
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unique aqueous self-assembly behavior of a thermoresponsive diblock copolymer†
Sarah J.
Byard
a,
Cate T.
O'Brien
a,
Matthew J.
Derry
 a,
Mark
Williams
a,
Oleksandr O.
Mykhaylyk
a,
Adam
Blanazs
b and
Steven P.
Armes
*a
a,
Mark
Williams
a,
Oleksandr O.
Mykhaylyk
a,
Adam
Blanazs
b and
Steven P.
Armes
*a
aDepartment of Chemistry, University of Sheffield, Dainton Building, Brook Hill, Sheffield, South Yorkshire S3 7HF, UK. E-mail: s.p.armes@sheffield.ac.uk
bBASF SE, GMV/P-B001, 67056 Ludwigshafen, Germany
First published on 12th November 2019
Abstract
It is well-recognized that block copolymer self-assembly in solution typically produces spheres, worms or vesicles, with the relative volume fraction of each block dictating the copolymer morphology. Stimulus-responsive diblock copolymers that can undergo either sphere/worm or vesicle/worm transitions are also well-documented. Herein we report a new amphiphilic diblock copolymer that can form spheres, worms, vesicles or lamellae in aqueous solution. Such self-assembly behavior is unprecedented for a single diblock copolymer of fixed composition yet is achieved simply by raising the solution temperature from 1 °C (spheres) to 25 °C (worms) to 50 °C (vesicles) to 70 °C (lamellae). Heating increases the degree of hydration (and hence the effective volume fraction) of the core-forming block, with this parameter being solely responsible for driving the sphere-to-worm, worm-to-vesicle and vesicle-to-lamellae transitions. The first two transitions exhibit excellent reversibility but the vesicle-to-lamellae transition exhibits hysteresis on cooling. This new thermoresponsive diblock copolymer provides a useful model for studying such morphological transitions and is likely to be of significant interest for theoretical studies.
Introduction
Block copolymer self-assembly has been extensively studied because it offers a wide range of potential applications, including thermoplastic elastomers,1 catalysts,2 toughening agents for epoxy resins,3 ultrafiltration membranes,4 lithographic materials,5 high-density arrays6,7 and drug delivery.8–10 It is well-known that the self-assembly of diblock copolymers in the solid state can result in a wide range of morphologies.7,11–15 Similarly, diblock copolymers can self-assemble in solution when the solvent is selective for one of the two blocks. In this case, the final morphology depends on the relative volume fractions of each block,16 the selectivity of the solvent(s) for each block,17 and the copolymer concentration.18,19 It is well-documented that the three main copolymer morphologies formed in solution are spheres,16 worms19,20 and vesicles21 but other morphologies such as rods,22–24 toroids25 and lamellae26 have also been reported.Traditionally, block copolymer self-assembly in solution has been achieved via various post-polymerization processing strategies.16,20–23 However, over the past decade increasing attention has been paid to polymerization-induced self-assembly (PISA).19,24,27–30 Most PISA syntheses reported in the literature are based on reversible addition–fragmentation chain transfer (RAFT) polymerization.29,31–44 The versatility of this radical-based chemistry has enabled the convenient preparation of a wide range of well-defined functional block copolymers in the form of concentrated dispersions.27–29 More specifically, PISA syntheses based on RAFT dispersion polymerization has enabled the synthesis of diblock copolymer nano-objects that can exhibit thermoresponsive behavior.19,45–50 For example, cooling an aqueous amphiphilic diblock copolymer worm gel below ambient temperature induces a worm-to-sphere transition (and concomitant degelation).19 Alternatively, heating the analogous hydrophobic copolymer worms in non-aqueous media can induce the same change in copolymer morphology.48 In each case, this change in morphology can be rationalized in terms of surface plasticization of the nano-objects by solvent molecules, which leads to a subtle change in the fractional packing parameter for the copolymer chains.51–53 There have been a couple of reports of diblock copolymers that can form more than one morphology in a given solvent18 or a binary mixture of solvents under certain conditions.17 However, as far as we are aware, there have been no reports of a single (i.e. fixed composition) diblock copolymer in any solvent that is capable of crossing three phase boundaries to form spheres, worms, vesicles or lamellae.
Such rich phase behavior is demonstrated herein for a new poly(N,N-dimethylacrylamide)-block-poly(4-hydroxybutyl acrylate-stat-diacetone acrylamide) [PDMAC–P(HBA-stat-DAAM)] amphiphilic diblock copolymer that is prepared directly in water under mild conditions (see Fig. 1). The sphere/worm and worm/vesicle phase transitions can be achieved by simply varying the temperature of the resulting aqueous copolymer dispersion and proceed both rapidly and reversibly even when conducted at copolymer concentrations as low as 0.10% w/w. Moreover, a vesicle-to-lamellae transition is also observed for this system, although this latter thermal transition is characterized by significant hysteresis during the cooling cycle.
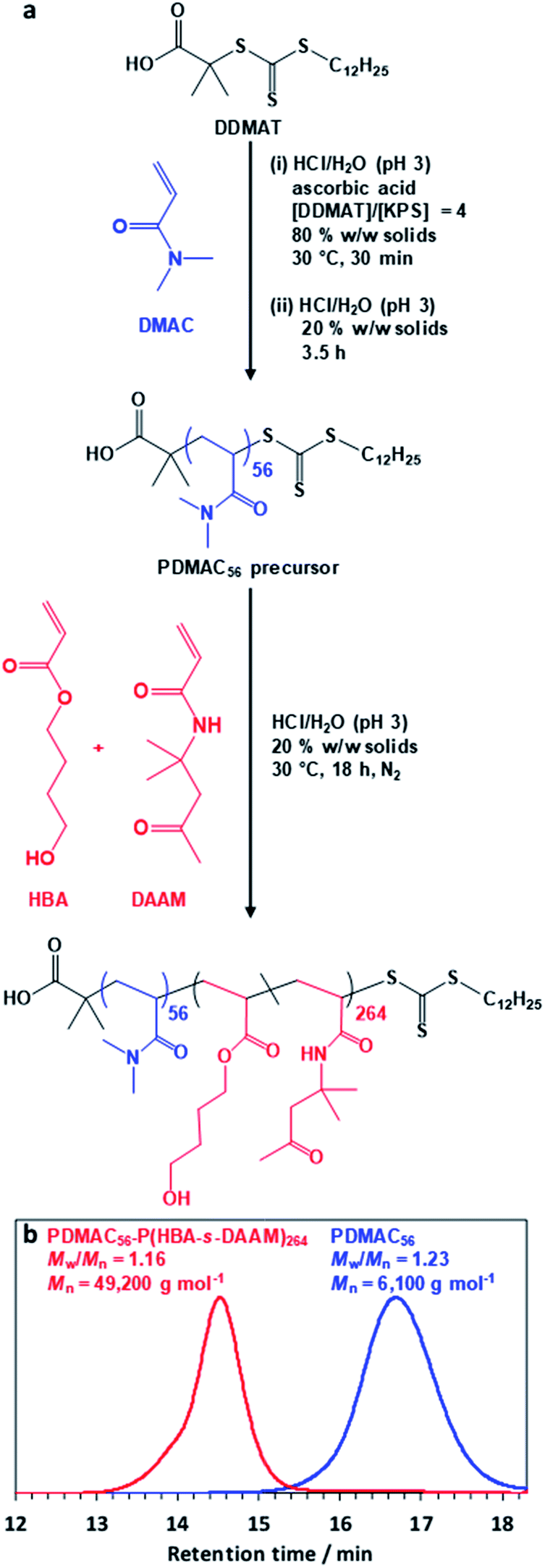 | ||
| Fig. 1 (a) Block copolymer synthesis via polymerization-induced self-assembly (PISA). Reaction scheme for the synthesis of the PDMAC56 precursor via RAFT solution polymerization of DMAC at 30 °C using a DDMAT chain transfer agent and a redox initiator [potassium persulfate (KPS) plus ascorbic acid]. Subsequent PDMAC56 chain extension with a binary mixture of HBA (80 mol%) and DAAM (20 mol%) via RAFT aqueous dispersion copolymerization at pH 3 produced a well-defined PDMAC56–P(HBA-stat-DAAM)264 diblock copolymer. (b) DMF GPC data obtained for the PDMAC56 precursor and final PDMAC56–P(HBA-stat-DAAM)264 diblock copolymer (refractive index detector; expressed relative to a series of poly(methyl methacrylate) calibration standards). | ||
Results and discussion
The diblock copolymer used in this study was prepared via PISA using a highly convenient one-pot RAFT aqueous dispersion polymerization formulation (Fig. 1a). First, N,N′-dimethylacrylamide (DMAC) was polymerized in an 80% w/w aqueous solution at 30 °C using a trithiocarbonate-based RAFT agent combined with a low-temperature redox initiator. The initial highly concentrated aqueous solution was required to ensure solubility of the RAFT agent. After 30 min, the reaction mixture was diluted to 20% w/w in order to lower its solution viscosity. After 4 h, a small aliquot of the resulting water-soluble PDMAC precursor was removed for analysis. Gel permeation chromatography (GPC; DMF eluent, 60 °C) analysis indicated a relatively narrow molecular weight distribution (Mw/Mn = 1.23, see Fig. 1b) while 1H NMR spectroscopy studies confirmed that more than 99% DMAC conversion had been achieved (Fig. S1, ESI†). The mean degree of polymerization (DP) of this PDMAC precursor was determined to be 56 by end-group analysis using UV spectroscopy (see Fig. S2, ESI†).This PDMAC56 precursor was then chain-extended in situ by statistical copolymerization of a mixture of 4-hydroxybutyl acrylate (HBA; 80 mol%) and diacetone acrylamide (DAAM; 20 mol%) at 20% w/w solids to produce a PDMAC56–P(HBA-stat-DAAM)264 diblock copolymer, where the subscripts refer to the mean DPs of each block. GPC analysis indicated efficient chain extension and a relatively narrow molecular weight distribution (Mw/Mn < 1.20) for the final diblock copolymer (see Fig. 1b). 1H NMR spectroscopy studies confirmed essentially full conversion for the HBA and DAAM comonomers (>99%; see Fig. S1, ESI†) and also that the expected composition for the core-forming P(HBA-stat-DAAM)264 block (78 ± 2 mol% HBA) was obtained within experimental error.
Recent in silico studies and preliminary experimental data suggest that HBA should be a suitable core-forming block for aqueous PISA.54 Indeed, the 20% w/w dispersion of PDMAC56–P(HBA-stat-DAAM)264 diblock copolymer nano-objects prepared herein formed a free-standing gel at 25 °C (Fig. 2). On cooling to 1 °C, degelation occurred to produce a transparent free-flowing dispersion. On heating to 50 °C, a turbid, free-flowing dispersion was formed, while a turbid paste was formed at 70 °C. To examine the copolymer morphologies associated with these thermal transitions, transmission electron microscopy (TEM) studies were performed on the PDMAC56–P(HBA-stat-DAAM)264 diblock copolymer nano-objects. Crosslinking of the P(HBA-stat-DAAM)264 block was conducted at the desired temperature using adipic acid dihydrazide (ADH) at pH 3, as previously reported for PDMAC–PDAAM diblock copolymer nano-objects.55 Such covalent stabilization was essential to obtain high-quality images: in the absence of any cross-linking, the relatively low glass transition temperature of the core-forming block simply led to film formation on the TEM grid. Moreover, crosslinking also eliminated the thermoresponsive behavior of this diblock copolymer, hence preserving the copolymer morphology at any desired temperature. Thus this cross-linking protocol enabled visualization of pure spheres, worms, vesicles or lamellae after covalent stabilization at 1, 25, 50 or 70 °C, respectively (Fig. 2f–i).
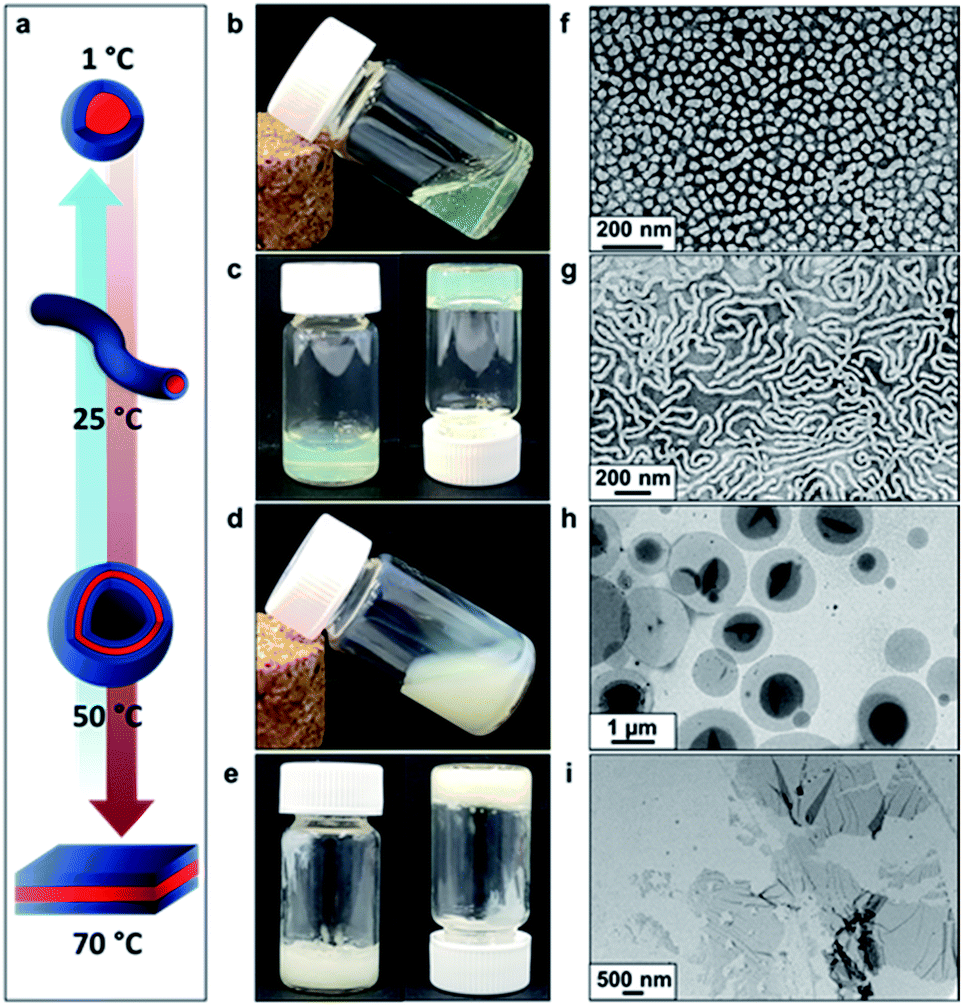 | ||
| Fig. 2 Thermally-induced diblock copolymer morphology transitions in aqueous solution. (a) Schematic representation of the reversible morphological transitions that occur for a 20% w/w aqueous dispersion of PDMAC56–P(HBA-stat-DAAM)264 diblock copolymer nano-objects on varying the temperature from 1° to 70 °C. Digital images show the physical appearance of this aqueous dispersion: (b) on cooling to 1 °C for 30 min, (c) at ambient temperature (25 °C), (d) on heating to 50 °C for 30 min and (e) on heating to 70 °C for 30 min. TEM images recorded for 0.10% w/w aqueous dispersions of PDMAC56–P(HBA-stat-DAAM)264 using uranyl formate as a negative stain after covalent stabilization at the desired temperature using adipic acid dihydrazide (ADH) at a DAAM/ADH molar ratio of 1.0: (f) spheres (crosslinked at 1 °C), (g) worms (crosslinked at 25 °C), (h) vesicles (crosslinked at 50 °C) and (i) lamellae (crosslinked at 70 °C). | ||
Rheology studies were conducted on a 20% w/w aqueous dispersion of the linear (i.e. non-crosslinked) PDMAC56–P(HBA-stat-DAAM)264 nano-objects as a function of temperature (Fig. 3 and S3, ESI†). As expected, a low-viscosity fluid was formed at 1 °C owing to the presence of free-flowing spherical nano-objects. Warming to ambient temperature induced a sol–gel transition, producing a soft, transparent free-standing gel. This indicates the formation of highly anisotropic worms, with multiple inter-particle contacts producing a 3D network.56 The storage modulus (G′) exceeds the loss modulus (G′′) at 17 °C, which corresponds to the critical gelation temperature (CGT) (Fig. S3, ESI,† for G′ and G′′ data). A maximum gel viscosity was observed at 25 °C. Further heating led to a significant reduction in viscosity (and a concomitant increase in turbidity) owing to the formation of vesicles. These sphere-to-worm and worm-to-vesicle transitions proved to be remarkably reversible, with relatively little hysteresis being observed at heating/cooling rates of 1 °C min−1 (Fig. 3a). Heating the turbid, free-flowing vesicular dispersion above 50 °C initially caused a further reduction in the complex viscosity (see Fig. 3b). However, the dispersion became a turbid paste between 63 °C and 70 °C and the complex viscosity increased by approximately two orders of magnitude, which corresponds to the formation of lamellae (see Fig. 2i). Significant hysteresis was observed for the lamellae-to-vesicle transition on cooling, but good reversibility was observed below approximately 22 °C.
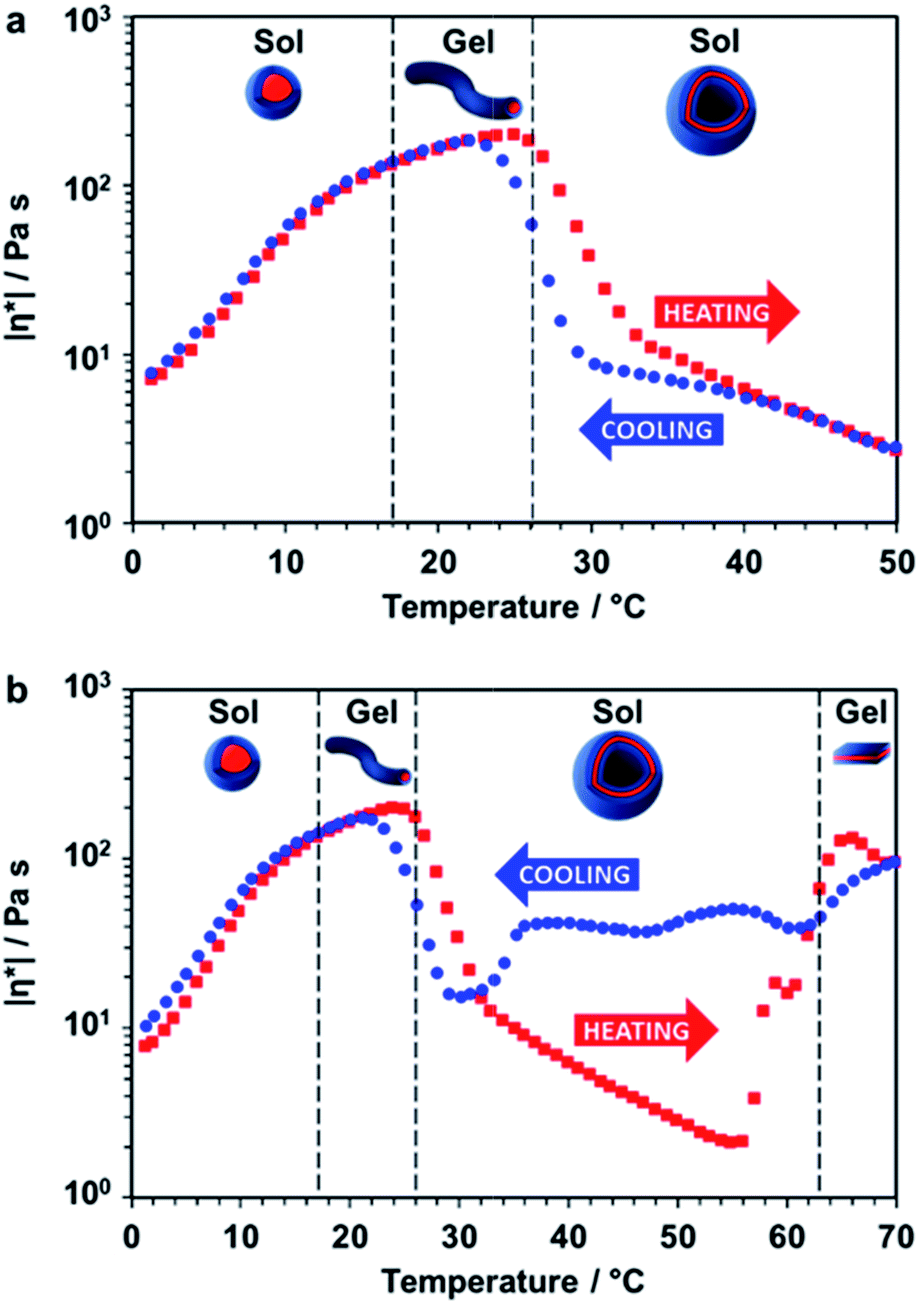 | ||
| Fig. 3 Thermally-induced change in complex viscosity for an aqueous dispersion of linear diblock copolymer nano-objects. Temperature-dependent rheological studies for a 20% w/w aqueous dispersion of PDMAC56–P(HBA-stat-DAAM)264 nano-objects at an applied strain of 1.0% and an angular frequency of 1.0 rad s−1. The dispersion was equilibrated at 1 °C for 15 min prior to heating. The black dashed lines indicate the sol–gel transitions observed on heating as determined from the G′ and G′′ values (Fig. S3, ESI†). (a) Complex viscosity (|η*|) vs. temperature data obtained for a thermal cycle from 1 °C to 50 °C to 1 °C at a heating/cooling rate of 1 °C min−1 [■ heating curve, ● cooling curve]. (b) Complex viscosity vs. temperature data for a thermal cycle over an expanded temperature range of 1 °C to 70 °C to 1 °C at a heating/cooling rate of 1 °C min−1 [■ heating curve, ● cooling curve]. | ||
Further evidence for these four types of diblock copolymer nano-objects was obtained using shear-induced polarized light imaging (SIPLI), see Fig. S4.† This opto-rheological technique has been recently used to demonstrate the alignment of anisotropic nano-objects such as block copolymer worms and lamellae at a certain critical rate of applied shear.57 Thus, the SIPLI image recorded under continuous shear flow at 3 °C appears dark (see Fig. S4†); this is consistent with the presence of isotropic spheres, which cannot be aligned. A characteristic Maltese cross is observed under this shear flow at approximately 20 °C, indicating the formation (and shear-induced alignment) of highly anisotropic worms. This distinctive pattern disappears at 33 °C owing to the formation of isotropic vesicles while a new Maltese cross (with associated multiple colors arising from strong birefringence) is obtained at 45 °C. The latter feature indicates the presence of anisotropic lamellae that have either a perpendicular (with the lamellar normal parallel to the neutral direction of the flow) or transverse (with the lamellar normal parallel to the velocity direction of the flow) orientation.58 [N.B. The above characteristic temperatures required to induce formation of block copolymer worms, vesicles and lamellae do not match those indicated by the oscillatory rheology data shown in Fig. 3. This is because the applied (continuous) shear is significantly greater in the latter case, which promotes the formation of worms, vesicles and lamellae under milder conditions, i.e. lower temperatures].
Small-angle X-ray scattering (SAXS) studies were conducted on a 1.0% w/w aqueous dispersion of linear PDMAC56–P(HBA-stat-DAAM)264 nano-objects as a function of temperature at pH 3 (Fig. 4). The gradient in the low q region of an I(q) vs. q plot (where I(q) is the scattering intensity and q is the scattering vector) is characteristic of the predominant copolymer morphology.59 This gradient is close to zero at 1 °C, which suggests the presence of spheres. At 25 °C, the gradient shifts towards −1, indicating the formation of highly anisotropic worms.19,48 On raising the temperature to 50 °C, the low q gradient increases to −2, which is characteristic of bilayer (or vesicle) formation. At 70 °C, the diffraction peak observed at q = 0.019 Å−1 suggests stacked lamellae sheets (see Fig. 4a, black arrow). Analysis of the SAXS data shown in Fig. 4a indicated a core diameter of 22.8 nm for the spheres, which is consistent with an overall hydrodynamic DLS diameter of 33 nm recorded at the same temperature. A core cross-section diameter of 23.0 nm was determined for the worms; this is consistent with TEM studies (26 ± 3 nm) if the highly deformable nature of the core-forming block is taken into consideration. The mean vesicle membrane thickness was 17.2 nm, which indicates significant inter-digitation of the structure-directing hydrophobic P(HBA-stat-DAAM)264 chains.60
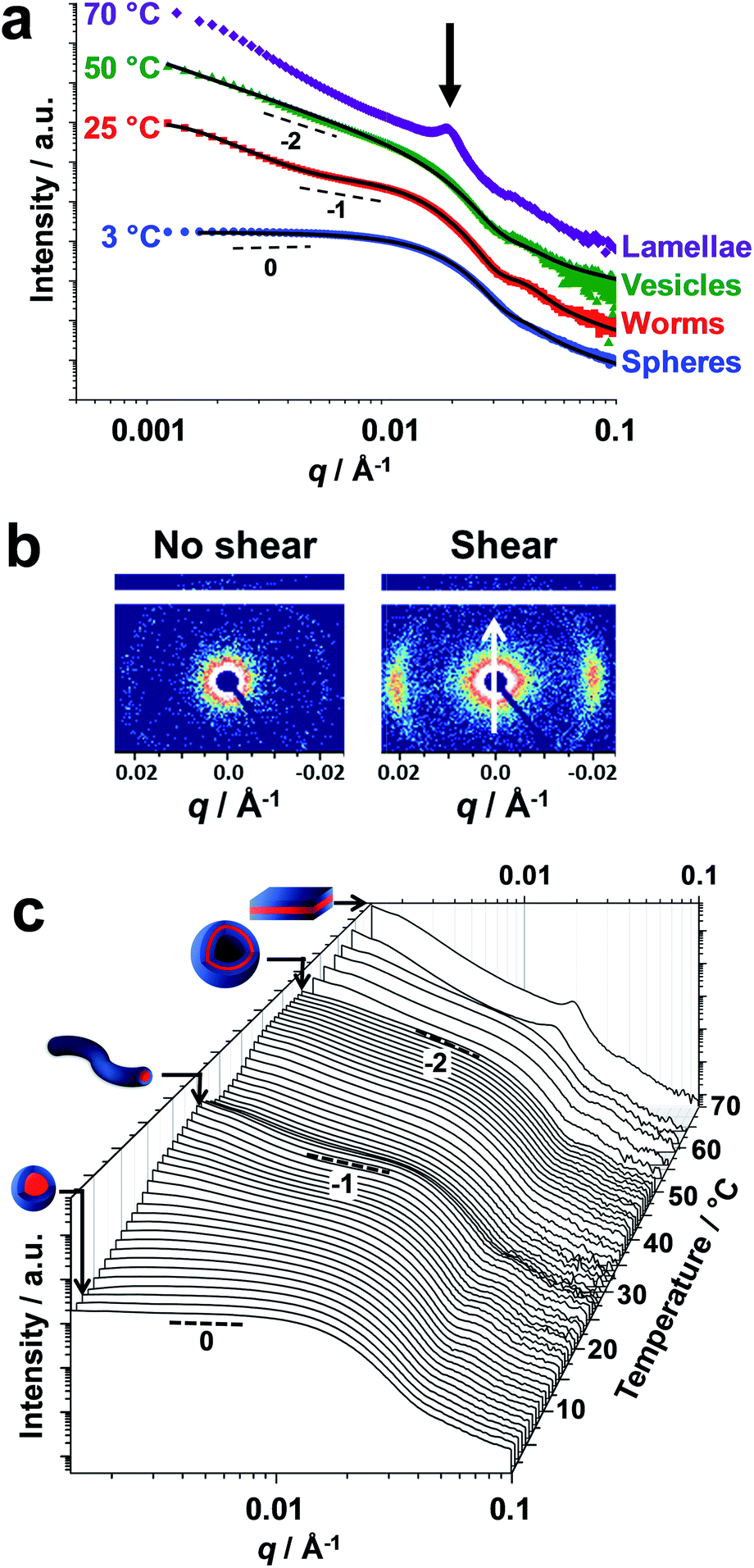 | ||
| Fig. 4 Small-angle X-ray scattering studies of linear diblock copolymer nano-objects. (a) Double-logarithmic plot of SAXS patterns recorded for a 1.0% w/w aqueous dispersion of thermoresponsive PDMAC56–P(HBA-stat-DAAM)264 nano-objects at 3 °C (blue data), 25 °C (red data), 50 °C (green data) and 70 °C (purple data). The black lines indicate the data fits obtained using appropriate scattering models. For guidance, black dashed lines indicate zero, −1 and −2 gradients, while the blue, green and purple data sets are offset by arbitrary factors to aid clarity. (b) 2D SAXS patterns recorded either at zero shear or under applied shear (direction indicated by the white arrow) during rheo-SAXS experiments conducted on a 20% w/w aqueous dispersion of PDMAC56–P(HBA-stat-DAAM)264 nano-objects at 63 °C. (c) SAXS patterns recorded for a 1.0% w/w aqueous dispersion of thermoresponsive PDMAC56–P(HBA-stat-DAAM)264 nano-objects between 1 and 70 °C using a heating rate of 1 °C min−1. For guidance, black dashed lines indicate zero, −1 and −2 gradients. | ||
Finally, the mean distance between the stacked lamellae sheets was determined to be 33 nm as calculated from the diffraction peak observed at q = 0.019 Å−1. Fig. 4b shows 2D SAXS patterns recorded under applied shear and also at zero shear during rheo-SAXS experiments conducted at 63 °C (see ESI† for further details). The distinctly anisotropic pattern obtained under applied shear clearly indicates the presence of lamellae in a perpendicular orientation.58Fig. 4c shows a series of SAXS patterns recorded for a 1.0% w/w aqueous dispersion of PDMAC56–P(HBA-stat-DAAM)264 nano-objects on heating from 1 °C to 70 °C at a heating rate of 1 °C min−1. Clearly, there is a gradual increase in the low q gradient as the initial spheres are converted into first worms and then vesicles. The diffraction peak assigned to stacked lamellae is observed at 64 °C and 70 °C (Fig. 4c). This data set confirms that the interconversion between these four copolymer morphologies occurs rapidly on relatively short time scales, even at rather low copolymer concentration. In summary, the TEM images (Fig. 2), oscillatory rheology studies (Fig. 3), SIPLI measurements (Fig. S4†) and SAXS experiments (Fig. 4) provide compelling evidence for inter-conversion between spheres, worms, vesicles and lamellae for this PDMAC56–P(HBA-stat-DAAM)264 diblock copolymer. Variable temperature 1H NMR spectroscopy studies were conducted to examine the mechanism for this unique thermoresponsive behavior. Accordingly, a 20% w/w aqueous copolymer dispersion was heated from 5 °C to 70 °C with spectra being recorded at 5 °C intervals and normalized relative to an external standard (pyridine). Partial 1H NMR spectra are shown in Fig. 5 (see Fig. S5, ESI,† for the full spectra). 1H NMR signals assigned to the core-forming P(HBA-stat-DAAM)264 chains become more prominent at higher temperature, indicating progressively greater hydration for this weakly hydrophobic structure-directing block (Fig. 5a). Such spectral changes can be quantified by normalizing the integrated intensity of the two C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–OH protons assigned to the HBA repeat units relative to that of the external standard. This approach enables the apparent degree of hydration of the HBA repeat units within the P(HBA-stat-DAAM)264 core-forming block to be calculated at any given temperature. This parameter is expressed as a percentage of the maximum value determined by 1H NMR spectroscopy using CD3OD (the PDMAC56–P(HBA-stat-DAAM)264 chains are molecularly dissolved in this solvent); it increases from 62% to 83% on heating a 20% w/w aqueous copolymer dispersion from 5 °C to 70 °C (Fig. 5b).
2–OH protons assigned to the HBA repeat units relative to that of the external standard. This approach enables the apparent degree of hydration of the HBA repeat units within the P(HBA-stat-DAAM)264 core-forming block to be calculated at any given temperature. This parameter is expressed as a percentage of the maximum value determined by 1H NMR spectroscopy using CD3OD (the PDMAC56–P(HBA-stat-DAAM)264 chains are molecularly dissolved in this solvent); it increases from 62% to 83% on heating a 20% w/w aqueous copolymer dispersion from 5 °C to 70 °C (Fig. 5b).
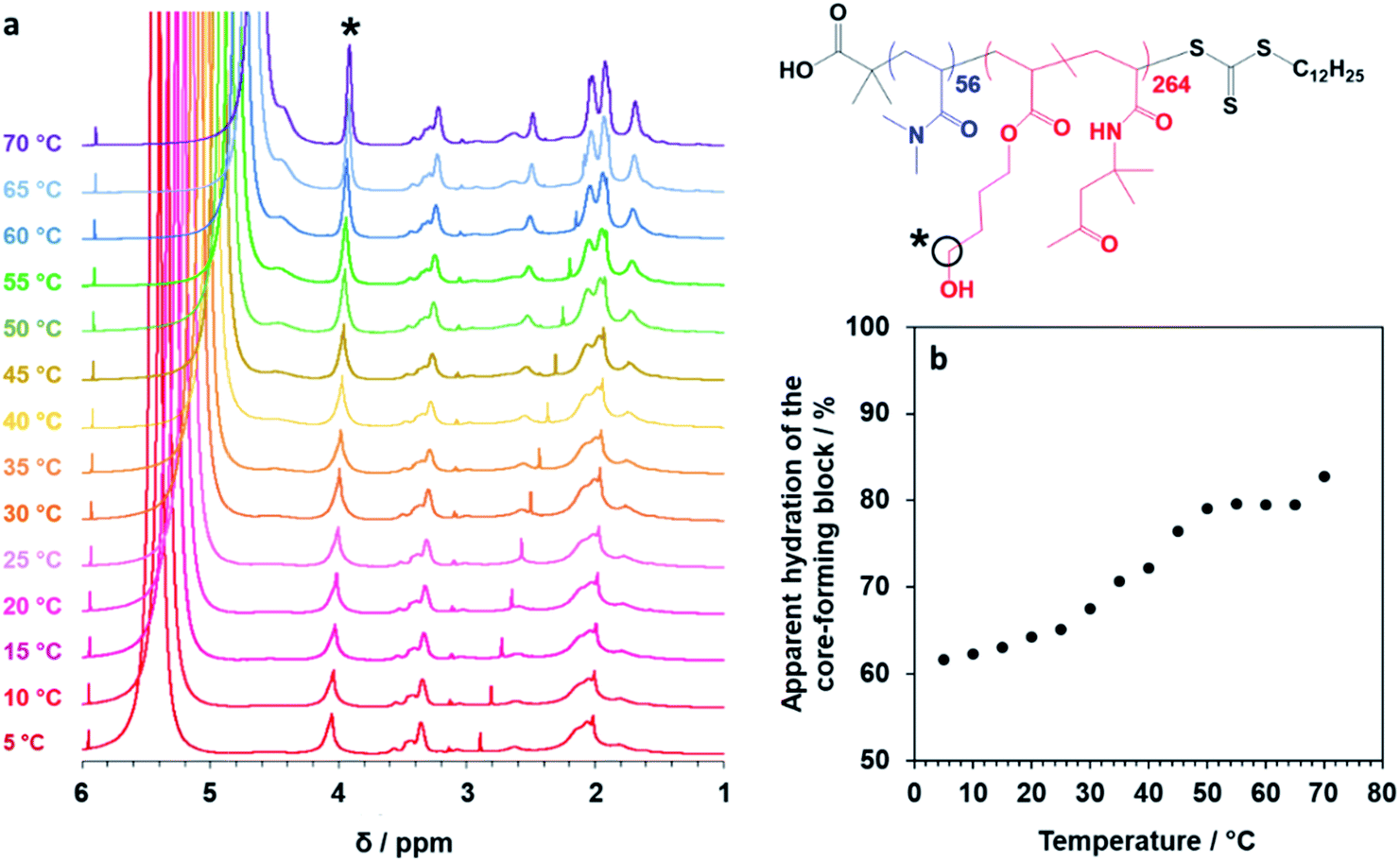 | ||
| Fig. 5 Variable temperature NMR studies of linear thermoresponsive diblock copolymer nano-objects. (a) Normalized 1H NMR spectra recorded for a 20% w/w aqueous dispersion of PDMAC56–P(HBA-stat-DAAM)264 nano-objects on heating from 5 °C to 70 °C. For clarity, only partial spectra in the 1–6 ppm range are shown (see Fig. S5, ESI,† for the full spectra). The signal marked with an asterisk is assigned to the two C2–OH protons on the HBA residues: the gradual increase in its intensity on heating indicates progressively greater hydration of the core-forming block at higher temperature. (b) Apparent degree of hydration of the hydrophobic P(HBA-stat-DAAM)264 block as a function of temperature (with 100% hydration corresponding to the true composition of this structure-directing block, as calculated by 1H NMR spectroscopy studies of the molecularly-dissolved copolymer chains in CD3OD). | ||
Clearly, the P(HBA-stat-DAAM)264 block is partially hydrated at all temperatures and its degree of hydration increases at higher temperature. This is attributed to the temperature dependence of the Flory–Huggins χ parameter for the interaction between the P(HBA-stat-DAAM)264 chains and water. As this hydrophobic block becomes progressively more hydrated, its volume fraction increases relative to that of the hydrophilic PDMAC stabilizer block. This leads to a subtle increase in the fractional packing parameter, P, for the copolymer chains,51,52 which accounts for the evolution in morphology from spheres to worms to vesicles to lamellae that is observed on heating. These morphological transitions are ultimately reversible on cooling, although significant hysteresis is observed for the initial lamellae-to-vesicle transformation (see Fig. 3b). Interestingly, similar thermoresponsive behavior has been recently predicted for a single diblock copolymer composition by Borisov and co-workers.61
Dynamic light scattering (DLS) was used to determine the sphere-equivalent diameter for a 0.10% w/w PDMAC56–P(HBA-stat-DAAM)264 aqueous dispersion during a thermal cycle from 2 °C to 50 °C to 2 °C (Fig. S6, ESI†). These DLS data suggest that both the sphere-to-worm and worm-to-vesicle thermal transitions occur rapidly and reversibly, with minimal hysteresis being observed even at copolymer concentrations as low as 0.10% w/w. The excellent reversibility observed under such conditions is attributed to the relatively high mobility of the acrylic-rich core-forming block, which has a relatively low glass transition temperature.
Finally, raising the solution pH from pH 3 to pH 7 at 20 °C causes a reversible reduction in the sphere-equivalent particle diameter from 333 nm (worms) to 38 nm (spheres) as judged by DLS (see Fig. S7, ESI†). This morphological transition is attributed to end-group ionization owing to deprotonation of the single carboxylic acid group located at the end of each PDMAC steric stabilizer block.62 Thus this remarkable thermoresponsive diblock copolymer also exhibits pH-responsive behavior.
Conclusions
In summary, we report the one-pot PISA synthesis of a new amphiphilic PDMAC56–P(HBA-stat-DAAM)264 diblock copolymer that can form spheres, worms or vesicles rapidly and reversibly in aqueous solution at copolymer concentrations as low as 0.10% w/w. In addition, a vesicle-to-lamellae transition also occurs on heating to around 70 °C, but the complementary lamellae-to-vesicle transition suffers from significant hysteresis on cooling. This new system is expected to provide a suitable model for studying the kinetics and mechanism(s) of block copolymer self-assembly in solution, as well as offering a highly convenient model system for theoretical studies of thermoresponsive block copolymer nano-objects.63 In addition, the excellent thermoreversibility exhibited by this new system is expected to facilitate the in situ loading of vesicles with either nanoparticles or globular proteins/enzymes, with subsequent release of such payloads being feasible simply by lowering the solution temperature to induce a vesicle-to-worm (or vesicle-to-sphere) morphological transition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank EPSRC (EP/L016281) for a CDT PhD studentship for S. J. B. and BASF SE (Ludwigshafen, Germany) for partial funding of this project. S. P. A. thanks EPSRC for a Particle Technology Fellowship (EP/R003009). BASF SE is also thanked for permission to publish this work. We are grateful to the European Synchrotron Radiation Facility (ESRF) for providing synchrotron beam-time (SC-4601, SC-4864) and thank Dr T. Zinn and the personnel of ID02 for their assistance. Scott Bader Ltd. is thanked for supplying the HBA monomer. O. O. M. and S. P. A. thank EPSRC for a capital equipment grant to purchase the Xenocs Xeuss 2.0/Excillum SAXS instrument used to perform the rheo-SAXS experiments (EP/M028437/1).Notes and references
- A. V. Ruzette and L. Leibler, Nat. Mater., 2005, 4, 19–31 CrossRef CAS.
- M. Seo and M. A. Hillmyer, Science, 2012, 336, 1422–1426 CrossRef CAS PubMed.
- P. M. Lipic, F. S. Bates and M. A. Hillmyer, J. Am. Chem. Soc., 1998, 120, 8963–8970 CrossRef CAS.
- H. Ahn, S. Park, S.-W. Kim, P. J. Yoo, D. Y. Ryu and T. P. Russell, ACS Nano, 2014, 8, 11745–11752 CrossRef CAS.
- C. Tang, E. M. Lennon, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Science, 2008, 322, 429–433 CrossRef CAS.
- S. Ouk Kim, H. H. Solak, M. P. Stoykovich, N. J. Ferrier, J. J. de Pablo and P. F. Nealey, Nature, 2003, 424, 411–414 CrossRef.
- S. H. Kim, M. J. Misner, T. Xu, M. Kimura and T. P. Russell, Adv. Mater., 2004, 16, 226–231 CrossRef CAS.
- S. W. Kim, B. Jeong, Y. H. Bae and D. S. Lee, Nature, 1997, 388, 860–862 CrossRef.
- A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428 CrossRef CAS PubMed.
- Y. A. N. Geng, P. Dalhaimer, S. Cai, R. Tsai, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS.
- J. T. Chen, E. L. Thomas, C. K. Ober and G. Mao, Science, 1996, 273, 343–346 CrossRef CAS PubMed.
- F. S. Bates, Science, 1991, 251, 898–905 CrossRef CAS.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- A. K. Khandpur, S. Ftirster, F. S. Bates, I. W. Hamley, A. J. Ryan, W. Bras, K. Almdal and K. Mortensen, Macromolecules, 1995, 28, 8796–8806 CrossRef CAS.
- M. W. Matsen and F. S. Bates, Macromolecules, 1996, 29, 1091–1098 CrossRef CAS.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS.
- S. Abbas, Z. Li, H. Hassan and T. P. Lodge, Macromolecules, 2007, 40, 4048–4052 CrossRef CAS.
- S. Y. Kim, K. E. Lee, S. S. Han and B. Jeong, J. Phys. Chem. B, 2008, 112, 7420–7423 CrossRef CAS PubMed.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- Y.-Y. Won, H. T. Davis and F. S. Bates, Science, 1999, 283, 960–963 CrossRef CAS PubMed.
- B. M. Discher, Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS.
- J. B. Gilroy, T. Gädt, G. R. Whittell, L. Chabanne, J. M. Mitchels, R. M. Richardson, M. A. Winnik and I. Manners, Nat. Chem., 2010, 2, 566–570 CrossRef CAS.
- C. E. Boott, J. Gwyther, R. L. Harniman, D. W. Hayward and I. Manners, Nat. Chem., 2017, 9, 785–792 CrossRef CAS.
- D. J. Pochan, Z. Chen, H. Cui, K. Hales, K. Qi and K. L. Wooley, Science, 2004, 306, 94–97 CrossRef CAS.
- P. Yang, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2013, 46, 8545–8556 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS.
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- S. Sugihara, A. H. Ma'Radzi, S. Ida, S. Irie, T. Kikukawa and Y. Maeda, Polymer, 2015, 76, 17–24 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- X. Wang and Z. An, Macromol. Rapid Commun., 2019, 40, 1–14 Search PubMed.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS.
- S. Perrier and P. Takolpuckdee, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5347–5393 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- J. Tan, H. Sun, M. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 CrossRef CAS.
- Y. Jiang, N. Xu, J. Han, Q. Yu, L. Guo, P. Gao, X. Lu and Y. Cai, Polym. Chem., 2015, 6, 4955–4965 RSC.
- D. Zhou, S. Dong, R. P. Kuchel, S. Perrier and P. B. Zetterlund, Polym. Chem., 2017, 8, 3082–3089 RSC.
- S. Y. Khor, J. F. Quinn, M. R. Whittaker, N. P. Truong and T. P. Davis, Macromol. Rapid Commun., 2019, 40, 1–22 CrossRef.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- W. Zhou, Q. Qu, Y. Xu and Z. An, ACS Macro Lett., 2015, 4, 495–499 CrossRef CAS.
- X. Zhang, S. Boissé, W. Zhang, P. Beaunier, F. D'Agosto, J. Rieger and B. Charleux, Macromolecules, 2011, 44, 4149–4158 CrossRef CAS.
- J. Rieger, C. Grazon, B. Charleux, D. Alaimo and C. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2373–2390 CrossRef CAS.
- X. Wang, J. Zhou, X. Lv, B. Zhang and Z. An, Macromolecules, 2017, 50, 7222–7232 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS.
- Y. Ma, P. Gao, Y. Ding, L. Huang, L. Wang, X. Lu and Y. Cai, Macromolecules, 2019, 52, 1033–1041 CrossRef.
- C. A. Figg, A. Simula, K. A. Gebre, B. S. Tucker, D. M. Haddleton and B. S. Sumerlin, Chem. Sci., 2015, 6, 1230–1236 RSC.
- J. N. Israelachvil, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans., 1975, 72, 1525–1568 RSC.
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS.
- J. C. Foster, S. Varlas, B. Couturaud, J. R. Jones, R. Keogh, R. T. Mathers and R. K. O'Reilly, Angew. Chem., Int. Ed., 2018, 57, 15733–15737 CrossRef CAS.
- S. J. Byard, M. Williams, B. E. Mckenzie, A. Blanazs and S. P. Armes, Macromolecules, 2017, 50, 1482–1493 CrossRef CAS.
- J. R. Lovett, M. J. Derry, P. Yang, F. L. Hatton, N. J. Warren, P. W. Fowler and S. P. Armes, Chem. Sci., 2018, 9, 7138–7144 RSC.
- O. O. Mykhaylyk, N. J. Warren, A. J. Parnell, G. Pfeifer and J. Laeuger, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 2151–2170 CrossRef CAS.
- O. O. Mykhaylyk, A. J. Parnell, A. Pryke and J. P. A. Fairclough, Macromolecules, 2012, 45, 5260–5272 CrossRef CAS.
- O. Glatter and O. Kratky, Small Angle X-ray Scattering, Academic Press, London, 1982, vol. 130 Search PubMed.
- G. Battaglia and A. J. Ryan, J. Am. Chem. Soc., 2005, 127, 8757–8764 CrossRef CAS.
- A. M. Rumyantsev, F. A. M. Leermakers, E. B. Zhulina, I. I. Potemkin and O. V. Borisov, Langmuir, 2019, 35, 2680–2691 CrossRef CAS.
- J. R. Lovett, N. J. Warren, L. P. D. Ratcliffe, M. K. Kocik and S. P. Armes, Angew. Chem., Int. Ed., 2015, 54, 1279–1283 CrossRef CAS.
- L. P. D. Ratcliffe, M. J. Derry, A. Ianiro, R. Tuinier and S. P. Armes, Angew. Chem., Int. Ed., 2019, 58 DOI:10.1002/anie.201909124.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, polymer characterization techniques, SAXS models, supporting figures: GPC chromatograms, assigned 1H NMR spectra, calibration plot for determining the mean DP of the PDMAC precursor, temperature-dependent rheological studies, SIPLI rheology data, full spectra for the variable temperature 1H NMR spectroscopy study, apparent z-average diameter as a function of temperature as determined by DLS studies and apparent z-average diameter as a function of pH as determined by DLS studies. See DOI: 10.1039/c9sc04197d |
| This journal is © The Royal Society of Chemistry 2020 |