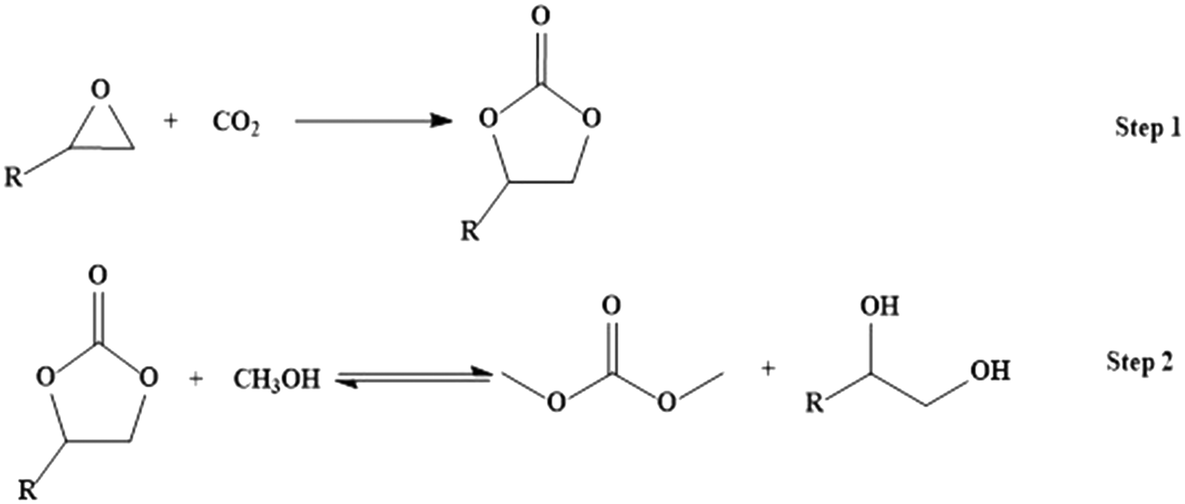
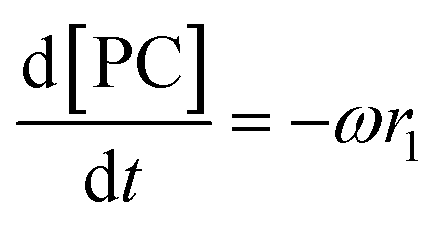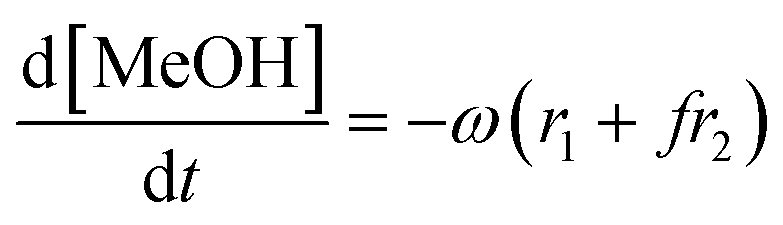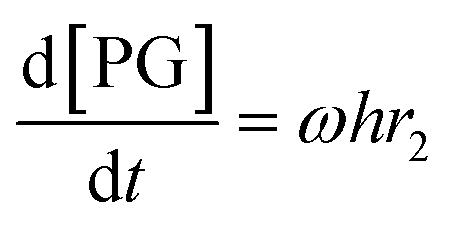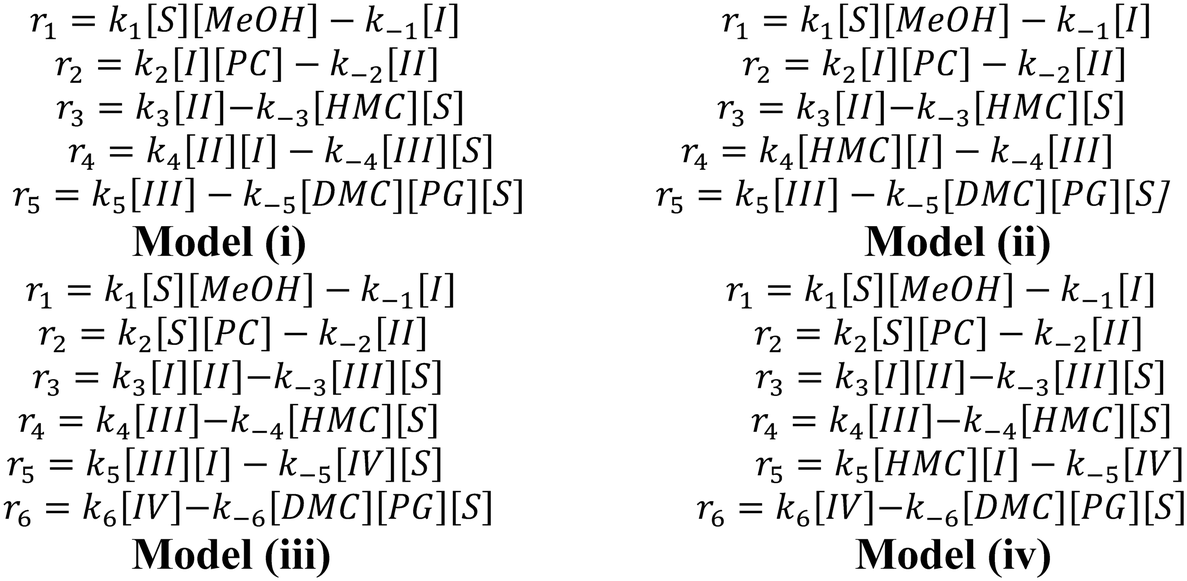Kinetic modeling and mechanistic investigations of transesterification of propylene carbonate with methanol over an Fe–Mn double metal cyanide catalyst†
Received
15th September 2019
, Accepted 24th October 2019
First published on 24th October 2019
Abstract
Kinetic modeling of transesterification of propylene carbonate with methanol using an Fe–Mn double metal cyanide catalyst has been investigated based on experimental data obtained under kinetically controlled conditions in a batch slurry reactor in the 140–200 °C range. A simple two-step power law model was found to represent the experimental data well. In addition, a detailed kinetic model based on a molecular level description of the reaction mechanism is also evaluated to provide better insight into the reaction mechanism. It is found that a kinetic model based on the following mechanistic steps provides the best description of the experimental data: (a) activation of methanol by the catalyst to form a methoxy intermediate, (b) activation of propylene carbonate by interaction with the methoxy intermediate to form a second intermediate, (c) reaction of the second intermediate with methoxy species to form a third intermediate which decomposes into the final products dimethyl carbonate and propylene glycol with regeneration of the catalyst precursor. The kinetic model along with mechanistic insights from the present study provides rational guidance for catalyst redesign and process optimization.
Introduction
Various strategies for CO2 utilization have attracted significant interest in the last decade, including catalytic conversion of CO2 to value added chemicals and fuels.1–5 CO2 can be catalytically converted to various important industrial products, such as hydrocarbons, cyclic carbonates,6–9 organic carbonates,10,11 and carboxylic acids12,13 as well as fuel range compounds including methane and methanol.14
Dimethyl carbonate (DMC) is an environmentally benign substitute for phosgene, dimethyl sulfate and methyl halide, and can be produced through a two-step process using CO2 as a starting material (Scheme 1).15,16 Transesterification of cyclic carbonate with methanol has drawn much attention as a phosgene-free process for making DMC.
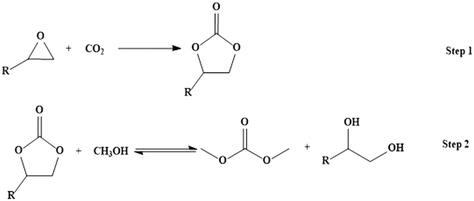 |
| | Scheme 1 Reaction scheme for production of dimethyl carbonate from CO2. | |
Previous studies have focused on the development of various catalysts for the transesterification of cyclic carbonates with methanol, including sodium methoxide,17 alkali metals,18 basic metal oxides,15,19,20 ion exchange resins,21 and hydrotalcites.22 Although homogeneous catalysts display high activity under mild reaction conditions,17 a major drawback is the usual difficulty in separation and recovery of products and catalysts. Therefore, the development of heterogeneous catalysts for transesterification continues to attract research interest.
In the 1960s, the General Tire & Rubber Company discovered that a double metal cyanide complex has high activity for the ring-opening polymerization of epoxy compounds, cyclic anhydrides and cyclic sulfides.23 Since then, companies like Arco Chemical Technology,24 the Dow Chemical Company,25 and Shell Oil Company26 have put huge effort into the development of this type of catalyst. To date, this kind of catalyst has been successfully used industrially in various ring-opening reactions, such as in epoxide polymerization reactions27 and copolymerization reaction of epoxide with CO2.27–33 Recently, an Fe–Zn double metal cyanide catalyst has been successfully used in the transesterification reaction for the manufacture of biodiesel.34 Additionally, Srivastava and coworkers have found Fe–Zn to be an excellent heterogeneous catalyst for transesterification of propylene carbonate with methanol.35 However, the cited reports focus on the structural properties of different double metal cyanides along with activity testing with very little information on the kinetics and reaction engineering aspects, which are essential for rational process development. Although several reports have provided insights into the possible mode of activation of cyclic carbonates and methanol on metal catalysts,35,36 there is a lack of reliable information on the mechanistic and kinetic aspects of heterogeneously catalyzed transesterification of cyclic carbonates.
In our previous work, we reported that an Fe–Mn double metal cyanide complex shows a significantly higher activity for the transesterification of cyclic carbonates with methanol compared with other double metal cyanide complexes.37 In this work, detailed kinetic modeling of the transesterification of propylene carbonate (PC) with methanol over Fe–Mn double metal cyanide is reported. The effects of several experimental parameters, including temperature (140–200 °C), PC initial concentration (1.1–4.3 kmol m−3), and methanol initial concentration (1.2–4.8 kmol m−3) on the intrinsic conversion and product formation profiles were studied. The data were analyzed with empirical power law models as well as detailed kinetic models based on the molecular description of the various reaction steps of the proposed mechanisms. A kinetic model involving sequential activation of reactants and intermediates with the catalyst was found to best represent the experimental data. The results have thus provided better insights into the reaction mechanism and kinetics of this industrially important reaction system.
Experimental
Materials
Chemicals such as methanol (99.9%), PC (99.7%), DMC (extra dry, 98+%), propylene glycol (PG, 99.5+%) and mesitylene (98%) were purchased from Sigma-Aldrich. K4Fe(CN)6·3H2O (98%) was obtained from Alfa Aesar. All chemicals were used as received without further treatment. The Fe–Mn double metal cyanide catalysts were prepared using the method described in previous work.35
Reactor setup and procedures
The kinetic experiments were conducted in a stirred slurry reactor (Parr reactor). The reactor system consists of a 300 mL reactor equipped with a thermocouple, pressure transducer, gas inlet and stirring impeller. The temperature, pressure and stirring speed are independently controlled and monitored with Labview® software through the control module of the reactor system. In a typical reaction experiment, a methanol-based slurry was charged in an autoclave with a known amount of catalyst and mesitylene (internal standard). The reactor was sealed and loaded into the setup. Following this step, 0.69 MPa N2 gas was introduced into the reactor while stirring slowly (∼50 rpm). The mixture was then heated to the desired temperature, after which another reactant, PC, was pumped into the reactor using a HPLC pump [Scientific Systems, Inc. (SSI) Series I pump] to the desired initial concentration. After the mixture reaches the desired temperature, the reaction was initiated by increasing the stirrer speed to 1000 rpm. During the reaction, liquid samples were withdrawn at different times to develop concentration–time profiles for the reactants and products. The liquid samples were analyzed using a GC (GC-7860A) equipped with a ZB-FFAP capillary column of 30 m in length and a flame-ionization detector (FID); the detailed information is shown in the ESI† Part 2.
Results and discussion
Reaction kinetics
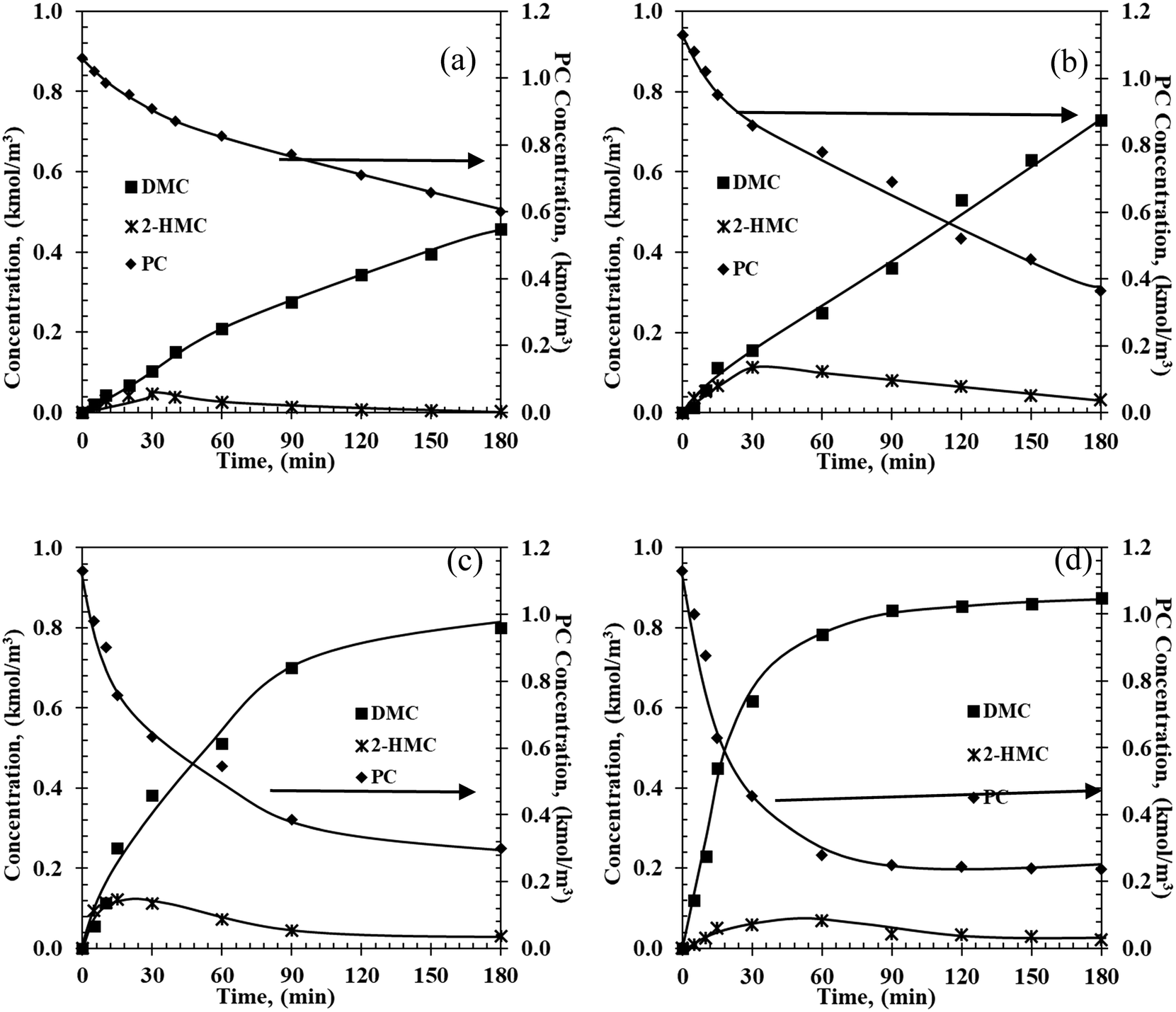
Detailed kinetic models for PC transesterification were investigated using experimentally measured concentration–time profiles at different temperatures and under different reaction conditions (Fig. 1). These profiles capture the temporal evolution of various products including intermediates. Typical experiments were repeated twice and values of substrate/product concentration were measured (see Fig. S3 in the ESI†). The standard deviation was 3.92%.
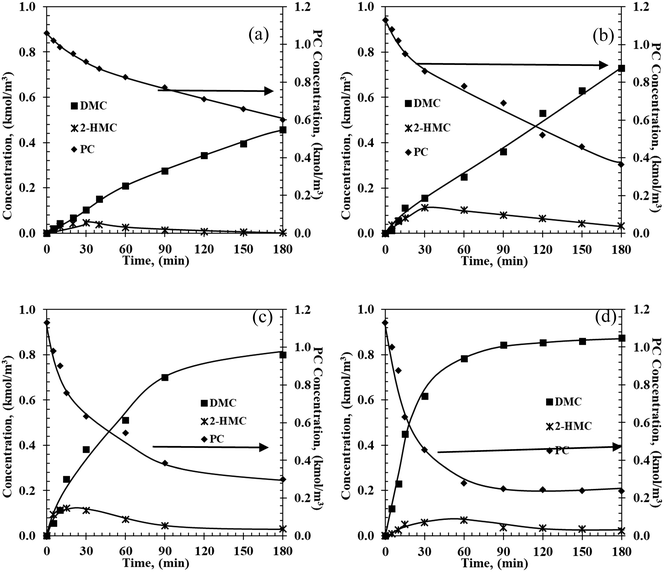 |
| | Fig. 1 Temporal concentration–time profiles of PC conversion over the Fe–Mn catalyst at (a) 140 °C, (b) 160 °C, (c) 180 °C, and (d) 200 °C. Reaction conditions: PC: 1.1 kmol m−3; methanol: 22.2 kmol m−3; catalyst: 5 kg m−3; 0.69 MPa initial N2. | |
Concentration–time profiles.
It is clear from the concentration–time profiles that the major products are DMC and PG, with a smaller but significant amount of the mono transesterified product, 2-hydroxypropyl methyl carbonate (2-HMC). At 140 °C (Fig. 1a), the concentration of reactant PC decreased from 1.1 kmol m−3 to 0.56 kmol m−3 in 3 h. Concentrations of DMC and PG increased gradually with reaction time, while the formation of 2-HMC first increased and then decreased to nearly none. As expected, when the temperature is increased from 140 °C to 200 °C, the formation rates of products show a significant increase. The various temporal reaction profiles are provided in the ESI† (Part 3, Fig. S4–S7). Data from sixteen temporal profiles at four different temperatures were used for model discrimination and parameter estimation.
Analysis of mass transfer effects.
Since the transesterification reaction involves a liquid–solid catalytic reaction, liquid–solid and intraparticle mass transfer limitations for the limiting reactant, PC, were considered (since methanol is in large excess). To assess these limitations, the criteria described by Ramachandran and Chaudhari were used.38 In these criteria, experimentally measured rates were compared with maximum rates of the mass transfer steps: liquid–solid and intraparticle mass transfer steps. Details of calculations for a few cases are shown in the ESI† (Part 5). The analysis showed that both liquid–solid and intraparticle mass transfer limitations were negligible, confirming that the experimental data were obtained in the kinetic regime.
Effect of initial substrate concentration.
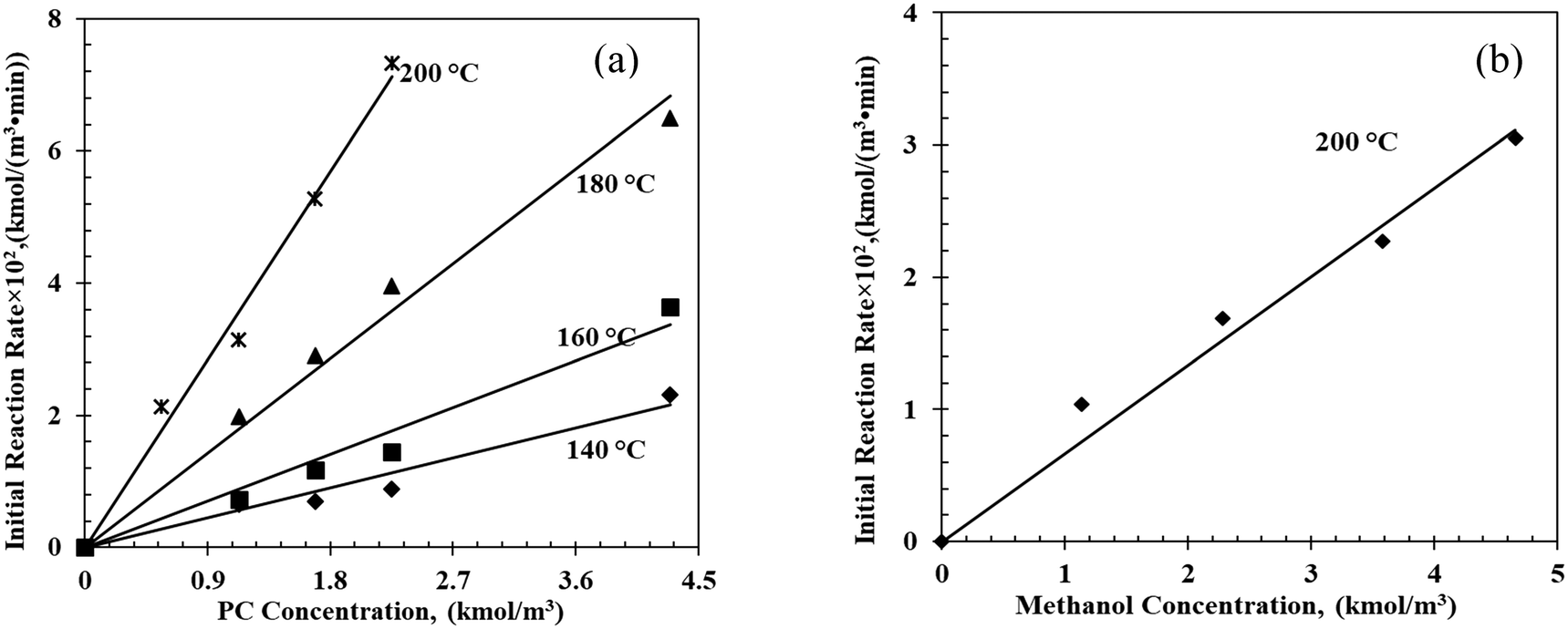
The effect of the initial substrate (PC) concentration on initial reaction rates is shown in Fig. 2a. As the PC concentration increased from 1.1 to 4.3 kmol m−3, the initial reaction rate increased from 0.65 (10−2) to 2.32 (10−2) kmol m−3 min−1 at 140 °C suggesting first-order dependence of the initial reaction rate on PC concentration. Similar trends were also observed at higher reaction rates at 160 °C, 180 °C and 200 °C. The effect of the methanol concentration on the initial reaction rate shows a similar trend at 200 °C (Fig. 2b).
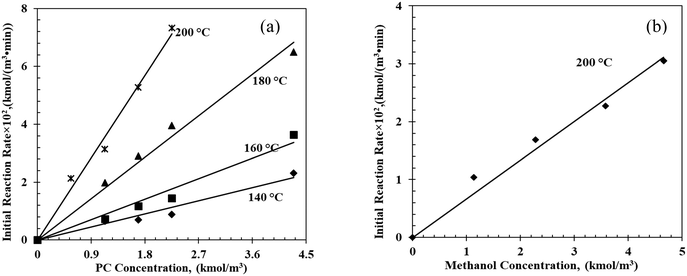 |
| | Fig. 2 Effects of (a) PC initial concentration and (b) methanol initial concentration on initial reaction rates. Reaction conditions: (a) methanol, 17.9–22.8 kmol m−3; catalyst, 5 kg m−3; T = 140 °C; 0.69 MPa N2; (b) PC, 9.3–10.8 kmol m−3; catalyst, 5 kg m−3; T = 200 °C; 0.69 MPa N2. | |
Apparent activation energy.
The estimated first-order forward reaction rate constants for PC transesterification at 140 °C, 160 °C, 180 °C and 200 °C, estimated from the slopes of the regression line in Fig. 2a, were 0.50 min−1, 0.79 min−1, 1.60 min−1 and 3.17 min−1, respectively. The activation energy was estimated to be 50.4 kJ mol−1, with a pre-exponential factor of 1.07 × 104 min−1 (see Fig. S9 in the ESI†).
Two-step power law model.
Given that a significant amount of intermediate 2-HMC was formed during the reaction, the following reaction scheme (Scheme 2) was considered for the power law model. Since the initial reaction rate is first order in both PC and methanol concentrations, the exponents a and b are fixed at a value of unity (a = b = 1). The rate expressions and batch reactor mass balance equations of each component are given by eqn (1)–(7).| | | r1 = k1CPCCMeOH − k−1C2-HMCc | (1) |
| | | r2 = k2C2-HMCeCMeOHf − k−2CDMCgCPGh | (2) |
| | 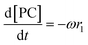 | (3) |
| | 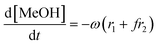 | (4) |
| | 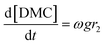 | (5) |
| | 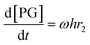 | (6) |
| | 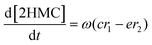 | (7) |
where r1 and r2 are the specific reaction rates for step 1 and step 2, k1, k−1 and k2, k−2 are the temperature dependent forward and reversible reaction rate constants for step 1 and step 2, respectively, and ω is the catalyst concentration.
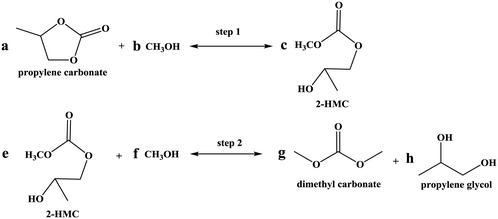 |
| | Scheme 2 Two-step reaction scheme. | |
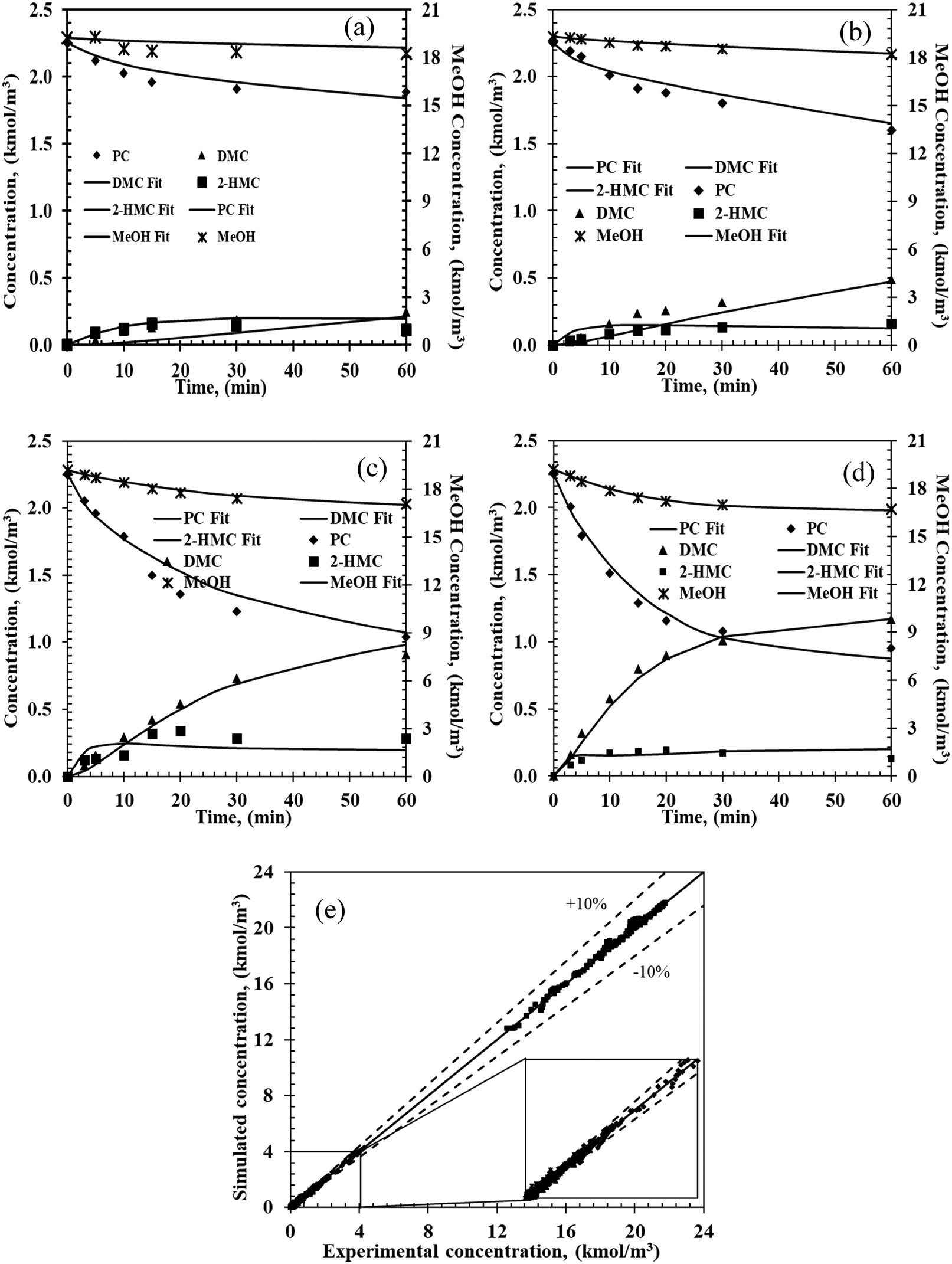
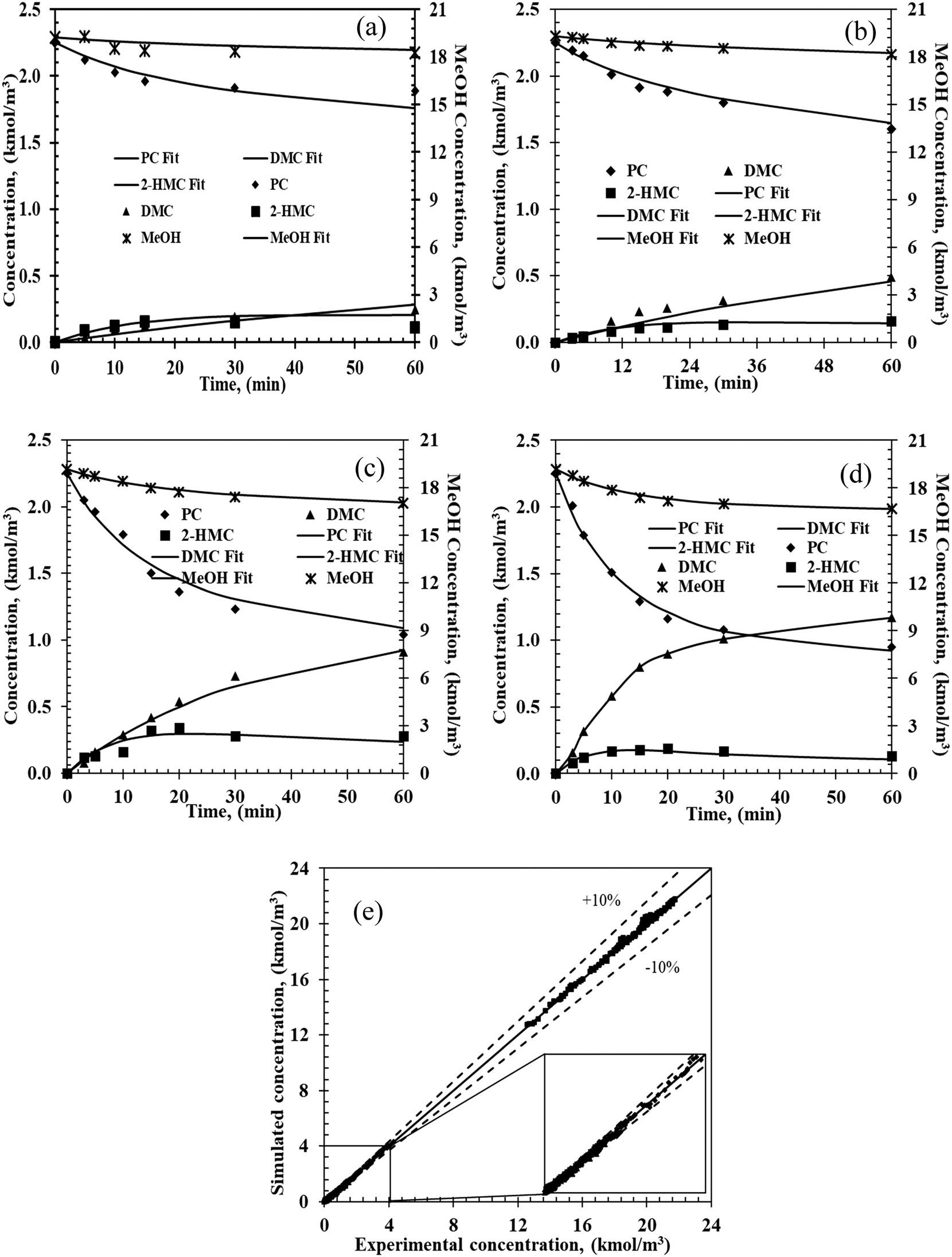
To simulate the experimental concentration–time data, eqn (1)–(7) were solved with an optimization routine using Athena Visual Studio software.39–42 For the parameter estimation and model discrimination, several convergence criteria were used, as summarized in the ESI† (Part 7). Through the parameter estimation, it is found that when c = d = e = f = g = h = 1, the power law fits the experimental data well. Typical results of experimental and predicted concentration–time profiles for various chemical components are shown in Fig. 3.
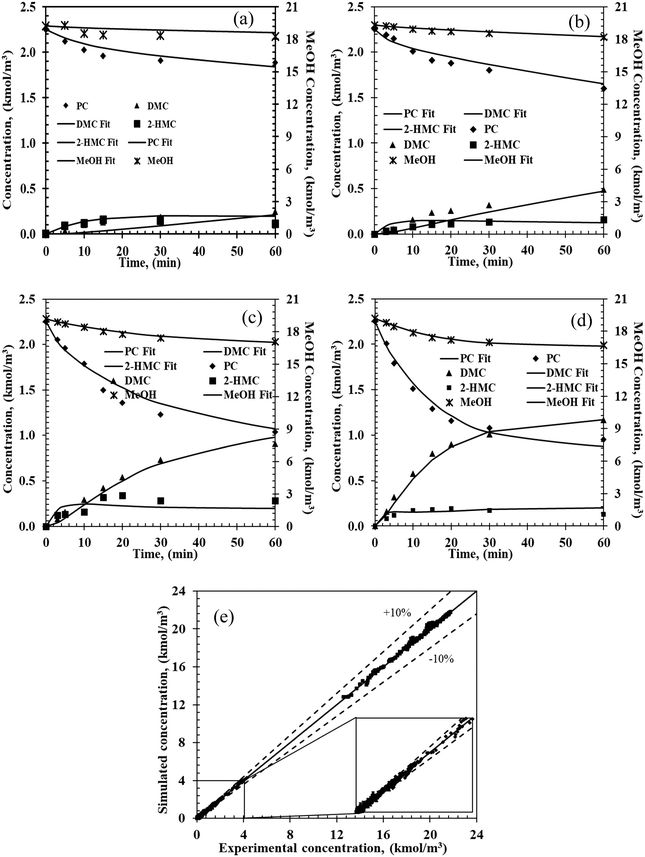 |
| | Fig. 3 Experimental and predicated concentration–time profiles from the two-step power law model. Reaction conditions: PC, 2.25 kmol m−3; methanol, 20.5 kmol m−3; catalyst, 5 kg m−3; 0.69 MPa N2; (a) 140 °C, (b) 160 °C, (c) 180 °C, (d) 200 °C. (e) Parity plot of the simulated and experimental concentrations for the four chemicals. Markers: experimental values; lines: estimated values from power law. | |
A similar procedure was followed for modeling the data from all the 16 experiments at four different temperatures. As shown in Fig. 3e, the predicted concentrations are well within ±10% of the experimental concentrations demonstrating that the empirical power law model can be applied to predict the temporal concentration–time profiles for reactor design and optimization in the range of the experimental conditions studied.
The optimized values of the rate parameters are shown in the Arrhenius plot in Fig. S10† while the reaction rate constants and activation energies are presented in Table 1. An activation energy of 47.0 ± 20.2 kJ mol−1 was observed for the ring opening reaction, the mono-transesterification of PC with methanol using the Fe–Mn catalyst. In contrast, a much higher activation energy (79.6 ± 9.8 kJ mol−1) was observed for the second step transesterification of 2-HMC with methanol. The higher energy barrier of the second step is attributed to the release of ring strain energy when the ring of PC was broken.43
Table 1 Values of activation energy and rate constants derived from power law
| Rate constants |
140a °C |
160a °C |
180a °C |
200a °C |
E
a
|
|
Rate constants with 95% confidence level.
Unit of Ea: kJ mol−1, detailed error analysis is in the ESI.†
|
|
k
1 × 101, (m6 min−1 kmol−2) |
1.26 ± 0.47 |
2.42 ± 0.57 |
5.50 ± 0.61 |
6.52 ± 0.59 |
47.0 ± 20.2 |
|
k
−1 × 10−1, (m3 min−1 kmol−1) |
1.76 ± 0.88 |
4.64 ± 1.47 |
4.49 ± 0.63 |
4.57 ± 0.72 |
23.7 ± 27.2 |
|
k
2, (m6 min−1 kmol−2) |
0.26 ± 0.024 |
0.78 ± 0.049 |
1.60 ± 0.12 |
5.38 ± 0.97 |
79.6 ± 9.8 |
|
k
−2, (m6 min−1 kmol−2) |
0.28 ± 0.06 |
0.60 ± 0.16 |
4.29 ± 0.43 |
12.9 ± 2.89 |
108.9 ± 14.6 |
Mechanistic modelling
Reaction mechanism and proposed models.
The transesterification reaction between cyclic carbonates and methanol generally follows the following main steps: 1) the formation of an –M–O–CH3 intermediate through the interaction between CH3O− from methanol and the active metal center;44–46 2) the formation of mono-transesterified products (2-HMC and 1-HP-2-MC) from activated methanol with PC or with activated PC;36,47–51 3) further transesterification of mono-transesterified products/activated mono-transesterified products with activated methanol to form the final products DMC and PG. However, a specific activation sequence of methanol, PC and 2-HMC is not obvious from the general trends observed.
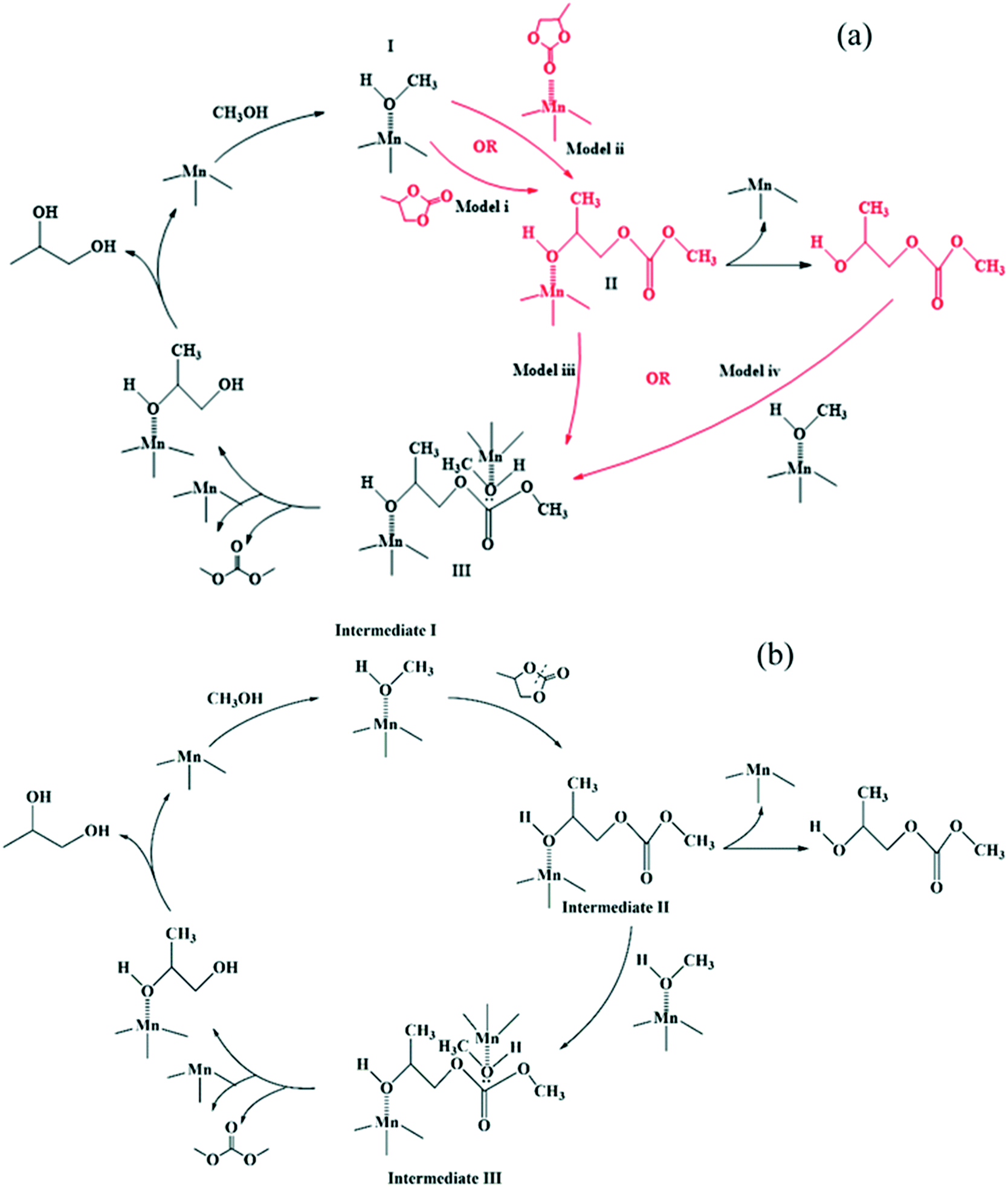
In our previous work, we found that Mn is the active species while Fe functions as a metal-dispersing agent and a stabilizer of the cyano-bridged complex ensuring a truly heterogeneous catalyst.35,37 Additionally, based on the reaction orders estimated from the power law model, it is clear that the reaction rate shows a first order dependence on PC, methanol, 2-HMC, DMC and PG concentrations. Furthermore, from our previous FTIR test,37 PC is known to be adsorbed on the catalyst. However, it is unclear whether PC is adsorbed on the catalyst directly or on an active intermediate. To better understand the reaction activation sequence and the overall mechanism, four kinetic models were evaluated (Scheme 3) based on different sequential reaction steps. The possible catalytic cycles are shown in Scheme 5a. Each model was fitted with all the experimental data and discriminated based on the predictive ability of the models and whether the values of the regressed reaction parameters make physical sense.
 |
| | Scheme 3 Proposed kinetic models for transesterification of PC with methanol on the Fe–Mn catalyst. | |
For model (i), methanol is first activated by the Fe–Mn complex (S stands for the active sites) to form intermediate I.30,35 Then, PC reacts with intermediate I to form intermediate II,30,35 followed by decomposition of intermediate II into the mono-transesterified product, 2-HMC, while releasing the catalytic site for further reaction. Further, intermediate II reacts with another activated methanol (intermediate I) to form intermediate III followed by decomposition into products DMC and PG. The only difference between model (ii) and model (i) is in step (4), where 2-HMC is assumed to react with activated methanol (intermediate I).
For model (iii), methanol is first activated by the Fe–Mn complex to form intermediate I;30,35 simultaneously, PC is activated to form intermediate II.20 Then, intermediates I and II react to form intermediate III followed by its decomposition into mono-transesterified product 2-HMC and the catalyst. Intermediate III further reacts with another activated methanol (intermediate I) species to form intermediate IV which decomposes into products DMC and PG. The only difference between model (iv) and model (iii) is in step (5), where 2-HMC is assumed to react with activated methanol (intermediate I).
Rate equations and batch reactor equations.
The rate equations for each of the individual steps of the four models are summarized in Scheme 4. The corresponding batch reactor model equations are summarized in Table S1 in the ESI.†
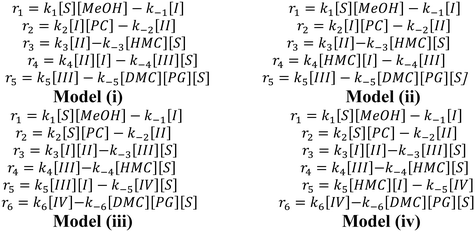 |
| | Scheme 4 Rate equations for various steps in each model. | |
For all four kinetic models, no specific step was assumed as rate-determining, which allows the evaluation of rate parameters for each reaction step including those involving catalytic intermediate species.
The initial conditions are:
| at t = 0, [PC] = [PC]0, [MeOH] = [MeOH]0, [DMC] = [PG] = [2-HMC] = 0, [S] = C0, [I] = [II] = [III] = [IV] = 0, |
where [PC]
0, [MeOH]
0, and
C0 represent the initial concentrations of PC, methanol and the catalyst precursor, respectively. For solving these ODEs, reasonable initial guesses were provided for the rate constants with ranges determined from the power law model. The ODEs were solved using Athena Visual Studio. Identical convergence criteria and constraints were used as those used in the power law models.
Model validation.
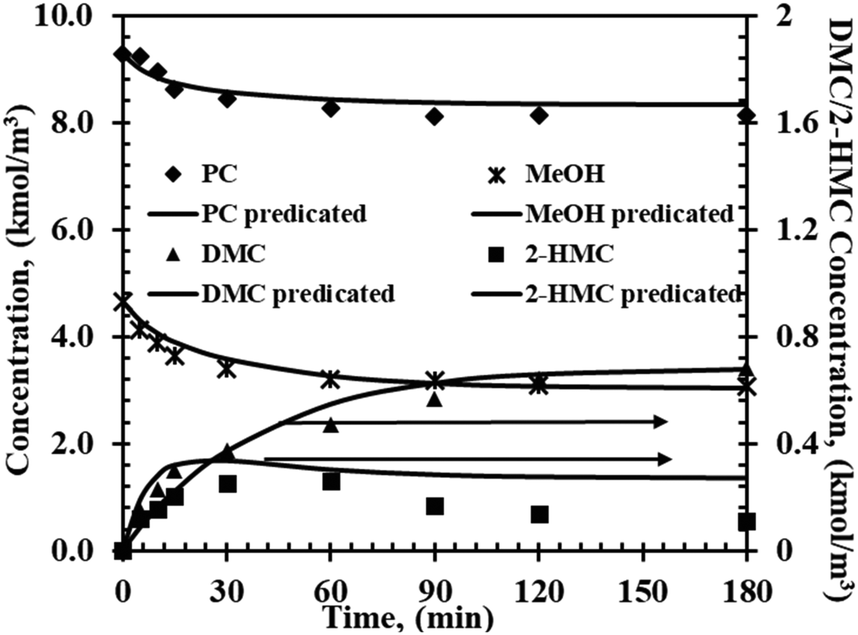
To show the feasibility of the mechanistic model for the description of the transesterification of PC with methanol developed above, an additional set of data was used to compare with predictions by the model. The operating conditions of this experiment differed from those of other experiments in this work used for fitting parameters. The aim of this experiment was to demonstrate that model (i) can be used to predict concentration–time profiles over wider range of conditions. Fig. 4 shows temporal concentration–time profiles for the additional experiment, with the symbols representing the experimental results and lines representing the predicted results using model (i). A comparison between the experimental and predicated data indicates a high accuracy of model (i).
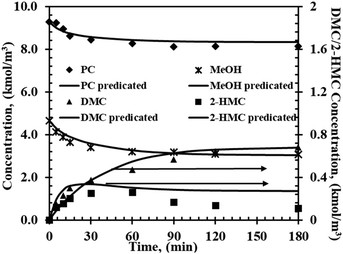 |
| | Fig. 4 Experimental and predicted concentration–time profiles for the validation experiment. Reaction conditions: T = 200 °C, PC, 9.8 kmol m−3; methanol, 3.58 kmol m−3; catalyst, 5 kg m−3; 0.69 MPa N2. | |
Proposed mechanism based on model discrimination.
The main observations of the data regression using models (i)–(iv) are summarized in Table 2. The estimated parameters are shown in Table 3 [for model (i)] and in the ESI† Tables S2–S4 [for models (ii)–(iv)]. Model (i) parameters were deemed to be acceptable based not only on the quality of fit with the experimental data but also on realistic parameter values. As summarized in Table 2, the fit with other models yielded parameter values whose signs and/or magnitudes cannot be reconciled with typical values for these parameters.
Table 2 Basis for discrimination of models (i)–(iv)
| Models |
Model discrimination basis |
| Model i |
Best fit with experimental data |
| Model ii |
Rate constants (k−3 and k−4) decrease with increasing temperature, and 95% confidence level >120% for k2 and k−2 |
| Model iii |
Negative k−1 at 160 °C, the constants do not agree with the Arrhenius equation |
| Model iv |
95% confidence level >120% for k4 at 160 °C, activation energy of step 4 > 400 kJ mol−1 |
Table 3 Rate constants and activation energy for each reaction step
| Rate constants |
140a °C |
160a °C |
180a °C |
200a °C |
E
a
|
|
Rate constants with 95% confidence level.
Unit of Ea: kJ mol−1, the detailed error calculation is in the ESI.†
|
|
k
1 × 10−2, (m3 min−1 kmol−1) |
0.51 ± 0.01 |
1.22 ± 0.06 |
2.79 ± 0.06 |
4.81 ± 0.11 |
61.7 ± 2.7 |
|
k
−1 × 10−4, (min−1) |
2.15 ± 0.31 |
5.11 ± 0.20 |
8.15 ± 0.52 |
14.0 ± 1.04 |
49.6 ± 7.8 |
|
k
2 × 10−4, (m3 min−1 kmol−1) |
2.80 ± 0.26 |
7.18 ± 0.27 |
1.24 ± 0.82 |
17.20 ± 0.85 |
49.1 ± 5.1 |
|
k
−2 × 10−6, (min−1) |
0.48 ± 0.10 |
2.11 ± 0.0.06 |
4.28 ± 0.50 |
6.26 ± 0.17 |
68.8 ± 11.3 |
|
k
3 × 10−2, (min−1) |
1.21 ± 0.10 |
2.56 ± 0.04 |
8.13 ± 1.25 |
9.50 ± 0.70 |
60.0 ± 8.4 |
|
k
−3, (m3 min−1 kmol−1) |
2.32 ± 0.19 |
3.98 ± 0.69 |
6.85 ± 1.36 |
8.34 ± 0.67 |
35.8 ± 10.8 |
|
k
4 × 10−5, (m3 min−1 kmol−1) |
1.84 ± 0.25 |
5.06 ± 1.30 |
8.17 ± 0.46 |
16.80 ± 2.39 |
58.0 ± 13.9 |
|
k
−4 × 10−4, (m3 min−1 kmol−1) |
0.68 ± 0.20 |
1.66 ± 1.36 |
1.99 ± 0.33 |
2.48 ± 0.39 |
33.4 ± 44.2 |
|
k
5 × 10−3, (min−1) |
0.23 ± 0.06 |
0.90 ± 0.21 |
2.96 ± 0.48 |
5.16 ± 0.68 |
86.2 ± 14.8 |
|
k
−5, (m6 kmol−2 min−1) |
3.17 ± 0.62 |
8.23 ± 5.67 |
9.20 ± 1.59 |
18.68 ± 2.78 |
44.29 ± 37.3 |
A schematic description of the mechanism represented in model (i) is presented in Scheme 5b as a complete catalytic cycle. The kinetic parameters obtained from fitting model (i) with experimental data are listed in Table 3. The activation energy values for each step are estimated from the Arrhenius equation (ESI† Fig. S11). Additionally, a comparison of predicted and experimental data at different temperatures is also shown in Fig. 5. The excellent match between the experimental and predicted results under various reaction conditions indicates that the proposed reaction mechanism in Scheme 5b represents the transesterification of PC and methanol satisfactorily.
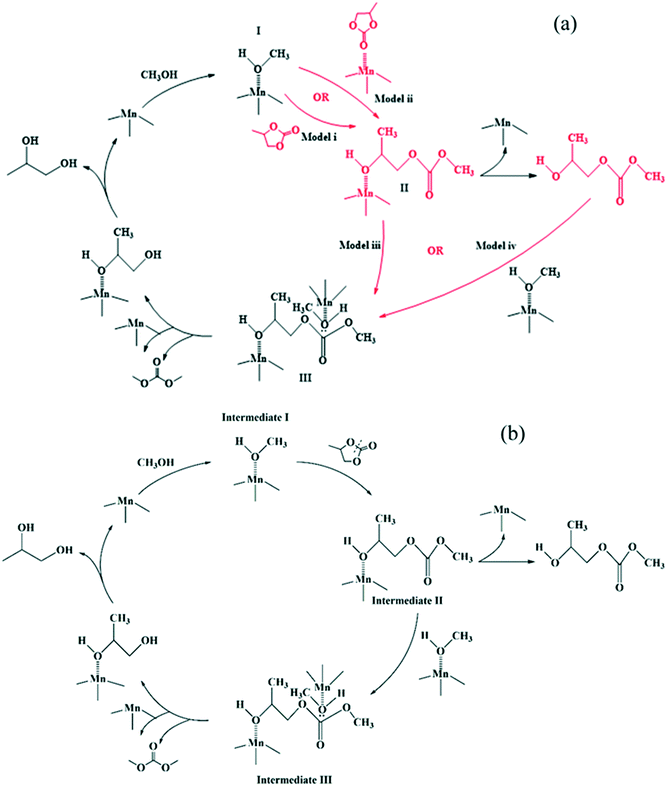 |
| | Scheme 5 (a) Possible activation sequences in a catalytic cycle and (b) the proposed reaction mechanism based on model discrimination. | |
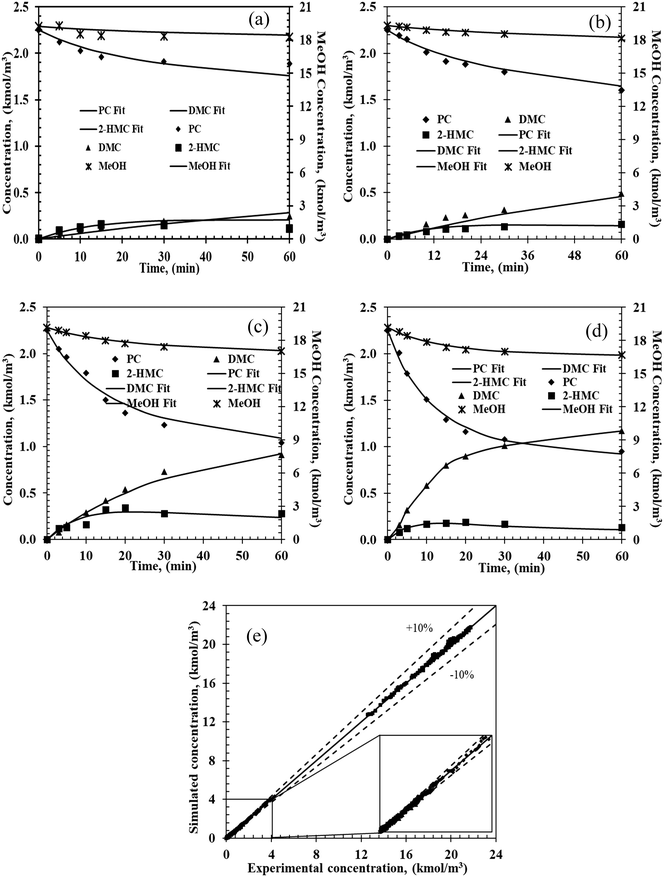 |
| | Fig. 5 Experimental and predicted concentration–time profiles from the micro kinetic model. Reaction conditions: PC, 2.25 kmol m−3; methanol, 20.5 kmol m−3; catalyst, 5 kg m−3; 0.69 MPa N2; (a) 140 °C, (b) 160 °C, (c) 180 °C, (d) 200 °C; (e) parity plot. | |
Conclusion
A detailed kinetic study for transesterification of PC with methanol using Fe–Mn double metal cyanide revealed that both a two-step empirical power law model and a mechanistic model based on the transesterification mechanism were found to fit the experimental data well. Four mechanistic models based on different activation sequences of methanol, PC and 2-HMC were also considered. Through model discrimination based on the quality of fit between experimental and model-predicted data as well as the realistic of the parameter values, it was concluded that methanol was first activated by the catalyst to form intermediate I, which then reacts with PC to form intermediate II. Next, intermediate II reacts with activated methanol (intermediate I) to form intermediate III, which decomposed into DMC and PG, the major products. These results provide valuable insights into the reaction pathways underlying transesterification. The kinetic model is useful for rational reactor design and further optimization of this industrially significant reaction system.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The authors gratefully acknowledge support to this work from the following grants: the joint National Science Foundation and Environmental Protection Agency program (NSF-EPA 1339661) and the Yanshan University funding BL19012.
References
- D. J. Darensbourg, Chemistry of carbon dioxide relevant to its utilization: a personal perspective, Inorg. Chem., 2010, 49(23), 10765–10780 CrossRef CAS PubMed.
- C. Song, Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing, Catal. Today, 2006, 115(1–4), 2–32 CrossRef CAS.
-
M. Aresta, Carbon dioxide as chemical feedstock, John Wiley & Sons, 2010 Search PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Catalysis for the valorization of exhaust carbon: from CO2 to chemicals, materials, and fuels. Technological use of CO2, Chem. Rev., 2013, 114(3), 1709–1742 CrossRef PubMed.
- I. Omae, Recent developments in carbon dioxide utilization for the production of organic chemicals, Coord. Chem. Rev., 2012, 256(13–14), 1384–1405 CrossRef CAS.
- J. Sun, S. Zhang, W. Cheng and J. Ren, Hydroxyl-functionalized ionic liquid: a novel efficient catalyst for chemical fixation of CO2 to cyclic carbonate, Tetrahedron Lett., 2008, 49(22), 3588–3591 CrossRef CAS.
- M. North and R. Pasquale, Mechanism of cyclic carbonate synthesis from epoxides and CO2, Angew. Chem., 2009, 121(16), 2990–2992 CrossRef.
- J.-L. Jiang, F. Gao, R. Hua and X. Qiu, Re(CO)5Br-catalyzed coupling of epoxides with CO2 affording cyclic carbonates under solvent-free conditions, J. Org. Chem., 2005, 70(1), 381–383 CrossRef CAS.
- H. Yasuda, L.-N. He and T. Sakakura, Cyclic carbonate synthesis from supercritical carbon dioxide and epoxide over lanthanide oxychloride, J. Catal., 2002, 209(2), 547–550 CrossRef CAS.
- C. J. Whiteoak, A. Nova, F. Maseras and A. W. Kleij, Merging sustainability with organocatalysis in the formation of organic carbonates by using CO2 as a feedstock, ChemSusChem, 2012, 5(10), 2032–2038 CrossRef CAS PubMed.
- Y. Yoshida, Y. Arai, S. Kado, K. Kunimori and K. Tomishige, Direct synthesis of organic carbonates from the reaction of CO2 with methanol and ethanol over CeO2 catalysts, Catal. Today, 2006, 115(1–4), 95–101 CrossRef CAS.
- A. Polyzos, M. O'Brien, T. P. Petersen, I. R. Baxendale and S. V. Ley, The continuous-flow synthesis of carboxylic acids using CO2 in a tube-in-tube gas permeable membrane reactor, Angew. Chem., Int. Ed., 2011, 50(5), 1190–1193 CrossRef CAS.
- M. D. Greenhalgh and S. P. Thomas, Iron-catalyzed, highly regioselective synthesis of α-aryl carboxylic acids from styrene derivatives and CO2, J. Am. Chem. Soc., 2012, 134(29), 11900–11903 CrossRef CAS.
- G. Centi and S. Perathoner, Opportunities and prospects in the chemical recycling of carbon dioxide to fuels, Catal. Today, 2009, 148(3–4), 191–205 CrossRef CAS.
- B. M. Bhanage, S.-I. Fujita, Y. Ikushima and M. Arai, Synthesis of dimethyl carbonate and glycols from carbon dioxide, epoxides, and methanol using heterogeneous basic metal oxide catalysts with high activity and selectivity, Appl. Catal., A, 2001, 219(1–2), 259–266 CrossRef CAS.
- B. M. Bhanage, S.-I. Fujita, Y. Ikushima, K. Torii and M. Arai, Synthesis of dimethyl carbonate and glycols from carbon dioxide, epoxides and methanol using heterogeneous Mg containing smectite catalysts: effect of reaction variables on activity and selectivity performance, Green Chem., 2003, 5(1), 71–75 RSC.
- J. Holtbruegge, M. Leimbrink, P. Lutze and A. Górak, Synthesis of dimethyl carbonate and propylene glycol by transesterification of propylene carbonate with methanol: Catalyst screening, chemical equilibrium and reaction kinetics, Chem. Eng. Sci., 2013, 104, 347–360 CrossRef CAS.
-
H.-J. Buysch, H. Krimm and H. Rudolph, Process for the preparation of dialkyl carbonates, US4181676A, 1980.
-
Y. Urano, M. Kirishiki, Y. Onda and H. Tsuneki, Process for preparing dialkyl carbonates, US5430170A, 1995.
- Z. Song, X. Jin, Y. Hu, B. Subramaniam and R. V. Chaudhari, Intriguing catalyst (CaO) pretreatment effects and mechanistic insights during propylene carbonate transesterification with methanol, ACS Sustainable Chem. Eng., 2017, 5(6), 4718–4729 CrossRef CAS.
- A. Pyrlik, W. F. Hoelderich, K. Müller, W. Arlt, J. Strautmann and D. Kruse, Dimethyl carbonate via transesterification of propylene carbonate with methanol over ion exchange resins, Appl. Catal., B, 2012, 125, 486–491 CrossRef CAS.
- P. Unnikrishnan and D. Srinivas, Calcined, rare earth modified hydrotalcite as a solid, reusable catalyst for dimethyl carbonate synthesis, Ind. Eng. Chem. Res., 2012, 51(18), 6356–6363 CrossRef CAS.
-
B. R. Joseph, Method of making a polyether using a double metal cyanide complex compound, US3278458A, 1966.
-
B. Le-Khac, Double metal cyanide complex catalysts, US5470813A, 1995.
-
W. J. Kruper Jr and D. J. Swart, Carbon dioxide oxirane copolymers prepared using double metal cyanide complexes, US4500704A, 1985.
-
J. Kuyper and P. van Schaik-Struykenkamp, Process for the polymerization of epoxides, US4472560A, 1984.
-
M. Jack, Method of making a polyether using a double metal cyanide complex compound, US3278457, Google Patents, 1966.
- Y. Dienes, W. Leitner, M. G. Müller, W. K. Offermans, T. Reier, A. Reinholdt, T. E. Weirich and T. E. Müller, Hybrid sol–gel double metal cyanide catalysts for the copolymerisation of styrene oxide and Co2, Green Chem., 2012, 14(4), 1168–1177 RSC.
-
J. M. O'connor, D. L. Lickei and R. L. Grieve, Preparing polyether polyols with DMC catalysts, US6539101, 2002.
- I. Kim, J.-T. Ahn, C. S. Ha, C. S. Yang and I. Park, Polymerization of propylene oxide by using double metal cyanide catalysts and the application to polyurethane elastomer, Polymer, 2003, 44(11), 3417–3428 CrossRef CAS.
- I. Kim, J.-T. Ahn, S.-H. Lee, C.-S. Ha and D.-W. Park, Preparation of multi-metal cyanide catalysts and ring-opening polymerization of propylene oxide, Catal. Today, 2004, 93, 511–516 CrossRef.
- I. Kim, M. J. Yi, K. J. Lee, D.-W. Park, B. U. Kim and C.-S. Ha, Aliphatic polycarbonate synthesis by copolymerization of carbon dioxide with epoxides over double metal cyanide catalysts prepared by using ZnX2 (X= F, Cl, Br, I), Catal. Today, 2006, 111(3), 292–296 CrossRef CAS.
-
M. B. Eleveld, R. A. W. Grotenbreg and R. Van Kempen, Preparation of a double metal cyanide catalyst, US 6977236, 2005.
-
D. Srinivas, R. Srivastava and P. Ratnasamy, Transesterification catalyst and a process for the preparation thereof, US 7754643, 2010.
- R. Srivastava, D. Srinivas and P. Ratnasamy, Fe–Zn double-metal cyanide complexes as novel, solid transesterification catalysts, J. Catal., 2006, 241(1), 34–44 CrossRef CAS.
- C. Murugan and H. Bajaj, Transesterification of propylene carbonate with methanol using Mg-Al-CO3 hydrotalcite as solid base catalyst. Indian journal of chemistry. Section A, Inorganic, bio-inorganic, physical, theoretical, Anal. Chem., 2010, 49(9), 1182 Search PubMed.
- Z. Song, B. Subramaniam and R. V. Chaudhari, Transesterification of propylene carbonate with methanol using Fe–Mn double metal cyanide catalyst, ACS Sustainable Chem. Eng., 2019, 7(6), 5698–5710 CrossRef CAS.
-
P. Ramachandran and R. Chaudhari, Three-phase Catalytic Reactors, Gordon & Breach Science Pub, 1983, vol. 2 Search PubMed.
-
A. V. Studio, Modeling, Optimization and Nonlinear Parameter Estimation for Scientists and Engineers, 2011 Search PubMed.
- J. P. Bernacki and R. M. Murphy, Model discrimination and mechanistic interpretation of kinetic data in protein aggregation studies, Biophys. J., 2009, 96(7), 2871–2887 CrossRef CAS.
- Z. Song, B. Subramaniam and R. V. Chaudhari, Kinetic study of CaO-catalyzed transesterification of cyclic carbonates with methanol, Ind. Eng. Chem. Res., 2018, 57(44), 14977–14987 CrossRef CAS.
- X. Jin, P. Bobba, N. Reding, Z. Song, P. S. Thapa, G. Prasad, B. Subramaniam and R. V. Chaudhari, Kinetic modeling of carboxylation of propylene oxide to propylene carbonate using ion-exchange resin catalyst in a semi-batch slurry reactor, Chem. Eng. Sci., 2017, 168, 189–203 CrossRef CAS.
-
B. Sirjean, P. A. Glaude, M. F. Ruizlopez and R. Fournet, Theoretical kinetic study of the ring opening of cyclic alkanes, Physics, 2009 Search PubMed.
-
A. Kumar, D. Some and K. H. Kiriamiti, in Pretreatment of CaO catalyst for transesterification of Croton megalocarpus oil, Proceedings of sustainable research and innovation conference, 2014, pp. 150–153 Search PubMed.
- M. Kouzu, T. Kasuno, M. Tajika, S. Yamanaka and J. Hidaka, Active phase of calcium oxide used as solid base catalyst for transesterification of soybean oil with refluxing methanol, Appl. Catal., A, 2008, 334(1–2), 357–365 CrossRef CAS.
-
K. Tanabe, M. Misono, Y. Ono, H. Hattori, K. Tanabe, M. Misono, Y. Ono and H. Hattori, New solid acids and bases studies in the surface science and catalysis, Elsevier, Amsterdam, 1989 Search PubMed.
- U. Schuchardt, R. Sercheli and R. M. Vargas, Transesterification of vegetable oils: a review, J. Braz. Chem. Soc., 1998, 9(3), 199–210 CrossRef CAS.
- S. Gryglewicz, Rapeseed oil methyl esters preparation using héterogeneous catalysts, Bioresour. Technol., 1999, 70(3), 249–253 CrossRef CAS.
- X. Liu, H. He, Y. Wang and S. Zhu, Transesterification of soybean oil to biodiesel using SrO as a solid base catalyst, Catal. Commun., 2007, 8(7), 1107–1111 CrossRef CAS.
- M. Kouzu, T. Kasuno, M. Tajika, Y. Sugimoto, S. Yamanaka and J. Hidaka, Calcium oxide as a solid base catalyst for transesterification of soybean oil and its application to biodiesel production, Fuel, 2008, 87(12), 2798–2806 CrossRef CAS.
- H. Masood, R. Yunus, T. S. Y. Choong, U. Rashid and Y. H. Taufiq Yap, Synthesis and characterization of calcium methoxide as heterogeneous catalyst for trimethylolpropane esters conversion reaction, Appl. Catal., A, 2012, 425–426, 184–190 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9re00372j |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 abc,
Bala
Subramaniam
abc,
Bala
Subramaniam