
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZirconium metal–organic frameworks incorporating tetrathiafulvalene linkers: robust and redox-active matrices for in situ confinement of metal nanoparticles†
Jian
Su‡
 a,
Shuai
Yuan‡
b,
Tao
Wang
a,
Christina Tori
Lollar
b,
Jing-Lin
Zuo
*a,
Jiangwei
Zhang
*c and
Hong-Cai
Zhou
*b
a,
Shuai
Yuan‡
b,
Tao
Wang
a,
Christina Tori
Lollar
b,
Jing-Lin
Zuo
*a,
Jiangwei
Zhang
*c and
Hong-Cai
Zhou
*b
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210093, P. R. China. E-mail: zuojl@nju.edu.cn
bDepartment of Chemistry, College Station, TX 77843, USA. E-mail: zhou@chem.tamu.edu
cState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences (CAS), Dalian 116023, China. E-mail: jwzhang@dicp.ac.cn
First published on 9th January 2020
Abstract
Redox-active metal–organic frameworks (MOFs), with highly ordered porous structures and redox tunability, have attracted research interest in the fields of catalysis, energy storage, and electrochemical sensing. However, the chemical lability has limited the application scope of many redox-active MOFs. Herein, we selected stable Zr6 inorganic nodes and redox-active tetrathiafulvalene (TTF)-based linkers to construct two robust, redox-active MOFs, namely compounds 1 ([Zr6(TTFTB)2O8(OH2)8]) and 2 ([Zr6(Me-TTFTB)1.5O4(OH)4(C6H5COO)6]) (TTFTB = tetrathiafulvalene tetrabenzoate; Me-TTFTB = tetrathiafulvalene tetramethylbenzoate). The structure and topology of the MOFs were controlled by tuning the linker conformation through steric effects, resulting in a variety of pore structures from microporous channels (compound 1) to hierarchically micro/mesoporous cages (compound 2). Compound 2 shows high porosity with a BET surface area of 1932 m2 g−1 and strong chemical stability in aqueous solutions with pH ranging from 1 to 12. Furthermore, the reductive TTF moieties allow for in situ generation and stabilization of ultra-small noble metal (Ag, Pd, and Au) nanoparticles by incubating MOFs in the respective metal salt solution. Single crystal structures, TEM images, and pore size distribution data from N2 adsorption measurements indicated that the metal nanoparticles were mostly placed in the small cubic cavities of hierarchically porous compound 2, leaving the large cages open for substrate diffusion. As a proof of concept, Pd NPs@compound 2 was utilized as a heterogeneous catalyst for aerobic oxidation of alcohols, showing noteworthy activity and recyclability.
Introduction
Metal–organic frameworks (MOFs) are crystalline porous materials constructed from metal ions/clusters and organic linkers showing high surface areas and tunable pore environments. They have attracted considerable research interest in the fields of gas storage,1,2 separations,2,3 energy storage,4 catalysis5 and chemical sensing.6 While many MOFs remain inert under application conditions, recent studies have highlighted their redox activity potential arising from metal ions,7–15 organic linkers,16–25 or the guest molecules.18,26 Redox-active MOFs represent a unique class of switchable solid materials in which the different redox states with distinct physical properties can be achieved by redox modulation.27 This will expand the application of MOFs to exciting new areas including gas adsorption,28,29 microporous conducting materials,7,16,17 catalysis,30 magnetism,31–33 energy storage,34–36 and electrochemical sensing.37 For example, Long and coworkers reported a redox-active MOF, Fe2(BDP)3 (BDP = 1,4-benzenedipyrazolate), displaying a nearly 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold enhancement in conductivity upon reduction by potassium naphthalenide.7 Yoshikawa, Awaga and coworkers studied the electrochemistry of Cu(2,7-anthraquinone dicarboxylate) in lithium batteries and revealed the coexistence of metal and linker redox activities within the MOF.38 Studies on redox-active MOFs not only provide new insights into charge transfer mechanisms but also highlight potential applicability in electrochemical devices.
000-fold enhancement in conductivity upon reduction by potassium naphthalenide.7 Yoshikawa, Awaga and coworkers studied the electrochemistry of Cu(2,7-anthraquinone dicarboxylate) in lithium batteries and revealed the coexistence of metal and linker redox activities within the MOF.38 Studies on redox-active MOFs not only provide new insights into charge transfer mechanisms but also highlight potential applicability in electrochemical devices.
Tetrathiafulvalene (TTF) and its derivatives have been widely investigated as important electron donor components for photo-thermal conversion,39 supramolecular switching,40 solid catalysis,30 magnetic materials,33 and conductive materials.41–45 TTF motif has also been adopted as a linker fragment in the synthesis of redox-active MOFs with intriguing conductivity16,17,40,46–49 and redox switchable adsorption properties.28,29 The highly electron donating nature of the TTF moiety makes TTF-based MOFs highly reductive. Indeed, the redox potential of TTF is lower than most noble metal cations including Ag+, Au3+, Pt2+, Pd2+, Ru3+, Ir3+, etc. Therefore, we propose that TTF-based MOFs can act as suitable matrices to generate metal nanoparticles (MNPs) in situ from metal cation solutions and stabilize them from aggregation by pore confinement.
Usually, MNPs are immobilized into MOFs following a two-step process involving the intrusion of metal cation precursors followed by treatment with reducing reagents (such as NaBH4 or H2) to form MNPs.50–53 Compared with redox-inert MOFs, redox-active TTF-based MOFs allows for in situ reduction of metal cation precursors by the framework. This will favor the formation of well-dispersed, ultra-small metal nanoparticles by avoid inhomogeneity of reducing reagents. In fact, the idea of generating nanoparticles in redox-active MOFs has been demonstrated by Suh and coworkers using a redox-active macrocyclic nickel(II)-based MOF or organic redox centers MOF (triphenylamine-based linker) as a matrix to produce and stabilize MNPs without external reducing agent.8,10,14,15,54 Despite these initial works, the application of MNPs@MOF materials in catalysis are often hindered by their poor chemical stability. To address these problems, we selected stable Zr6 inorganic nodes and TTF-based linkers to construct robust and redox-active MOFs. Two TTF-based linkers with the same connectivity but different conformations were selected, giving rise to two MOFs with different pore architectures (compounds 1 and 2). Taking advantage of the reductive TTF moieties, ultra-small noble metal (Ag, Pd, and Au) nanoparticles were generated in situ and stabilized in the cavities of compounds 1 and 2 by incubating the MOFs in solutions of the respective metal salts. With highly accessible metal nanoparticles in the hierarchically porous framework, Pd NPs@2 was utilized as a heterogeneous catalyst for aerobic oxidation of alcohols.
Results and discussion
Controlling pore architecture by steric tuning of linkers
Current Zr-tetracarboxylate-based MOFs in the literature provide a blueprint for the design of our MOF system.55 Topologically, TTF-based linkers, such as tetrathiafulvalene tetrabenzoate (TTFTB),16,17,29,30,46,47,49 can be simplified into 4-connected square planar nodes, whereas Zr6 clusters have variable connection numbers and can act as 12-connected octahedral nodes, 8-connected cubic nodes, 6-connected hexagonal nodes, etc. Theoretically, the combination of Zr6 clusters with tetracarboxylate linkers will give rise to 4,8-connected scu, 4,8-connected csq, and 4,6-connected she topology, depending on the connectivity of Zr6 clusters and the conformation of tetracarboxylate linkers. Previous studies have demonstrated that the framework topology can be controlled through conformational tuning of the tetracarboxylate linkers by introducing bulky functional groups.56 For TTF-based linkers, the substituents on the 3-position of the phenyl rings will affect the dihedral angle between the TTF and the phenyl carboxylate plane. A smaller dihedral angle favors the scu network whereas a larger angle promotes the formation of a she network. Based on the known TTFTB, a new linker with methyl-substituents on the 3-position of the phenyl rings was designed, namely Me-TTFTB (Fig. 1a and S1†). As expected, the reaction of Zr4+ with TTFTB and Me-TTFTB give rise to two MOFs exhibiting scu and she topology, compounds 1 and 2, respectively.Compound 1 was obtained as needle-shaped microcrystals with an average size of 3 × 1 × 0.5 μm. The structure of compound 1 was determined by Rietveld refinement against PXRD patterns (Table S1 and Fig. S2†). It crystalizes in the orthorhombic space group Cmmm, similar to the structures of previously reported NU-90157 and PCN-606.58 The Zr6 cluster in compound 1 is 8-connected (Fig. 1b), therefore the overall structure can be simplified into a 4,8-connected net with scu topology (Fig. 1c and d). It possesses uniform channels along the b-axis direction with a diameter of 13.7 Å according to the crystal structure (Fig. S3 and S4†).
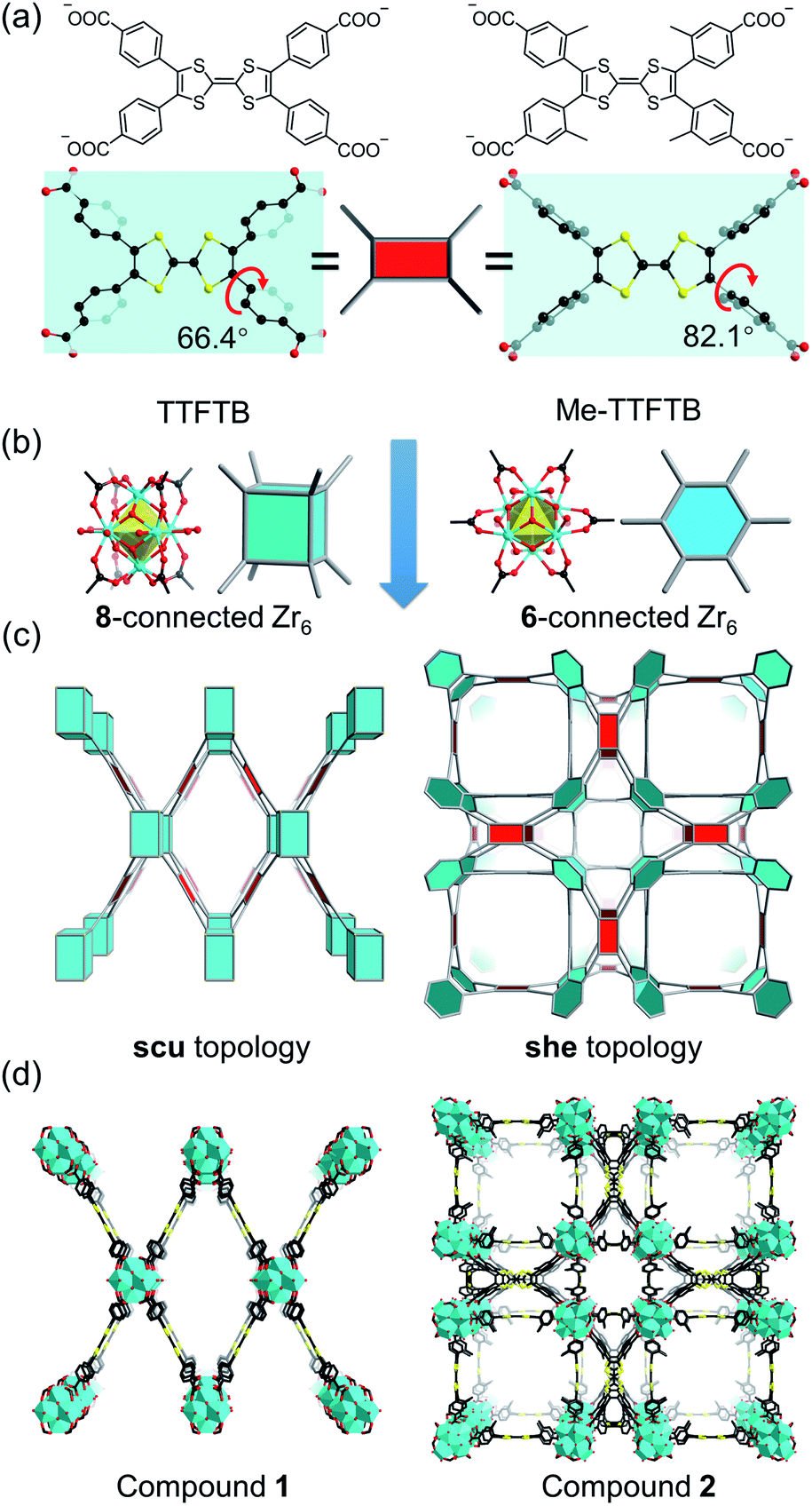 | ||
| Fig. 1 Topologically guided steric tuning strategy for MOF design (a) TTFTB and Me-TTFTB linkers with different conformations. (b) 8-connected and 6-connected Zr6 clusters. (c) Targeted topologies formed by Zr6 clusters and conformationally differing linkers. (d) Crystal structures of compounds 1 and 2. Color scheme: Zr, cyan; O, red; C, black; S, yellow. | ||
Single crystals of compound 2 were successfully obtained. Single crystal X-ray diffraction (SC-XRD) analysis reveals that compound 2 crystallizes in the cubic space group P![[m with combining macron]](https://www.rsc.org/images/entities/i_char_006d_0304.gif) 3m (Table S2†). The asymmetric unit contains one Zr4+ ion, a quarter of a Me-TTFTB linker, two thirds of an O2− ion, two thirds of an OH− ion and one terminal carboxylate anion (Fig. S5†). The TTF unit is two-fold disordered with an average central C
3m (Table S2†). The asymmetric unit contains one Zr4+ ion, a quarter of a Me-TTFTB linker, two thirds of an O2− ion, two thirds of an OH− ion and one terminal carboxylate anion (Fig. S5†). The TTF unit is two-fold disordered with an average central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond length of 1.28 (3) Å (Fig. 3a), comparable to the neutral TTF moiety.28,33,59 Each Zr6 cluster is connected to six carboxylates from Me-TTFTB, leaving the other six sites terminated by carboxylate anions (Fig. S6†). The terminal carboxylate anions capping on the Zr6 clusters cannot be precisely refined because of the disorder, but they were determined to be benzoate anion (PhCOO−) by 1H-NMR spectra of digested samples (Fig. S7†). The overall framework can be simplified into she topology if regarding the [Zr6O4(OH)4(PhCOO)6(COO)6] clusters as 6-connected hexagonal nodes and Me-TTFTB as 4-connected planar nodes (Fig. 1c). This topology is identical to PCN-224,60 a Zr-MOF based on tetratopic porphyrin-based linkers. The topological analysis can be different when TTFTB and Me-TTFTB were regarded as two 3-connected nodes (Fig. S8†). Compound 2 shows a hierarchically porous structure with a tetragonal cage and two cubic cages (Fig. 1d and S9†). The large cubic cages are closely packed forming an interconnected channel along the a, b and c-direction.
C bond length of 1.28 (3) Å (Fig. 3a), comparable to the neutral TTF moiety.28,33,59 Each Zr6 cluster is connected to six carboxylates from Me-TTFTB, leaving the other six sites terminated by carboxylate anions (Fig. S6†). The terminal carboxylate anions capping on the Zr6 clusters cannot be precisely refined because of the disorder, but they were determined to be benzoate anion (PhCOO−) by 1H-NMR spectra of digested samples (Fig. S7†). The overall framework can be simplified into she topology if regarding the [Zr6O4(OH)4(PhCOO)6(COO)6] clusters as 6-connected hexagonal nodes and Me-TTFTB as 4-connected planar nodes (Fig. 1c). This topology is identical to PCN-224,60 a Zr-MOF based on tetratopic porphyrin-based linkers. The topological analysis can be different when TTFTB and Me-TTFTB were regarded as two 3-connected nodes (Fig. S8†). Compound 2 shows a hierarchically porous structure with a tetragonal cage and two cubic cages (Fig. 1d and S9†). The large cubic cages are closely packed forming an interconnected channel along the a, b and c-direction.
The different framework structures of compounds 1 and 2 are attributed to the steric effects of the methyl groups, which force the phenyl rings to rotate off the TTF plane. Indeed, the dihedral angle between the TTF and phenyl carboxylate motif were measured to be 66.4° in compound 1 and 82.1° in compound 2 (Fig. 1a). The single crystal structure of the TTFTB linker terminated by ethoxy groups (i.e. TTFTB-OEt, Tables S3 and S4†) was obtained to show the dihedral angle between the TTF and phenyl carboxylate motif in small molecules. The angle in TTFTB-OEt crystals were measured to be 68.2°, which matches well with the structure of compound 1 (Fig. S1†). The steric effects of the methyl groups increase the dihedral angle and thus favor the formation of compound 2.
Stability and porosity
The phase purity of compounds 1 and 2 bulk samples was confirmed by matching the PXRD patterns with simulations based on structural models (Fig. 2a and d). The PXRD patterns of compounds 1 and 2 are unaltered in conventional organic solvents and aqueous solution of various pH (1–12) after 24 hours' exposure (10 mg of MOF per mL of solvent), demonstrating good chemical stability (Fig. S10–S14†). N2 adsorption isotherms also indicate that compound 2 remains porous after pH 1 and 12 treatment, although a slight decrease in total N2 uptake was observed (Fig. S12†). TGA curves of compounds 1 and 2 under N2 atmosphere show good thermal stability until 400 °C (Fig. S15†). To assess the porosity and surface area of compounds 1 and 2, the N2 sorption measurements were conducted at 77 K. Compound 1 shows a type-I isotherm with a total uptake of 393 cm3 g−1 and a BET surface area of 1353 m2 g−1, indicative of a typical microporous structure (Fig. 2b). Pore size distribution derived from the adsorption isotherms shows a uniform pore size at around 12.7 Å, matching well with the structural model (Fig. 2c). In contrast, type-IV isotherms were observed for compound 2 (Fig. 2e). The total N2 uptake and BET surface area reaches 536 cm3 g−1 and 1932 m2 g−1 for compound 2. Pore size distribution indicates a hierarchically porous structure with two types of micropores at 12.6 and 15.8 Å, and a mesopore of 20.7 Å (Fig. 2f), corresponding to the small cubic cages, tetragonal cages, and large cubic cages observed in single crystal structures.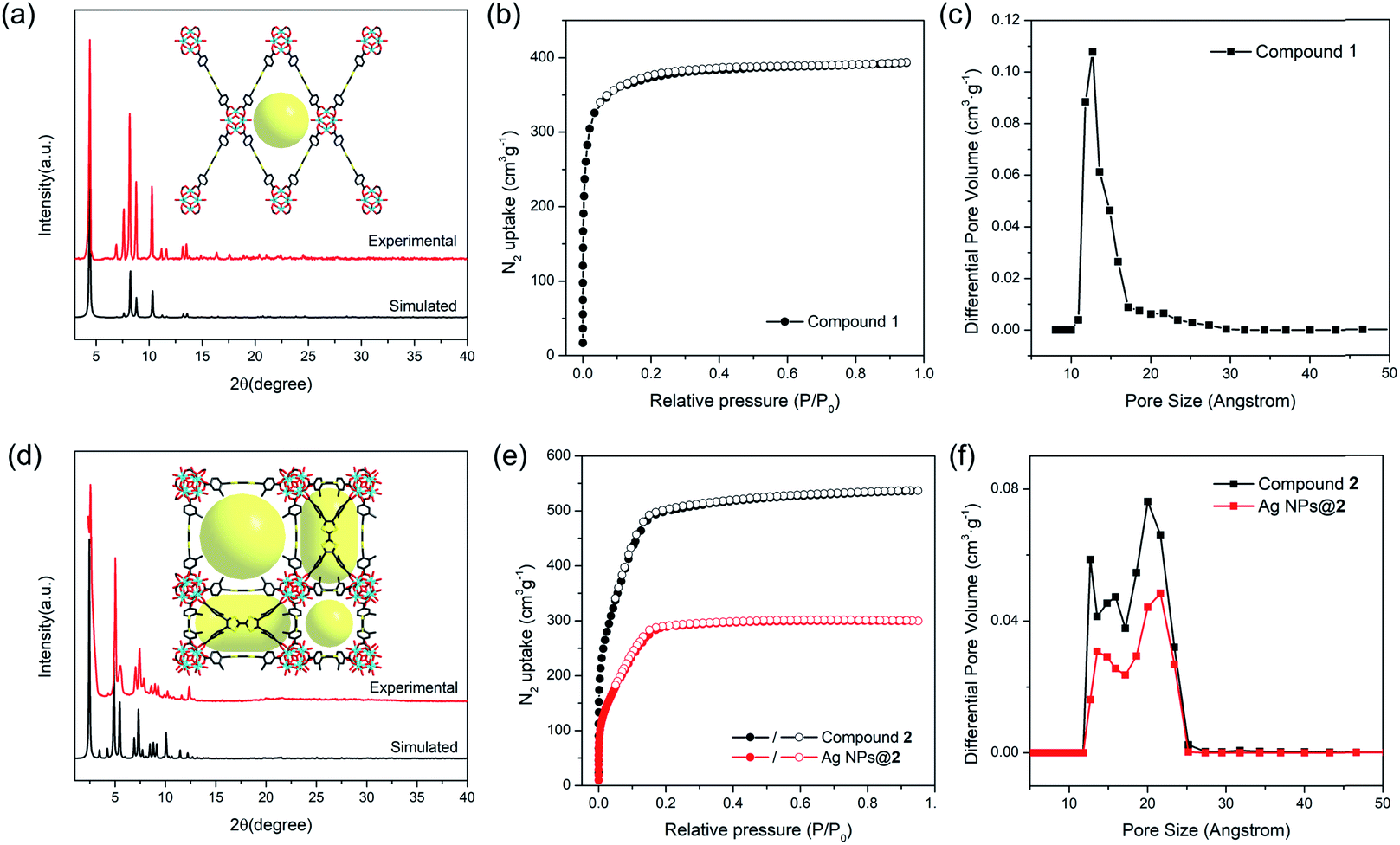 | ||
| Fig. 2 The experimental and simulated PXRD patterns for compounds 1 (a) and 2 (d) after solvent removal. The N2 adsorption–desorption isotherms of compounds 1 (b), 2 and Ag NPs@2 (e) at 77 K. Pore size distribution of compounds 1 (c), 2 and Ag NPs@2 (f) calculated from N2 adsorption isotherms using a density functional theory (DFT) model. | ||
Probing redox activity of compounds 1 and 2
Cyclic voltammetry (CV) of TTF-based linker, Me-H4TTFTB, showed the expected redox process of TTF with two one-electron oxidation waves. The redox peaks centered at E1/2 = 0.16 and 0.38 V (vs. Fc/Fc+) for Me-H4TTFTB can be assigned to the TTF/TTF˙+ and TTF˙+/TTF2+ redox couples (Fig. S16†), which is comparable to that of H4TTFTB (E1/2 = 0.07 and 0.30 V vs. Fc/Fc+).61 CV curves of compounds 1 and 2 also showed the expected redox process of TTF with two one-electron oxidation waves. The redox center was observed at E1/2 = 0.40 and 0.93 V (vs. Fc/Fc+) for compound 1 and E1/2 = 0.03 and 0.96 V (vs. Fc/Fc+) for compound 2, respectively (Fig. S17–S22†). The redox activity of compound 2 was further revealed by the I2 treatment. After incubating compound 2 in the I2 solution in cyclohexane, an obvious color change from orange to black was observed, which can be attributed to the formation of TTF˙+ and the adsorption of I3− species (Fig. S23†). In the crystal structure of oxidized compound 2 (namely I3−@2), I3− was clearly observed as a product of the oxidation reaction (Fig. 3e and S24†). The central C–C distance increased to 1.401 (8) Å (Fig. 3b), corresponding to the conversion of double bonds in neutral TTF to single bonds in the radical cationic state TTF˙+.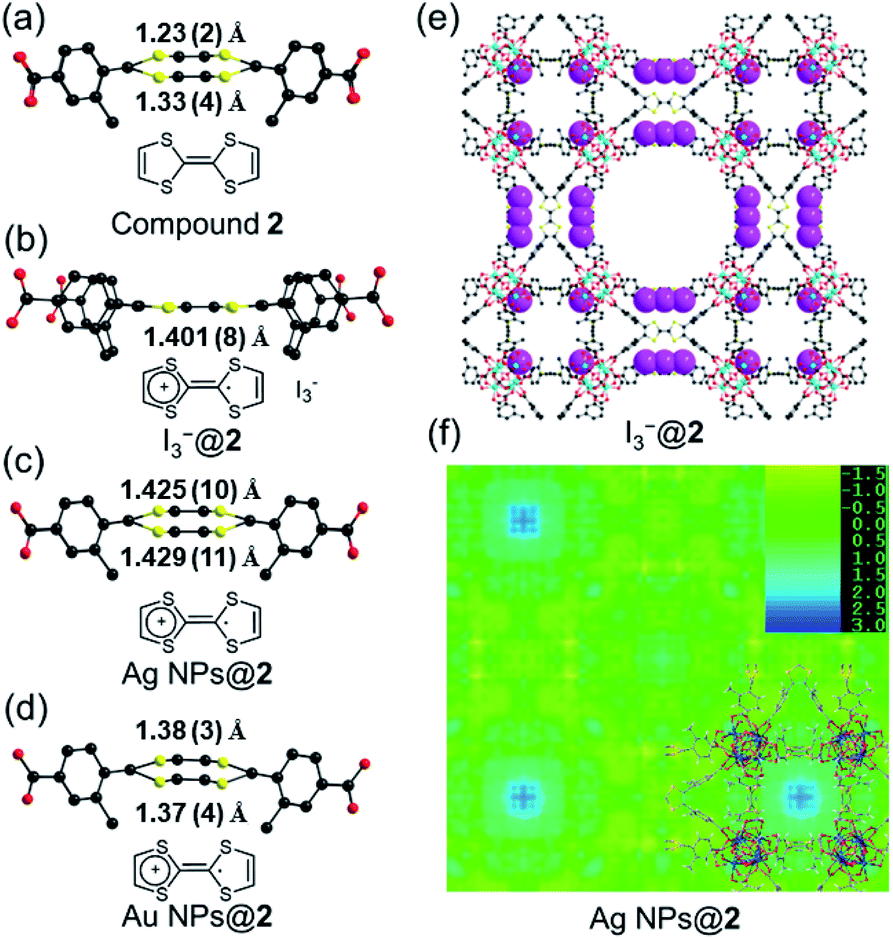 | ||
| Fig. 3 The conformation of Me-TTFTB in the single crystal structure of 2 (a), I3−@2 (b), Ag NPs@2 (c) and Au NPs@2 (d); (e) I3− locations in the single crystal structure I3−@2. (f) Electron density map for Ag NPs@2 single crystal structure showing strong residual electron density in the small cavity. | ||
The maintained crystallinity after oxidation was also confirmed by PXRD patterns of bulk I3−@2 samples (Fig. S25†). The incorporation of I3− was further confirmed by IR and TGA (Fig. S26–S27†). The coordination environment and oxidation state of Zr are not affected by oxidation, as confirmed by X-ray photoelectron spectroscopy (XPS) (Fig. S28†). Electron paramagnetic resonance (EPR) studies were performed at 110 K to confirm the generation of the TTF˙+ radical (Fig. S29†). The EPR spectra showed g = 2.002 for I3−@2, matching well with those of typical TTF˙+ cation radicals.28,62–64 Compound 1 shows redox behavior similar to compound 2 and the generation of the TTF˙+ radical upon I2 treatment was confirmed by the EPR study (Fig. S30†). The foregoing results unambiguously confirm the redox activity of the TTF motif in compounds 1 and 2.
In situ generation of metal nanoparticles
Inspired by their redox activity, we propose that the redox reaction between noble metal ions (Ag+, Pd2+, and Au3+) and TTF-moieties will generate ultra-small metal nanoparticles in MOFs. The sulfur-rich pore environment the MOF will act as a support to stabilize the in situ generated nanoparticles from aggregation through a confinement effect which was also reported by Pardo and coworkers.65 The solution of Me-H4-TTFTB linker can react with the methanol solution of Ag+, Pd2+, and Au3+, as indicated by the color change (Fig. S31†). Since the TTF moieties are well-separated within the MOF lattice, the aggregation of MNPs can be minimized.The loading of metal-nanoparticles was realized by soaking crystals of compound 2 in a solution of AgNO3, PdCl2 or KAuCl4, respectively. The resulting products, namely Ag NPs@2, Pd NPs@2, and Au NPs@2, exhibit different shades of green under an optical microscope, indicative of the generation of MNPs and TTF˙+ (Fig. 4a and S32†). High resolution transmission electron microscopy (HRTEM) images show the uniformly distributed MNPs throughout the MOF matrix. A narrow size distribution was observed with a peak at 1.6 nm for all three samples (i.e. Ag, Pd, and Au), in line with the size of the framework's small cavity (Fig. S33†). Note that the calculated average sizes of NPs might be overestimated because of nanoparticle overlap in the TEM images. The distance between lattice fringes were measured to be 0.236, 0.243, and 0.245 nm for Ag, Pd, and Au, corresponding to the spacing of (111),66 1/3(422) (face-centered cubic Pd),67 and (111)66 lattice planes (Fig. 5). The noble metal species were uniformly distributed throughout the MOF particle, as indicated by elemental mapping under a scanning electron microscope with energy-dispersive X-ray spectroscopy (SEM/EDX) (Fig. 4b and S34–S36†).
 | ||
| Fig. 4 Optical microscope images (a), and element mapping of Ag NPs@2, Pd NPs@2, and Au NPs@2 under SEM/EDX (b). | ||
 | ||
| Fig. 5 HRTEM images of 2 (a), Ag NPs@2 (b), Pd NPs@2 (c), and Au NPs@2 (d). | ||
The position of MNPs within the MOF structures were investigated by single crystal synchrotron X-ray diffraction study and N2 adsorption isotherms. The single crystal structure of Ag NPs@2 (Table S5†) shows a strong electron density at the center of the cubic cavity, which is attributed to the Ag NPs (Fig. 3f). An elongated C–C bond distance (1.427 (10) Å) was observed in TTF moieties, corresponding to the partial oxidation of TTF into TTF˙+ radicals by Ag+ (Fig. 3c). Similar behavior was also observed in Au NPs@2 with an average C–C bond distance of 1.38 (3) Å (Fig. 3d and S37†). Meanwhile, the reduced C–S distance of Ag NPs@2 (1.676 (7) Å) and Au NPs@2 (1.690 (8) Å) compared to the original compound 2 (1.773 (7) Å), also suggested the partial oxidation of the TTF (Table S7†).68 Furthermore, pore size distributions derived from N2 adsorption isotherms show a significant reduction in the amount of 12.6 Å micropores while the larger pores at 15.6 and 21.4 Å are maintained (Fig. 2f). These results imply that Ag NPs occupy the small cavities.
The crystallinity of compound 2 after metal nanoparticle (MNP) loading was confirmed by PXRD studies (Fig. S25 and S38†). The diffraction peaks of MNPs are not observed due to their small size.14,15,20 The total metal loadings of Ag, Pd, and Au in compound 2 were determined to be 3.4 wt%, 3.4 wt%, and 4.2 wt%, respectively, by inductively coupled plasma atomic emission spectroscopy (ICP-AES, Table S7†). X-ray photoelectron spectroscopy (XPS) further revealed that all the Ag within Ag NPs@2 exist as Ag0 whereas only half of the Pd and Au species exist as Pd0 and Au0 (Fig. S39–S42†). The Pd2+ and Au3+ were attributed to the metal salts trapped in the MOF cavity. Therefore, the loadings of MNPs (excluding metal cations) were calculated to be 3.4 wt%, 1.7 wt%, and 2.2 wt% for Ag, Pd, and Au. Accordingly, they are formulated as Ag00.75[Zr6(Me-TTFTB)1.5O4(OH)4(C6H5COO)6]·(NO3)0.75, Pd00.38Pd0.382+[Zr6(Me-TTFTB)1.5O4(OH)4(C6H5COO)6]·Cl1.5, and Au00.25Au0.253+[Zr6(Me-TTFTB)1.5O4(OH)4(C6H5COO)6]·Cl1.5. Based on the loading of MNPs, about half of the TTF molecules was oxidized by Ag+, Pd2+, and Au3+ within compound 2. EPR studies of MNPs@2 samples at 110 K further confirm the formation of TTF˙+ radicals (Fig. S43–S45†). Notably, the anisotropic signal of MNPs@2 as well as MNPs@1 (Fig. S30†) can be originated from the hyperfine coupling of sulfur and carbon atoms in the TTF molecule which exist in all the samples. Owing to the overlap of multiple peaks, this hyperfine coupling was not obvious in some samples such as Ag NPs@2 and Au NPs@2. To balance the charge, Ag NPs@2 includes free NO3− ions as revealed by the unsplit 1382 cm−1 peak in the IR spectra (Fig. S46†);14,15,20 Pd NPs@2 and Au NPs@2 include free Cl− ions as indicated by the SEM-EDX (Fig. S35 and S36†).
Following a similar synthetic procedure, MNPs can also be loaded into compound 1. The successful synthesis of MNPs@1 (M = Ag, Pd, and Au) was evidenced by optical images (Fig. S47†), PXRD (Fig. S48†), TGA (Fig. S49†), XPS (Fig. S50–S54†), SEM/EDX (Fig. S55–S61†), and TEM (Fig. S62–S64†). The total metal loadings were determined to be 4.6 wt%, 4.5 wt%, and 5.5 wt% for Ag, Pd, and Au by ICP-AES.
Compared to the traditional syntheses of MNPs@MOF materials, the presented redox-active MOF systems offer facile control of nanoparticle sizes. Ultra-small nanoparticles (<2 nm) with uniform sizes can be easily obtained under a wide range of synthetic conditions. Similar concept has been demonstrated by Pardo et al.69 For comparison, the size of nanoparticles synthesized by external reductants are highly sensitive to the concentration of reductant and metal ions, temperature, and reaction time. Furthermore, the hierarchically porous structure of compound 2 allows for the immobilization of MNPs within the small cavity, leaving the large mesopores and interconnected channels open for the diffusion of metal salts/substrates.
Aerobic oxidation of alcohols by Pd NPs@2
Ultra-small metal nanoparticles immobilized in a stable and hierarchically porous framework afford an ideal platform for heterogeneous catalysis. As a proof of concept, Pd NPs@2 was selected as a catalyst for aerobic alcohol oxidation (Fig. S65†), a reaction widely used in organic synthesis and chemical industry. The oxidation reaction of benzyl alcohol was carried out with Pd NPs@2 under 0.1 MPa O2 at 90 °C in cyclohexane. Pd NPs@2 shows good activity with nearly 100% conversion and more than 50% selectivity toward benzaldehyde (Table S8†). A broad substrate scope of substrates bearing electron-rich and electron-deficient functional groups were converted to corresponding aldehyde in good yields (Table S9†). Control experiments using compound 2 without Pd NPs do not show any activity, suggesting Pd NPs to be the active sites (Fig. S66, S67 and Table S10†). To confirm the heterogeneous nature of catalysis, hot filtration was carried out by removing the MOF catalysts from the reaction mixture, after which no further reaction was observed (Fig. S66†). As a heterogeneous catalyst, Pd NPs@2 can be easily recycled from the reaction system by centrifugation and reused in successive runs. Recycling experiments demonstrated that the Pd NPs@2 catalyst can be reused three times without significant loss of conversion and selectivity (Fig. S68 and Table S8†). Considering the large pore size of compound 2 and diverse metal species that can be incorporated, a wide range of catalytic reactions can be envisioned in this MOF system.Conclusions
In conclusion, we designed two stable and redox-active MOFs incorporating robust Zr6 clusters and redox-active TTF-based linkers. Among them, compound 2 shows hierarchical porosity with both microporous and mesoporous cages. Taking advantage of the stable framework and redox-active TTF-moieties, noble metal (Ag, Pd, and Au) nanoparticles can be generated in situ and stabilized in the MOF cavity by the reduction of respective metal cations. Single crystal structures, TEM images, and pore size distributions derived from N2 adsorption measurements indicate ultra-small metal nanoparticles located in the microporous cavity, leaving the larger cages open for guest diffusion. A heterogeneous catalyst for aerobic oxidation of alcohols was synthesized by incorporating Pd nanoparticles in compound 2. The high chemical stability, hierarchical porosity, and redox activity of the MOF matrices promise future applications in heterogeneous catalysis and beyond. Further research activities are undergoing to enrich the structures and compositions of the MNPs@redox-MOF systems and to extend their applications into the field of conductive porous materials and electrocatalysis.Experimental section
Synthesis of compound H4TTFTB
H4TTFTB were prepared adopting the previously reported procedure.28Synthesis of compound Me-H4TTFTB
Me-H4TTFTB were synthesized similar to H4TTFTB with methyl-4-bromo-3-methylbenzoate instead of ethyl-4-bromobenzoate. Me-H4TTFTB in red solid was obtained with a yield of 36%. 1H NMR (400 MHz, d6-DMSO) δ 13.06 (br, 4H), 7.73 (s, 4H), 7.66–7.64 (d, 4H), 7.40–7.39 (d, 4H), 2.32 (s, 12H); ESI-MS m/z calcd for C38H28O8S4 [M − H−] 739.08, found 739.07. FT-IR (KBr pellet, cm−1): 2971 w, 2532 w, 1692 vs., 16606 s, 1563 m, 1417 m, 1288 vs., 1257 s, 1190 s, 1125 s, 1048 m, 903 m, 839 m, 758 s, 559 m, 445 m.
Synthesis of compound 1: [Zr6(TTFTB)2O8(OH2)8]·2(C3H7ON)
To a solution of H4TTFTB (0.010 g, 0.015 mmol) and ZrCl4 (0.020 g, 0.086 mmol) in 3 mL DMF, benzoic acid (0.6 g, 4.91 mmol) was added. The mixture was capped and heated to 120 °C for 72 h then cooled to room temperature at a rate of 5 °C h−1. Red microcrystals (0.013 g) of 1 were obtained by filtration and washed with DMF and acetone three times. Yield 74.5% (based on H4TTFTB). Calcd for C74H62N2O34S8Zr6 (Mr = 2327.14 g mol−1): C, 38.19; H, 2.69; N, 1.20%. Found: C, 37.50; H, 2.83; N, 0.98%. FT-IR (KBr pellet, cm−1): 3667 w, 2360 m, 1662 s, 1601 s, 1558 s, 1418 vs., 1180 m, 1101 m, 1019 m, 866 m, 807 m, 768 m, 649 s, 569 m, 471 m.Synthesis of compound 2: [Zr6(Me-TTFTB)1.5O4(OH)4 (C6H5COO)6]·20(H2O)·4(C3H7ON)
To a solution of Me-H4TTFTB (0.010 g, 0.013 mmol), ZrCl4 (0.030 g, 0.129 mmol) and benzoic acid (0.6 g, 4.91 mmol) in 3 mL DMF was added 0.1 mL water. The mixture was capped and heated to 120 °C for 72 h, then cooled to room temperature at a rate of 5 °C h−1. Orange-yellow cubic crystals (0.007 g) of 2 were obtained by filtration and washed with DMF and acetone three times. Yield 25.5% (based on Me-H4TTFTB). Calcd for C111H138N4O56S6Zr6 (Mr = 3164.01 g mol−1): C, 42.14; H, 4.40; N, 1.77%. Found: C, 40.06; H, 4.10; N, 1.98%. FT-IR (KBr pellet, cm−1): 3421 w, 1653 s, 1598 s, 1544 vs., 1419 vs., 1243 m, 1028 m, 777 m, 721 s, 644 m, 460 m.Synthesis of compound I3−@1: [Zr6(TTFTB)2O8(OH2)8]·(I3)2·8(C6H12)
Iodine oxidation to produce I3−@1 was undertaken using a solid-solution diffusion technique. Crystals of 1 were soaked in 0.1 M iodine in cyclohexane solution for 12 hours at room temperature. Over time, the crystals' color deepened until becoming black. The obtained crystals were washed by cyclohexane three times. The quantities of iodine incorporated were confirmed by TGA (Fig. S39†) and elemental analysis: two I3− for I3−@1 per unit cell. Anal. Calcd. For C116H144O32S8Zr6I6 (Mr = 3615.66 g mol−1): C, 38.53; H, 4.01%. Found: C, 36.24; H, 3.35%. Selected IR data (KBr, cm−1): 3423 w, 2362 w, 1604 s, 1543 s, 1420 vs., 1285 w, 1185 m, 1144 w, 1108w, 1017 m, 865 w, 809 m, 768 w, 704 w, 654 s, 568 w, 465 m.Synthesis of compound I3−@2: [Zr6(Me-TTFTB)1.5O4(OH)4 (C6H5COO)6]·(I3)1.5·1.5(C6H12)
The iodine oxidation of I3−@2 was undertaken using a solid-solution diffusion technique. Crystals of 2 were soaked in 0.1 M iodine in cyclohexane solution for 12 hours at room temperature. The color of crystals changed from orange to black. The obtained crystals were washed by cyclohexane for three times. The quantities of iodine incorporated were confirmed by TGA (Fig. S16†) and elemental analysis: a quarter of I3− for I3−@2 per unit cell. Anal. Calcd. For C108H88O32S6Zr6I4.5 (Mr = 3208.64 g mol−1): C, 40.43; H, 2.76%. Found: C, 38.89; H, 2.31%. Selected IR data (KBr, cm−1): 3344 w, 2921 m, 1598 s, 1550 s, 1491 w, 1421 vs., 842 w, 777 m, 721 s, 651 s, 489 w, 461 m, 407 w.Synthesis of MNPs@1 and MNPs@2 (M = Ag, Pd, and Au)
Crystals of compound 1 were immersed in 0.1 mol L−1 AgNO3, PdCl2 or KAuCl4 methanol solutions for 24 h in the dark. The color of crystals changed from orange to black. The obtained products were washed by fresh methanol for three times to remove the metal salts. Ag NPs@1: Ag0[Zr6(TTFTB)2O8(OH2)8]·(NO3); Pd NPs@1: Pd00.5Pd0.52+[Zr6(TTFTB)2O8(OH2)8]·Cl2; Au NPs@1: Au00.33Au0.333+[Zr6(TTFTB)2O8(OH2)8]·Cl2. MNPs@2 were synthesized by a similar method except that compound 2 were used. Ag NPs@2: Ag00.75[Zr6(Me-TTFTB)1.5O4(OH)4(C6H5COO)6]·(NO3)0.75; Pd NPs@2: Pd00.38Pd0.382+[Zr6(Me-TTFTB)1.5O4(OH)4(C6H5COO)6]·Cl1.5; Au NPs@2: Au00.25Au0.253+[Zr6(Me-TTFTB)1.5O4(OH)4 (C6H5COO)6]·Cl1.5.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Basic Research Program of China (2018YFA0306004), the National Natural Science Foundation of China (No. 21631006, 21875099 and 21701168), and the National Science Foundation Graduate Research Fellowship under Grant No. DGE: 1252521. The gas adsorption–desorption studies of this research were supported by the Center for Gas Separations, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001015. Structural analyses were funded by the Robert A. Welch Foundation through a Welch Endowed Chair to HCZ (A-0030). The authors also acknowledge financial support from the U.S. Department of Energy Office of Fossil Energy, National Energy Technology Laboratory (DE-FE0026472). Prof. Deanna M. D'Alessandro and Dr Ryuichi Murase in School of Chemistry, the University of Sydney are acknowledged for their assistance in solid state electrochemical measurements. Prof. Zhuangzhi Shi, Dr Chendan Zhu and Ziqi Zhu in School of Chemistry, Nanjing University are acknowledged for their assistance in GC-MS measurements. We thank the staff from the BL17B beamline of the National Facility for Protein Science in Shanghai (NFPS) at the Shanghai Synchrotron Radiation Facility for assistance during data collection.Notes and references
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- W. Zhao, J. Peng, W. Wang, S. Liu, Q. Zhao and W. Huang, Coord. Chem. Rev., 2018, 377, 44–63 CrossRef CAS.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- M. L. Aubrey, B. M. Wiers, S. C. Andrews, T. Sakurai, S. E. Reyes-Lillo, S. M. Hamed, C. J. Yu, L. E. Darago, J. A. Mason, J. O. Baeg, F. Grandjean, G. J. Long, S. Seki, J. B. Neaton, P. Yang and J. R. Long, Nat. Mater., 2018, 17, 625–632 CrossRef CAS PubMed.
- H. J. Choi and M. P. Suh, J. Am. Chem. Soc., 2004, 126, 15844–15851 CrossRef CAS.
- G. Férey, F. Millange, M. Morcrette, C. Serre, M.-L. Doublet, J.-M. Grenèche and J.-M. Tarascon, Angew. Chem., 2007, 119, 3323–3327 CrossRef.
- H. R. Moon, J. H. Kim and M. P. Suh, Angew. Chem., Int. Ed., 2005, 44, 1261–1265 CrossRef CAS.
- B. Tu, Q. Pang, H. Xu, X. Li, Y. Wang, Z. Ma, L. Weng and Q. Li, J. Am. Chem. Soc., 2017, 139, 7998–8007 CrossRef CAS.
- Y. Tulchinsky, C. H. Hendon, K. A. Lomachenko, E. Borfecchia, B. C. Melot, M. R. Hudson, J. D. Tarver, M. D. Korzynski, A. W. Stubbs, J. J. Kagan, C. Lamberti, C. M. Brown and M. Dinca, J. Am. Chem. Soc., 2017, 139, 5992–5997 CrossRef CAS PubMed.
- Y. M. Litvinova, Y. M. Gayfulin, K. A. Kovalenko, D. G. Samsonenko, J. van Leusen, I. V. Korolkov, V. P. Fedin and Y. V. Mironov, Inorg. Chem., 2018, 57, 2072–2084 CrossRef CAS PubMed.
- M. P. Suh, H. R. Moon, E. Y. Lee and S. Y. Jang, J. Am. Chem. Soc., 2006, 128, 4710–4718 CrossRef CAS PubMed.
- Y. E. Cheon and M. P. Suh, Chem.–Eur. J., 2008, 14, 3961–3967 CrossRef CAS PubMed.
- S. S. Park, E. R. Hontz, L. Sun, C. H. Hendon, A. Walsh, T. Van Voorhis and M. Dinca, J. Am. Chem. Soc., 2015, 137, 1774–1777 CrossRef CAS PubMed.
- T. C. Narayan, T. Miyakai, S. Seki and M. Dinca, J. Am. Chem. Soc., 2012, 134, 12932–12935 CrossRef CAS PubMed.
- Q. Chen, J. Sun, P. Li, I. Hod, P. Z. Moghadam, Z. S. Kean, R. Q. Snurr, J. T. Hupp, O. K. Farha and J. F. Stoddart, J. Am. Chem. Soc., 2016, 138, 14242–14245 CrossRef CAS PubMed.
- R. Murase, B. F. Abrahams, D. M. D'Alessandro, C. G. Davies, T. A. Hudson, G. N. L. Jameson, B. Moubaraki, K. S. Murray, R. Robson and A. L. Sutton, Inorg. Chem., 2017, 56, 9025–9035 CrossRef CAS PubMed.
- Y. E. Cheon and M. P. Suh, Angew. Chem., Int. Ed., 2009, 48, 2899–2903 CrossRef CAS PubMed.
- A. F. Cozzolino, C. K. Brozek, R. D. Palmer, J. Yano, M. Li and M. Dinca, J. Am. Chem. Soc., 2014, 136, 3334–3337 CrossRef CAS PubMed.
- J. Y. Ha, J. Y. Koo, H. Ohtsu, Y. Yakiyama, K. Kim, D. Hashizume and M. Kawano, Angew. Chem., Int. Ed., 2018, 57, 4717–4721 CrossRef CAS PubMed.
- B. A. Johnson, A. Bhunia, H. Fei, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2018, 140, 2985–2994 CrossRef CAS PubMed.
- M. E. Ziebel, L. E. Darago and J. R. Long, J. Am. Chem. Soc., 2018, 140, 3040–3051 CrossRef CAS.
- I. Hod, W. Bury, D. M. Gardner, P. Deria, V. Roznyatovskiy, M. R. Wasielewski, O. K. Farha and J. T. Hupp, J. Phys. Chem. Lett., 2015, 6, 586–591 CrossRef CAS PubMed.
- A. Van Wyk, T. Smith, J. Park and P. Deria, J. Am. Chem. Soc., 2018, 140, 2756–2760 CrossRef CAS PubMed.
- D. M. D'Alessandro, Chem. Commun., 2016, 52, 8957–8971 RSC.
- J. Su, S. Yuan, H. Y. Wang, L. Huang, J. Y. Ge, E. Joseph, J. Qin, T. Cagin, J. L. Zuo and H. C. Zhou, Nat. Commun., 2017, 8, 2008 CrossRef PubMed.
- M. Souto, J. Romero, J. Calbo, I. J. Vitorica-Yrezabal, J. L. Zafra, J. Casado, E. Orti, A. Walsh and G. Minguez Espallargas, J. Am. Chem. Soc., 2018, 140, 10562–10569 CrossRef CAS PubMed.
- M. Souto, A. Santiago-Portillo, M. Palomino, I. J. Vitorica-Yrezabal, B. J. C. Vieira, J. C. Waerenborgh, S. Valencia, S. Navalon, F. Rey, H. Garcia and G. Minguez Espallargas, Chem. Sci., 2018, 9, 2413–2418 RSC.
- K. S. Pedersen, P. Perlepe, M. L. Aubrey, D. N. Woodruff, S. E. Reyes-Lillo, A. Reinholdt, L. Voigt, Z. Li, K. Borup, M. Rouzieres, D. Samohvalov, F. Wilhelm, A. Rogalev, J. B. Neaton, J. R. Long and R. Clerac, Nat. Chem., 2018, 10, 1056–1061 CrossRef CAS.
- F. Pointillart, B. le Guennic, O. Cador, O. Maury and L. Ouahab, Acc. Chem. Res., 2015, 48, 2834–2842 CrossRef CAS.
- H. Y. Wang, J. Y. Ge, C. Hua, C. Q. Jiao, Y. Wu, C. F. Leong, D. M. D'Alessandro, T. Liu and J. L. Zuo, Angew. Chem., Int. Ed., 2017, 56, 5465–5470 CrossRef CAS PubMed.
- H. Wang, Q.-L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52–80 CAS.
- Y. Zhang, S. N. Riduan and J. Wang, Chem.–Eur. J., 2017, 23, 16419–16431 CrossRef CAS PubMed.
- H. B. Wu and X. W. D. Lou, Sci. Adv., 2017, 3, eaap9252 CrossRef PubMed.
- P. M. Usov, C. Fabian and D. M. D'Alessandro, Chem. Commun., 2012, 48, 3945–3947 RSC.
- Z. Zhang, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2014, 136, 16112–16115 CrossRef CAS PubMed.
- Y. Wang, W. Zhu, W. Du, X. Liu, X. Zhang, H. Dong and W. Hu, Angew. Chem., Int. Ed., 2018, 57, 3963–3967 CrossRef CAS.
- J. S. Park, J. Park, Y. J. Yang, T. T. Tran, I. S. Kim and J. L. Sessler, J. Am. Chem. Soc., 2018, 140, 7598–7604 CrossRef CAS PubMed.
- J. Ferraris, D. O. Cowan, V. Walatka and J. H. Perlstein, J. Am. Chem. Soc., 1973, 95, 948–949 CrossRef CAS.
- E. Coronado, J. R. Galan-Mascaros, C. J. Gomez-Garcia and V. Laukhin, Nature, 2000, 408, 447–449 CrossRef CAS PubMed.
- T. Enoki and A. Miyazaki, Chem. Rev., 2004, 104, 5449–5478 CrossRef CAS PubMed.
- C. Rovira, Chem. Rev., 2004, 104, 5289–5318 CrossRef CAS PubMed.
- R. P. Shibaeva and E. B. Yagubskii, Chem. Rev., 2004, 104, 5347–5378 CrossRef CAS PubMed.
- S. S. Park, A. J. Rieth, C. H. Hendon and M. Dinca, J. Am. Chem. Soc., 2018, 140, 2016–2019 CrossRef CAS.
- S. S. Park, C. H. Hendon, A. J. Fielding, A. Walsh, M. O'Keeffe and M. Dinca, J. Am. Chem. Soc., 2017, 139, 3619–3622 CrossRef CAS PubMed.
- L. Sun, M. G. Campbell and M. Dinca, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- L. S. Xie, E. V. Alexandrov, G. Skorupskii, D. M. Proserpio and M. Dinca, Chem. Sci., 2019, 10, 8558–8565 RSC.
- Q.-L. Zhu and Q. Xu, Chem, 2016, 1, 220–245 CAS.
- S. Dang, Q.-L. Zhu and Q. Xu, Nat. Rev. Mater., 2017, 3, 17075 CrossRef.
- Z. Li and Q. Xu, Acc. Chem. Res., 2017, 50, 1449–1458 CrossRef CAS.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- H. R. Moon and M. P. Suh, Eur. J. Inorg. Chem., 2010, 2010, 3795–3803 CrossRef.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H. C. Zhou, Adv. Mater., 2018, 30, e1704303 CrossRef.
- J. Pang, S. Yuan, J. Qin, C. Liu, C. Lollar, M. Wu, D. Yuan, H. C. Zhou and M. Hong, J. Am. Chem. Soc., 2017, 139, 16939–16945 CrossRef PubMed.
- C.-W. Kung, T. C. Wang, J. E. Mondloch, D. Fairen-Jimenez, D. M. Gardner, W. Bury, J. M. Klingsporn, J. C. Barnes, R. Van Duyne, J. F. Stoddart, M. R. Wasielewski, O. K. Farha and J. T. Hupp, Chem. Mater., 2013, 25, 5012–5017 CrossRef.
- J. Pang, S. Yuan, D. Du, C. Lollar, L. Zhang, M. Wu, D. Yuan, H. C. Zhou and M. Hong, Angew. Chem., Int. Ed., 2017, 56, 14622–14626 CrossRef.
- J. Su, T. H. Hu, R. Murase, H. Y. Wang, D. M. D'Alessandro, M. Kurmoo and J. L. Zuo, Inorg. Chem., 2019, 58, 3698–3706 CrossRef.
- D. Feng, W. C. Chung, Z. Wei, Z. Y. Gu, H. L. Jiang, Y. P. Chen, D. J. Darensbourg and H. C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 CrossRef PubMed.
- C. F. Leong, C.-H. Wang, C. D. Ling and D. M. D'Alessandro, Polyhedron, 2018, 154, 334–342 CrossRef.
- R. Berridge, P. J. Skabara, C. Pozo-Gonzalo, A. Kanibolotsky, J. Lohr, J. J. McDouall, E. J. McInnes, J. Wolowska, C. Winder, N. S. Sariciftci, R. W. Harrington and W. Clegg, J. Phys. Chem. B, 2006, 110, 3140–3152 CrossRef PubMed.
- E. Gomar-Nadal, L. Mugica, J. Vidal-Gancedo, J. Casado, J. T. L. Navarrete, J. Veciana, C. Rovira and D. B. Amabilino, Macromolecules, 2007, 40, 7521–7531 CrossRef.
- H.-Y. Wang, L. Cui, J.-Z. Xie, C. F. Leong, D. M. D'Alessandro and J.-L. Zuo, Coord. Chem. Rev., 2017, 345, 342–361 CrossRef.
- M. Mon, M. A. Rivero-Crespo, J. Ferrando-Soria, A. Vidal-Moya, M. Boronat, A. Leyva-Perez, A. Corma, J. C. Hernandez-Garrido, M. Lopez-Haro, J. J. Calvino, G. Ragazzon, A. Credi, D. Armentano and E. Pardo, Angew. Chem., Int. Ed., 2018, 57, 6186–6191 CrossRef.
- Y. Wei, S. Han, D. A. Walker, P. E. Fuller and B. A. Grzybowski, Angew. Chem., Int. Ed., 2012, 51, 7435–7439 CrossRef PubMed.
- X. Huang, S. Tang, J. Yang, Y. Tan and N. Zheng, J. Am. Chem. Soc., 2011, 133, 15946–15949 CrossRef PubMed.
- F. Pointillart, Y. Le Gal, S. Golhen, O. Cador and L. Ouahab, Chem. Commun., 2009, 3777–3779, 10.1039/b901932d.
- F. R. Fortea-Perez, M. Mon, J. Ferrando-Soria, M. Boronat, A. Leyva-Perez, A. Corma, J. M. Herrera, D. Osadchii, J. Gascon, D. Armentano and E. Pardo, Nat. Mater., 2017, 16, 760–766 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1850014–18500181974366. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06009j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |