
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRh(II)-catalyzed branch-selective C–H alkylation of aryl sulfonamides with vinylsilanes†
Supriya
Rej
and
Naoto
Chatani
 *
*
Department of Applied Chemistry, Faculty of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: chatani@chem.eng.osaka-u.ac.jp
First published on 11th November 2019
Abstract
Rhodium(II)-catalyzed unusual branch-selective ortho-C–H alkylation of aryl sulfonamides with vinylsilanes was achieved using an 8-aminoquinoline directing group. Notably, the para-substituted aryl sulfonamides gave mono-(branched)alkylated products exclusively without the formation of any double C–H alkylated byproducts. The results of deuterium labeling experiments suggest that both hydrometalation and carbometalation pathways are involved in this conversion.
Introduction
The transition metal catalyzed directed C–H activation strategy is one of the most straightforward and site-selective approaches in organic chemistry for constructing C–C bonds. A variety of C–H functionalization reactions have been achieved to date by using a directing group strategy.1 In particular, directed C–H alkylation with alkenes provides an atom economic protocol because all of the atoms of the starting materials are incorporated into the products. In 1993, Murai reported a ketone-directed strategy for Ru-catalyzed ortho-C–H alkylation of aromatic ketones with alkenes.2 Following this pioneering reaction, numerous directing groups have been designed for use in regio-selective C–H alkylation reactions.3 It is noteworthy that most of the reports deal with linear-selective alkylation reactions. However, only a limited number of studies that deal with branch-selective C–H alkylation with alkenes have been reported (Fig. 1A).4,5 In this respect, non-directed strategies were discussed for the alkylation of 1,3,4-oxadiazoles (a),4a indoles (b and c),4b,c benzimidazoles (d, e, and j),4d,e,h benzoxazoles (f),4i benzothiazoles (g),4j pyridines (h),4f and azines (i)4g with either styrenes or acrylate esters as coupling partners. A few directed strategies were also demonstrated: Kuninobu and Takai reported a Re-catalyzed branch-selective alkylation of para-substituted phenols (I),5a Yoshikai reported a Co-catalyzed branched alkylation of 2-arylpyridine with styrene derivatives (II),5b Ramana reported a Ru-catalyzed ketone-directed C3-alkylation of 2-aroylbenzofurans with α,β-unsaturated carbonyl derivatives (III),5c Bower reported a carbonyl-directed Ir-catalyzed ortho-alkylation of aromatic ketones (IV),5d Nishimura reported an Ir-catalyzed alkylation of 2-phenylpyridine derivatives with vinyl ethers (V),5e Bower reported an Ir-catalyzed branch-selective ortho-alkylation of acetanilides (VI),5f and an yttrium-catalyzed ortho-alkylation of N,N-dimethylaniline with alkenes was demonstrated by Hou (VII).5g Dong reported an Ir-catalyzed branch selective α-alkylation of ketones with styrenes and unactivated alkenes via the use of an enamine directing strategy (VIII).5h In 2017, Ackermann reported a Co-catalyzed branch-selective alkylation of indole using unactivated alkenes with a detailed mechanistic explanation (IX).5i Yoshikai recently presented a Co-catalyzed N–H imine-directed branch selective alkylation of aromatic imine derivatives with styrenes (X).5j All of these branch-selective alkylation reactions were achieved using styrenes, acrylate esters, vinyl ethers, and in a few cases unactivated 1-alkenes as coupling partners. However, branch-selective alkylation with vinylsilanes has not been achieved to date, although linear selective alkylation with vinylsilanes with the aid of a directing group strategy has been widely explored (Fig. 1B).2,6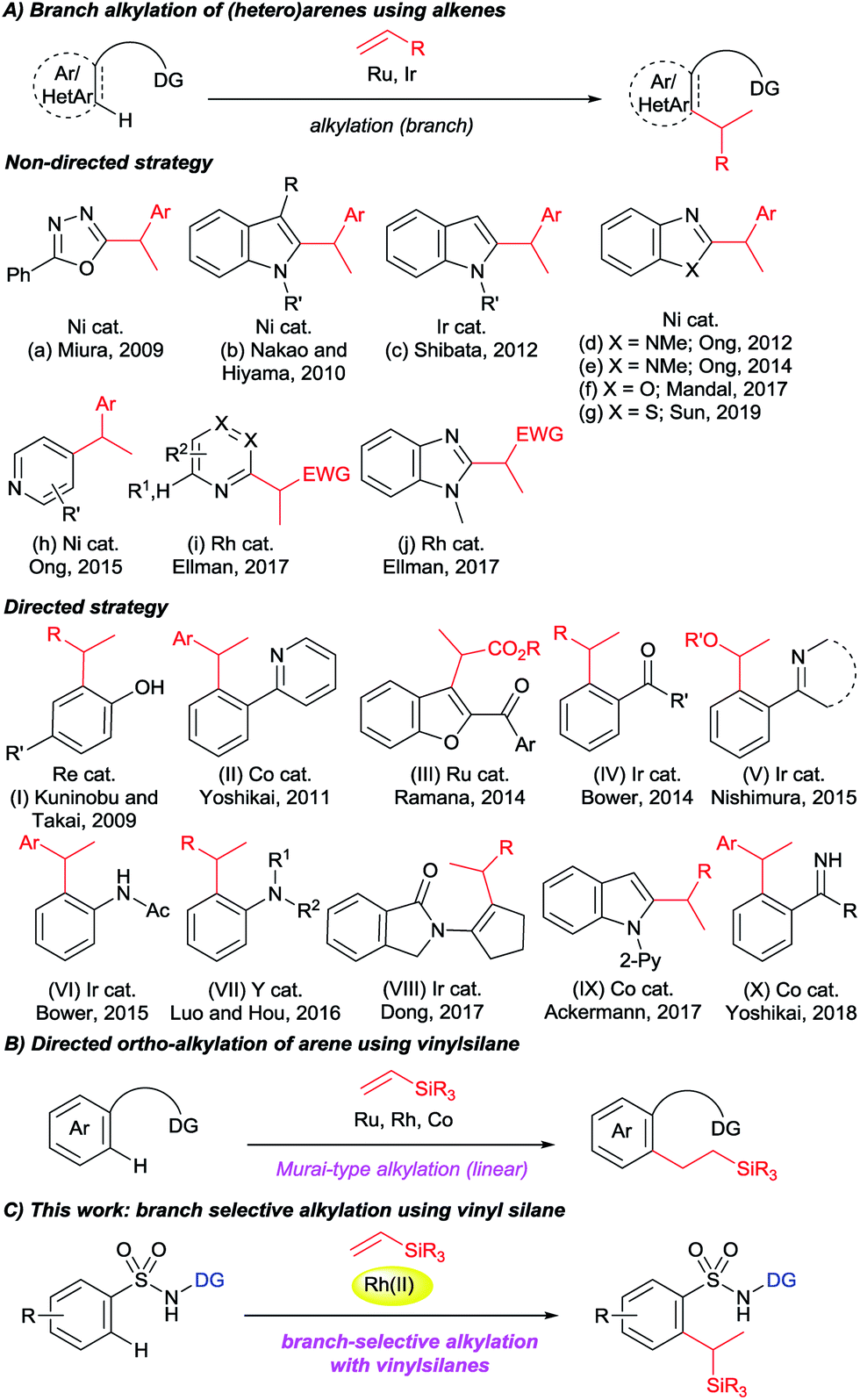 | ||
| Fig. 1 (A) Literature overview of the branched alkylation of (hetero)arenes with alkenes, (B) directed ortho-alkylation of arenes with vinylsilanes, and (C) this work: Rh(II)-catalyzed branched alkylation of sulfonamides with vinylsilanes. | ||
Our group recently reported a series of linear selective C–H alkylations of benzamides,6m,7a–c,g,h naphthylamides,8 and sulfonamides9 with vinyl ketones, acrylate esters, styrenes, N-vinylphthalimides, unactivated 1-alkenes, and vinylsilanes using an 8-aminoquinoline or picolinamide directing group, which was first introduced by Daugulis in 2005.10 Having continuous interest in alkylation reactions,7–9 we were very interested in achieving a branch selective C–H alkylation. Herein, we report on an unusual branch-selective ortho-C–H alkylation of biologically and medicinally important aryl sulfonamides11 with vinylsilanes by taking advantage of an 8-aminoquinoline directing group (Fig. 1C).
Results and discussion
We began our studies by investigating suitable directing groups for the branch-selective alkylation of aryl sulfonamides with triethylvinylsilane in the presence of a [Rh(OAc)2]2 catalyst.12 The reaction of benzenesulfonamide with triethylvinylsilane in the presence of [Rh(OAc)2]2 and 3-chloro-2-methylbenzoic acid remained unreactive (Table 1). The use of weak coordinating N-acetyl and N-phenyl substituted sulfonamides as substrates failed to give the desired product. These observations prompted us to use a strongly coordinating chelation system. However, the use of 2-pyridinylmethylamine and oxazoline-based aniline as directing groups failed to give the desired product. The breakthrough came when 8-aminoquinoline was used as an auxiliary group, giving a 49% yield of the expected product with a decent 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 branch selectivity (Table 1).
:8 branch selectivity (Table 1).
| a Reaction conditions: sulfonamide (0.2 mmol, 1 equiv.), triethylvinylsilane (0.5 mmol, 2.5 equiv.), [Rh(OAc)2]2 (5.0 mol%), and 3-chloro-2-methylbenzoic acid (0.4 mmol) in toluene (0.5 mL) at 160 °C for 24 h. Yields and the ratio of branched and linear isomers were determined by 1H NMR of the crude mixture. N.d. refers to not detected. |
|---|
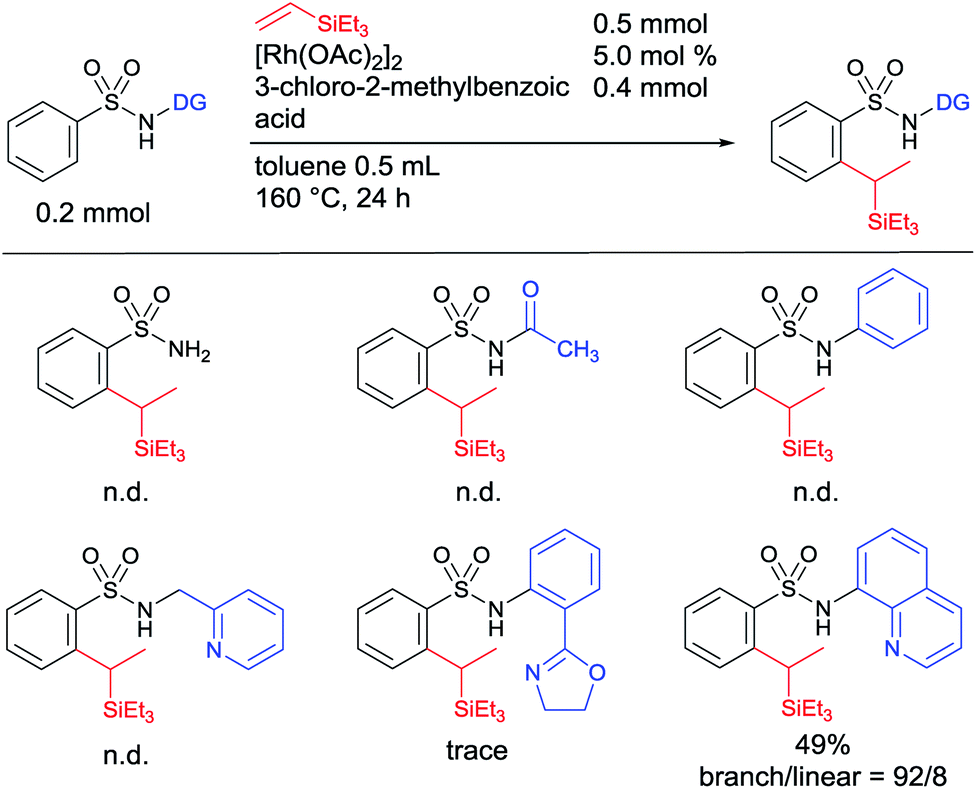
|
To obtain good yield and selectivity, we continued our optimization studies using 1a as a model substrate and triethylvinylsilane (Table 2). The use of other Rh(I) or Rh(III) catalysts failed to show impressive results (entry 1 vs. entries 2–4). Other acid additives were examined next. Although the use of ortho-toluic acid and pivalic acid slightly improved the product yield, the selectivity decreased (entries 6 and 7). The exact role of an acid additive in the selectivity of the reaction is unclear at this point. Among the carboxylic acid additives examined, 3-chloro-2-methylbenzoic acid was the choice of acid. Finally, we found that the use of 7.5 mol% of Rh(II) catalyst and 2 equiv. of 3-chloro-2-methylbenzoic acid in the reaction of amide 1a and 6 equiv. of triethylvinylsilane at 160 °C for 24 h produced the corresponding branched alkylated product 2aa in 72% isolated yield with a high branch selectivity (86:14) (entry 10). Under these optimized conditions, a trace amount of the inseparable alkenylated product 4aa was formed.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Rh cat. | Additive | 2aa + 3aa | 2aa : 3aa | 4aa |
| a Reaction conditions: sulfonamide (0.2 mmol, 1 equiv.), triethylvinylsilane (0.5 mmol, 2.5 equiv.), [Rh(OAc)2]2 (5.0 mol%), and 3-chloro-2-methylbenzoic acid (0.4 mmol, 2 equiv.) in toluene (0.5 mL) at 160 °C for 24 h. Yields and the ratio of branched and linear isomers were determined by 1H NMR of the crude mixture. Isolated yield is given in parentheses. N.d. refers to not detected. b 5.0 equiv. of triethylvinylsilane. c 6.0 equiv. of triethylvinylsilane. | |||||
| 1 | [Rh(OAc)2]2 (5.0 mol%) | 3-Chloro-2-methyl benzoic acid | 42 | 86:14 |
3 |
| 2 | [RhCp*Cl2]2 (5.0 mol%) | 3-Chloro-2-methyl benzoic acid | n.d. | n.d. | n.d. |
| 3 | RhCl(PPh3)3 (10.0 mol%) | 3-Chloro-2-methyl benzoic acid | 8 | 88:12 |
Trace |
| 4 | [Rh(OAc)cod]2 (5.0 mol%) | 3-Chloro-2-methyl benzoic acid | 27 | 82:18 |
4 |
| 5 | [Rh(OAc)2]2 (5.0 mol%) | 2,3-Difluorobenzoic acid | 32 | 85:15 |
n.d. |
| 6 | [Rh(OAc)2]2 (5.0 mol%) | o-Toluic acid | 48 | 80:20 |
5 |
| 7 | [Rh(OAc)2]2 (5.0 mol%) | Pivalic acid | 58 | 75:25 |
5 |
| 8b | [Rh(OAc)2]2 (5.0 mol%) | 3-Chloro-2-methyl benzoic acid | 57 | 86:14 |
4 |
| 9b | [Rh(OAc)2]2 (7.5 mol%) | 3-Chloro-2-methyl benzoic acid | 70 | 86:14 |
2 |
| 10c | [Rh(OAc)2]2 (7.5 mol%) | 3-Chloro-2-methyl benzoic acid | 82 (72) | 86:14 |
3 |
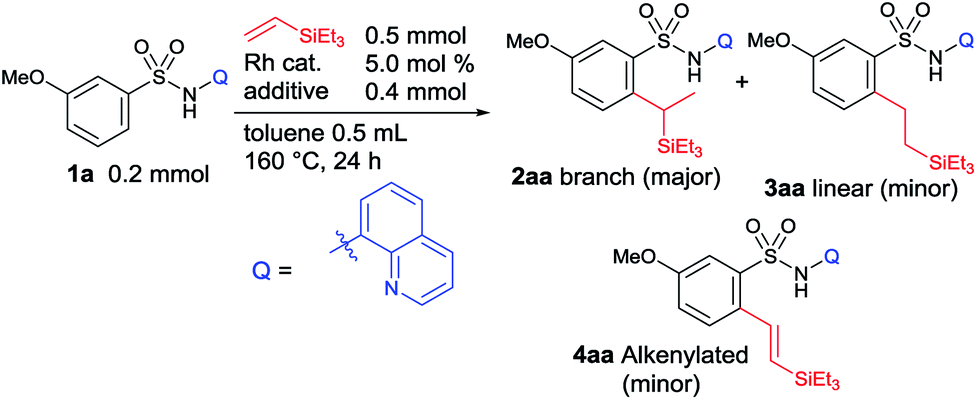
With the optimized conditions in hand, the substrate scope was examined for this branch-selective alkylation and the results are shown in Table 3. We observed that meta-substituted aryl sulfonamides produce the corresponding products in good yield with good selectivity (2aa and 2ba). A complete site-selectivity for less hindered C–H bonds was found. An ortho-Me substituted sulfonamide showed moderate reactivity, giving 2da in 46% yield with a selectivity of 88:12. Most importantly, when para-substituted aryl sulfonamides were used, the corresponding branched alkylated products were obtained in good yields with excellent branch-selectivity over 90:10 (2ca and 2ea–ma). Importantly, this branch selective alkylation reaction is well tolerable for various functional groups such as –OMe, –alkyl, –F, –Cl, –NHCOCH3, –CF3, and –benzylic chloride, giving the desired product without any decomposition of the starting materials. 2-Naphthyl sulfonamide (1na) and a Br-substituted substrate, 4-bromo-3-methylbenzenesulfonamide (1oa), reacted smoothly and produced the desired product in high yield with good selectivity. Higher branch-selectivity was obtained in the case of para-substituted sulfonamides than the ortho- or meta-substituted substrates, suggesting that steric effects play an important role in controlling the selectivity of the reaction. It should also be noted that no dialkylated products were observed in any of the cases. The use of other vinylsilanes such as trimethylvinylsilane, 1,1,1,3,5,5,5-heptamethyltrisiloxane, dimethylphenylvinylsilane, and diethoxymethylvinylsilane as coupling partners produced the corresponding branch-selective alkylation products in good yields (2ab–ae and 2pa–pc).
| a Reaction conditions: sulfonamide (0.2 mmol, 1 equiv.), triethylvinylsilane (1.2 mmol, 6 equiv.), [Rh(OAc)2]2 (7.5 mol%), and 3-chloro-2-methylbenzoic acid (0.4 mmol, 2 equiv.) in toluene (0.5 mL) at 160 °C for 24 h. The ratio of branched and linear isomers was determined by 1H NMR of the crude mixture. Yield of alkenylated product 4 is given in parentheses. b 10 mol% catalyst was used. c 10 equiv. of vinylsilane. |
|---|
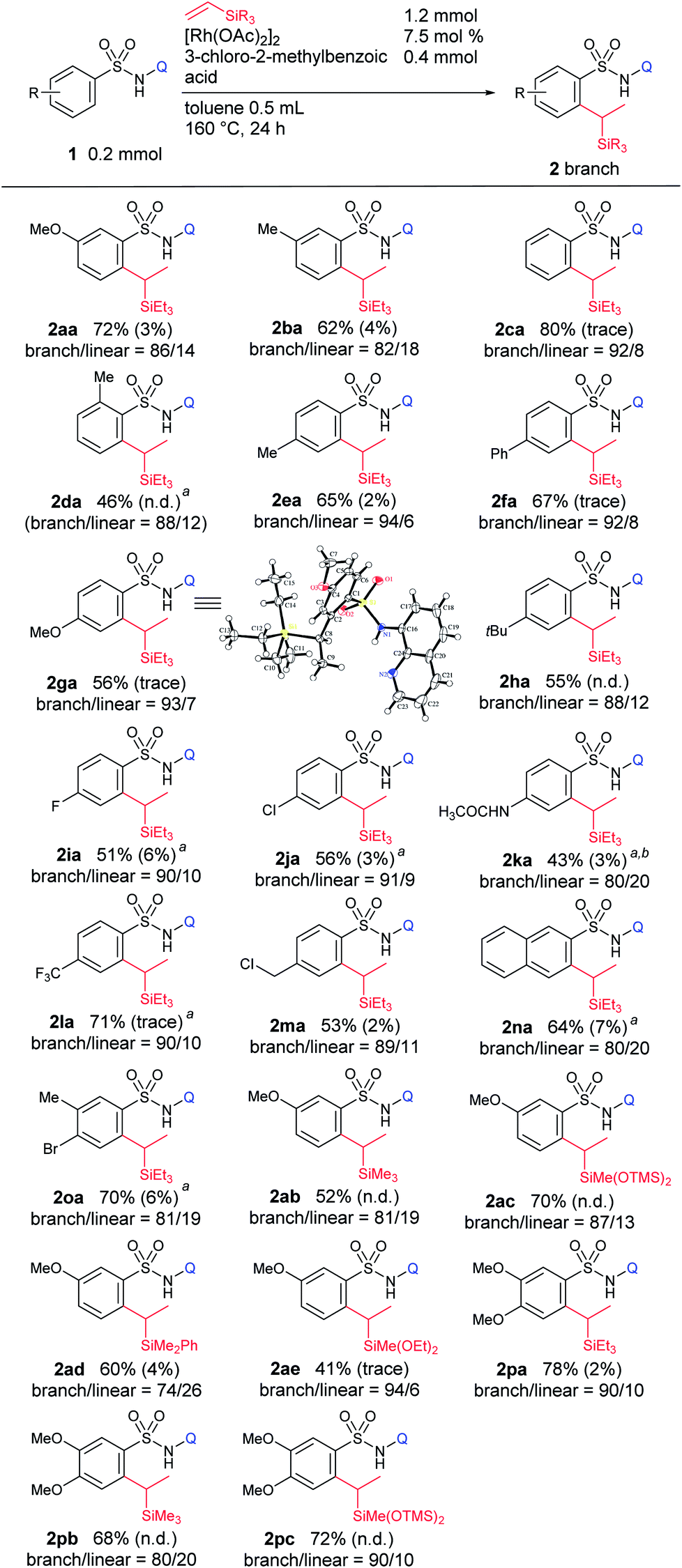
|
To gain insights into the mechanism for this reaction, a series of deuterium labelling experiments were performed (Fig. 2). A significant amount of H/D exchange took place, but only at the ortho-position, when the reaction of sulfonamide 1c with [Rh(OAc)2]2 in the presence of CD3COOD was carried out (Fig. 2a). This result indicates that C–H bond activation is reversible. To collect additional information regarding the mechanism, a reaction between the deuterated sulfonamide 1c-d5 and triethylvinylsilane was performed under the optimized reaction conditions, in which 0.34 D atom (2.66 H) was incorporated at the methyl position (β-position), while no D incorporation was detected at the tertiary carbon center (α-position) of product 6 (Fig. 2b). This observation suggests that a hydrometallation mechanism may be involved. The use of CD3COOD as the only deuterated reagent in a reaction of 1c and triethylvinylsilane gave product 8 in which 0.51 D atom (2.49 H) was incorporated only at the methyl position (β-position) (Fig. 2c). This result implies the involvement of a carbometallation pathway.
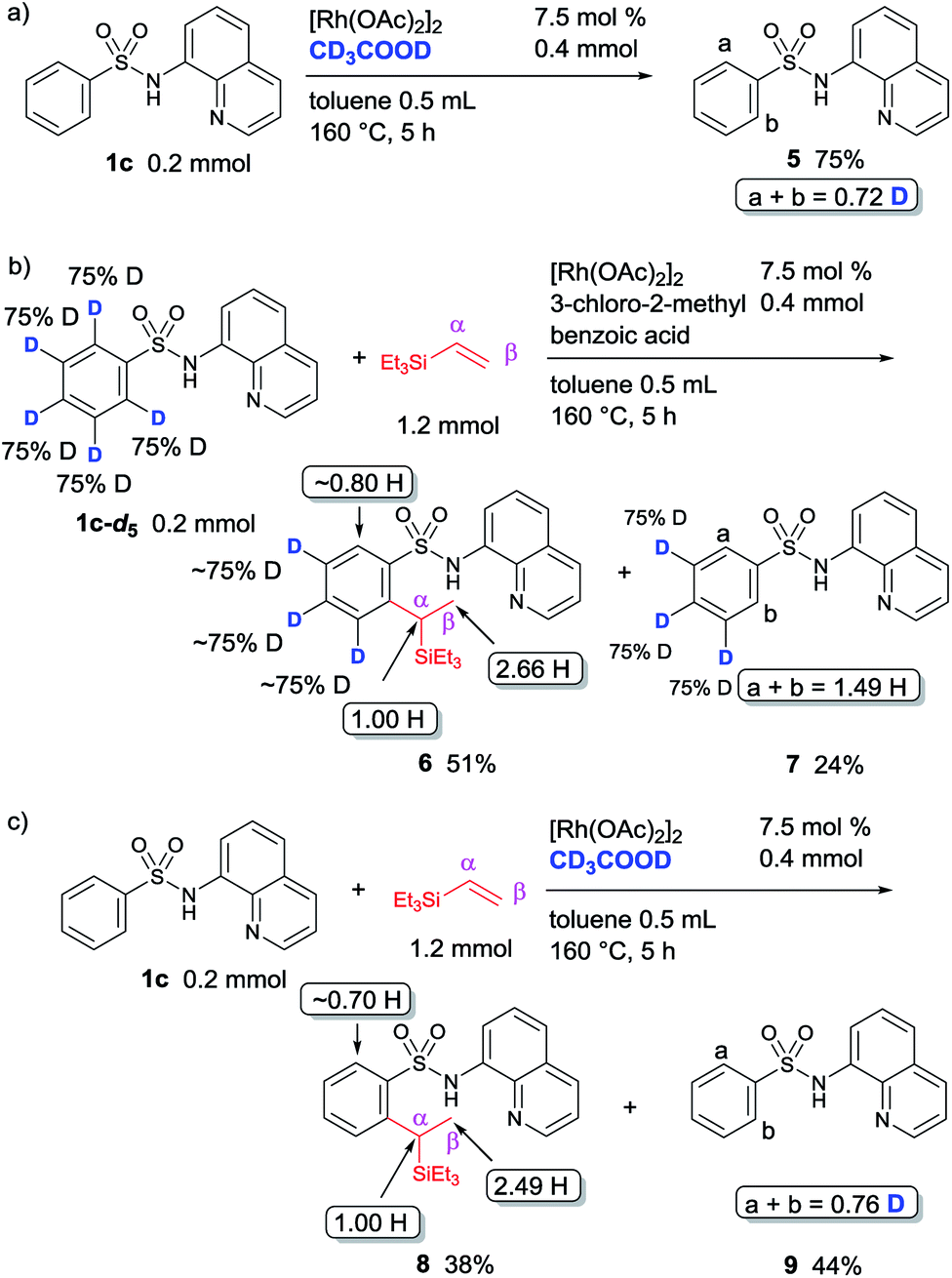 | ||
| Fig. 2 Deuterium labelling experiments. (a) Reaction of 1c in the presence of CD3COOD, (b) reaction of 1c-d5 with triethylvinylsilane, and (c) reaction of 1c with triethylvinylsilane in the presence of CD3COOD. | ||
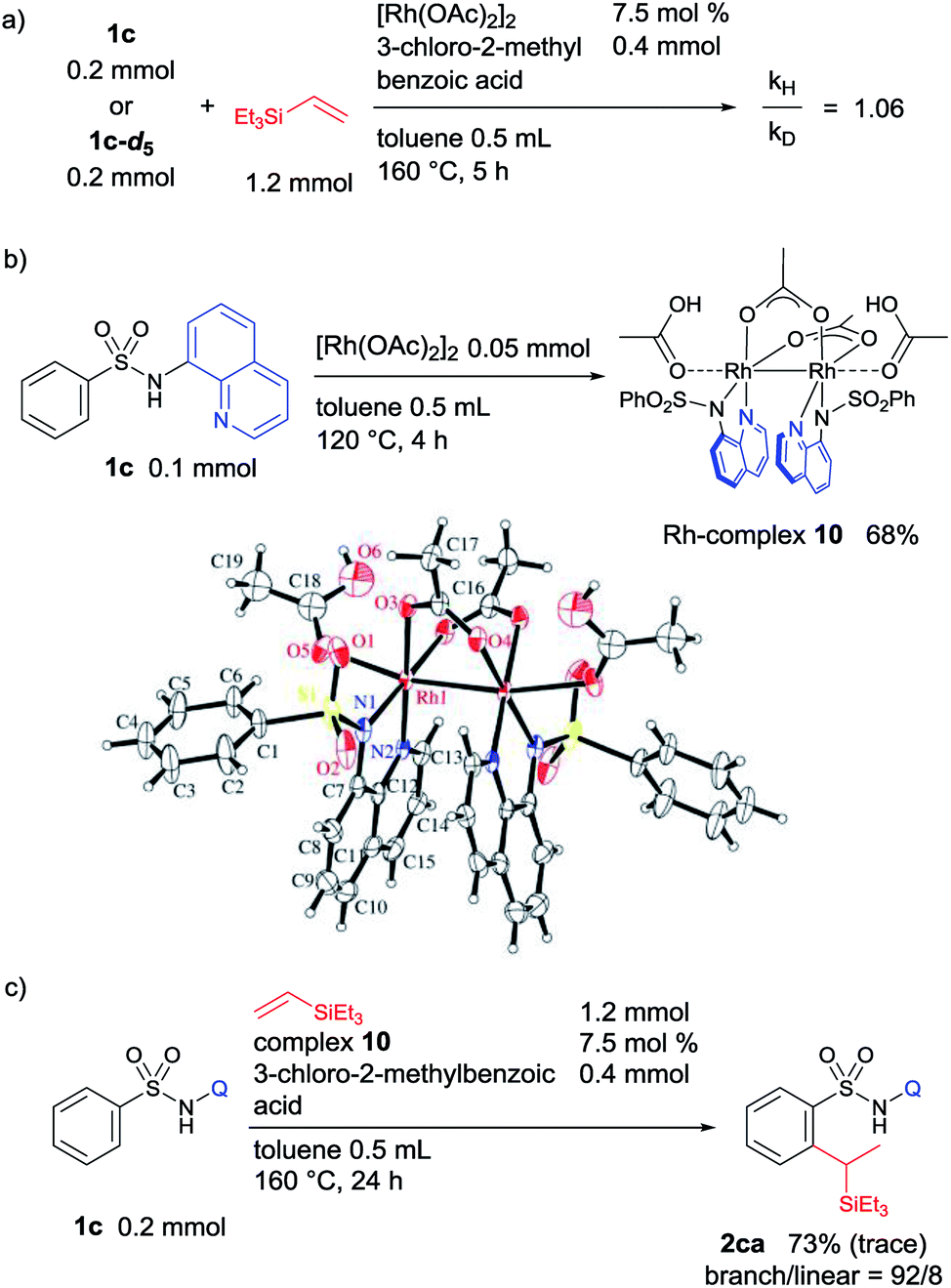 | ||
| Fig. 3 (a) KIE experiment, (b) synthesis of bimetallic Rh-complex 10, and (c) the reaction using complex 10 as a catalyst. | ||
The kinetic isotopic effect (KIE) for this reaction was determined in two parallel experiments using an equimolar amount of 1c or deuterated 1c-d5, and a kH/kD ratio of 1.06 was obtained. This observation indicates that the C–H activation step is not the rate limiting step (Fig. 3a). A stoichiometric reaction of 1c and [Rh(OAc)2]2 was performed, and it resulted in the formation of a dimeric Rh-complex, 10 (Fig. 3b). To trap any other intermediates, several control experiments were performed in the presence or absence of an acid additive with varying temperature; however, it was not possible to isolate the corresponding rhodacycle. A catalytic reaction of 1c and triethylvinylsilane catalyzed by complex 10 under optimized reaction conditions was performed, and it provides a comparable yield and selectivity of product 2ca (Fig. 3c). This result suggested that complex 10 is involved in the catalytic cycle as an intermediate.
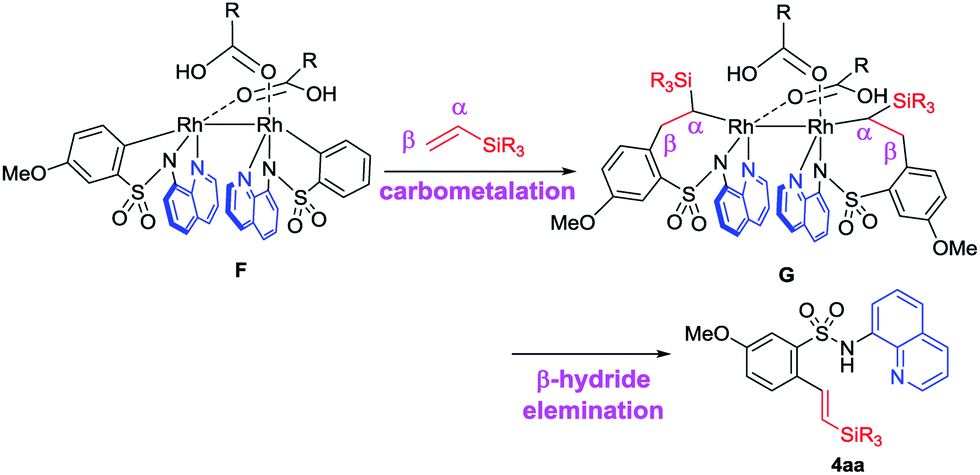
Based on the deuterium studies, we proposed a reaction mechanism that follows two major pathways as shown in Fig. 4. Complexation between Rh(II) and the bidentate sulfonamide initially occurs to form intermediate A, which was isolated as 10 and the structure was confirmed by an X-ray crystallographic analysis (Fig. 3b). Complex A then releases two equivalents of acid to produce B, which is detected in the 1H NMR spectrum (see the ESI†),13 followed by a subsequent oxidative addition of the ortho C–H bond to form a Rh-hydride complex, C. The insertion of a vinylsilane into the Rh–H bond in Cvia a hydrometalation pathway forms intermediate D,14 which then undergoes reductive elimination to generate E. Finally, the product is released from E in the presence of acid, along with the regeneration of the Rh(II) catalyst. According to this proposed pathway, a D-atom should be incorporated only into the β-position of the product when a deuterated sulfonamide is used. In fact, D-incorporation was observed only at the β-position and no D-incorporation was detected at the α-position (Fig. 2b). However, due to the low D-incorporation we concluded that an alternative mechanism could also be involved, as shown in cycle-II. After the formation of C, two molecules of carboxylic acid can be dissociated and covalently coordinated to the Rh-centre to afford a metallacycle, F. Direct formation of F from B could also be possible. The migratory insertion of an alkene into a Rh–C bond forms G,15 which could then react with two equivalents of acid to give the product via Rh-complex E. The results of a deuterium labelling experiment using CD3COOD suggest that D-incorporation took place only at the methyl position (β-position) of the product (Fig. 3c), which is consistent with this proposed catalytic cycle-II. We anticipated that the trace amount of alkenylated product had formed via the migratory insertion of an alkene into a Rh–C bond of F followed by β-hydride elimination.16 The stabilizing effect of two Rh-centers bonded through a single bond could be useful for facilitating double C–H activation at the same time. The exact reason for this unusual branch selective alkylation is currently under investigation in our laboratory.17
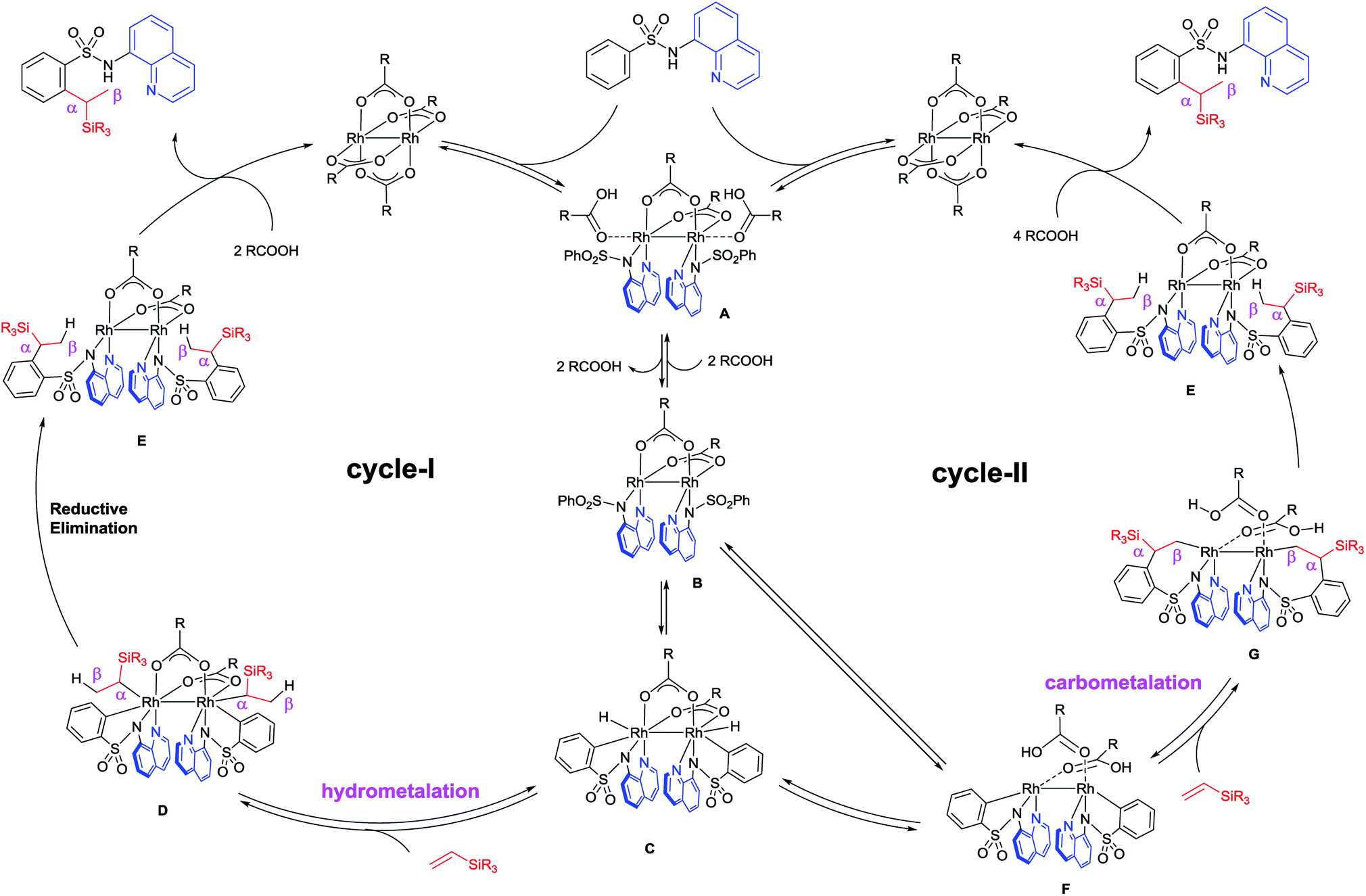 | ||
| Fig. 4 Plausible mechanistic pathways. | ||
Conclusions
In summary, we report the first example of Rh(II)-catalyzed branch-selective ortho-C–H alkylation of aryl sulfonamides with vinylsilanes using an 8-aminoquinoline auxiliary group. Benzenesulfonamide and para-substituted aryl sulfonamides produced selectively mono-(branched)alkylated products without any double C–H activated byproducts being produced. Based on deuterium labelling experiments, a reasonable catalytic cycle is proposed, in which two parallel catalytic pathways, i.e. hydrometalation and carbometalation pathways, are involved. An investigation of the reaction conditions for achieving other branch-selective C–H alkylation reactions is currently ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant in Aid for Specially Promoted Research by MEXT (17H06091).Notes and references
- For selected recent reviews on directing group assisted C–H functionalization, see: (a) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC; (b) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764–7786 RSC; (c) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC; (d) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS; (e) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS; (f) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS; (g) J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163–9227 CrossRef CAS; (h) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (i) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS; (j) S. Rej and N. Chatani, Angew. Chem., Int. Ed., 2019, 58, 8304–8329 CrossRef CAS; (k) A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 10820–10843 CrossRef CAS; (l) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS; (m) P. Gandeepan, N. Kaplaneris, S. Santoro, L. Vaccaro and L. Ackermann, ACS Sustainable Chem. Eng., 2019, 7, 8023–8040 CrossRef CAS; (n) S. M. Khake and N. Chatani, Trends in Chemistry, 2019, 1, 524–539 CrossRef.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS.
- For reviews on directing group assisted C–H alkylation reaction, see: (a) G. E. M. Crisenza and J. F. Bower, Chem. Lett., 2016, 45, 2–9 CrossRef CAS; (b) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS; (c) N. Chatani, Bull. Chem. Soc. Jpn., 2018, 91, 211–222 CrossRef CAS; (d) G. Evano and C. Theunissen, Angew. Chem., Int. Ed., 2019, 58, 7202–7236 CrossRef CAS.
- For papers on non-directed branch-selective alkylation, see: (a) T. Mukai, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2009, 74, 6410–6413 CrossRef CAS; (b) Y. Nakao, N. Kashihara, K. S. Kanyiva and T. Hiyama, Angew. Chem., Int. Ed., 2010, 49, 4451–4454 CrossRef CAS; (c) S. Pan, N. Ryu and T. Shibata, J. Am. Chem. Soc., 2012, 134, 17474–17477 CrossRef CAS; (d) W.-C. Shih, W.-C. Chen, Y.-C. Lai, M.-S. Yu, J.-J. Ho, G.-P. A. Yap and T.-G. Ong, Org. Lett., 2012, 14, 2046–2049 CrossRef CAS; (e) W.-C. Chen, Y.-C. Lai, W.-C. Shih, M.-S. Yu, G. P. A. Yap and T.-G. Ong, Chem.–Eur. J., 2014, 20, 8099–8105 CrossRef CAS; (f) W.-C. Lee, C.-H. Chen, C.-Y. Liu, M.-S. Yu, Y.-H. Lin and T.-G. Ong, Chem. Commun., 2015, 51, 17104–17107 RSC; (g) G. Tran, K. D. Hesp, V. Mascitti and J. A. Ellman, Angew. Chem., Int. Ed., 2017, 56, 5899–5903 CrossRef CAS; (h) G. Tran, D. Confair, K. D. Hesp, V. Mascitti and J. A. Ellman, J. Org. Chem., 2017, 82, 9243–9252 CrossRef CAS; (i) G. Vijaykumar, A. Jose, P. K. Vardhanapu, P. Sreejyothi and S. K. Mandal, Organometallics, 2017, 36, 4753–4758 CrossRef CAS; (j) R.-P. Li, Z.-W. Shen, Q.-J. Wu, J. Zhang and H.-M. Sun, Org. Lett., 2019, 21, 5055–5058 CrossRef CAS.
- For papers on directing group assisted branch-selective alkylation, see: (a) Y. Kuninobu, T. Matsuki and K. Takai, J. Am. Chem. Soc., 2009, 131, 9914–9915 CrossRef CAS; (b) K. Gao and N. Yoshikai, J. Am. Chem. Soc., 2011, 133, 400–402 CrossRef CAS; (c) Y. Kommagalla, K. Srinivas and C. V. Ramana, Chem.–Eur. J., 2014, 20, 7884–7889 CrossRef CAS; (d) G. E. M. Crisenza, N. G. McCreanor and J. F. Bower, J. Am. Chem. Soc., 2014, 136, 10258–10261 CrossRef CAS; (e) Y. Ebe and T. Nishimura, J. Am. Chem. Soc., 2015, 137, 5899–5902 CrossRef CAS; (f) G. E. M. Crisenza, O. O. Sokolova and J. F. Bower, Angew. Chem., Int. Ed., 2015, 54, 14866–14870 CrossRef CAS; (g) G. Song, G. Luo, J. Oyamada, Y. Luo and Z. Hou, Chem. Sci., 2016, 7, 5265–5270 RSC; (h) D. Xing and G. Dong, J. Am. Chem. Soc., 2017, 139, 13664–13667 CrossRef CAS; (i) D. Zell, M. Bursch, V. Müller, S. Grimme and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 10378–10382 CrossRef CAS; (j) W. Xu and N. Yoshikai, Org. Lett., 2018, 20, 1392–1395 CrossRef CAS.
- For papers on directing group assisted linear C–H alkylation using vinylsilanes, see: (a) F. Kakiuchi, T. Sato, M. Yamauchi, N. Chatani and S. Murai, Chem. Lett., 1999, 28, 19–20 CrossRef; (b) F. Kakiuchi, M. Sonoda, T. Tsujimoto, N. Chatani and S. Murai, Chem. Lett., 1999, 28, 1083–1084 CrossRef; (c) F. Kakiuchi, T. Sato, K. Igi, N. Chatani and S. Murai, Chem. Lett., 2001, 30, 386–387 CrossRef; (d) F. Kakiuchi, H. Ohtaki, M. Sonoda, N. Chatani and S. Murai, Chem. Lett., 2001, 30, 918–919 CrossRef; (e) F. Kakiuchi, T. Tsujimoto, M. Sonoda, N. Chatani and S. Murai, Synlett, 2002, 948–951 Search PubMed; (f) R. Martinez, M. O. Simon, R. Chevalier, C. Pautigny, J.-P. Genet and S. Darses, J. Am. Chem. Soc., 2009, 131, 7887–7895 CrossRef CAS; (g) M.-O. Simon, J.-P. Genet and S. Darses, Org. Lett., 2010, 12, 3038–3041 CrossRef CAS; (h) M.-O. Simon, R. Martinez, J.-P. Genet and S. Darses, J. Org. Chem., 2010, 75, 208–210 CrossRef CAS; (i) K. Gao and N. Yoshikai, Angew. Chem., Int. Ed., 2011, 50, 6888–6892 CrossRef CAS; (j) Z. Ding and N. Yoshikai, Beilstein J. Org. Chem., 2012, 8, 1536–1542 CrossRef CAS; (k) M. Schinkel, L. Wang, K. Bielefeld and L. Ackermann, Org. Lett., 2014, 16, 1876–1879 CrossRef CAS; (l) W. Xu and N. Yoshikai, Angew. Chem., Int. Ed., 2016, 55, 12731–12735 CrossRef CAS; (m) C. Wang, S. Rej and N. Chatani, Chem. Lett., 2019, 48, 1185–1187 CrossRef CAS.
- For Ru- or Rh-catalyzed 8-aminoquinoline directed C–H alkylation by our group, see: (a) G. Rouquet and N. Chatani, Chem. Sci., 2013, 4, 2201–2208 RSC; (b) K. Shibata and N. Chatani, Org. Lett., 2014, 16, 5148–5151 CrossRef CAS; (c) K. Shibata, T. Yamaguchi and N. Chatani, Org. Lett., 2015, 17, 3584–3587 CrossRef CAS; (d) K. Shibata and N. Chatani, Chem. Sci., 2016, 7, 240–245 RSC; (e) K. Shibata, S. Natsui, M. Tobisu, Y. Fukumoto and N. Chatani, Nat. Commun., 2017, 8, 1448 CrossRef; (f) Q. He, T. Yamaguchi and N. Chatani, Org. Lett., 2017, 19, 4544–4547 CrossRef CAS; (g) Q. He and N. Chatani, J. Org. Chem., 2018, 83, 13587–13594 CrossRef CAS; (h) T. Yamaguchi, S. Natsui, K. Shibata, K. Yamazaki, S. Rej, Y. Ano and N. Chatani, Chem.–Eur. J., 2019, 25, 6915–6919 CrossRef CAS.
- For a paper on picolinamide directed C(8)–H alkylation of 1-naphthylamine derivatives, see: S. Rej and N. Chatani, ACS Catal., 2018, 8, 6699–6706 CrossRef CAS.
- For a paper on 8-aminoquinoline directed C–H alkylation of sulfonamides, see: S. Rej and N. Chatani, Chem. Commun., 2019, 55, 10503–10506 RSC.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS.
- For the importance of sulfonamides, see: (a) J. D. Larsen and H. Bundgaard, Int. J. Pharm., 1987, 37, 87–95 CrossRef CAS; (b) J. Drews, Science, 2000, 287, 1960–1964 CrossRef CAS; (c) A. Scozzafava, T. Owa, A. Mastrolorenzo and C. T. Supuran, Curr. Med. Chem., 2003, 10, 925–953 CrossRef CAS.
- Besides carbene and nitrene transfer reactions, the use of a Rh(II) catalyst in C–H alkylation reactions is very rare in the literature. For a C–H arylation, see: J. Kwak, M. Kim and S. Chang, Rh(NHC)-catalyzed direct and selective arylation of quinolines at the 8-position, J. Am. Chem. Soc., 2011, 133, 3780–3783 CrossRef CAS.
- Complex 11, which is similar to B, was detected by 1H NMR when complex 10 was washed with hexane and then dried under vacuum (ESI†). This complex was also detected when the reaction of sulfonamide 1c and [Rh(OAc)2]2 was carried out in the presence of 3-chloro-2-methylbenzoic acid.
- For a paper on metal–hydride insertion into alkenes, see: J. Yu and J. B. Spencer, J. Am. Chem. Soc., 1997, 119, 5257–5258 CrossRef CAS.
- For papers on the migratory insertion of metal–C bonds into alkenes, see: (a) J. Ledford, C. S. Shultz, D. P. Gates, P. S. White, J. M. DeSimone and M. Brookhart, Organometallics, 2001, 20, 5266–5276 CrossRef CAS; (b) C. N. Iverson and W. D. Jones, Organometallics, 2001, 20, 5745–5750 CrossRef CAS; (c) A. Haynes, C. E. Haslam, K. J. Bonnington, L. Parish, H. Adams, S. E. Spey, T. B. Marder and D. N. Coventry, Organometallics, 2004, 23, 5907–5909 CrossRef CAS; (d) J. Vicente, J.-A. Abad, W. Förtsch, M.-J. López-Sáez and P. G. Jones, Organometallics, 2004, 23, 4414–4429 CrossRef CAS; (e) L. Li, Y. Jiao, W. W. Brennessel and W. D. Jones, Organometallics, 2010, 29, 4593–4605 CrossRef CAS; (f) Y. Jiao, M. E. Evans, J. Morris, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2013, 135, 6994–7004 CrossRef CAS PubMed.
- Alkenylated products such as 4aa were obtained from β-hydride elimination in complex G, which is responsible for the formation of a linear alkylated product
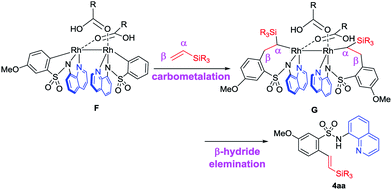 .
. - The intermediacy of mono-nuclear Rh species cannot be excluded..
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectroscopic data. CCDC 1910136, 1918726. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04308j |
| This journal is © The Royal Society of Chemistry 2020 |