dark excited states caused by arbitrary aggregation.
The spontaneously assembled G2–CB[8]2 complex satisfies all three requirements and facilitates the formation of preorganized π-stacked dimers. The two fluorophores inside a G2–CB[8]2 complex form a discrete dimeric stack with a significant overlap of π electrons and a restricted interplanar spacing defined by the CB[8] cavities. Steric hindrance from both CB[8] macrocycles facilitates “mechanical” separation between all dimers in aqueous solution ensuring pairwise fluorophores perform as a discrete entity. More importantly, the dimers are stabilized by CB[8] clamping and remain as such for a sufficiently long period of time. Finally, discrete preorganized dimers can be readily obtained through our strategy in aqueous solution at ambient temperatures, and therefore do not require formation of a specific crystal60,61 or crystalline solvent at extremely low temperature.62,63 Moreover, owing our modular design, a variety of fluorophores are incorporated to give the corresponding π-stacked dimers without any limitation on fluorophore size in direct contrast to other methods.64
2.9 Controlling photophysics by clamping modules
Radiative decay is practically prohibited in the case of Ant14Me when it is complexed with CB[8], either by dual clamping or by triple clamping, exhibiting negligible quantum yield in either case (Table 1). Fluorescence quenching in the G2–CB[8]3 complex of Ant14Me may be readily explained by triple clamping, however, it does not explain why complete quenching is also observed for its G2–CB[8]2 counterpart. One hypothesis is that the protruding anthracenyl moieties in one dimer may be long enough to interact with other protruding anthracenyl pairs located in another dimer, leading to some radiationless decay pathways that quickly deactivate the excited state. Interactions between protruding anthracenyl moieties is also supported by the diminished quantum yield observed for its G1–CB[7]2 complex compared to that of other anthracenyl homologues. Another possibility leading to radiationless decay may be a transition from a singlet to a triplet state through intersystem crossing, however, this requires substantial further study of the dynamics of Ant14Me complexes in their excited states.
Interestingly, the G2–CB[8]2 complex of Ant15Me also adopts a single H–H stacking configuration, as the spacing between its two off-line clamping modules is too large to allow for a feasible H–T configuration (Scheme S3†). This specific stacking configuration is also revealed in the 1H NMR spectrum of its G2–CB[8]2 complex, in which the protons of the 1,5-anthracenyl moieties exhibit sharp and well-resolved signals (Fig. S9†). In contrast, Np15Me with a smaller gap between its off-line clamping modules may allow for both H–H and H–T stacking configurations of the two fluorophores, which leads to a significant broadening of proton signals in the NMR of its G2–CB[8]2 complex. Moreover, all the proton signals are equally broadened suggesting a dynamic process that involves the entire complex, which very likely correlates to an interconversion between H–H and H–T stacking configurations with an exchanging rate on the intermediate NMR timescale (Fig. S7†). As a result, the aromatic fluorophores in the G2–CB[8]2 complexes Ant15Me and Np15Me both exhibit a substantial overlap of π-electrons in a less J aggregate-like fashion, leading to smaller bathochromic shifts in their emission maxima (Fig. 5) compared to other fluorescent molecules.65 Considering their red-shift in absorption, the smaller bathochromic shifts in emission maxima may also correlate to an anti-Kasha behavior as mentioned above, which requires further investigation.
Although both H–H and H–T stacking should be feasible by Np14Me and Ant14Me, the NMR spectra of their G2–CB[8]2 complexes suggest a preference towards head-to-head stacking. The protons residing on the protruding ring exhibit an upfield shift due to shielding of the aromatic ring current, which is best explained by a head-to-head overlapping of the fluorophores. This further suggests that the π–π interactions play a role in determining energy-favorable stacking configurations.
On the other hand, the photophysical properties of the dimeric stacked fluorophores are indeed affected by the size of the aryl substituents. As shown in Fig. 9, Np14H, a naphthyl fluorescent molecule without any substituent on its clamping module displays the same absorption and emission spectra as those of Np14Me. However, a significant difference of the emission maximum is observed for the G2–CB[8]2 complex of Np14CMe2. As both the absorption and emission spectra of G and G1–CB[7]2 of Np14CMe2 are similar to those of Np14H and Np14Me, this difference observed for the G2–CB[8]2 complex must stem from a certain variation in the stacking of the naphthyl pair, which is very likely caused by a significant volume exclusion between neighboring isopropyl substituents. The resultant stacking configuration in G2–CB[8]2 of Np14CMe2 still leads to a red-shifted absorption band corresponding to π-electron delocalization in the preorganized dimer. It seems that the preorganized dimer (in this case) does not result in an effective formation of an excimer-like state, as the emission maximum is very similar to that in pristine solution without an obvious bathochromic shift. This observation thus offers an additional opportunity to tune the photophysical properties of stacked fluorophores by choosing appropriate substituents.
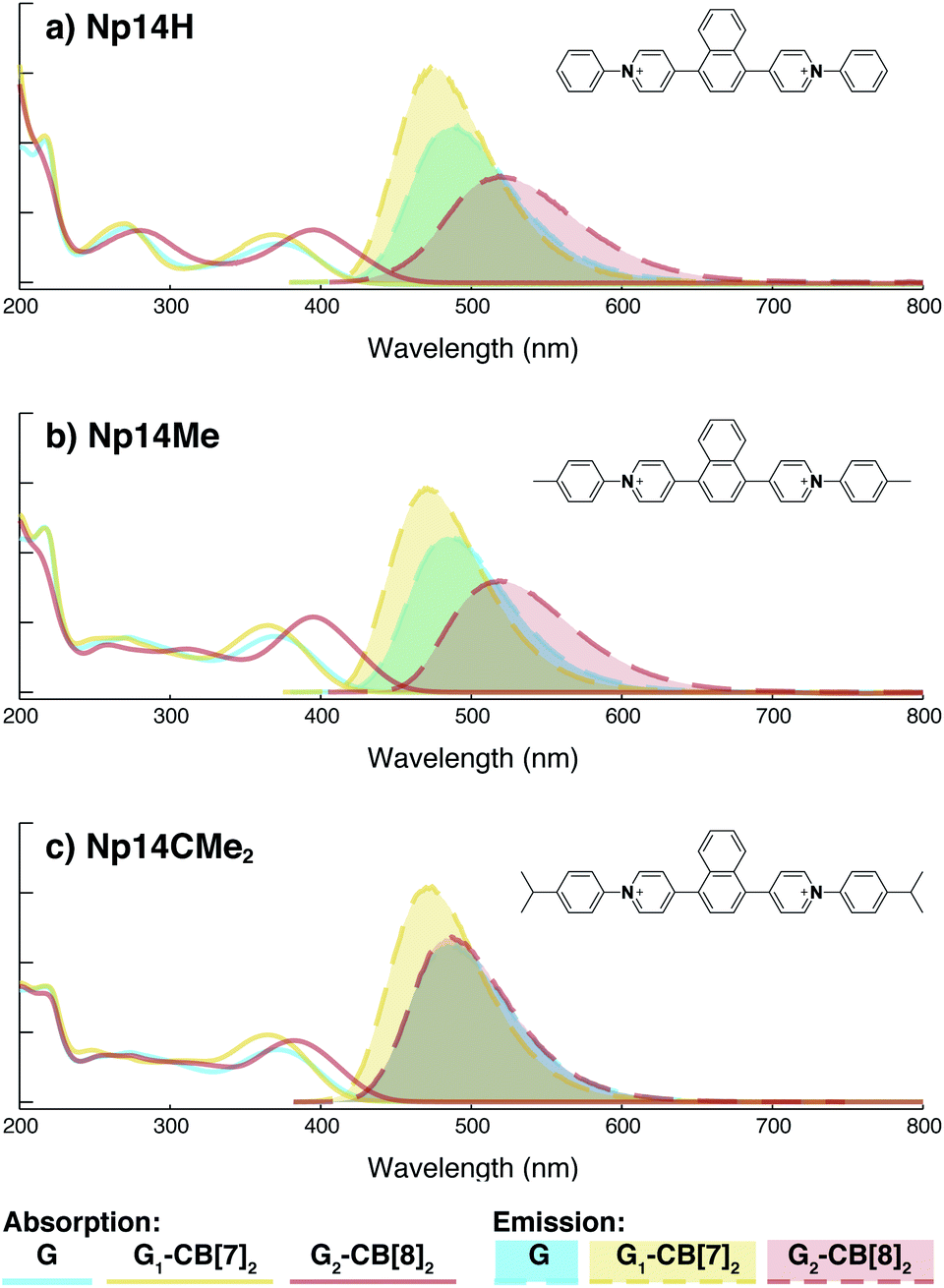 | ||
| Fig. 9 Absorption (solid line) and emission (dash line with filling color) spectra of 1,4-naphthyl based fluorescent molecules (blue) with variation in substituents including (a) Np14H, (b) Np14Me, (c) Np14CMe2, along with their G1–CB[7]2 complexes (yellow) and G2–CB[8]2 complexes (red). Aqueous solution of each species are tested under a uniform guest concentration at 298 K. The intensity is not normalized but scaled up by the same factor. The Cl− counterions are omitted for clarity. | ||
3 Conclusions
In summary, we have demonstrated a modular strategy to design a new class of fluorescent molecules that (i) generate discrete, dimeric stacked fluorophores in aqueous solution and (ii) are constrained by CB[8]-mediated multiple clamping. This modular design is surprisingly applicable and flexible and has been validated by testing nine different fluorophore cores ranging in size, shape, and geometric variation of their clamping modules. When complexed with CB[7], all fluorescent molecules are dispersed in aqueous solution as discrete monomers, exhibiting an impressively high fluorescence efficiency. On the other hand, complexation with CB[8] as 2:2 or 2:3 complexes leads to the immediate formation of discrete dimeric stacked fluorophores. Multiple CB[8] clamping results in stable, preorganized ground-state dimers, which can be readily photoexcited to excimer-like states, displaying significant bathochromic shifts in absorption and emission with elongated fluorescence lifetimes. Bathochromic shifts in the emission spectra can be readily tuned by controlling the stacking of fluorophores through specific variations in the clamping modules (through off-line alignment or altering substituents).
We demonstrate that intracomplex motion in the preorganized dimers is significantly restricted, which suppresses both radiative and non-radiative deactivation, resulting in a substantially high quantum yield (up to 1) despite formation of excimer-like states. Some complexes are further restricted through non-parallel or triple clamping, which slows down radiative relaxation to an even greater extent, leading to elongated excited-state lifetimes up to 37 ns in aqueous solution. Moreover, complexes stabilized by multiple non-parallel clamping exhibit self-sorting in the presence of excess CB[8], which facilitates the design and fabrication of hierarchical functional structures.
While only arylpyridinium moieties have been employed as the clamping module in this study, current investigations suggest other chemical motifs with rigid structure exhibit the same clamping feature. The high rigidity ensures intrinsically low conformational entropy change during complexation, thus facilitating the formation of a long-lived, multicomponent complex in aqueous solution.
From a fundamental point of view, this study offers a model system with explicitly stable dimeric structures and tuneable features that can be utilized as a platform to study various intermolecular processes including excimer formation, charge transfer, exciton coupling, and singlet fission. Moreover, such a modular molecular design towards quadrupolar fluorescent molecules may provide a feasible toolbox in pursuit of distinct features such as large two-photon cross-sections66,67 and non-Kasha behavior.68 On the practical side, CB[7]- and CB[8]-mediated fluorescent complexes developed here are promising candidates for various (biological) imaging applications on account of their emergent photophysical properties such as long lifetimes, high emission brightness, and red-shifted excitation bands.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Leverhulme Trust (project: “Natural material innovation for sustainable living”), the Marie Curie FP7 SASSYPOL ITN (607602) programme, EPSRC Programme Grant (NOtCH, EP/L027151/1), and ERC-2016 Consolidator Grant (CAM-RIG, 726470) (G. W., M. O., O. A. S.), the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award DE-FG02-99ER14999 (Y. J. B., M. R. W.), EPSRC (EP/M508007/1) (D. A. G.), and EPSRC (EP/R013012/1, EP/L027151/1 and EP/N020669/1), BBSRC (BB/N007700/1), ERC (project 757850 BioNet) (I. S., E. R.). The authors thank Dr Peter Grice and Mr Duncan Howe for their help on NMR analysis, and thank Prof Luisa De Cola, Dr Bruno Frka-Petesic, Dr Stefan Mommer, and Wiebke Schnettger for their suggestion.References
- M. A. Slifkin, Nature, 1963, 200, 766–767 CrossRef CAS
.
- Y. Wu, J. Zhou, B. T. Phelan, C. M. Mauck, J. F. Stoddart, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2017, 139, 14265–14276 CrossRef CAS PubMed
- J. Hoche, H.-C. Schmitt, A. Humeniuk, I. Fischer, R. Mitrić and M. I. S. Röhr, Phys. Chem. Chem. Phys., 2017, 19, 25002–25015 RSC
- H. Tamura, J. Phys. Chem. A, 2016, 120, 9341–9347 CrossRef CAS PubMed
- C. Kaufmann, D. Bialas, M. Stolte and F. Würthner, J. Am. Chem. Soc., 2018, 140, 9986–9995 CrossRef CAS PubMed
- C. E. Miller, M. R. Wasielewski and G. C. Schatz, J. Phys. Chem. C, 2017, 121, 10345–10350 CrossRef CAS
- Y. J. Bae, G. Kang, C. D. Malliakas, J. N. Nelson, J. Zhou, R. M. Young, Y.-L. Wu, R. P. Van Duyne, G. C. Schatz and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 15140–15144 CrossRef CAS PubMed
- A. Rao and R. H. Friend, Nat. Rev. Mater., 2017, 2, 17063 CrossRef CAS
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed
- B. Heyne, Photochem. Photobiol. Sci., 2016, 15, 1103–1114 RSC
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS PubMed
- S. Anderson and H. L. Anderson, Angew. Chem., Int. Ed., 1996, 35, 1956–1959 CrossRef CAS
- J. C. Barnes, M. Juríček, N. L. Strutt, M. Frasconi, S. Sampath, M. A. Giesener, P. L. McGrier, C. J. Bruns, C. L. Stern, A. A. Sarjeant and J. F. Stoddart, J. Am. Chem. Soc., 2012, 135, 183–192 CrossRef PubMed
- E. J. Dale, N. A. Vermeulen, M. Juricek, J. C. Barnes, R. M. Young, M. R. Wasielewski and J. F. Stoddart, Acc. Chem. Res., 2016, 49, 262–273 CrossRef CAS PubMed
- L. Yang, P. Langer, E. S. Davies, M. Baldoni, K. Wickham, N. A. Besley, E. Besley and N. R. Champness, Chem. Sci., 2019, 10, 3723–3732 RSC
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC
- A. L. Koner and W. M. Nau, Supramol. Chem., 2007, 19, 55–66 CrossRef CAS
- J. Mohanty and W. M. Nau, Angew. Chem., Int. Ed., 2005, 44, 3750–3754 CrossRef CAS PubMed
- M. Megyesi, L. Biczók and I. Jablonkai, J. Phys. Chem. C, 2008, 112, 3410–3416 CrossRef CAS
- M. Freitag, L. Gundlach, P. Piotrowiak and E. Galoppini, J. Am. Chem. Soc., 2012, 134, 3358–3366 CrossRef CAS PubMed
- K. Liu, Y. Yao, Y. Kang, Y. Liu, Y. Han, Y. Wang, Z. Li and X. Zhang, Sci. Rep., 2013, 3, 2372 CrossRef PubMed
- H.-J. Kim, J. Heo, W. S. Jeon, E. Lee, J. Kim, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 1526–1529 CrossRef CAS PubMed
- L. M. Heitmann, A. B. Taylor, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2006, 128, 12574–12581 CrossRef CAS PubMed
- S. Senler, W. Li, M. H. Tootoonchi, S. Yi and A. E. Kaifer, Supramol. Chem., 2014, 26, 677–683 CrossRef CAS
- P. Montes-Navajas and H. Garcia, J. Phys. Chem. C, 2010, 114, 2034–2038 CrossRef CAS
- G. Wu, M. Olesińska, Y. Wu, D. Matak-Vinkovic and O. A. Scherman, J. Am. Chem. Soc., 2017, 139, 3202–3208 CrossRef CAS PubMed
- H. Yin, Q. Cheng, R. Rosas, S. Viel, V. Monnier, L. Charles, D. Siri, D. Gigmes, O. Ouari and R. Wang,
et al.
, Chem.–Eur. J., 2019, 25, 12552–12559 CrossRef CAS PubMed
- S. Chakrabarti and L. Isaacs, Supramol. Chem., 2008, 20, 191–199 CrossRef CAS
- J. Mohanty, S. Dutta Choudhury, H. P. Upadhyaya, A. C. Bhasikuttan and H. Pal, Chem.–Eur. J., 2009, 15, 5215–5219 CrossRef CAS PubMed
- J. Mohanty, N. Thakur, S. Dutta Choudhury, N. Barooah, H. Pal and A. C. Bhasikuttan, J. Phys. Chem. B, 2012, 116, 130–135 CrossRef CAS PubMed
- B. Yang, S.-B. Yu, H. Wang, D.-W. Zhang and Z.-T. Li, Chem.–Asian J., 2018, 13, 1312–1317 CrossRef CAS PubMed
- K. Kotturi and E. Masson, Chem.–Eur. J., 2018, 24, 8670–8678 CrossRef CAS PubMed
- S. Schoder, H. V. Schröder, L. Cera, R. Puttreddy, A. Güttler, U. Resch-Genger, K. Rissanen and C. A. Schalley, Chem.–Eur. J., 2019, 25, 3257–3261 CAS
- T. K. Ronson, W. Meng and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 9698–9707 CrossRef CAS PubMed
- D. Bongard, M. Möller, S. N. Rao, D. Corr and L. Walder, Helv. Chim. Acta, 2005, 88, 3200–3209 CrossRef CAS
- M. Olesińska, G. Wu, S. Gómez-Coca, D. Antoón-Garciía, I. Szabó, E. Rosta and O. A. Scherman, Chem. Sci., 2019, 10, 8806–8811 RSC
- G. Wu, I. Szabó, E. Rosta and O. A. Scherman, Chem. Commun., 2019, 55, 13227–13230 RSC
- W. L. Mock and N. Y. Shih, J. Org. Chem., 1986, 51, 4440–4446 CrossRef CAS
- G. Wu, D. E. Clarke, C. Wu and O. A. Scherman, Org. Biomol. Chem., 2019, 17, 3514–3520 RSC
- Y. H. Ko, K. Kim, E. Kim and K. Kim, Supramol. Chem., 2007, 19, 287–293 CrossRef CAS
- S. Kohmoto, T. Chuko, S. Hisamatsu, Y. Okuda, H. Masu, M. Takahashi and K. Kishikawa, Cryst. Growth Des., 2015, 15, 2723–2731 CrossRef CAS
- H.-J. Kim, D. R. Whang, J. Gierschner and S. Y. Park, Angew. Chem., Int. Ed., 2016, 55, 15915–15919 CrossRef CAS PubMed
- X.-L. Ni, S. Chen, Y. Yang and Z. Tao, J. Am. Chem. Soc., 2016, 138, 6177–6183 CrossRef CAS PubMed
- R. Ye, Q. Cui, C. Yao, R. Liu and L. Li, Phys. Chem. Chem. Phys., 2017, 19, 31306–31315 RSC
- B. Zhang, Y. Dong, J. Li, Y. Yu, C. Li and L. Cao, Chin. J. Chem., 2019, 37, 269–275 CAS
- H. Osaki, C.-M. Chou, M. Taki, K. Welke, D. Yokogawa, S. Irle, Y. Sato, T. Higashiyama, S. Saito, A. Fukazawa and S. Yamaguchi, Angew. Chem., Int. Ed., 2016, 55, 7131–7135 CrossRef CAS PubMed
- J. V. Morris, M. A. Mahaney and J. R. Huber, J. Phys. Chem., 1976, 80, 969–974 CrossRef CAS
- G. M. Blackburn, G. Lockwood and V. Solan, J. Chem. Soc., Perkin Trans. 2, 1976, 1452–1456 RSC
- V. A. Kharlanov, M. I. Knyazhansky, N. I. Makarova and V. A. Lokshin, J. Photochem. Photobiol., A, 1993, 70, 223–227 CrossRef CAS
- F. C. Spano and C. Silva, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed
- A. Mulder, J. Huskens and D. N. Reinhoudt, Org. Biomol. Chem., 2004, 2, 3409–3424 RSC
-
J. K. M. Sanders and K. Brian, Modern NMR Spectroscopy, a Guide for Chemists, Oxford University. Oxford, GB, 1993 Search PubMed
- R. Joseph and E. Masson, Eur. J. Org. Chem., 2014, 2014, 105–110 CrossRef CAS
- M. Jin, T. S. Chung, T. Seki, H. Ito and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2017, 139, 18115–18121 CrossRef CAS PubMed
- S. Akine, T. Onuma and T. Nabeshima, New J. Chem., 2018, 42, 9369–9372 RSC
-
J. B. Birks, Photophysics of Aromatic Molecules, 1970 Search PubMed
- S. K. Pal, J. Peon, B. Bagchi and A. H. Zewail, J. Phys. Chem. B, 2002, 106, 12376–12395 CrossRef CAS
- H. Liu, L. Yao, B. Li, X. Chen, Y. Gao, S. Zhang, W. Li, P. Lu, B. Yang and Y. Ma, Chem. Commun., 2016, 52, 7356–7359 RSC
- Y. Shen, H. Liu, S. Zhang, Y. Gao, B. Li, Y. Yan, Y. Hu, L. Zhao and B. Yang, J. Mater. Chem. C, 2017, 5, 10061–10067 RSC
- E. A. Chandross, J. Ferguson and E. G. McRae, J. Chem. Phys., 1966, 45, 3546–3553 CrossRef CAS
- E. A. Chandross and J. Ferguson, J. Chem. Phys., 1966, 45, 3554–3564 CrossRef CAS
- L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, J. Am. Chem. Soc., 2005, 127, 3674–3675 CrossRef CAS PubMed
- Y. Gao, H. Liu, S. Zhang, Q. Gu, Y. Shen, Y. Ge and B. Yang, Phys. Chem. Chem. Phys., 2018, 20, 12129–12137 RSC
- M. Albota, D. Beljonne, J.-L. Brédas, J. E. Ehrlich, J.-Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCord-Maughon, J. W. Perry, H. Röckel, M. Rumi, G. Subramaniam, W. W. Webb, X.-L. Wu and C. Xu, Science, 1998, 281, 1653–1656 CrossRef CAS PubMed
- G. D'Avino, F. Terenziani and A. Painelli, J. Phys. Chem. B, 2006, 110, 25590–25592 CrossRef PubMed
- C. Zheng, C. Zhong, C. J. Collison and F. C. Spano, J. Phys. Chem. C, 2019, 123, 3203–3215 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, synthesis and characterization, DOSY results, complexation studies of nine fluorescent molecules, VT-NMR, transient absorption study, and MD simulation of the Ant910Me2–CB[8]2 complex in a water box: MD_Ant910Me_CB[8].avi. See DOI: 10.1039/c9sc04587b |
| This journal is © The Royal Society of Chemistry 2020 |