
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-selective redox isomerizations of furanosides using a combined arylboronic acid/photoredox catalyst system†
Victoria
Dimakos‡
a,
Daniel
Gorelik‡
a,
Hsin Y.
Su
a,
Graham E.
Garrett
 a,
Gregory
Hughes
b,
Hiromitsu
Shibayama
a and
Mark S.
Taylor
*a
a,
Gregory
Hughes
b,
Hiromitsu
Shibayama
a and
Mark S.
Taylor
*a
aDepartment of Chemistry, University of Toronto, 80 St. George St., Toronto, ON M5S 3H6, Canada. E-mail: mtaylor@chem.utoronto.ca
bGlobal Process Chemistry, Merck Research Laboratories, P. O. Box 2000, Rahway, NJ 07065, USA
First published on 3rd January 2020
Abstract
In the presence of an arylboronic acid and a hydrogen atom transfer mediator under photoredox conditions, furanoside derivatives undergo site-selective redox isomerizations to 2-keto-3-deoxyfuranosides. Experimental evidence and computational modeling suggest that the transformation takes place by abstraction of the hydrogen atom from the 2-position of the furanoside-derived arylboronic ester, followed by C3–O bond cleavage via spin-center shift. This mechanism is reminiscent of the currently accepted pathway for the formation of 3′-ketodeoxynucleotides by ribonucleotide reductase enzymes.
Introduction
The transformation of ribonucleotides to 2′-deoxyribonucleotides, catalyzed by ribonucleotide reductase enzymes, is a crucial process in DNA biosynthesis and repair. Detailed mechanistic studies have pointed towards a pathway involving abstraction of the 3′-hydrogen atom by an enzyme-derived radical, followed by dehydration and delivery of a hydrogen atom to the 2′-position, generating a 3′-ketodeoxynucleotide (Scheme 1a).1 Reduction of the latter intermediate delivers the 2′-deoxynucleotide. Synthetic methods for conversion of furanosides to ketodeoxynucleotides are of interest, both from the perspective of fundamental reactivity development and as a way to prepare useful derivatives of furanosides, nucleosides or nucleotides.2 Whereas furanosides equipped with a leaving group at the 2- or 3-position can be transformed to 3- or 2-ketodeoxy derivatives (by semipinacol-type rearrangement3 or by elimination to an enol derivative4), protocols that capitalize on radical reactivity to achieve redox isomerization of the diol group in a furanoside substrate have not been reported. Here, we show that the combined action of organoboron catalysis and photoredox hydrogen atom transfer (HAT) catalysis enables the synthesis of 2-ketodeoxyfuranosides from the corresponding furanosides. Although the regiochemical outcome differs from that of the enzyme-catalyzed process, the proposed mechanism is reminiscent of that of the ribonucleotide reductases, with the organoboron catalyst playing key roles in accelerating HAT from the furanoside as well as the dehydration of the resulting radical intermediate. The protocol provides access to useful furanoside derivatives that are challenging to generate by other means.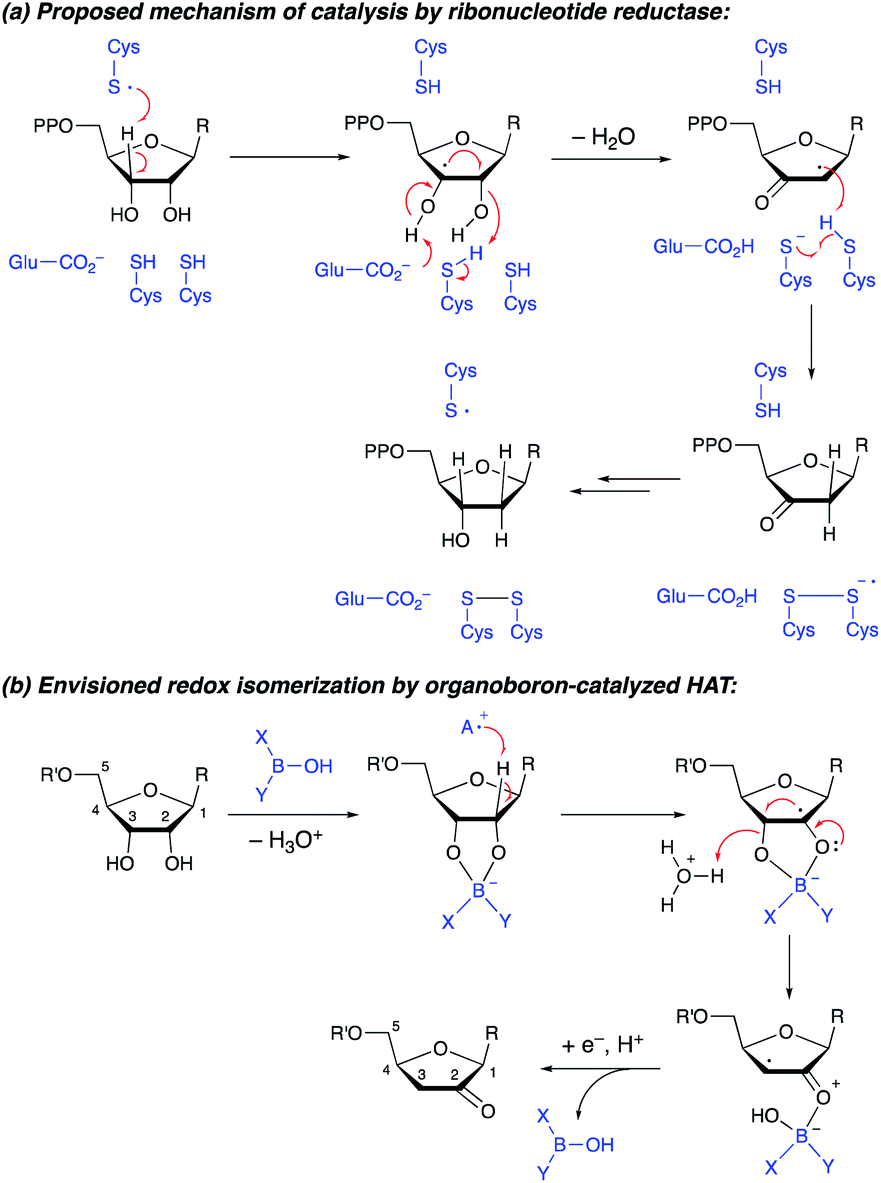 | ||
| Scheme 1 (a) Conversion of furanosides to ketodeoxyfuranosides by ribonucleotide reductase. (b) Envisioned organoboron-catalyzed redox isomerization. | ||
We recently showed that a diarylborinic acid co-catalyst could be used to achieve site-selective and stereoselective C–H alkylations of pyranoside derivatives in the presence of an iridium(III) photocatalyst and quinuclidine as an HAT mediator.5,6 Computational modeling suggested that the formation of a tetracoordinate borinic ester at a cis-1,2-diol group served both to weaken the α-C–H bonds and to accelerate HAT with the electron-deficient quinuclidine radical cation through polarity-matching and/or ion-pairing effects.7,8
We hypothesized that the ability to activate a carbohydrate derivative towards radical formation at a particular site could be exploited to achieve the transformation of furanosides to ketodeoxysugar derivatives (Scheme 1b). Considering the known activity of boronic acids as catalysts for substitution and rearrangement reactions of alcohols,9,10 we anticipated that organoboron complexation could promote C–O bond cleavage, accelerating the dehydration of the radical intermediate. Other important precedent for this proposal includes: (i) MacMillan and co-workers' discovery that benzylic alcohols can be employed as alkylating agents under photoredox conditions, via elimination of water from a radical intermediate (spin-center shift);11,12 and (ii) detailed studies of the reactivity of nucleoside-derived radicals.13
Results and discussion
Structurally diverse organoboron acids (3a–3m) were evaluated as catalysts for the redox isomerization of β-ribofuranoside 1a in the presence of Ir(III) photocatalyst 4 (ref. 14) and quinuclidine as HAT mediator (Scheme 2).8a The catalyst structure–activity relationships for this transformation were markedly different from those of our previously reported C–H alkylation reaction of carbohydrates;5 diphenylborinic acid 3a, the optimal co-catalyst for the latter reaction, was inactive, as was heterocyclic diarylborinic acid 3b,15 but the use of phenylboronic acid led to the formation of 2-keto-3-deoxyfuranoside 2a in 8% yield. Thus, diarylborinic acid catalysis is useful for promoting the coupling of the radical with an electron-deficient alkene via C–C bond formation, whereas arylboronic acid catalysis enables dehydration of a similar intermediate through C–O bond cleavage. Evaluation of a series of substituted arylboronic acids led to the identification of perfluorinated 3m as the optimal catalyst for this transformation. The structure–activity relationships for 3c–3m indicate that the best-performing catalysts possess relatively high acidities16—perhaps causing acceleration of the C–O bond-breaking step9—and that ortho-substitution tends to have a beneficial effect (e.g., catalysts 3e, 3j and 3m). The results of additional optimization experiments illustrating the effects of changing the solvent, HAT mediator and catalyst loadings, and control experiments demonstrating the importance of each element of the catalyst system, are provided in the ESI.† | ||
| Scheme 2 Evaluation of catalysts for the isomerization of ribofuranoside 1a to 2-keto-3-deoxyfuranoside 2a. Yields were determined by 1H NMR spectroscopic analysis of unpurified mixtures using a quantitative internal standard. | ||
A noteworthy aspect of the transformation of 1a to 2a is the regioselective formation of the 2-keto-3-deoxyfuranoside, in contrast to the pattern of selectivity observed for the ribonucleotide reductase-catalyzed process. The isomeric 3-keto-2-deoxyfuranoside 2a′ was observed as a minor side product in the reactions catalyzed by 3j and 3m (quinuclidine-mediated aldol dimerization of the ketodeoxysugar product was another observable side reaction – see the ESI for details†). Selectivity for the 2-keto-3-deoxy derivative, which presumably arises from hydrogen atom abstraction from the 2-position, was observed for a variety of furanoside substrates having cis-configured 2-OH and 3-OH groups, including α-lyxofuranosides, α-rhamnofuranosides, α-mannofuranosides, and β-allofuranosides (Scheme 3).17 Variation of the aglycon in β-ribofuranoside substrates was tolerated, including steroidal glycoside 1d.
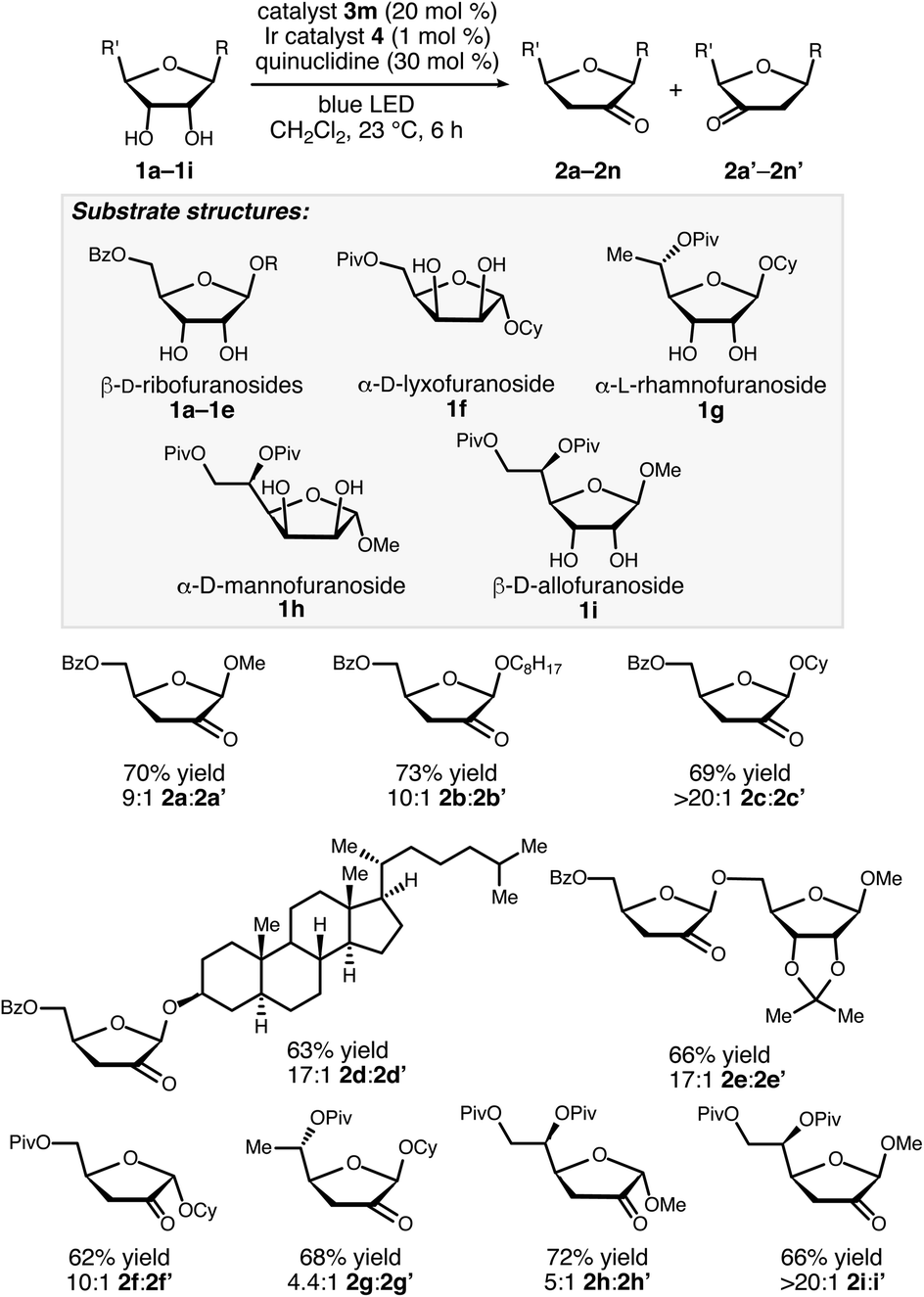 | ||
| Scheme 3 Redox isomerizations of furanosides to 2-keto-3-deoxyfuranosides. Combined yields of the 2-keto and 3-ketofuranoside products after purification by chromatography on silica gel are reported. Regioisomer ratios were determined by 1H NMR spectroscopic analysis of unpurified reaction mixtures. | ||
As shown in Scheme 4, the configuration and substitution pattern of the furanoside substrate influenced the efficiency and selectivity of the redox isomerization reaction. In contrast to the successful isomerizations of the β-ribofuranoside substrates, the reaction of α-ribofuranoside 1j was low-yielding and generated a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the 2-keto and 3-ketosugar products, indicating that a 1,2-trans-configuration is important for site-selectivity in the HAT step. We note that a product having this configuration can be accessed from the corresponding α-D-lyxofuranoside (Scheme 3, 1f → 2f). The nature of the protective group at the 5-position is also important: mixtures of isomers were obtained in low yields from 5-O-silylated ribofuranoside 1k and 5,6-O-isopropylidene mannofuranoside 1l. We propose that the inductive effects of the C5-substituents influence the relative rates of hydrogen atom abstraction from C-3 versus C-2, with the electron-withdrawing acyl protective groups deactivating the 3-position. N-Benzyluridine derivative 1m was transformed to ketodeoxy derivative 2m in a regioselective fashion, but in low yield. Loss of 3-benzyluracil, presumably via a competing spin-center shift from the C-2 radical,13b was the major side reaction. Substrates that did not undergo redox isomerization under the optimized conditions include an α-arabinofuranoside, which lacks the cis-1,2-diol group needed for binding to the boronic acid catalyst, a C-aryl β-ribofuranoside and a fucopyranoside (see the ESI†).
:1 mixture of the 2-keto and 3-ketosugar products, indicating that a 1,2-trans-configuration is important for site-selectivity in the HAT step. We note that a product having this configuration can be accessed from the corresponding α-D-lyxofuranoside (Scheme 3, 1f → 2f). The nature of the protective group at the 5-position is also important: mixtures of isomers were obtained in low yields from 5-O-silylated ribofuranoside 1k and 5,6-O-isopropylidene mannofuranoside 1l. We propose that the inductive effects of the C5-substituents influence the relative rates of hydrogen atom abstraction from C-3 versus C-2, with the electron-withdrawing acyl protective groups deactivating the 3-position. N-Benzyluridine derivative 1m was transformed to ketodeoxy derivative 2m in a regioselective fashion, but in low yield. Loss of 3-benzyluracil, presumably via a competing spin-center shift from the C-2 radical,13b was the major side reaction. Substrates that did not undergo redox isomerization under the optimized conditions include an α-arabinofuranoside, which lacks the cis-1,2-diol group needed for binding to the boronic acid catalyst, a C-aryl β-ribofuranoside and a fucopyranoside (see the ESI†).
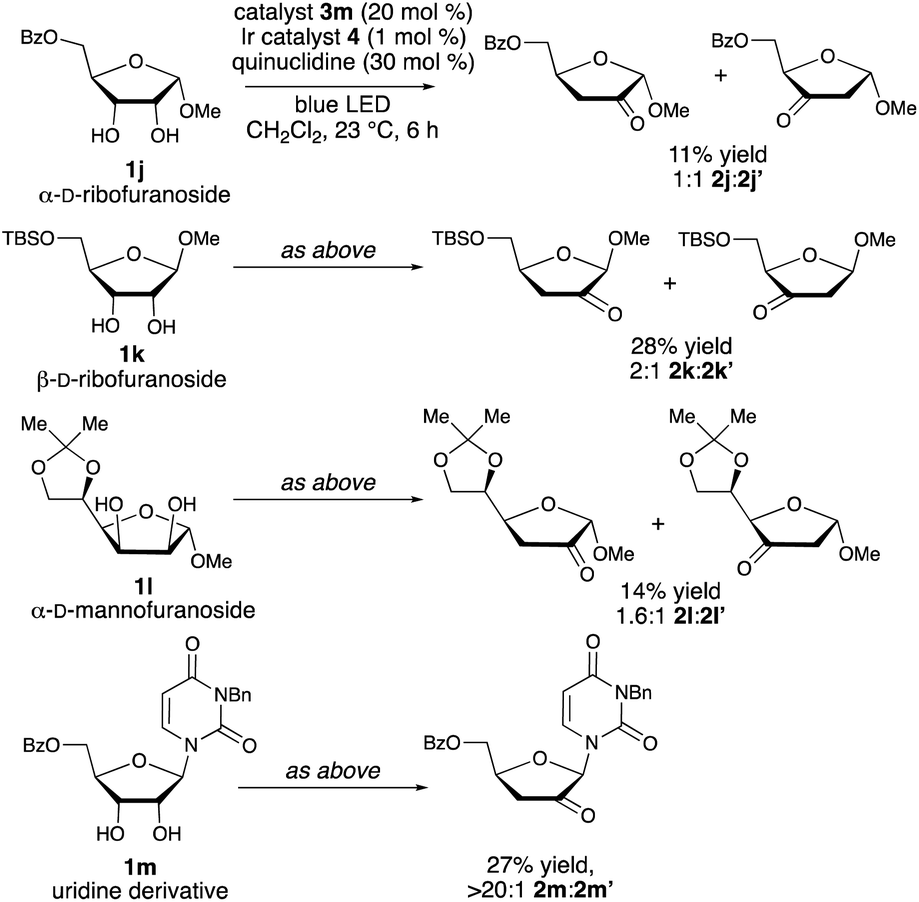 | ||
| Scheme 4 Limitations of the boronic acid/photoredox-catalyzed redox isomerization reaction. For 2k and 2l, yields were determined by 1H NMR spectroscopy. Regioisomer ratios were determined by 1H NMR spectroscopic analysis of unpurified reaction mixtures. | ||
Although protocol is presently limited in scope to a rather narrow class of substrates (furanosides having a cis-diol group at C2/C3 and a trans relationship between the anomeric substituent and the 2-OH group), it provides direct access to useful compounds that are challenging to generate by other means. Approaches based on semipinacol-type rearrangements3 require the site-selective introduction of a leaving group at the 3-position. This is challenging to achieve directly18—organotin-promoted sulfonylations of nucleosides are C2-selective, for example19—and necessitates multistep protection/deprotection sequences. Deoxygenation of furanosides has been achieved by conventional means (e.g., the Barton–McCombie protocol), but a site-selective variant20 has not been reported for such substrates. Furthermore, the transformation of the deoxy product to the corresponding ketodeoxy derivative would require an additional oxidation step. One of the most important applications of such ketodeoxyfuranosides is in the synthesis of nucleoside analogs, an important class of therapeutic agents. While additional optimization is needed to achieve the direct redox isomerization of a nucleoside derivative using the boronic acid/photoredox catalyst system (1m → 2m, Scheme 4), the alkyl furanoside-derived products can be employed in the stereoselective synthesis of such targets (Scheme 5). Thus, diastereoselective reduction of the carbonyl group of compound 2f (presumably dictated by the steric effect of the α-configured anomeric substituent),21 followed by hydrolysis of the glycoside and bis-benzoylation of the resulting hemiacetal, allowed for Lewis acid-catalyzed N-glycosylation to deliver protected 3-deoxyribonucleoside 5 in 95% yield. Product 6, having the arabino configuration, was accessed from 2avia one-pot diastereoselective reduction and ester saponification, bis-O-benzylation, glycoside hydrolysis and 1,2-cis-selective N-glycosylation under the conditions developed by Guindon and co-workers.22 We anticipate that the 2-keto-3-deoxyfuranosides will provide access to a variety of other nucleoside analogs by carbonyl addition23 and enolate functionalization reactions, and by installation of other anomeric substituents. In cases where analogs bearing a canonical nucleobase are needed, the direct transformation of the corresponding protected nucleosides (e.g., 1m → 2m, Scheme 4) may be the most efficient option, particularly if the yield can be improved through further optimization.
 | ||
| Scheme 5 Synthesis of uridine derivatives from alkyl 2-keto-3-deoxyfuranosides. | ||
A proposed co-catalytic cycle for the redox isomerization is depicted in Scheme 6. In line with previous mechanistic proposals,8 electron transfer to the photoexcited Ir complex serves to generate the quinuclidine radical cation. The latter engages the tetracoordinate quinuclidine–arylboronate complex via abstraction of the hydrogen atom at the 2-position, a kinetically favorable process due to the increased hydridic character of the α-C–H bonds.5,7 The resulting boronate-derived radical undergoes C3–O bond cleavage, releasing an α-keto radical that is subject to reduction and protonation (closing the photocatalytic cycle) or HAT (propagating a radical chain) to deliver the 2-keto-3-deoxyfuranoside product.
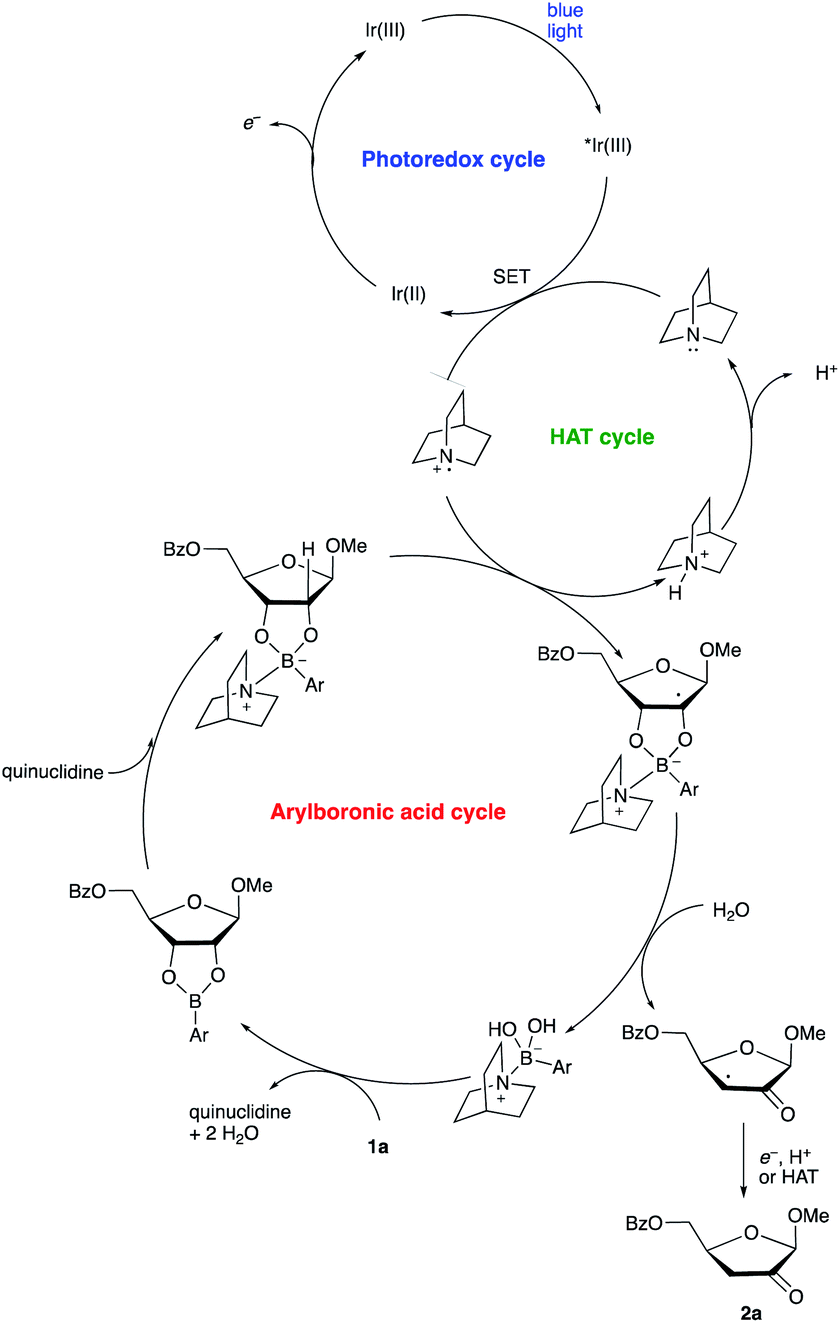 | ||
| Scheme 6 Proposed co-catalytic cycle. | ||
Results of experiments relevant to the mechanism of catalysis are summarized in Scheme 7. Upon consumption of the furanoside starting material, the formation of a precipitate identified as quinuclidinium borate was observed, implicating C–B bond cleavage as a catalyst decomposition pathway. Control experiments showed that this decomposition takes place upon irradiation of 3m in the presence of quinuclidine and complex 4. The formation of a perfluoroaryl radical via reduction of the quinuclidine–3m complex is likely involved.24 This catalyst decomposition pathway may be important to keep in mind when interpreting structure–activity relationship data and in further optimization studies.
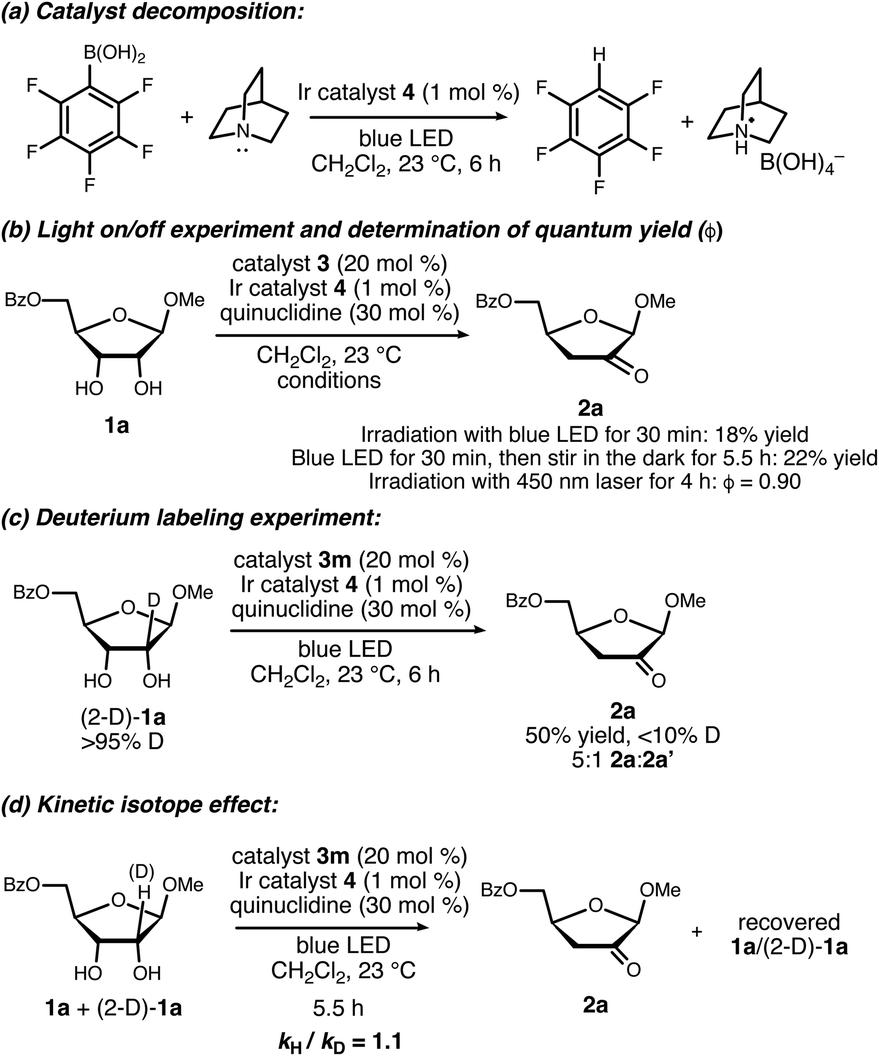 | ||
| Scheme 7 Experiments relevant to the mechanism of catalysis. | ||
Ceasing irradiation with the blue LED while the reaction of 1a was in progress largely halted the formation of the ketodeoxy product (18% yield of 2a after irradiation for 30 minutes, versus 22% yield after irradiation for 30 minutes followed by stirring in the dark for 5.5 hours). However, the quantum yield (ϕ) for the photoredox process using a 450 nm laser was determined to be 0.90.25 The fraction of quenching of the excited Ir(III) complex by quinuclidine was estimated to be 0.4–0.7, depending on the concentration of free quinuclidine versus the quinuclidine–boronate complex under the reaction conditions. Taking the quenching fraction into consideration, the value of ϕ is inconsistent with a closed photoredox cycle, pointing towards radical chain processes with average chain lengths on the order of 1.3–2.2.
When deuterated furanoside (2-D)-1a was irradiated in the presence of catalytic 3m, 4 and quinuclidine, the isomerization took place in diminished yield and site-selectivity in comparison to the non-deuterated isotopolog (50% yield, 5:1 2a:2a′). The deuterium content of the product was less than 10%; H/D exchange at the 3-position of 2a was observed under the reaction conditions (see the ESI†). An intermolecular competition experiment between 1a and (2-D)-1a gave an apparent KIE of 1.1 based on the deuterium content of the recovered starting material. Although the observation of a primary kinetic isotope effect in this type of experiment would not imply that the C–H cleavage step is turnover-limiting, its absence does rule out this possibility.26
Computational modeling with density functional theory (B97-D3/Def2-TZVP, implemented with Gaussian 16)27–29 was used to assess the feasibility of the organoboron-mediated steps of the proposed catalytic cycle. To reduce the number of basis functions, trimethylamine was used as a surrogate for quinuclidine in the tetracoordinate organoboron adduct. Gas-phase geometry optimizations and frequency calculations were conducted, enabling identification of intermediates and transition states corresponding to the proposed pathway, including the key spin-center shift step (Scheme 8). Intermolecular interactions involving the proposed radical cation intermediates were observed; for example, the formation of a pre-reactive complex from the quinuclidinium radical cation and the tetracoordinate boronate was calculated to be exergonic by 8.2 kcal mol−1. Whether such interactions are relevant to the mechanism of the solution-phase process (rather than in the gas phase) is not clear at this stage. In any case, the free energy profile, which implies that the proton transfer and spin-center shift steps are turnover-limiting, appears to be qualitatively consistent with the low intermolecular competition deuterium kinetic isotope effect of 1.1 at the 2-position (Scheme 7).
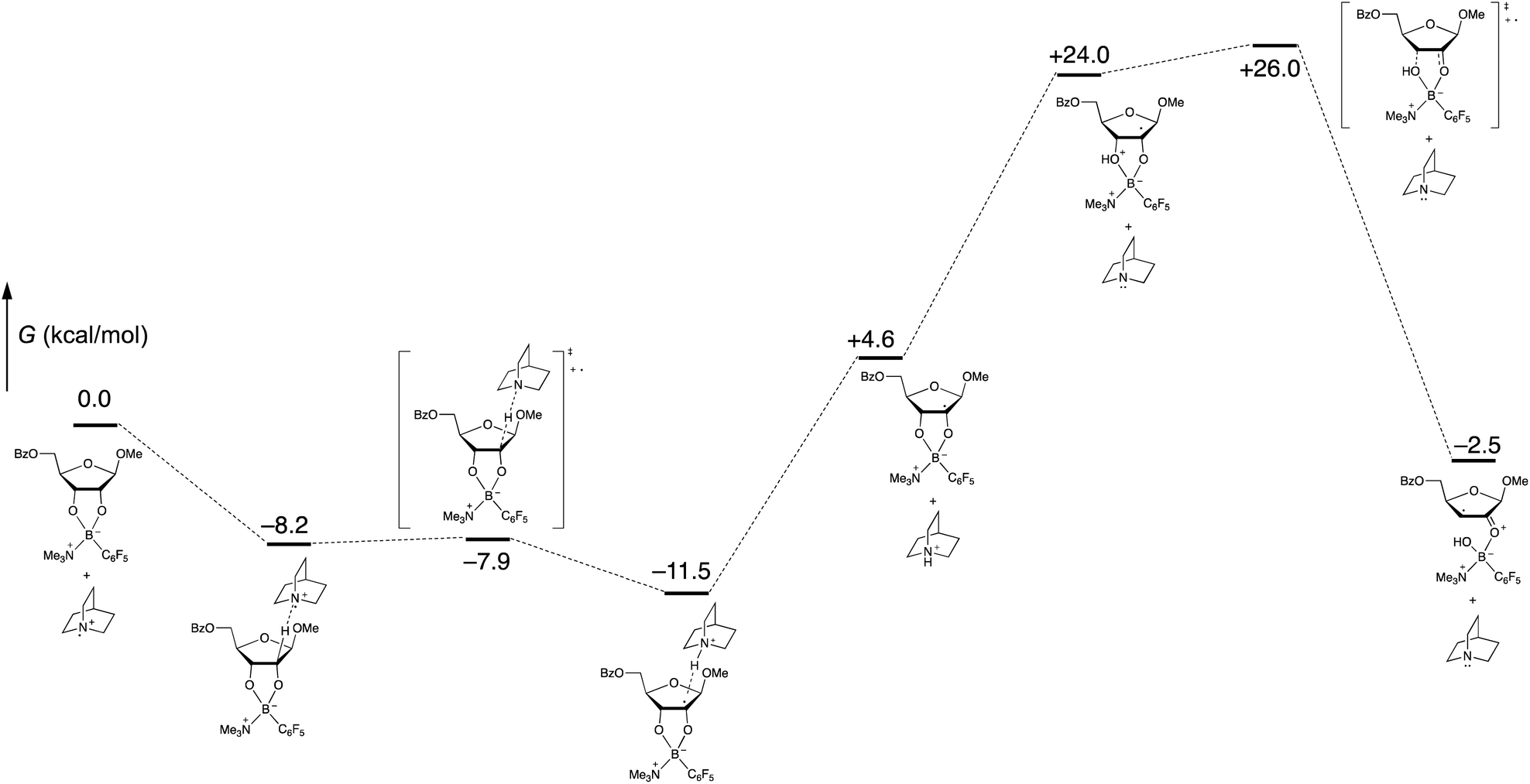 | ||
| Scheme 8 Calculated free energies of proposed organoboron intermediates and transition states (B97-D3/Def2-TZVP, gas phase). | ||
To probe the origin of site-selectivity, the transition state energies for HAT from C-2 and C-3 of the tetracoordinate boronate were compared (Fig. 1). In the gas phase, HAT from C-3 (leading to the minor isomer 2a′) was calculated to be kinetically preferred over C-2 by more than 3 kcal mol−1. However, the use of the SMD model for CH2Cl2 gave a difference in free energies of activation (ΔΔG‡) of 0.3 kcal mol−1 favoring HAT from the 2-position, in qualitative agreement with the experimentally observed result. The corresponding ΔΔG‡ value in acetonitrile was calculated to be 0.9 kcal mol−1; we note that a marginally higher selectivity—12:1 2a:2a′ in acetonitrile versus 9:1 in dichloromethane—was obtained experimentally, although the yield of the redox isomerization was low in the former solvent (see the ESI†). The results suggest that the transition states for HAT from C-2 and C-3 show distinct charge distributions, with moderately polar solvents playing a role in favoring the pathway that leads to the 2-keto-3-deoxyfuranoside.
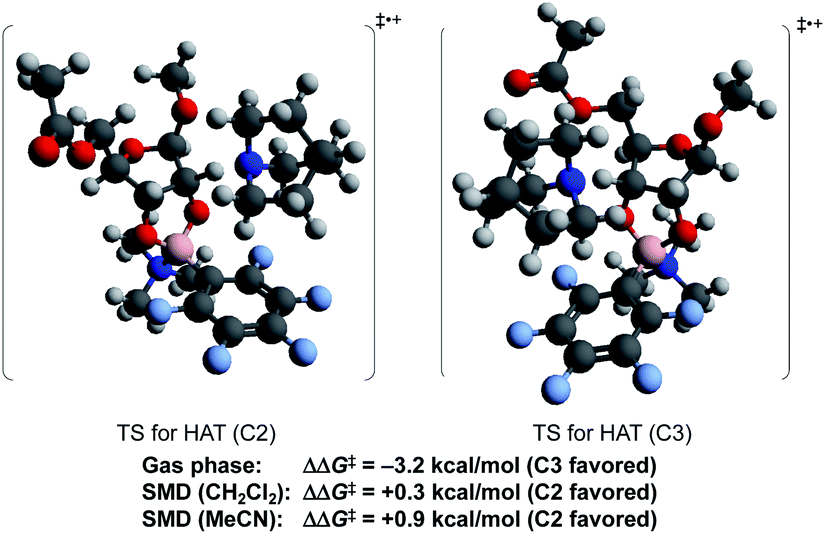 | ||
| Fig. 1 Calculated transition state energies for HAT from C-2 and C3 (B97-D3/Def2-TZVP). | ||
Conclusions
In conclusion, the merger of boronic acid catalysis and photoredox/HAT catalysis has enabled a direct transformation of furanosides to 2-keto-3-deoxyfuranosides. The formation of a boronic ester from a cis-1,2-diol group is proposed to accelerate the abstraction of the hydrogen atom from the 2-position (via a tetracoordinate amine–boronate complex) as well as the cleavage of the C3–O bond to trigger the key spin-center shift step. It is noteworthy that this combination of synthetic catalysts is able to emulate aspects of the proposed mechanism of action of the ribonucleotide reductase enzymes, but shows a distinct pattern of site-selectivity. The products are useful precursors to modified carbohydrate and nucleoside derivatives. The results further illustrate the utility of organoboron catalysts in triggering the site-selective formation of radicals from carbohydrate substrates.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NSERC (Discovery Grant and Canada Research Chairs programs, doctoral scholarship to V. D.), the McLean Foundation and University of Toronto (McLean Award to M. S. T.), the Canada Foundation for Innovation (project no. 17545 and 19119) and the Government of Ontario, and by a generous donation from Kennarshore Inc. H. S. acknowledges financial support from the Kyoto University ICR-iJURC exchange program. Computations were performed on resources and with support provided by Sharcnet, Compute Ontario and Compute Canada. Minxing Zhang is acknowledged for experimental assistance.Notes and references
-
(a) J. Stubbe and W. A. van der Donk, Chem. Biol., 1995, 2, 793–801 CrossRef CAS
; (b) J. Stubbe and W. A. van der Donk, Chem. Rev., 1998, 98, 705–762 CrossRef CAS PubMed
-
Chemical Synthesis of Nucleoside Analogues, ed. P. Merino, John Wiley & Sons, Hoboken, NJ, 2013 Search PubMed
-
(a) S. Jutonen, H. Essadiq, A. Grouiller and J. Chattopadhyaya, Nucleosides Nucleotides, 1985, 4, 187–189 CrossRef
-
(a) T. Sasaki, K. Minamoto and K. Hattori, Tetrahedron, 1974, 30, 2689–2694 CrossRef CAS
- V. Dimakos, H. Y. Su, G. E. Garrett and M. S. Taylor, J. Am. Chem. Soc., 2019, 141, 5149–5153 CrossRef CAS PubMed
- For other transformations of carbohydrate derivatives under conditions of visible light photocatalysis:
(a) R. Sangwan and P. K. Mandal, RSC Adv., 2017, 7, 26256–26321 RSC
- B. P. Roberts, Chem. Soc. Rev., 1999, 28, 25–35 RSC
-
(a) J. L. Jeffrey, J. A. Terrett and D. W. C. MacMillan, Science, 2015, 349, 1532–1536 CrossRef CAS
-
(a) J. A. McCubbin, H. Hosseini and O. Krokhin, J. Org. Chem., 2010, 75, 959–962 CrossRef CAS
- D. G. Hall, Chem. Soc. Rev., 2019, 48, 3475–3496 RSC
- E. D. Nacsa and D. W. C. MacMillan, J. Am. Chem. Soc., 2018, 140, 3322–3330 CrossRef CAS PubMed
- P. Wessig and O. Muehling, Eur. J. Org. Chem., 2007, 2219–2232 CrossRef CAS
-
(a) R. Lenz and B. Giese, J. Am. Chem. Soc., 1997, 119, 2784–2794 CrossRef CAS
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal Jr, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef CAS
- E. Dimitrijević and M. S. Taylor, Chem. Sci., 2013, 4, 3298–3303 RSC
- P. A. Cox, M. Reid, A. G. Leach, A. D. Campbell, E. J. King and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 13156–13165 CrossRef CAS PubMed
- S. Manhas and M. S. Taylor, J. Org. Chem., 2017, 82, 11406–11417 CrossRef CAS PubMed
-
(a) V. Dimakos and M. S. Taylor, Chem. Rev., 2018, 118, 11457–11517 CrossRef CAS PubMed
- D. Wagner, J. P. H. Verhayden and J. G. Moffatt, J. Org. Chem., 1974, 39, 24–30 CrossRef CAS
-
(a) M. Sánchez-Roselló, A. L. A. Puchlopek, A. J. Morgan and S. J. Miller, J. Org. Chem., 2008, 73, 1774–1782 CrossRef PubMed
- F. Hannske, D. Madej and M. J. Robins, Tetrahedron, 1994, 40, 125–135 CrossRef
- M. Prévost, O. St-Jean and Y. Guindon, J. Am. Chem. Soc., 2010, 132, 12433–12439 CrossRef PubMed
-
(a) A. Kakefuda, Y. Yoshimura, T. Sasaki and A. Matsuda, Tetrahedron, 1993, 49, 8513–8528 CrossRef CAS
- F. Lima, U. K. Sharma, L. Grunenberg, D. Saha, S. Johannsen, J. Sedelmeier, E. V. Van der Eycken and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 15136–15140 CrossRef CAS PubMed
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
-
J. J. Frisch
et al.
, Gaussian 16, Rev. B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and computational details, additional optimization data, characterization data for new compounds, copies of 1H and 13C NMR, 1H–1H COSY and 1H–13C HSQC spectra (PDF). See DOI: 10.1039/c9sc05173b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |