
Improvement of silver azide crystal morphology and detonation behavior by fast mixing using a microreaction system with an integrated static micromixer†
Cong
Chen
 a,
Shuangfei
Zhao
a,
Peng
Zhu
*a,
Jinyu
Shi
a,
Fanyuhui
Yan
a,
Huanming
Xia
b and
Ruiqi
Shen
a
a,
Shuangfei
Zhao
a,
Peng
Zhu
*a,
Jinyu
Shi
a,
Fanyuhui
Yan
a,
Huanming
Xia
b and
Ruiqi
Shen
a
aSchool of Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: zhupeng@njust.edu.cn
bSchool of Mechanical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China
First published on 15th November 2019
Abstract
A passive microreaction system with excellent mixing performance was used for the optimization of the size and shape of silver azide (SA) at the microscale. This safe and cost-effective method is characterized by high mixing efficiency, low reagent consumption, and rapid preparation. To exploit the difference, the preparation of SA at the microscale using the passive microreaction system was compared with that at the macroscale in a beaker. The results show that the preparation of SA using the passive microreaction system under microscale conditions has obvious advantages over its preparation in a beaker in terms of crystal morphology, particle size, particle size distribution and thermal stability. The shape of SA prepared in the system is spherical or spherical-like, while the morphology of SA synthesized in a beaker is mostly pyramidal with sharp points. The particle size of SA prepared in the microreaction system ranges from 712.4 nm to 1106.4 nm, while that of SA prepared in a beaker ranges from 255.0 nm to 825.0 nm. In addition, the detonation velocity of submicron-SA is 1850 m s−1, which is 150 m s−1 higher than that of confined SA published in the literature. This study demonstrates the feasibility of a safe and efficient method for fast preparation of SA with improved physical properties.
1 Introduction
Metal azides, consisting of a metal atom and an azide molecule, have attracted considerable attention because of their industrial applications as primary explosives, as gas generators, and even as monolayer crystal materials.1–4 Silver azide (SA), a typical metal azide, is a sensitive and powerful solid explosive that is used for initiating explosives.5,6 Conventionally, SA is synthesized from silver nitrate and sodium azide in a beaker.7 However, there is a great danger in the preparation and post-treatment of SA on a gram scale owing to the wide range of heat accumulation in the beaker. At the macroscale, insufficient mixing, nonhomogeneous temperature, and variations in the concentration distribution are challenges for reproducible preparation of SA and tend to produce products with inhomogeneous shapes and wide particle size distributions (PSDs),8,9 which affect its performance and applications in practice. Moreover, the raw materials are relatively expensive, and it is easy to waste resources when preparing SA in a beaker. Therefore, it is important to find a safe, fast and efficient method to synthesize SA with good crystal morphology and excellent flowability.Microfluidic technology provides a unique environment for the investigation of synthesis processes at the nano- or microscale. Recently, microfluidic reactors have emerged as an attractive technology in chemical synthesis to produce micro- and nanosized particles10–14 due to their precisely controlled reaction parameters, improved mixing and energy efficiency, effective mass and heat transfer and minimal reagent consumption.15,16 These effects are direct results of using microscale reactors. Under microscale conditions, the exquisite control of flow and mixing conditions led to the improved homogeneity of PSD and the control of particle size in a reproducible fashion. In virtually all cases, microfluidic procedures have been found to offer obvious advantages over macroscale synthesis – most notably in terms of their superior ability to fine-tune the physical properties of the final product.17 We have also done some research on the regulation and optimization mechanism of the crystal form and particle size of micro–nano explosives during the recrystallization/preparation process under microscale conditions.18–21 The passive microreaction system, as one of the microfluidic devices, utilizes no external energy input other than the pressure head used to drive the fluid flow,22–24 which makes the system safer for the synthesis of primary explosives. Compared with active micromixer platforms, passive microreaction systems have clear advantages in terms of energy consumption, fabrication and scope of applications. With controllable mixing efficiency, nucleation and crystal growth are easier to study, which provides the possibility of controlling the crystal shape and particle size. Therefore, passive microreaction systems have been widely used for tuning the particle size, PSD and crystal morphology.25–27 However, the current studies on SA mainly focus on its electronic band and crystal structure,28–30 and there have been no reports about the optimization of the size and shape of SA at the microscale.
In this paper, a passive microreaction system with excellent mixing performance was designed for optimizing the synthesis process of SA particles at the microscale. Rapid mixing of the reagents was realized using a passive micromixer, which resulted in a significantly faster synthesis rate of SA than that of SA prepared in a beaker. This is the first study to use microfluidic technology to optimize the crystal shape, particle size and PSD of SA on a microscale. Furthermore, a comparison of SA synthesized in the passive microreaction system and in a beaker was made, and the crystal morphology, PSD and heat release were investigated. The results show that SA synthesized at the microscale has better physical properties. Finally, the prepared samples were characterized by preliminary sensitivity and laser detonation tests.
2 Experimental section
2.1 Materials
In this study, the reactants for the synthesis of SA included silver nitrate (Sinopharm Chemical Reagent Co., Ltd, China, ≥99.8%) and sodium azide (Hubei Dongfang Chemical Industry Co., Ltd, China). Deionized water (EPED-10TJ, China, filter system) was used for all preparation and rinsing steps. Nitric acid (Sinopharm Chemical Reagent Co., Ltd, China) and ammonia water (Sinopharm Chemical Reagent Co., Ltd, China) played a role in adjusting the pH of sodium azide and silver nitrate solutions, respectively.2.2 Microfluidic devices
The mixing efficiency of the reagents is critical to the crystal form and PSD of SA, while also affecting the reaction efficiency. Therefore, a multi-cross-flow mixer (MCFM) was designed to enhance the initial mixing of the reagents. Fig. 1(a) shows a photograph of the one-to-four numbered-up microreaction system for the preparation of SA. This passive microreaction system includes two syringe pumps for continuous fluid drive, two sets of 4-fold parallelized fluid delivery manifolds, 4-fold parallelized MCFMs for rapid mixing of the reagents, a beaker for sample collection and connecting PTFE (polytetrafluoroethylene) tubes with an inner diameter of 0.8 mm and outer diameter of 1.6 mm. A schematic diagram of the single-channel passive microreaction system is shown in Fig. 1(b). The single-channel microreaction system is used for preliminary screening of the optimal experimental conditions, and the one-to-four numbered-up microreaction system is applied to achieve larger production volumes.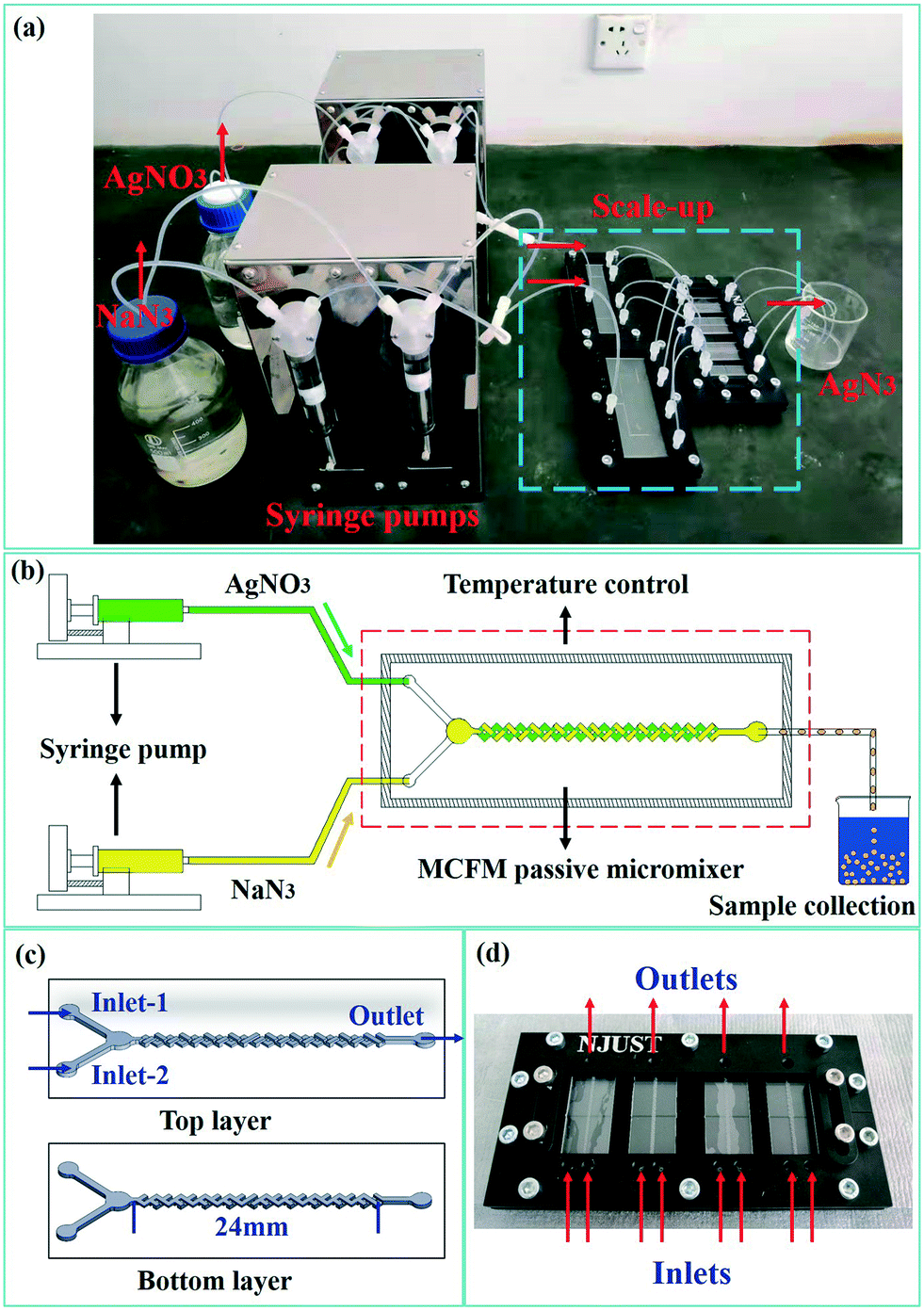 | ||
| Fig. 1 A photograph of the one-to-four numbered-up microreaction system for the synthesis of SA (a), schematic diagram of the single-channel passive microreaction system (b), the structures of an MCFM (c), and a photograph of the 4-fold parallelized MCFMs (d). | ||
The structures of the top and bottom layers are shown in Fig. 1(c), and a photograph of the 4-fold parallelized MCFMs is shown in Fig. 1(d). The passive micromixer is fabricated with 1.8 mm-thick transparent polymer (polymethylmethacrylate, PMMA) plates and can withstand temperatures up to 85 °C. CNC micro-milling (with end mill 7256 Φ0.25, DIXI) is used for machining of the microstructures. Then the PMMA plates are bonded layer by layer using acrylic glue to form the complete micromixer. The MCFM consists of a chamber and multiple three-dimensional crossing channels. The crossing channels are perpendicular to each other and have an angle of 45° with the principal axis of the MCFM. For materials with a faster nucleation rate, the chamber is designed to avoid channel blockage from fine precipitation. The diameter and depth of the chamber are 2.5 mm and 0.5 mm, respectively. Both the width and the depth of the channels are 0.5 mm. In addition, the length, mixing length, width and thickness of the MCFMs are 41 mm, 24 mm, 16 mm and 3.6 mm, respectively. Through continuous stretching, folding and splitting, reorientation and recombination, chaotic advection is generated and the contact material interface is greatly enlarged. Using this special structure, the two reagents can be mixed rapidly in the microchannel without any blockage.
2.3 Preparation of SA
Both silver nitrate and sodium azide were dissolved in deionized water at a concentration of 0.1 M and then drawn into syringes. The syringe pumps drove the solutions of silver nitrate (10 mL, pH = 7.5) and sodium azide (10 mL, pH = 7.0) in the syringes to flow along the tubes to the MCFMs. The flow rates of the two reactants were both 4.0 mL min−1, resulting in a total flow rate of 8.0 mL min−1. This was the optimum flow rate as screened by the experiments, and the details are provided in the ESI.† The MCFMs were kept in a water bath at 65 °C, and then the white colloidal liquid containing SA flowed along a 15 cm PTFE tube to the beaker. Finally, the crystals were collected by vacuum filtration and were rinsed with deionized water. The time required to complete a single experiment using the single-channel microreaction system was approximately 2.5 min (10 mL). Therefore, the efficiency was increased by three times using the one-to-four numbered-up microreaction system, and the time required was only 2.5 min (40 mL).To prove the superiority of the passive microreaction system in the synthesis of SA, a comparison between using the passive microreaction system and a beaker was made under the same experimental parameters. SA particles were synthesized by adding dropwise sodium azide (40 mL, 0.1 M, pH = 7.0) into a solution of silver nitrate (40 mL, 0.1 M, pH = 7.5) at 65 °C over a period of 30 min and continuously stirred. The reaction was kept at 65 °C for an additional 5 min after the addition was completed. The crystals were collected by vacuum filtration and were rinsed with deionized water. The time for a single experiment was approximately 35 min, which is much longer than the time for the synthesis of SA using the one-to-four numbered-up microreaction system.
2.4 Characterization methods
The composition and morphology of SA were characterized by powder X-ray diffraction (XRD, Bruker D8-advanced diffractometer equipped with a Cu-Kα X-ray source operating at 40 kV and 40 mA) and scanning electron microscopy (SEM, Zeiss Merlin at 10 kV). Particle size distributions were determined by dynamic light scattering (DLS) using a Zetasizer Nano ZS9 (Malvern Instruments GmbH, Germany). In addition, the thermal properties were characterized by differential scanning calorimetry (DSC, DSC-823e, Mettler Toledo) at a heating rate of 5 K min−1. To evaluate the safety performance of the SA synthesized using the passive microreaction system, the impact sensitivity (BAM Fallhammer) and electrostatic sensitivity (JGY-50III) were investigated. In addition, the SA was directly ignited with a pulsed laser to investigate its explosive properties. The wavelength, pulse width and incident laser energy were 1064 nm, 6.5 ns and 126.9 mJ, respectively. The whole explosion process was recorded using a high-speed camera (Phantom VEO 710L) that runs at 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 frames per second.
000 frames per second.
3 Results and discussion
3.1 Mixing performance of the MCFM
The mixing performance of the MCFM was studied by numerical simulation using ANSYS FLUENT 19.0. Liquid water with a density of 9.998 × 102 kg m−3 and a viscosity of 9 × 10−4 kg m−1 s−1 was selected as the working fluid for mixing characterization because the feed solutions have similar properties to water. All meshes in the simulations were composed of uniform quadrilateral cells, and the element size of each mesh was set to 0.2 mm. The wall surface of the channel was stationary and had no slip. All the fluids were assumed to be incompressible and isothermally constant in the numerical calculation process. In addition, it was verified by the encrypted mesh that the obtained data were mesh independent. The width of the inlet channels was 0.5 mm. The flow rates of inlet-1 and inlet-2 were both fixed at 4.0 mL min−1, and the corresponding residence time was 230 ms. Therefore, the Re value at this flow rate was 296 (Re = ρdu/μ, where ρ is the density of the solutions, d is the hydrodynamic diameter of the channel, which is 0.5 mm in the micromixer, u is the total flow rate of the solutions, and μ is the viscosity). The mass fraction of inlet-1 was set to 0, and the corresponding color was red. The mass fraction of inlet-2 was set to 1, and the corresponding color was blue.Fig. 2 shows the simulation results of the mixing performance of the MCFM at Re = 296. The results show that complete mixing is achieved at a distance of 7.19 mm from the inlet, and the corresponding time for complete mixing is approximately 57.5 ms. As the fluids are driven downstream, they are gradually subdivided into thin stream layers. This greatly enlarges the contact material interface and promotes mixing.
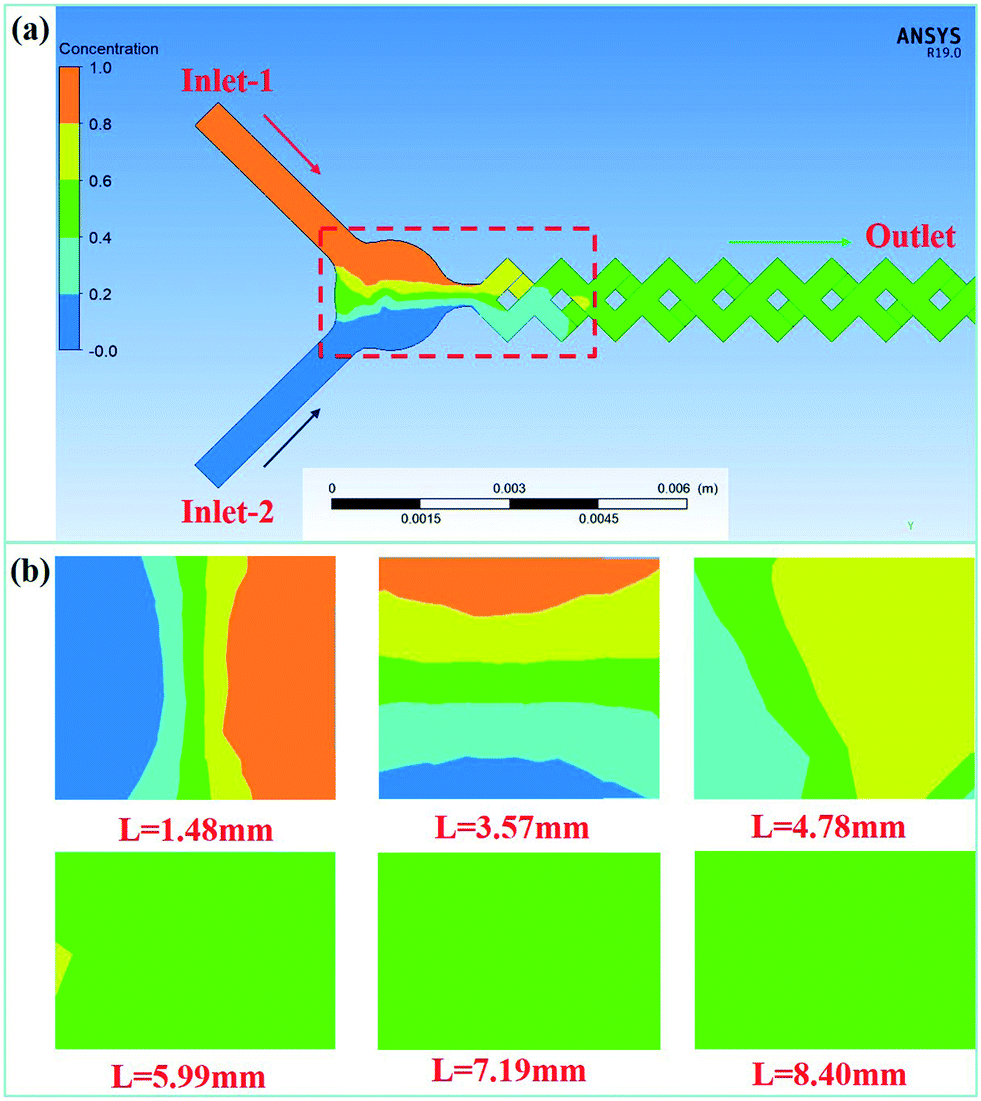 | ||
| Fig. 2 (a) Mixing results of the MCFM at Re = 296; (b) mixing results at the sampled planes. | ||
To further study the mixing performance of the MCFM at different flow rates, theoretical calculations with flow rates of 2.0 mL min−1, 3.0 mL min−1, 5.0 mL min−1 and 6.0 mL min−1 were also conducted. The results and relevant details are provided in the ESI.† Due to the excellent performance of the MCFM, rapid mixing can be achieved when the flow rate ranges from 2.0 mL min−1 to 6.0 mL min−1.
3.2 Comparison between the passive microreaction system and a beaker
The mixing efficiency and homogeneity are critical to the crystal shape, particle size and PSD. The preparation of SA is a fast reaction process that requires a higher mixing efficiency to synthesize SA with a more uniform crystal shape and narrow PSD. It is worth noting that this passive microreaction system is mainly suitable for substances with fast nucleation and low solubility in reaction solutions at a specific temperature. For example, lead azide, another common primary explosive, can also be quickly synthesized using this microreaction system with an integrated passive micromixer. For comparison, the preparation of SA was conducted with the two methods, and the details are in the Experimental section.The XRD patterns of the SA particles prepared in the passive microreaction system and in a beaker are summarized in Fig. 3(a). The diffraction peaks of the two samples are located at 2θ = 21.76°, 30.06°, 31.94°, 37.28°, 42.82°, 44.37°, 48.93°, 51.28°, 54.58°, 58.36°, 60.46°, 61.80° and 66.23°, which are in good agreement with the values in the standard card (PDF# 03-0906). The results show that the SA prepared using the passive microreaction system has a similar crystal structure to that synthesized by the literature method. However, the intensities of the diffraction peaks of the two samples are different. Some peaks of the SA obtained by the literature method are very weak or even disappear, which implies that the growth of some crystal planes is very weak, such as the (132), (312), (004) and (114) planes. It can be predicted that the particle size of SA prepared by the batch method may be smaller than that of SA synthesized using the passive microreaction system.
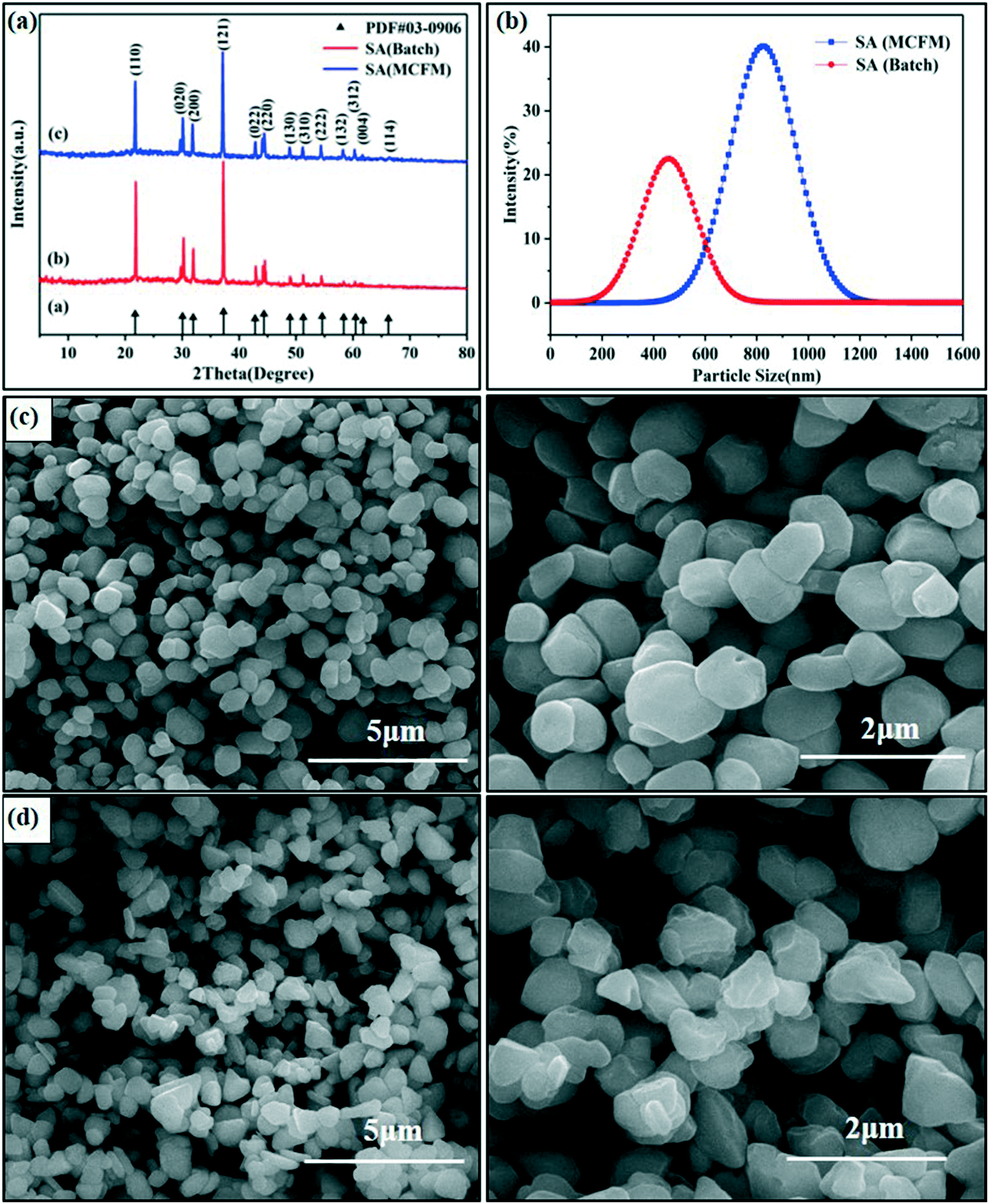 | ||
| Fig. 3 XRD patterns of SA crystals (a), PSDs (b), SEM images of SA prepared using the passive microreaction system (c) and SEM images of SA synthesized by the batch method (d). | ||
To further compare the crystal morphologies and particle sizes of SA synthesized by these two methods, SEM tests and particle size analyses of SA were conducted. It can be clearly seen that the morphology of SA obtained using the passive microreaction system is spherical or spherical-like and has good free-flowing properties (see Fig. 3c), while the morphology of SA synthesized by the batch method is mostly pyramidal with sharp points (see Fig. 3d). In addition, the average particle size of SA prepared in the passive microreaction system is 871.6 nm, and the particle size ranges from 712.4 nm to 1106.4 nm. In contrast, the average particle size of SA prepared by the batch method is 465.1 nm, and the particle size ranges from 255.0 nm to 825.0 nm (see Fig. 3b). The particle size of SA synthesized using the passive microreaction system is much larger than that of SA prepared in the beaker, and the PSD is narrower, which is beneficial to its flowability and initiation efficiency. The reason for the differences in the particle size between the two samples is discussed further below.
The thermal behaviors of SA synthesized using the passive microreaction system and batch method were determined by thermogravimetry (TG) and differential scanning calorimetry (DSC), as shown in Fig. 4. During the testing process, the heating rate was set to 5 K min−1 under the conditions of flowing Ar gas, and the heating temperature ranged from 50 °C to 500 °C. The masses of the two samples were both 0.23 mg. The weight loss and DSC curves for SA prepared by the batch method are shown in Fig. 4(a). The results show that the weight loss exhibits two major regimes: a melting process followed by the decomposition of SA at 307.6 °C to 317.2 °C, which is in agreement with the corresponding DSC curve. At a temperature above 307.6 °C, the compound melts decompose rapidly to silver and nitrogen gas, and the heat release of SA synthesized by the batch method is 1714 J g−1. Fig. 4(b) shows the mass loss and DSC curves for SA obtained with the passive microreaction system. Both SA particles prepared using the passive microreaction system and a beaker experience the melting and thermal decomposition processes. The difference is that the explosion process of SA synthesized using the passive microreaction system has a maximum at 320.7 °C and has a spread from 317.5 °C to 323.9 °C. In addition, the heat release is 1698 J g−1, which is 16 J g−1 lower than that of SA prepared in the beaker. The reason for the reduction in the heat release is that the surface energy of submicron-SA prepared with the passive microreaction system is lower than that of nano-SA synthesized in the beaker. However, the initial thermal decomposition temperature of SA obtained by the batch method is 307.6 °C, 9.9 °C lower than that of SA synthesized using the passive microreaction system. This is because submicron-SA prepared with the microreaction system has a larger particle size and a narrower PSD. Therefore, the heat transfer of submicron-SA is less efficient, which ensures higher safety and better thermal stability and compatibility.
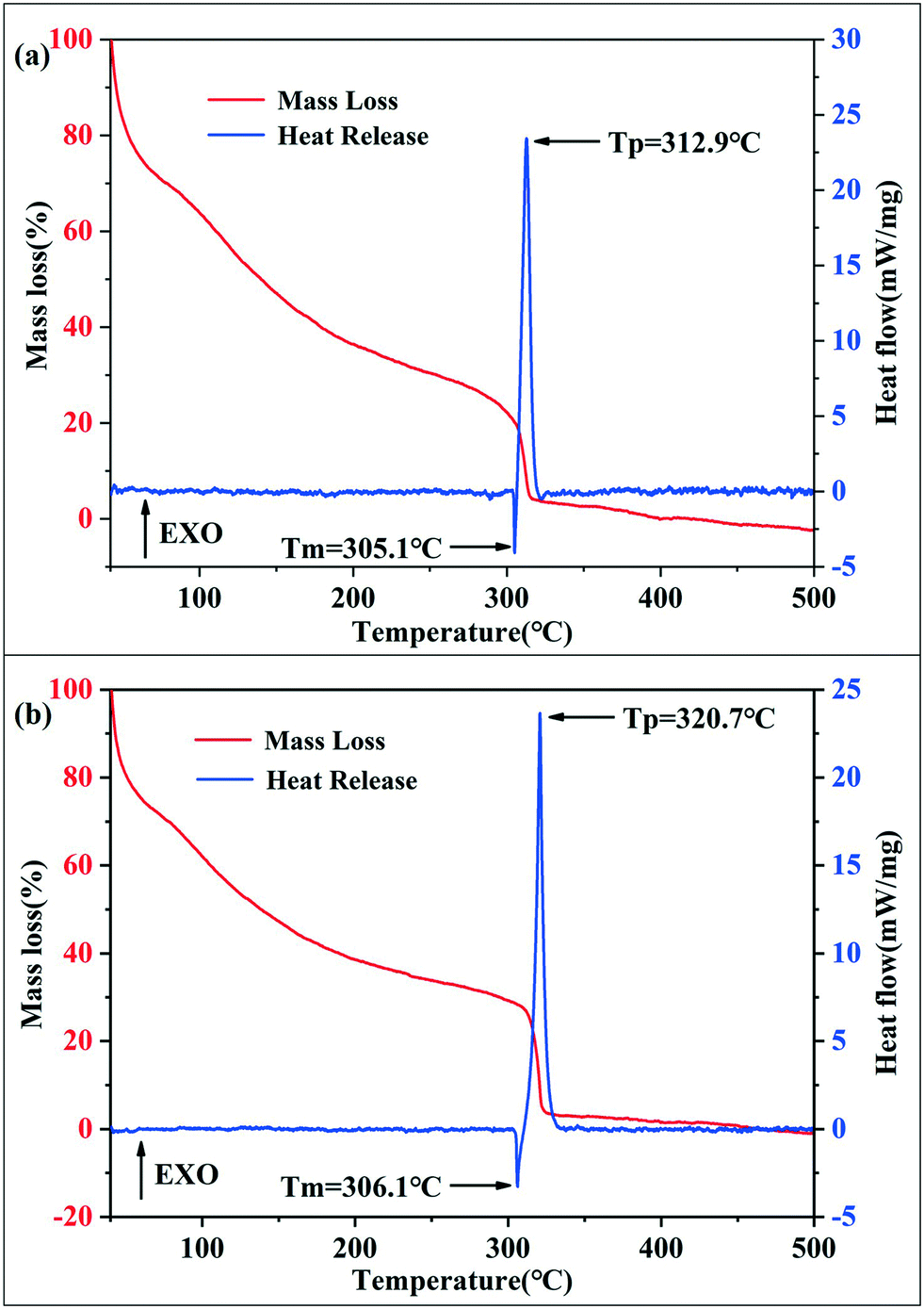 | ||
| Fig. 4 TG-DSC analysis of SA synthesized by two methods: (a) batch method and (b) passive microreaction system. | ||
3.3 Theoretical basis for the differences in particle size
The preparation of SA is a fast reaction process consisting of a nucleation process and a crystal growth process. The particle size of SA is determined by the nucleation rate and growth rate.31 Under microscale conditions, the transport rate of SA molecules can be easily improved by increasing the mixing efficiency. In the microfluidic platform, the diffusion includes free diffusion and convection. The mixing in the microchannel takes place by free diffusion and convection depending on the flow geometry and the operating conditions used. In simple channels (i.e., with smooth walls), the flow is almost always laminar and the mixing occurs purely via diffusion. The mixing time can be calculated by eqn (1). Obviously, the lower the τ value is, the better the mixing is.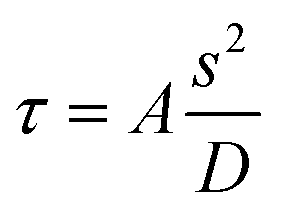 | (1) |
In this microreaction system, a passive micromixer was introduced and the convection–diffusion was greatly enhanced. The simulation results above show that the corresponding time for complete mixing is approximately 57.5 ms, which is more than two orders of magnitude faster than the free diffusion. In addition, the Péclet number is too large (Pe = Ul/D > 100, where Pe is the ratio between advection and diffusion and l is the characteristic length). Even on the scale of a microchannel, the diffusive mixing is slow compared with the convection along the channel. Therefore, the mixing in the system can be regarded as the convection of materials caused by the MCFMs.
Both the nucleation and growth processes can be divided into two processes: diffusion process and precipitation reaction. The diffusion process is determined by the mixing efficiency, and the precipitation reaction is determined by relative supersaturation. In the nucleation stage of the SA crystal, the nucleation rate equation can be expressed in the form of the Arrhenius reaction rate equation (eqn (2)):
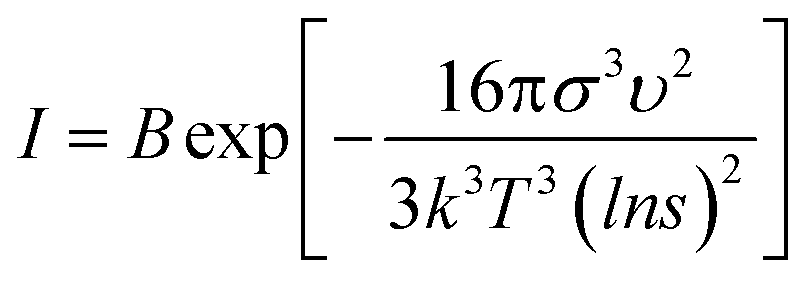 | (2) |
The relative supersaturation can be calculated by eqn (3).
| s = C/C0 | (3) |
According to eqn (2), the factors affecting the nucleation rate are σ, υ, T and s. For the synthesis of SA, υ and T are determined values. The concentration and pH values of the reagents are the same for the two methods, so σ can be regarded as a determined value. Therefore, the nucleation rate depends on the relative supersaturation s. For the passive microreaction system, the droplets of silver nitrate and sodium azide flow along the tubes to the MCFMs, followed by the formation of white colloidal droplets containing SA. For the preparation in the beaker, SA particles were synthesized by adding dropwise sodium azide into the solution of silver nitrate at 65 °C, which results in the concentration of SA in the solution being much smaller than that of SA in the droplets. Therefore, the value of C in the passive microreaction system is greater than that of s in the beaker. However, the solubility of SA is extremely low in aqueous solutions. After integration, we can clearly see that the nucleation rate of SA in the passive microreaction system is approximately equal to that of SA in the beaker.
Generally, the growth rate of SA is determined by both the diffusion process and the surface reaction process. Thus, the growth rate of SA determined by the diffusion process and the surface reaction process can be indicated as eqn (4) and (5), respectively. On the basis of eqn (4) and (5), the growth rate of SA can be calculated by eqn (6)
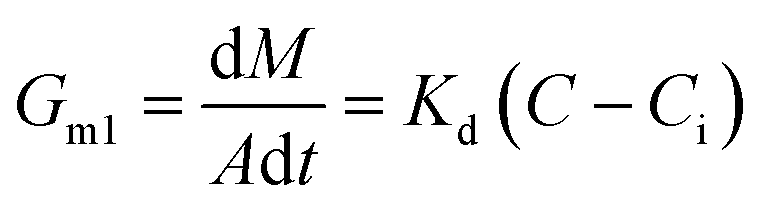 | (4) |
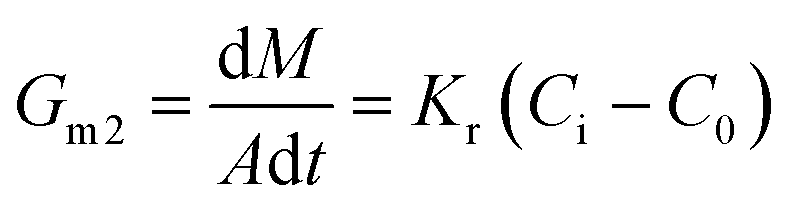 | (5) |
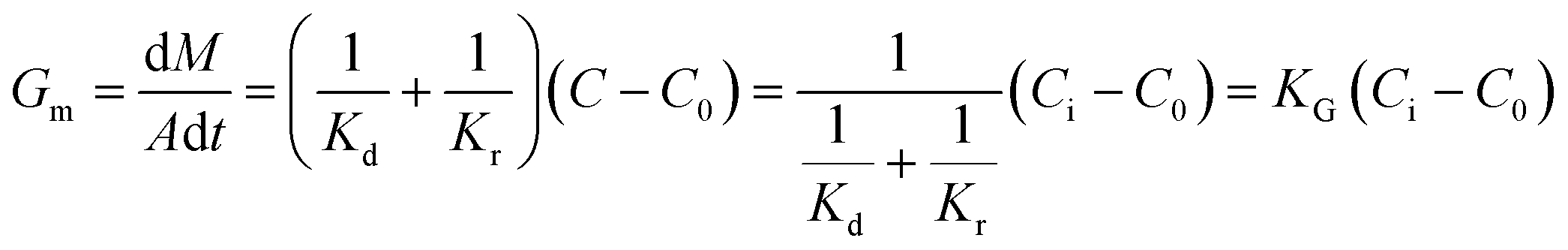 | (6) |
Compared with the preparation in the beaker, the mixing efficiency of the passive microreaction system is improved, which leads to an increase in the diffusion coefficient Kd. Therefore, higher mixing efficiency is beneficial for improving the growth rate of SA. Based on eqn (2) and (6), a relationship between the mixing performance of the passive microreaction system and the final particle size of SA can be built. A higher mixing efficiency has little effect on the nucleation rate of SA, but increases the crystal growth rate, which results in an increase in the ratio of the growth rate to the nucleation rate.
To better observe the mixing/reaction process in the device, sequential images of the SA synthesis process using the passive microreaction system were captured using a high-speed camera. The images of the mixing/reaction process are shown in Fig. 5. The field of view is too small to completely record the mixing/reaction process in the MCFM. Therefore, three different zones of the MCFM were selected to capture the desired events. The syringe pumps drive the reaction solutions to flow along the tubes to the MCFM, and no fine precipitation is observed in zone 1 within 250 ms. As the fluids are driven towards zone 2, fine precipitates are initially observed at a distance of 8.67 mm from the inlet, and the corresponding time is approximately 60 ms, which means that the time required for nucleation is much less than 60 ms. Due to limited equipment, the nucleation process had not been captured. Additionally, clearly visible precipitates are observed in zone 3. In conclusion, the precipitation process of SA takes place after the solutions are fully mixed, and the time interval is approximately 2.5 ms. Furthermore, higher mixing efficiency increases the growth rate of SA, which leads to the particle size of SA synthesized in the passive microreaction system being larger than that of SA synthesized in the beaker.
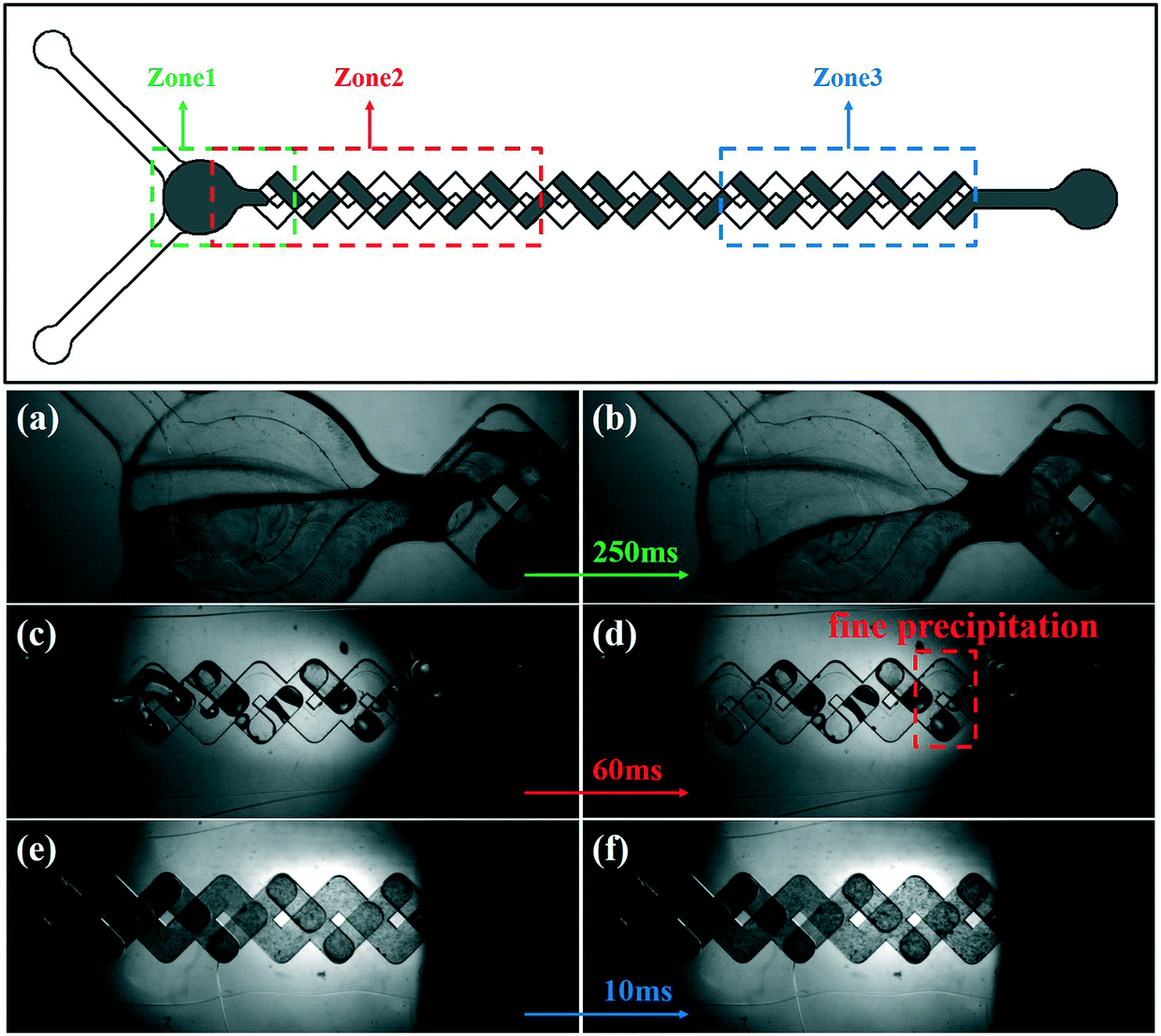 | ||
| Fig. 5 High-speed camera observations during the mixing/reaction process of SA in zone 1 within 250 ms (a and b), observations in zone 2 within 60 ms (c and d), observations in zone 3 within 10 ms (e and f). | ||
3.4 Sensitivity testing of submicron-SA
The sensitivities of energetic materials are critical to their practical applications. To evaluate the safety performance of the SA synthesized using the passive microreaction system, the impact sensitivity (IS) and electrostatic sensitivity were investigated. A BAM Fallhammer apparatus was used to measure the sensitivity of SA to the drop-weight impact, and an image of the experimental device is shown in Fig. 6(a and b). The impact energy was calculated from the mass of the drop weight (0.5, 1 or 2 kg) and the fall height (0.1 m to 1.0 m). Each sample was approximately 5 mg. We measured the material synthesized using the passive microreaction system five times at 20 J without detonation, so the IS value of SA obtained using the system is higher than 20 J. This result indicates that submicron-SA synthesized with the passive microreaction system is insensitive to the impact and safe to use. The reason is that the submicron-SA prepared using the passive microreaction system has also characteristics of nano-insensitivity.32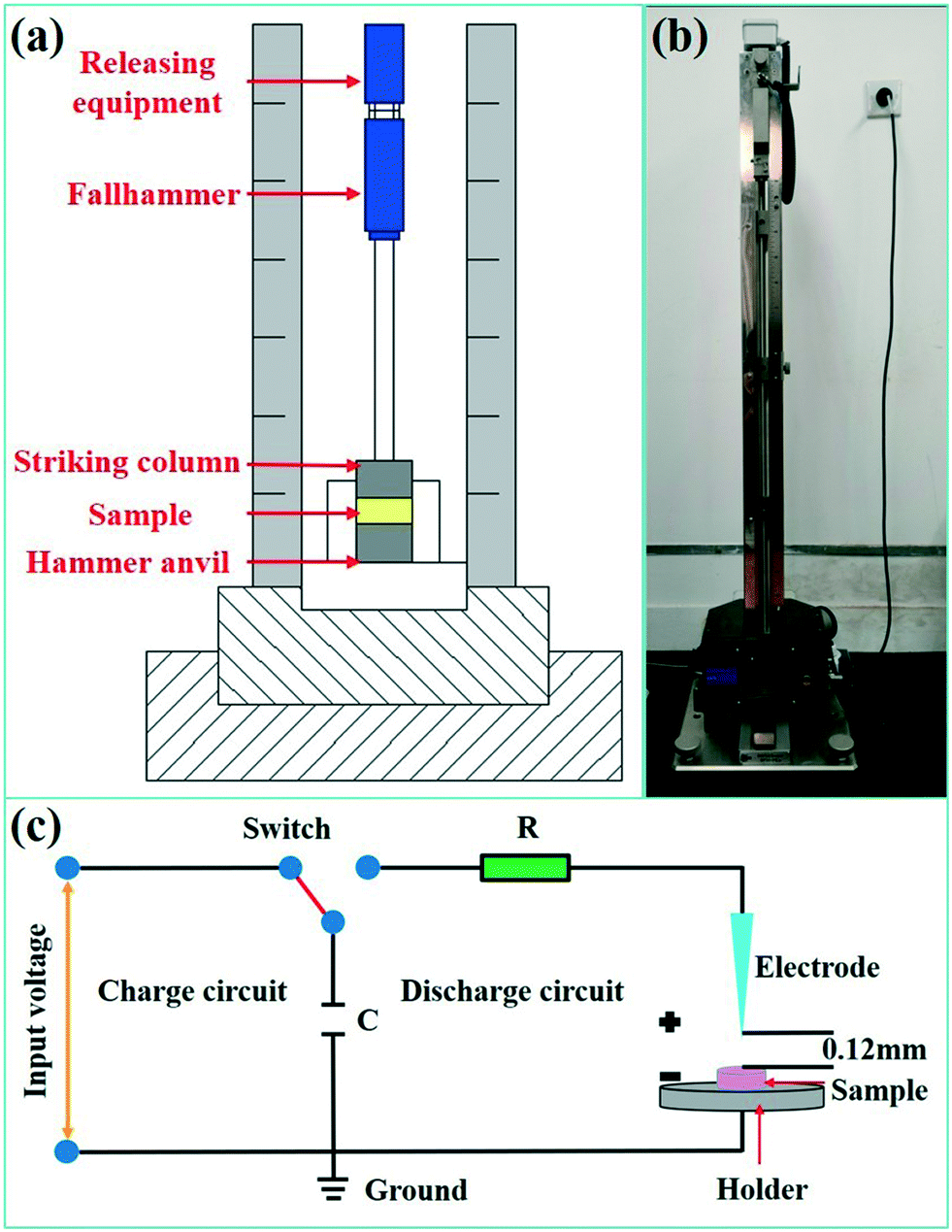 | ||
| Fig. 6 (a) Schematic diagram of the BAM Fallhammer tester, (b) image of the BAM Fallhammer tester and (c) schematic diagram of the ESD sensitivity tester. | ||
In addition, an electrostatic discharge (ESD) test was performed to evaluate the electrostatic safety of SA synthesized in the passive microreaction system, and a schematic diagram of the relevant experimental device (JGY-50III) is shown in Fig. 6(c). The test system consists of a charge circuit and a discharge circuit. In particular, the distance between the electrode and the sample holder was set as 0.12 mm. The test energy, E, was taken to be the total energy stored on the charged capacitor, which was calculated from the capacitance value in farads (C) and the charge voltage (V) according to the formula 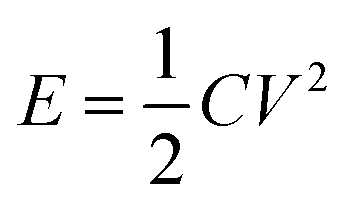 . Relatively small variations in discharge energy were obtained by varying the charging voltage of a given capacitor of 500 pF, and the capacitor was set to allow measurements in the voltage interval from 0 to 20 kV. A series of 5–10 samples were tested for SA at a charge voltage of 20 kV, and each sample was approximately 10 mg. All the samples were not ignited, which illustrates that the energy for ignition is higher than 0.1 J. The ESD value of SA prepared using the passive microreaction system is extremely high, implying that the submicron-SA is insensitive and thus is safe for handling.
. Relatively small variations in discharge energy were obtained by varying the charging voltage of a given capacitor of 500 pF, and the capacitor was set to allow measurements in the voltage interval from 0 to 20 kV. A series of 5–10 samples were tested for SA at a charge voltage of 20 kV, and each sample was approximately 10 mg. All the samples were not ignited, which illustrates that the energy for ignition is higher than 0.1 J. The ESD value of SA prepared using the passive microreaction system is extremely high, implying that the submicron-SA is insensitive and thus is safe for handling.
3.5 Explosive properties of submicron-SA
To better observe the explosion process and evaluate the explosive properties, sequential images of the SA synthesized using the passive microreaction system exploding in open air were captured with a high-speed camera. A 5.0 mg sample was pressed into a cylindrical grain with a diameter of 2.0 mm and a thickness of 0.5 mm under a pressure of 98 N for laser detonation. The images of the laser detonation process are shown in Fig. 7. After the laser pulse excites the SA, a twinkling white cone-shaped flash bursts out immediately. Then, the splattered products continue to expand outward in the form of explosion. The most intensive and luminous explosive reaction is observed for SA, indicating that the initiation efficiency of the SA prepared in the passive microreaction system is very high. However, some of the captured images have a problem at high frame rates that the field of view is too small to completely record the explosion process. The height of the flame reaches 18.5 mm at 10 μs, and the whole explosion process lasts approximately 220 μs. As a rough estimate, the detonation velocity of the submicron-SA prepared in the passive microreaction system can reach 1850 m s−1, while the detonation velocity of confined-SA with a 0.5 mm thickness is 1700 m s−1, as published by Bowden.33 | ||
| Fig. 7 High-speed camera observations during a laser detonation test for the SA synthesized using the passive microreaction system. | ||
4 Conclusions
The mixing efficiency and homogeneity are critical to the crystal form, particle size and PSD of SA and affect its performance and applications in practice. A passive microreaction system was developed to improve the mixing performance and optimize the crystal shape and PSD of SA particles. Testing results show that the passive micromixer (MCFM) works well at relatively low flow rates. For comparison, SA was also prepared by a literature method with the same operating conditions on a macroscale. Compared with the preparation in a beaker, the efficiency of SA synthesized in the passive microreaction system was increased by 13 times. Additionally, the two samples were analyzed, and the results showed that SA synthesized using the passive microreaction system has a good crystal morphology, larger particle size and narrower PSD. Thermal analyses revealed that the heat release of SA synthesized in the system is approximately equal to that of SA obtained in the beaker. However, the initial thermal decomposition temperature of SA prepared at the macroscale is 9.9 °C lower than that of SA synthesized at the microscale, which means that the SA prepared at the microscale has better thermal stability and compatibility. In addition, IS and ESD tests were performed to evaluate the safety performance of the SA prepared at the microscale. The results indicate that the SA synthesized at the microscale has also characteristics of nano-insensitivity and thus is safe for handling. Finally, the detonation velocity of the SA prepared using the passive system is 150 m s−1 higher than that of confined SA published in the literature, reaching 1850 m s−1. All the reported findings provide useful references for the regulation and control at the microscale over the crystal shape and particle size of SA and other explosives.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Shanghai Aerospace Science and Technology Innovation Fund (No. SAST2017-124) and the National Natural Science Foundation of China (No. 51575282).References
- B. L. Evans, A. D. Yoffe, P. Gray and P. Gray, Chem. Rev., 1959, 59(4), 515–568 CrossRef CAS.
- C. M. Pereira and M. M. Chaudhri, J. Energ. Mater., 1989, 7(4–5), 297–322 CrossRef.
- B. P. Aduev, É. D. Aluker, G. M. Belokurov, Y. A. Zakharov and A. G. Krechetov, J. Exp. Theor. Phys., 1999, 89(5), 906–915 CrossRef CAS.
- M. Yagmurcukardes, H. Sahin, J. Kang, E. Torun, F. M. Peeters and R. T. Senger, J. Appl. Phys., 2015, 118(10), 104303 CrossRef.
- M. M. Chaudhri, Nature, Phys. Sci., 1973, 242(120), 110–111 CrossRef CAS.
- T. B. Tang and M. M. Chaudhri, Proc. R. Soc. London, Ser. A, 1979, 369, 83 CrossRef CAS.
- J. C. Baliar and A. F. Trotman-Dickenson, Comprehensive Inorganic Chemistry, 1973 Search PubMed.
- Z. Yongxu, L. Dabin and L. Chunxu, Propellants, Explos., Pyrotech., 2005, 30(6), 438–441 CrossRef.
- X. Song, Y. Wang, C. An, X. Guo and F. Li, J. Hazard. Mater., 2008, 159, 222–229 CrossRef CAS PubMed.
- S. Li, S. Meierott and J. M. Köhler, Chem. Eng. J., 2010, 165, 958–965 CrossRef CAS.
- M. Thiele, J. Z. E. Soh, A. Knauer, D. Malsch, O. Stranik, R. Müller, A. Csáki, T. Henkel, J. M. Köhler and W. Fritzsche, Chem. Eng. J., 2016, 288, 432–440 CrossRef CAS.
- A. Tanimu, S. Jaenicke and K. Alhooshani, Chem. Eng. J., 2017, 327, 792–821 CrossRef CAS.
- A. Abou-Hassan, O. Sandre and V. Cabuil, Angew. Chem., Int. Ed., 2010, 49, 6268–6286 CrossRef CAS PubMed.
- Y. Liu and X. Jiang, Lab Chip, 2017, 17, 3960–3978 RSC.
- R. Wohlgemuth, I. Plazl, P. Žnidaršič-Plazl, K. V. Gernaey and J. M. Woodley, Trends Biotechnol., 2015, 33, 302–314 CrossRef CAS.
- D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384–6389 CrossRef CAS PubMed.
- A. Jahn, J. E. Reiner, W. N. Vreeland, D. L. DeVoe, L. E. Locascio and M. Gaitan, J. Nanopart. Res., 2008, 10, 925–934 CrossRef CAS.
- S. Zhao, J. Wu, P. Zhu, H. Xia, C. Chen and R. Shen, Ind. Eng. Chem. Res., 2018, 57, 13191–13204 CrossRef CAS.
- S. Zhao, C. Chen, P. Zhu, H. Xia, J. Shi, F. Yan and R. Shen, Ind. Eng. Chem. Res., 2019, 58, 16709–16718 CrossRef CAS.
- S. Zhao, F. Yan, P. Zhu, Y. Yang, H. Xia, R. Shen and Y. Ye, Propellants, Explos., Pyrotech., 2017, 43, 286–293 CrossRef.
- N. Zhou, P. Zhu, Y. Rong, H. Xia, R. Shen, Y. Ye and S. Lv, Propellants, Explos., Pyrotech., 2016, 41, 899–905 CrossRef CAS.
- C. Y. Lee, W. T. Wang, C. C. Liu and L. M. Fu, Chem. Eng. J., 2016, 288, 146–160 CrossRef CAS.
- V. Hessel, H. Löwe and F. Schönfeld, Chem. Eng. Sci., 2005, 60, 2479–2501 CrossRef CAS.
- G. Cai, L. Xue, H. Zhang and J. Lin, Micromachines, 2017, 8, 274 CrossRef PubMed.
- J. Wang, F. Zhang, Y. Wang, G. Luo and W. Cai, Chem. Eng. J., 2016, 293, 1–8 CrossRef CAS.
- A. Polyzoidis, T. Altenburg, M. Schwarzer, S. Loebbecke and S. Kaskel, Chem. Eng. J., 2016, 283, 971–977 CrossRef CAS.
- H. Trzenschiok, M. Distaso and W. Peukert, Chem. Eng. J., 2019, 361, 428–438 CrossRef CAS.
- D. Hou, F. Zhang, C. Ji, T. Hannon, H. Zhu, J. Wu, V. I. Levitas and Y. Ma, J. Appl. Phys., 2011, 110, 023524 CrossRef.
- W. Zhu and H. Xiao, J. Solid State Chem., 2007, 180, 3521–3528 CrossRef CAS.
- P. Jain, J. Sahariya, H. S. Mund, M. Sharma and B. L. Ahuja, Comput. Mater. Sci., 2013, 72, 101–106 CrossRef CAS.
- Z. Wu, S. Yang and W. Wu, Nanoscale, 2016, 8, 1237–1259 RSC.
- F. P. Bowden and K. Singh, Nature, 1953, 172, 378–380 CrossRef CAS.
- F. P. Bowden and H. T. Williams, Proc. R. Soc. London, Ser. A, 1951, 208, 176–188 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9re00393b |
| This journal is © The Royal Society of Chemistry 2020 |