
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibition of K-Ras4B-plasma membrane association with a membrane microdomain-targeting peptide†
Fang-Yi
Li
a,
Zhen-Feng
Zhang
b,
Stephanie
Voss
cd,
Yao-Wen
Wu
 cde,
Yu-Fen
Zhao
a,
Yan-Mei
Li
a and
Yong-Xiang
Chen
*a
cde,
Yu-Fen
Zhao
a,
Yan-Mei
Li
a and
Yong-Xiang
Chen
*a
aKey Laboratory of Bioorganic Phosphorus Chemistry and Chemical Biology (Ministry of Education), Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: chen-yx@mail.tsinghua.edu.cn
bState Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, No. 1 West Beichen Road, Chaoyang District, Beijing 100101, China
cChemical Genomics Centre of the Max Planck Society, Otto-Hahn-Str. 15, 44227 Dortmund, Germany
dMax-Planck-Institute of Molecular Physiology, Otto-Hahn-Str. 11, 44227 Dortmund, Germany
eDepartment of Chemistry, Umeå University, 90187 Umeå, Sweden
First published on 3rd December 2019
Abstract
The association of K-Ras4B protein with plasma membrane (PM) is required for its signaling activity. Thus, direct inhibition of K-Ras4B–PM interaction could be a potential anti-Ras therapeutic strategy. However, it remains challenging to modulate such protein–PM interaction. Based on Ras isoform-specific PM microdomain localization patterns, we have developed a potent and isoform-selective peptide inhibitor, Memrasin, for detachment of K-Ras4B from the PM. Memrasin is one of the first direct inhibitors of K-Ras4B–PM interaction, and consists of a membrane ld region-binding sequence derived from the C-terminal region of K-Ras4B and an endosome-escape enhancing motif that can aggregate on membrane. It forms peptide-enriched domains in the ld region, abrogates the tethering of K-Ras4B to the PM and accordingly impairs Ras signaling activity, thereby efficiently decreasing the viability of several human lung cancer cells in a dose-responsive and K-Ras dependent manner. Memrasin provides a useful tool for exploring the biological function of K-Ras4B on or off the PM and a potential starting point for further development into anti-Ras therapeutics.
Introduction
Mutational activation of RAS proto-oncogenes (HRAS, NRAS and KRAS) occurs in 20–30% of human cancers, rendering four encoded Ras proteins (H-Ras, N-Ras, two splice variants K-Ras4A and K-Ras4B) among the most powerful drivers of cancers. The KRAS gene is the most frequently mutated RAS gene, particularly in some lethal cancers (pancreatic, lung and colorectal tumors).1 K-Ras4B, the predominant splice variant, is thus considered a major drug target in cancer therapy, and has attracted intense attention for more than three decades.2,3 However, no effective anti-Ras therapeutics have been taken into clinic. Ras proteins function as membrane-bound molecular switches in vital signaling pathways by cycling between an active GTP-bound state and an inactive GDP-bound state.4 They possesses globular G-domains, which are challenging to be targeted by small molecules. With advances in technology and knowledge in recent years, a series of promising therapeutic approaches has been developed for inhibiting Ras signaling,5–9 including preventing the formation of active Ras,10–15 impairing Ras-effector interactions,16–20 and perturbing Ras localization.21,22 Nevertheless, in order to ultimately conquer K-Ras4B, diverse and novel strategies remain to be developed.Despite containing a highly conserved globular G-domain similar to the other Ras isoforms, K-Ras4B possesses a unique flexible poly-lysine motif upstream of a farnesylated and methylated cysteine at its C-terminus.23 These structural features determine K-Ras4B's enrichment on the inner leaflet of the plasma membrane (PM), and particularly formation of clusters in membrane liquid-disordered (ld) microdomains, which are prerequisites for its signaling activity.23–27 Thus, blocking the association of K-Ras4B with the PM is a promising strategy for impairing its signaling activity, as proven by recent progress.9,28 Fendiline has been shown to be capable of mislocalizing K-Ras from the PM by reducing membrane phosphatidylserine content.29,30 Small molecules that target the prenyl-binding chaperon protein PDEδ can disrupt the attachment of K-Ras4B to the PM.21 However, although inhibition of the protein–membrane interaction (PMI) represents a potential therapeutic strategy for related human diseases,31,32 direct inhibitors of the K-Ras4B–PM interaction remain forthcoming, most likely due to the lack of a defined binding pocket on this large PMI interface for canonical ligand development.
Here, inspired by isoform-specific PM microdomain localization pattern of K-Ras4B,24–27 we report a rationally-designed hybrid peptide named Memrasin that possesses a membrane ld region-binding sequence deriving from the C-terminal of K-Ras4B and an endosome-escape enhancing motif. It efficiently associates with the specific ld region of the PM and directly blocks its interaction with K-Ras4B, thereby impairing Ras signaling and suppressing the proliferation of cancer cells (Fig. 1).
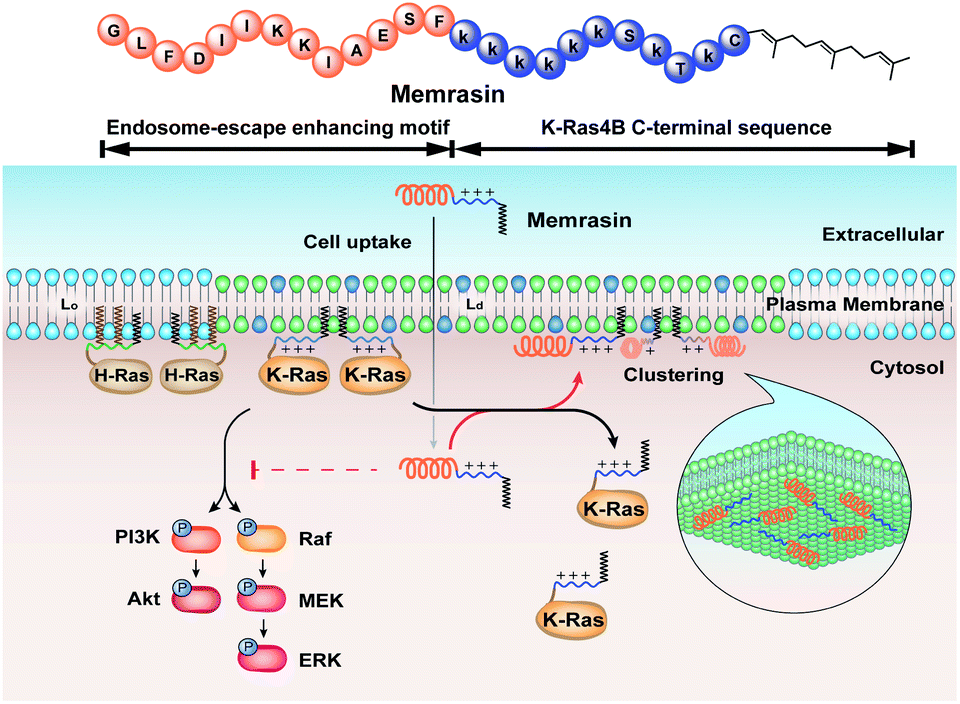 | ||
| Fig. 1 Illustration of the blockage of Ras signaling by disruption of the association of K-Ras4B with the plasma membrane (PM) using Memrasin. Lowercase letters stand for D-amino acids. | ||
Results and discussion
Design and synthesis of K-Ras4B–PM interaction inhibitors
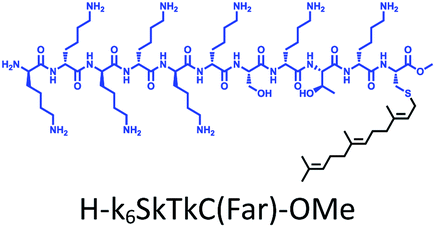
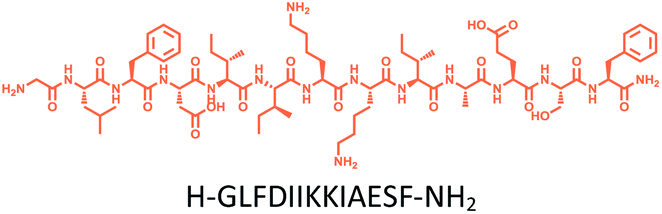
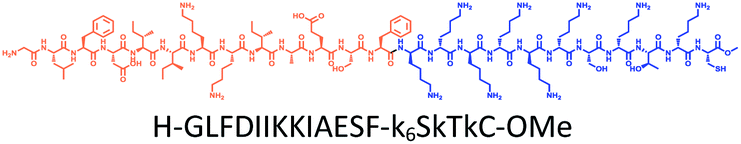
The distribution of Ras proteins in distinct intracellular membrane microdomains is mainly determined by their different C-terminal structures.24–27 We, and others, have reported that K-Ras4B mainly localizes in the ld microdomains of the PM via its unique C-terminal tail.24–27,33 We thus speculate that a fully modified K-Ras4B C-terminal peptide sequence may be a good leading structure for specifically inhibiting this PMI. Interestingly, the farnesylated K-Ras4B peptide and protein display excellent cell membrane permeability due to their poly-lysine motif and lipid moiety.33,34 Based on the minimal PM-targeting motif of K-Ras4B,23 the cell penetrating capability and synthetic consideration, we chose an 11-mer farnesylated and methylated sequence as the C-terminal part of this peptide inhibitor. To enhance the binding potency of the farnesylated K-Ras4B peptide with the PM, we conjugated it with a highly efficient endosomal-escape enhancing peptide with low cellular toxicity, aurein 1.2, which has been identified by screening 36 chosen anti-microbial peptides (≤25 amino acids) for application in protein delivery.35 As a membrane active peptide, it displays a α-helical structure and then forms aggregates on the membrane surface, which can induce membrane perturbation.35–37 As clustering of K-Ras4B on the PM is important for its signaling activity, the aggregating property of aurein 1.2 on the membrane might facilitate Memrasin to form aggregates on the PM and enhance its inhibitory potency to this PMI. In addition, to improve the in vivo stability of Memrasin while maintaining the α-helical structure of its N-terminal part and the electrostatic interaction properties of the anchor segment to the maximum extent, we replaced all lysine residues within the flexible anchor segment with D-lysine. To evaluate the necessity of each part of Memrasin, we designed C1–C3 as corresponding control peptides (Table 1).| Name | Peptide structure and sequence |
|---|---|
| Memrasin |
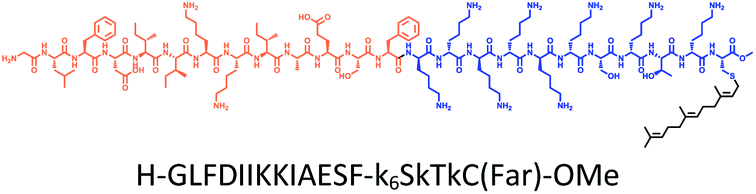
|
| C1 |
|
| C2 |
|
| C3 |
|
Memrasin was synthesized according to a previously reported strategy (Fig. 2).33,38 First, Fmoc-Cys-OMe was anchored to 2-chlorotrityl chloride resin via a thioether bond. The peptide chain was elongated using standard Fmoc-based SPPS strategy. The released pre-farnesylated peptide precursor was then farnesylated in a zinc catalyzed farnesylation reaction with excess farnesyl bromide in a DMF/BuOH/H2O organic cocktail solution. The control peptides and fluorescein-labeled ones were prepared using similar approaches. The HPLC and CD analysis displayed that the substitution of (L)-Lys by (D)-Lys improved the stability of Memrasin in serum without apparently changing its secondary structure (Fig. S4 and S5†).
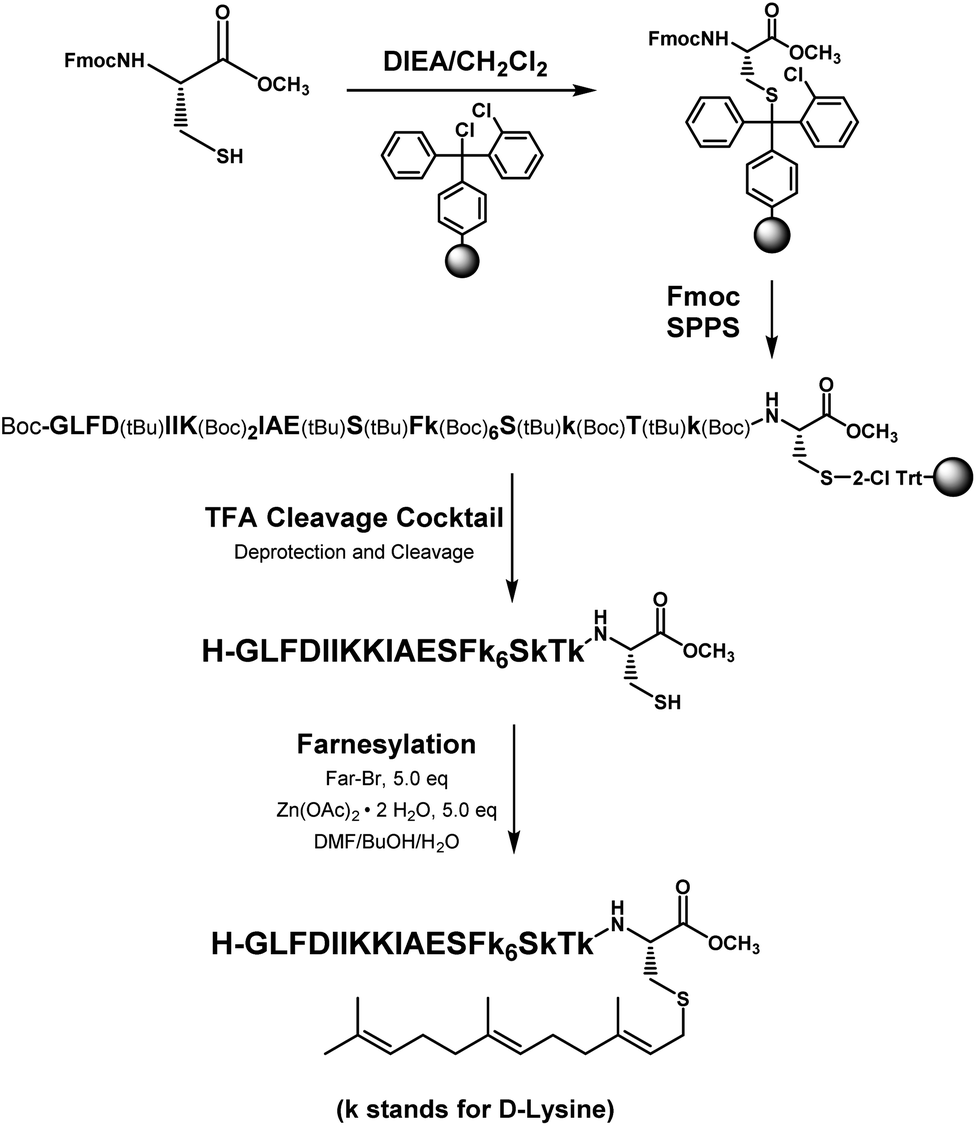 | ||
| Fig. 2 The synthetic strategy of Memrasin. | ||
Memrasin mislocalized K-Ras4B but not H-Ras from the PM
To evaluate the mislocalization activity of Memrasin on K-Ras4B, we constructed a MDCK cell line (Madin–Darby Canine Kidney Cells, chosen for the imaging purpose) stably overexpressing mCitrine tagged K-Ras4B, in which mCitrine-K-Ras4B displayed a clear basolateral PM localization. The cells were treated with various concentrations of Memrasin for 10 minutes, during which mCitrine-K-Ras4B rapidly lost its PM localization and redistributed to the cytosol in a dose-dependent manner (Fig. 3a and S6†). We further performed fractionation assay for the cells treated with Memrasin at lower concentrations (≤10 μM) by western blot. The results indicated that Memrasin elevated the ratio of cytosolic (C) mCitrine-K-Ras to membrane-bound (M) mCitrine-K-Ras slightly, although the changes were difficult to be detected by cell images probably due to a large distribution area in cytosol (Fig. 3a). The redistribution of K-Ras4B was not attenuated after an extended incubation time (Fig. S7†). In contrast, C1–C3 did not cause detectable mislocalization of K-Ras4B under the same conditions, indicating the indispensability of each part of Memrasin. Moreover, a combination treatment of C1 and C2 also resulted in fewer changes in the distribution of K-Ras4B, thereby demonstrating the necessity of a covalent linkage between the two parts of Memrasin (Fig. 3b). As mentioned above, the membrane ld region-binding sequence was introduced into Memrasin to specifically inhibit the interaction between K-Ras4B and the PM. We thus constructed an MDCK cell line stably overexpressing mCitrine tagged H-Ras, an isoform that binds to distinct PM microdomains in comparison with K-Ras4B.24,39 mCitrine-H-Ras showed a predominant PM localization. However, after treatment of the cells with Memrasin, mCitrine-H-Ras maintained its enrichment in the PM, distinct from mCitrine-K-Ras4B (Fig. 3c). This result infers that Memrasin has isoform-specific inhibitory effects on the association of K-Ras4B with the PM.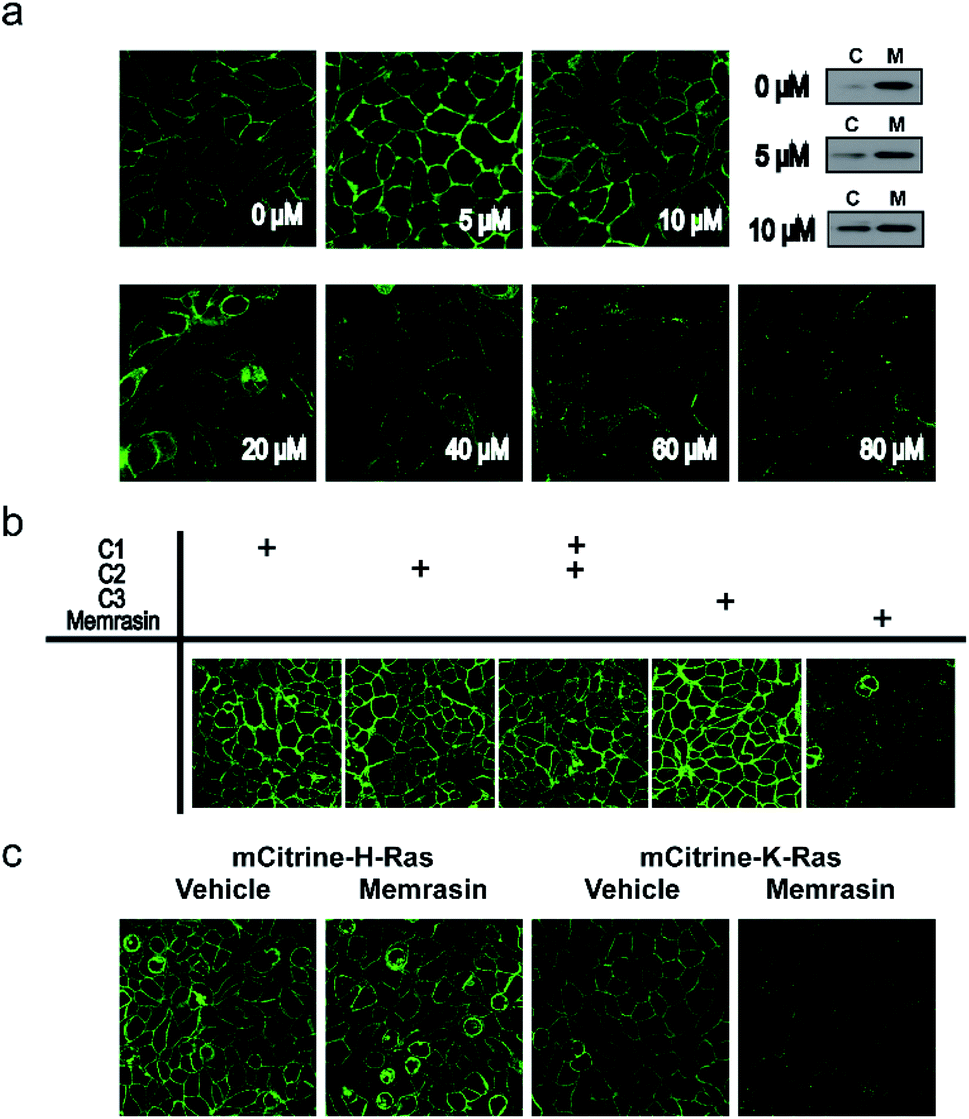 | ||
| Fig. 3 Memrasin mislocalizes mCitrine-K-Ras4B, but not mCitrine-H-Ras, from the PM. (a) Confocal images of mCitrine-K-Ras4B-overexpressing MDCK cells treated with Memrasin (0–80 μM, 10 min) and fractionation assay of mCitrine-K-Ras in MDCK cells treated with Memrasin (0, 5 and 10 μM, 10 min). C: cytosolic fraction; M: membrane fraction. (b) Confocal images of mCitrine-K-Ras4B-overexpressing MDCK cells treated with Memrasin or control peptides C1–C3 (80 μM, 12 h). (c) Confocal images of mCitrine-K-Ras4B or mCitrine-H-Ras-overexpressing MDCK cells treated with vehicle (DMSO) or Memrasin (80 μM, 1 h). mCitrine, green. | ||
Exploration of the mechanism by which Memrasin disassociated K-Ras4B from the PM
We first validated Memrasin's ability to enter cells and interact with the PM using fluorescein-labeled Memrasin. Results from flow cytometry assays indicated that FAM-Memrasin showed higher cell permeability than a common cell penetrating peptide (oligo-Arg7) labeled with fluorescein and the control peptides (Fig. S8†). Confocal images of A549 cells treated with inhibitors further demonstrate that FAM-Memrasin displays a clearer tethering on the PM in contrast to C1 and C3 (Fig. 4a and S9†). Besides, we noticed that Memrasin displayed an intracellular vesicles localization, too, which might result from its partially trapping in endosomes or its maintaining cycling between PM and endomembrane like K-Ras4B protein.40 We next designed a competitive fluorescence polarization (FP) assay to validate the blockage of the K-Ras4B-Membrane interaction by Memrasin (Fig. 4b). We constructed an in vitro system to evaluate the association of K-Ras4B with membranes using a fluorescein-labeled C-terminal peptide of K-Ras4B (FAM-C1) and a well-established anionic artificial PM model (DOPC/DOPG/DPPC/DPPG/Chol, 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:40:10:25, molar ratio), that can segregate into liquid-ordered (lo) and ld microdomains.26 The FP signal of FAM-C1 increased to ∼160 mP upon addition of the artificial membrane, indicating their efficient association. When the pre-incubated complexes were treated with increasing concentrations of Memrasin, the resultant FP signal gradually decreased (Fig. 4b), showing the successful breakdown of the K-Ras4B-membrane interaction. In contrast, C2 exhibited almost no influence on this interaction at tested concentrations, suggesting that the membrane anchor from K-Ras4B is very essential for the inhibition effect of Memrasin. Meanwhile, both C1 and C3 displayed weaker inhibition than Memrasin, suggesting that the endosome-escape enhancing motif and the farnesyl group, together with the polybasic fragment, contributes to the disruption of K-Ras4B-Membrane association in vitro. Besides, the effective inhibition concentration of Memrasin toward K-Ras-Membrane association rises up with the increase of FAM-C1 and liposome's concentrations (Fig. 4a and S10†).
:10:40:10:25, molar ratio), that can segregate into liquid-ordered (lo) and ld microdomains.26 The FP signal of FAM-C1 increased to ∼160 mP upon addition of the artificial membrane, indicating their efficient association. When the pre-incubated complexes were treated with increasing concentrations of Memrasin, the resultant FP signal gradually decreased (Fig. 4b), showing the successful breakdown of the K-Ras4B-membrane interaction. In contrast, C2 exhibited almost no influence on this interaction at tested concentrations, suggesting that the membrane anchor from K-Ras4B is very essential for the inhibition effect of Memrasin. Meanwhile, both C1 and C3 displayed weaker inhibition than Memrasin, suggesting that the endosome-escape enhancing motif and the farnesyl group, together with the polybasic fragment, contributes to the disruption of K-Ras4B-Membrane association in vitro. Besides, the effective inhibition concentration of Memrasin toward K-Ras-Membrane association rises up with the increase of FAM-C1 and liposome's concentrations (Fig. 4a and S10†).
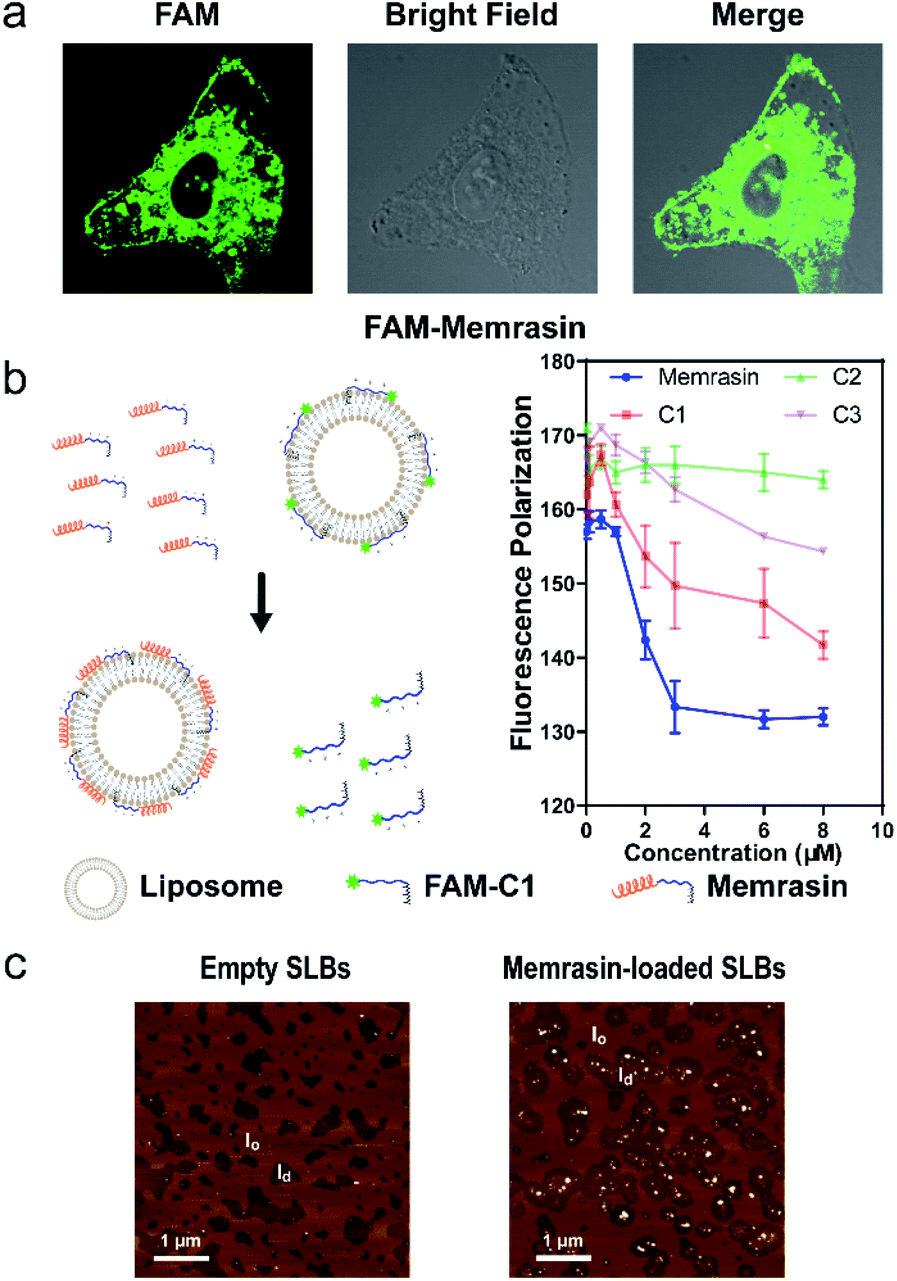 | ||
| Fig. 4 Exploration of the mechanism by which Memrasin disassociates K-Ras4B from the PM. (a) Confocal images of A549 cells treated with FAM-Memrasin (1 μM, 10 minutes) showing clear PM tethering. FAM, green. (b) Schematic illustration of the competitive FP assay of Memrasin and control peptides with pre-incubated complexes composed of the K-Ras4B peptide (0.1 μM FAM-C1) and anionic liposomes (60 μM, left panel). FP signals measured in the presence of Memrasin are shown in the right panel. (c) AFM image of Memrasin in supported lipid bilayers (SLBs). Surface morphology was detected before (left panel) and after (right panel) incubation with Memrasin (2 μM, 5 min). Dark brown: ld, light brown: lo. | ||
To investigate the membrane microdomains where Memrasin localizes and its membrane partitioning behavior, we analyzed the interaction between Memrasin and anionic model membranes (DOPC/DOPG/DPPC/DPPG/Chol, 20:5:45:5:25, molar ratio) using atomic force microscopy (AFM). Unilamellar lipid vesicles prepared by extrusion were flattened on a mica surface to generate phase-separated supported lipid bilayers (SLBs), which were then detected by AFM (Fig. 4c). The difference in height between lo phase (light brown) and ld phase (dark brown) was ∼1.0 nm, in agreement with previous reports (Fig. S11†).26 After addition of Memrasin, AFM measurements indicated exclusive localization of Memrasin in the ld phase, in which K-Ras4B proteins localized preferentially (Fig. 4c). In addition, Memrasin formed 4–8 nm-tall peptide-enriched domains in the ld phase (Fig. S11†), indicating the occurrence of peptide aggregates or clustering. In contrast, the addition of C1 into SLBs caused shape changes of the ld phase but did not form detectable enrichment domains (Fig. S10b and S12†), demonstrating the significance of this endosome-escape enhancing motif for Memrasin's aggregation on membranes.
Memrasin impaired Ras signaling activity
The Raf/MEK/ERK and PI3K/Akt signaling cascades are two vital pathways downstream of Ras.4 To verify whether Memrasin impairs K-Ras signaling activity, we examined the phosphorylation levels of MEK, ERK and Akt kinases in several lung cancer cell lines (NCI-H358, A549, and NCI-H441) in the absence or the presence of Memrasin. Western blot results (Fig. 5a and S13†) indicated that Memrasin inhibited hEGF-stimulated phosphorylation of MEK, ERK and Akt in a dose dependent manner in tested cell lines while corresponding total protein levels maintained constant. In contrast, C1 and C2 had no significant inhibitory effects on Ras signal output at the same concentrations of Memrasin (Fig. S14†), in line with their effects on K-Ras4B cellular distribution. In addition, the total amount of K-Ras was not influenced by Memrasin treatment (Fig. 5a). We thus performed cell fractionation assay to verify the effects of Memrasin on the C/M ratio of endogenous K-Ras in tested cell lines. Western blot results indicate that K-Ras was mostly localized in the membrane fractions in the absence of Memrasin while the C/M ratio of K-Ras significantly increased upon Memrasin treatment (Fig. 5b). When 4 μM Memrasin was applied, nearly all K-Ras were found in the cytosolic fractions. These results suggested that the disassociation of K-Ras from the PM caused by Memrasin treatment is consistent with the impairment of Ras signal output.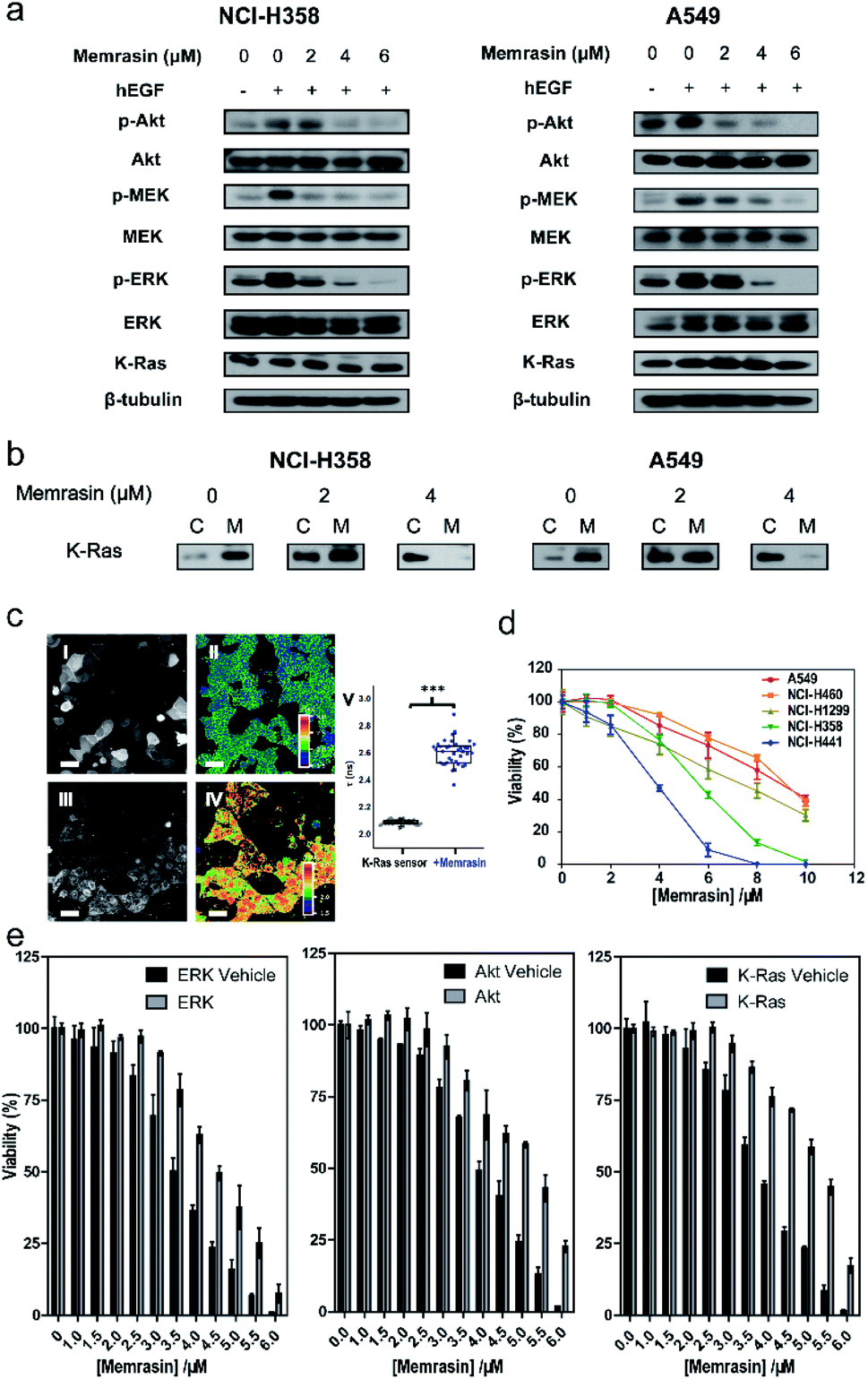 | ||
| Fig. 5 Memrasin inhibits Ras signaling activity and the viability of several lung cancer cell lines. (a) Dose-dependent inhibition of MEK, ERK and Akt phosphorylation in NCI-H358 and A549 cell lines by Memrasin. (b) Fractionation assay of K-Ras in NCI-H358 and A549 cell lines treated with Memrasin. C: cytosolic fraction; M: membrane fraction. (c) Imaging of intracellular K-Ras4B activity with or without Memrasin treatment by FLIM-FRET assay using mCitrine-K-RasE31C-TF4-CVIM sensor. Confocal mCitrine (I, III) and FLIM (II, IV) images were recorded before (I, II) or after (III, IV) Memrasin treatment (60 μM). Scale bar: 20 μm. Quantification of mCitrine fluorescence lifetime before (N = 40 cells, lifetime: 2.092 ± 0.017 ns) and after Memrasin treatment (N = 38 cells, lifetime: 2.608 ± 0.091 ns) are shown in graph V. ***: p < 0.001 (two-tailed t test). (d) Memrasin inhibits the viability of K-Ras related lung tumor cell lines. Incubation time: 24 hours. (e) Viability recovery assays of H441 cells against Memrasin treatments through transient expression of a constitutively active ERK, Akt or wt K-Ras4B protein. | ||
In addition, we found that Memrasin hardly affects the C/M ratio of Na+/K+ ATPase α1 subunit41,42 and transferrin receptor,43–45 two typical marker proteins of membrane non-raft regions (ld),46–48 within the same concentration range (0–4 μM) (Fig. S15a and b†). Besides, Memrasin at the concentration of 0–4 μM exhibited weak influence on the C/M ratio of a small GTPase Rab35 (Fig. S15c†), which contains a polybasic and prenylated C-terminal as well as exhibits cellular localization on PM and endomembranes as previously reported.49 We estimated that three tested proteins might have distinct precise localization from K-Ras4B within PM microdomains. These data reflect the selectivity of Memrasin toward anti-K-Ras-PM association to some extent, but we cannot rule out its potential off-target effects to all of the proteins in the PM. Besides, the intracellular chaperone proteins of farnesylated Ras proteins such as PDEδ and gelactin-3 (ref. 21) have the potential to bind Memrasin. However, neither farnesylated control peptide C1 solely nor it along with C2 obviously change the localization of mCitrine-K-Ras4B in MDCK cells, indicating that the interaction between Memrasin and chaperone proteins mentioned above might barely contribute to K-Ras redistribution.
We further applied Memrasin to MDCK cells containing an established K-Ras4B activity sensor (COSGA sensor), that can report the conformational changes in K-Ras4B on GTP or GDP binding by intramolecular Förster resonance energy transfer (FRET) signals.50 Live cell imaging results indicate that Memrasin efficiently dissociated the sensor from the PM (Fig. 5c). Interestingly, the FRET-FLIM (fluorescence lifetime imaging microscopy) measurements revealed that Memrasin caused significant increase in lifetime of COSGA sensor (Fig. 5c), suggesting the enhanced activity of K-Ras4B upon dissociation from the PM. These results suggest that K-Ras4B activity might be out of control by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs) when K-Ras is dissociated from the PM. The “active” K-Ras dissociated from the PM is not active in signaling, since K-Ras can transduce signals to downstream effectors only when it localizes to PM.23
Memrasin decreased the viability of several lung cancer cells in a dose-dependent manner
Inhibition of Ras/Raf/MEK/ERK signaling pathway can lead to a decrease in cell viability.4 As Memrasin possesses the ability to impair Ras signal output, we further examined the viability of several lung cancer cells bearing wild-type or mutated K-Ras treated with a 0–10 μM range of Memrasin using ATP-based CellTiter-Glo cell viability assay. Memrasin efficiently decreased the viability of these lung cancer cells in a dose-dependent manner (Fig. 5d). Moreover, Memrasin worked more effectively in K-Ras dependent cell lines (NCI-H358, NCI-H441, K-Ras dependency refers to the requirement of cell lines for K-Ras to maintain their viability51) than in K-Ras independent cell lines (A549, NCI-H460), particularly below the concentration of 6 μM (Fig. 5d), proving its propensity to regulate K-Ras signal output. In contrast, the control peptides showed minimal anti-cancer activities among all tested cell lines within the same concentration range and did not exhibit obvious K-Ras dependency toxicities (Fig. S16†). Moreover, NCI-H441 cells were respectively transfected with a plasmid containing a constitutively active form of ERK, Akt52,53 or a wild type (wt) K-Ras (Fig. S17†), resulting in their less sensitivities toward the growth inhibition effects of Memrasin, whereas the cells transfected with empty plasmids exhibited similar response as non-transfected cells toward Memrasin (Fig. 5e). Thus, our data suggest that Memrasin manifests a dose-responsive and Ras signal dependent anticancer activity across several lung cancer cell lines within tested concentrations (0–10 μM).Besides, we examined the cell viability of several normal cells (MDCK, Beas-2b, and COS-7) treated with Memrasin. Since K-Ras4B is expressed in most normal cell lines and acts as molecular switches in vital signaling pathways to regulate cell growth,23 the toxicity of Memrasin to normal cells can hardly be avoided. The results indicate that Memrasin could inhibit the growth of tested normal cells, too (Fig. S18†). However, these tested cells, particularly two epithelial cells MDCK and Beas-2b, are less sensitive to the growth inhibition of Memrasin within 0–4 μM concentrations than NCI-H441, which suggests a possible concentration range for further applications. Moreover, considering that a general toxicity of Memrasin to tested cell lines at higher concentrations might be partially resulted from its membrane-perturbation capability, we thus tested the hemolytic ability of Memrasin. As shown in Fig. S19,† Memrasin displayed a hemolytic activity of 3.57% or 5.68% at the concentration of 4 or 6 μM, at which it inhibits Ras signaling and the viability of tested K-Ras-dependent lung cancer cells. We further determined that serum albumin proteins (BSA) could help to reduce the hemolytic activity of Memrasin. In addition, further optimization of the peptide sequence and the combination with pharmaceutical delivery systems would benefit future application of Memrasin in animal models.
Conclusions
In summary, we have developed a potent and isoform-selective peptide inhibitor named Memrasin for mislocalization of K-Ras4B from the PM. Being the first direct inhibitor of the K-Ras4B-PM interaction, Memrasin possesses a membrane ld region-binding sequence deriving from the C-terminal of K-Ras4B and an endosome-escape enhancing motif, with which we have proved the concept of our strategy. It displays excellent cell permeability and can form peptide-enriched domains after association with the membrane ld regions, resulting in isoform-specific blockage of the tethering of K-Ras4B on the PM. Accordingly, Memrasin impairs downstream Ras signaling activity, efficiently suppressed the proliferation of a series of human lung cancer cells at a concentration of a few micromolar. Moreover, we further confirmed that the inhibition of Memrasin to cell growth was dependent on Ras signaling pathway. Thus, these results demonstrate the feasibility of our strategy to abrogate K-Ras4B signaling activity by direct inhibition of K-Ras4B-PM interaction. Certainly, Memrasin still has the in vivo off-target risk of interacting with a more precise region within ld microdomains distinct from the localization of K-Ras4B or even binding to non-ld microdomains at high concentrations, which could be reduced by further structural optimization of Memrasin or utilization of biologics delivery systems for future application. Memrasin provides a useful tool for exploring the biological function of K-Ras4B on or off the PM and a potential starting point for further development into anti-Ras therapeutics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from National Key R&D Program of China (2018YFA0507600) and the National Natural Science Foundation of China (91753122, 21672125) to Y. X. C. and by the Deutsche Forschungsgemeinschaft, DFG (grant No. SPP 1623), European Research Council, ERC (ChemBioAP) and Knut and Alice Wallenberg Foundation to Y. W. W.Notes and references
- I. A. Prior, P. D. Lewis and C. Mattos, Cancer Res., 2012, 72, 2457–2467 CrossRef CAS PubMed.
- Y. Pylayeva-Gupta, E. Grabocka and D. Bar-Sagi, Nat. Rev. Cancer, 2011, 11, 761–774 CrossRef CAS.
- D. K. Simanshu, D. V. Nissley and F. McCormick, Cell, 2017, 170, 17–33 CrossRef CAS.
- A. Wittinghofer and H. Waldmann, Angew. Chem., Int. Ed., 2000, 39, 4192–4214 CrossRef CAS.
- S. Lu, H. Jang, S. Gu, J. Zhang and R. Nussinov, Chem. Soc. Rev., 2016, 45, 4929–4952 RSC.
- J. M. L. Ostrem and K. M. Shokat, Nat. Rev. Drug Discovery, 2016, 15, 771–785 CrossRef CAS.
- P. M. Cromm, J. Spiegel, T. N. Grossmann and H. Waldmann, Angew. Chem., Int. Ed., 2015, 54, 13516–13537 CrossRef CAS.
- B. Sautier, C. F. Nising and L. Wortmann, Angew. Chem., Int. Ed., 2016, 55, 15982–15988 CrossRef CAS.
- B. Papke and C. J. Der, Science, 2017, 355, 1158–1163 CrossRef CAS.
- A. Patgiri, K. K. Yadav, P. S. Arora and D. Bar-Sagi, Nat. Chem. Biol., 2011, 7, 585–587 CrossRef CAS PubMed.
- I. C. Rosnizeck, M. Spoerner, T. Harsch, S. Kreitner, D. Filchtinski, C. Herrmann, D. Engel, B. König and H. R. Kalbitzer, Angew. Chem., Int. Ed., 2012, 51, 10647–10651 CrossRef CAS PubMed.
- J. M. Ostrem, U. Peters, M. L. Sos, J. A. Wells and K. M. Shokat, Nature, 2013, 503, 548–551 CrossRef CAS.
- E. S. Leshchiner, A. Parkhitko, G. H. Bird, J. Luccarelli, J. A. Bellairs, S. Escudero, K. Opoku-Nsiah, M. Godes, N. Perrimon and L. D. Walensky, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1761–1766 CrossRef CAS.
- M. R. Janes, J. Zhang, L.-S. Li, R. Hansen, U. Peters, X. Guo, Y. Chen, A. Babbar, S. J. Firdaus, L. Darjania, J. Feng, J. H. Chen, S. Li, S. Li, Y. O. Long, C. Thach, Y. Liu, A. Zarieh, T. Ely, J. M. Kucharski, L. V. Kessler, T. Wu, K. Yu, Y. Wang, Y. Yao, X. Deng, P. P. Zarrinkar, D. Brehmer, D. Dhanak, M. V. Lorenzi, D. Hu-Lowe, M. P. Patricelli, P. Ren and Y. Liu, Cell, 2018, 172, 578–589 CrossRef CAS.
- R. C. Hillig, B. Sautier, J. Schroeder, D. Moosmayer, A. Hilpmann, C. M. Stegmann, N. D. Werbeck, H. Briem, U. Boemer and J. Weiske, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2551–2560 CrossRef CAS.
- A. Cruz-Migoni, P. Canning, C. E. Quevedo, C. J. Bataille, N. Bery, A. Miller, A. J. Russell, S. E. Phillips, S. B. Carr and T. H. Rabbitts, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 2545–2550 CrossRef CAS.
- P. Upadhyaya, Z. Qian, N. G. Selner, S. R. Clippinger, Z. Wu, R. Briesewitz and D. Pei, Angew. Chem., Int. Ed., 2015, 54, 7602–7606 CrossRef CAS.
- M. E. Welsch, A. Kaplan, J. M. Chambers, M. E. Stokes, P. H. Bos, A. Zask, Y. Zhang, M. Sanchez-Martin, M. A. Badgley, C. S. Huang, T. H. Tran, H. Akkiraju, L. M. Brown, R. Nandakumar, S. Cremers, W. S. Yang, L. Tong, K. P. Olive, A. Ferrando and B. R. Stockwell, Cell, 2017, 168, 878–889 CrossRef CAS.
- C. E. Quevedo, A. Cruz-Migoni, N. Bery, A. Miller, T. Tanaka, D. Petch, C. J. R. Bataille, L. Y. W. Lee, P. S. Fallon, H. Tulmin, M. T. Ehebauer, N. Fernandez-Fuentes, A. J. Russell, S. B. Carr, S. E. V. Phillips and T. H. Rabbitts, Nat. Commun., 2018, 9, 3169 CrossRef.
- D. Kessler, M. Gmachl, A. Mantoulidis, L. J. Martin, A. Zoephel, M. Mayer, A. Gollner, D. Covini, S. Fischer, T. Gerstberger, T. Gmaschitz, C. Goodwin, P. Greb, D. Häring, W. Hela, J. Hoffmann, J. Karolyi-Oezguer, P. Knesl, S. Kornigg, M. Koegl, R. Kousek, L. Lamarre, F. Moser, S. Munico-Martinez, C. Peinsipp, J. Phan, J. Rinnenthal, J. Sai, C. Salamon, Y. Scherbantin, K. Schipany, R. Schnitzer, A. Schrenk, B. Sharps, G. Siszler, Q. Sun, A. Waterson, B. Wolkerstorfer, M. Zeeb, M. Pearson, S. W. Fesik and D. B. McConnell, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15823–15829 CrossRef CAS PubMed.
- G. Zimmermann, B. Papke, S. Ismail, N. Vartak, A. Chandra, M. Hoffmann, S. A. Hahn, G. Triola, A. Wittinghofer, P. I. H. Bastiaens and H. Waldmann, Nature, 2013, 497, 638–642 CrossRef CAS PubMed.
- C. J. Novotny, G. L. Hamilton, F. McCormick and K. M. Shokat, ACS Chem. Biol., 2017, 12, 1956–1962 CrossRef CAS.
- I. M. Ahearn, K. Haigis, D. Bar-Sagi and M. R. Philips, Nat. Rev. Mol. Cell Biol., 2011, 13, 39–51 CrossRef.
- Y. Zhou and J. F. Hancock, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 841–849 CrossRef CAS.
- Y.-X. Chen, S. Koch, K. Uhlenbrock, K. Weise, D. Das, L. Gremer, L. Brunsveld, A. Wittinghofer, R. Winter, G. Triola and H. Waldmann, Angew. Chem., Int. Ed., 2010, 49, 6090–6095 CrossRef CAS PubMed.
- K. Weise, S. Kapoor, C. Denter, J. Nikolaus, N. Opitz, S. Koch, G. Triola, A. Herrmann, H. Waldmann and R. Winter, J. Am. Chem. Soc., 2011, 133, 880–887 CrossRef CAS PubMed.
- Y. Zhou, P. Prakash, H. Liang, K.-J. Cho, A. A. Gorfe and J. F. Hancock, Cell, 2017, 168, 239–251 CrossRef CAS PubMed.
- A. D. Cox, C. J. Der and M. R. Philips, Clin. Cancer Res., 2015, 21, 1819–1827 CrossRef CAS PubMed.
- K.-j. Cho, D. van der Hoeven, Y. Zhou, M. Maekawa, X. Ma, W. Chen, G. D. Fairn and J. F. Hancock, Mol. Cell. Biol., 2016, 36, 363–374 CrossRef CAS.
- D. van der Hoeven, K.-j. Cho, X. Ma, S. Chigurupati, R. G. Parton and J. F. Hancock, Mol. Cell. Biol., 2013, 33, 237–251 CrossRef CAS.
- K. Segers, O. Sperandio, M. Sack, R. Fischer, M. A. Miteva, J. Rosing, G. A. Nicolaes and B. O. Villoutreix, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12697–12702 CrossRef CAS.
- H. Yin and A. D. Flynn, Annu. Rev. Biomed. Eng., 2016, 18, 51–76 CrossRef CAS.
- S.-Y. Zhang, B. Sperlich, F.-Y. Li, S. Al-Ayoubi, H.-X. Chen, Y.-F. Zhao, Y.-M. Li, K. Weise, R. Winter and Y.-X. Chen, ACS Chem. Biol., 2017, 12, 1703–1710 CrossRef CAS.
- J. W. Wollack, N. A. Zeliadt, D. G. Mullen, G. Amundson, S. Geier, S. Falkum, E. V. Wattenberg, G. Barany and M. D. Distefano, J. Am. Chem. Soc., 2009, 131, 7293–7303 CrossRef CAS.
- M. Li, Y. Tao, Y. Shu, J. R. LaRochelle, A. Steinauer, D. Thompson, A. Schepartz, Z.-Y. Chen and D. R. Liu, J. Am. Chem. Soc., 2015, 137, 14084–14093 CrossRef CAS.
- M.-A. Sani and F. Separovic, Acc. Chem. Res., 2016, 49, 1130–1138 CrossRef CAS.
- M. Shahmiri, M. Enciso and A. Mechler, Sci. Rep., 2015, 5, 16378 CrossRef CAS.
- V. Diaz-Rodriguez, D. G. Mullen, E. Ganusova, J. M. Becker and M. D. Distefano, Org. Lett., 2012, 14, 5648–5651 CrossRef CAS.
- L. Janosi, Z. Li, J. F. Hancock and A. A. Gorfe, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 8097–8102 CrossRef CAS.
- M. Schmick, N. Vartak, B. Papke, M. Kovacevic, D. C. Truxius, L. Rossmannek and P. I. Bastiaens, Cell, 2014, 157, 459–471 CrossRef CAS.
- I. A. Prior, A. Harding, J. Yan, J. Sluimer, R. G. Parton and J. F. Hancock, Nat. Cell Biol., 2001, 3, 368–375 CrossRef CAS.
- G. Santos, M. Díaz and N. V. Torres, Front. Physiol., 2016, 7, 90 Search PubMed.
- R. C. Franson, L. K. Harris, S. S. Ghosh and M. D. Rosenthal, Biochim. Biophys. Acta, Mol. Cell Res., 1992, 1136, 169–174 CrossRef CAS.
- M. Sankarshanan, Z. Ma, T. Iype and U. Lorenz, J. Immunol., 2007, 179, 483–490 CrossRef CAS.
- K. Simons and J. L. Sampaio, Cold Spring Harbor Perspect. Biol., 2011, 3, a004697 Search PubMed.
- S. N. Ahmed, D. A. Brown and E. London, Biochemistry, 1997, 36, 10944–10953 CrossRef CAS.
- D. Brown and E. London, J. Membr. Biol., 1998, 164, 103–114 CrossRef CAS.
- J. K. Zehmer and J. R. Hazel, Biochim. Biophys. Acta, Biomembr., 2004, 1664, 108–116 CrossRef CAS.
- K. Klinkert and A. Echard, Traffic, 2016, 17, 1063–1077 CrossRef CAS.
- S. Voss, D. M. Krüger, O. Koch and Y.-W. Wu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14348–14353 CrossRef CAS.
- A. Singh, P. Greninger, D. Rhodes, L. Koopman, S. Violette, N. Bardeesy and J. Settleman, Cancer Cell, 2009, 15, 489–500 CrossRef CAS.
- M. J. Robinson, S. A. Stippec, E. Goldsmith, M. A. White and M. H. Cobb, Curr. Biol., 1998, 8, 1141–1152 CrossRef CAS.
- A. D. Kohn, F. Takeuchi and R. A. Roth, J. Biol. Chem., 1996, 271, 21920–21926 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc04726c |
| This journal is © The Royal Society of Chemistry 2020 |