
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntroducing random bio-terpene segments to high cis-polybutadiene: making elastomeric materials more sustainable†
José Luis González-Zapataa,
Francisco Javier Enríquez-Medranoa,
Héctor Ricardo López González*a,
Javier Revilla-Vázquezb,
Ricardo Mendoza Carrizalesa,
Dimitrios Georgouvelasc,
Luis Valencia *d and
Ramón Enrique Díaz de León Gómez*a
*d and
Ramón Enrique Díaz de León Gómez*a
aResearch Center for Applied Chemistry, Blvd. Enrique Reyna 140, San José de los Cerritos, 25294, Saltillo, Coahuila, Mexico. E-mail: Ricardo.lopez@ciqa.edu.mx; Ramon.diazdeleon@ciqa.edu.mx
bDepartamento de Ingeniería y Tecnología, División Ciencias Químicas, Facultad de Estudios Superiores Cuautitlán, UNAM, Av. Primero de Mayo s/n, Cuautitlán Izcalli, C.P. 54740, Estado de México, México
cDivision of Materials and Environmental Chemistry, Stockholm University, Frescativägen 8, 10691 Stockholm, Sweden
dMaterials Technology and Chemistry, Alfa Laval Tumba AB, SE-14782, Tumba, Sweden. E-mail: Luisalexandro.valencialopez@alfalaval.com; luisalex_val@hotmail.com
First published on 15th December 2020
Abstract
In this work, we explore the statistical copolymerization of 1,3-butadiene with the terpenic monomers myrcene and farnesene, carried out via coordination polymerization using a neodymium-based ternary catalytic system. The resultant copolymers, poly(butadiene-co-myrcene) and poly(butadiene-co-farnesene), were synthesized at different monomer ratios, elucidating the influence of the bio-based monomer content over the kinetic variables, molecular and thermal properties, and the reactivity constants (Fineman–Ross and Kelen–Tüdös methods) of the resultant copolymers. The results indicate that through the herein employed conditions, it is possible to obtain “more sustainable” high-cis (≈95%) polybutadiene elastomers with random and tunable content of bio-based monomer. Moreover, the polymers exhibit fairly high molecular weights and a rather low dispersity index. Upon copolymerization, the Tg of high-cis PB can be shifted from −106 to −75 °C (farnesene) or −107 to −64 °C (myrcene), without altering the microstructure control. This work contributes to the development of more environmentally friendly elastomers, to form “green” rubber materials.
Introduction
The need to abate the current environmental issues, such as the “undeniable” global warming, and the massive amount of fossil-based plastic pollution that wind up in the oceans every year, urges a reduction in the usage of fossil-based materials, and their partial replacement with more sustainable (bio-based) alternatives. At the same time, the accelerated technological progress, as well as the high demands in the manufacturing industry, requires the development of high-performance elastomers that meet the needs of the upcoming markets.1 An ideal material selection should then find a good balance between performance and eco-friendliness.A good example of a material that does not meet the aforementioned criteria is polybutadiene (PB). PB is one of the most industrially used polymers due to its good abrasion and low rolling resistance. However, 1,3-butadiene monomer (Bd), which is required to produce PB, is extracted from steam cracking and therefore represents a growing environmental concern. The performance of PB depends largely on their microstructure (here referring to the geometrical isomerism). For instance, for PB, 1,4-cis insertion leads to higher-breaking strength and wear resistance.2 To obtain such control in PB microstructure it is necessary to use coordination polymerization as a synthesis route, employing Ziegler – Natta catalytic systems, which are mainly carboxylate and allyl derivates based on transition metals such as titanium (Ti), cobalt (Co), nickel (Ni) or lanthanide metals such as neodymium (Nd), lanthanum (La), and praseodymium (Pd) activated by alkylaluminums. These catalytic systems have repeatedly demonstrated their ability to yield PB with a high 1,4-cis structure (>90%),3–5 and, under specific reaction conditions, to yield narrow molecular weight distributions by promoting the coordinative chain transfer polymerization (CCTP). CCTP depends mainly on the appropriate amount of chain transfer agent (CTA), such as the alkylaluminum, which must be enough to promote chain transfer reactions to be reversible and faster than the chain growth, thus avoiding chain termination reactions. Under this scenario, polymers with narrow distributions can be obtained.6
A key alternative to minimize the environmental impact of PB is replacing it (at least partially) with more sustainable monomer alternatives, such as bio-sourced terpenes. Terpenes are a vast family of aliphatic and cycloaliphatic substances, mostly obtained from pine tree turpentine, which consists of hydrocarbons with a hemiterpene moiety, and therefore a conjugated double bond in their structure that allows their polymerization.7–14 An important group of terpenes is the monoterpenes, from which, perhaps the one with the greatest potential in the elastomer industry is β-myrcene (My), which is a compound that occurs naturally in many types of foods and beverages, such as citrus peel oils, corn, coriander, etc. Its extraction is mainly obtained through the pyrolysis of β-pinene.15,16 The sesquiterpenes, such as the trans-β-farnesene (Fa), are extracted from essential oils, but in the last years, industrial processes have been developed for the synthesis of this monomer through the yeast fermentation of sugar syrups.17,18 Fa has been reported to have a mixture of α- and β-isomers but only the β-isomer can be polymerized.19
Several authors have previously realized the importance of copolymerizing terpenic monomers to industrially relevant elastomers, obtaining partially bio-based elastomeric materials. For instance, Lamparelli et al.12 reported the synthesis of poly(myrcene-co-butadiene), poly(farnesene-co-butadiene), and poly(ocimene-co-butadiene) copolymers by using a titanium-based catalyst system, in which MMAO was used as an activator. Copolymers with a wide range of compositions were obtained in each system, displaying a 47, 52, and 85% maximum incorporation of polymyrcene (PMy), polyfarnesene (PFa), and polyocimene (POc) segments, respectively. The evaluated catalytic system provided a good stereoselectivity for both components in each series of copolymers, reporting up to 95, 71, and 86% of 1,4-trans structure for PB, PMy, and PFa, respectively, and up to 92% of 1,2-structure for POc. Moreover, Yu et al.,20 Li et al.,21 and Liu et al.22,23 studied the copolymerization of My with isoprene employing different rare-earth-based catalytic systems, under varied reactions conditions. However, despite the numerous reports, little attention has been centered on randomly incorporating terpenes into PB with high cis-1,4 unit content, which shows superior mechanical performance, in terms of higher tensile strength and elongation at break, as well as improved fatigue and crack resistance. Moreover, high cis-1,4 content promotes the strain-induced crystallization of rubbers.24–26
In this work, we report the copolymerization of 1,3-butadiene with the bio-based terpenes β-myrcene and trans-β-farnesene, via CCTP using a tertiary catalytic system based on Nd. With this approach, we can obtain more sustainable elastomers with a quasi-linear relationship between the composition of the terpenic monomer and the glass transition temperature (Tg). Furthermore, through the calculation of reactivity relationships, we can establish a random arrangement of the monomers in the copolymer chains.
Experimental section
Materials
The trans-β-farnesene monomer (purchased from AMIRYS with a purity of 98%) was washed with sulfuric acid and distilled under reduced pressure in the presence of metallic sodium. The β-myrcene monomer (purchased from VENTOS with a purity of 95%) was distilled under reduced pressure in the presence of metallic sodium. The 1,3-butadiene monomer was purchased from Aldrich, with 99% purity, and it was purified by passing it through an alumina packed column and molecular mesh before being fed to the polymerization reactor. The industrial-grade cyclohexane solvent was previously washed with concentrated sulfuric acid, cleaned with distilled water until obtaining a neutral pH, and was double distilled in the presence of lithium aluminum hydride and metallic sodium, all this in an inert nitrogen atmosphere. The catalytic system, comprised of neodymium versatate (NdV3) (0.54 M solution, from Solvay), diisobutylaluminum hydride (DIBAH), and dimethyldichlorosilane (DMDCS), was purchased from Sigma Aldrich, and all chemicals were used as received.Preparation of catalytic system
The preparation of the NdV3/DIBAH/DMDCS catalytic system was carried out in a glove box under a nitrogen atmosphere and room temperature. DIBAH was added dropwise to a glass vial at a concentration of 1 M (at a 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with respect to NdV3), immediately afterward the catalytic precursor NdV3 was added at a concentration of 0.5 M, and left stirring for 5 min. Then, the halide donor (DMDCS) was added in a 0.22 M cyclohexane solution, in a 1:1 molar ratio with respect to the catalytic precursor. The system was let to stir for 30 min and then injected into the reactor to start the polymerization.
:1 ratio with respect to NdV3), immediately afterward the catalytic precursor NdV3 was added at a concentration of 0.5 M, and left stirring for 5 min. Then, the halide donor (DMDCS) was added in a 0.22 M cyclohexane solution, in a 1:1 molar ratio with respect to the catalytic precursor. The system was let to stir for 30 min and then injected into the reactor to start the polymerization.
(Co)polymerizations
For the synthesis of the elastomers, a 2 L stainless steel Parr reactor was utilized, which was equipped with a double turbine agitation system and coupled with a PID temperature controller. Initially, the reactor was cured at 125 °C and, via multiple Schlenk cycles, decontaminated of humidity traces or any other volatile substance that may influence the synthesis. Then, the reaction temperature (at 70 °C for butadiene-co-myrcene copolymer and 60 °C for butadiene-co-farnesene copolymer) was set and the solvent (cyclohexane) and monomers are added to the desired compositions with 15% of solids, maintaining constant stirring at 100 rpm. Then, the pre-aged catalytic system (NdV3/DIBAH/DMDCS) was incorporated, and immediately the reactor was pressurized to 40 psi of nitrogen. Upon the addition of the catalytic system, the reaction initiated and ran until reaching 100% conversion (samples were taken at different times to evaluate the %-conversion gravimetrically). The reaction was finally killed by injecting acidified methanol, and the copolymers were obtained by precipitation in methanol, after the addition of antioxidants. The resultant polymers were then vacuum-dried at 50 °C prior to characterization.Characterization
The molecular weight characteristics of the synthesized elastomers were studied by Size Exclusion Chromatography (SEC) using an Agilent Technologies model PL-GPC 50, equipped with a 5 μm mixed type column at a pressure of 2.34 MPa and index detector refraction calibrated with polystyrene standards. The thermal transitions were elucidated via Differential Scanning Calorimetry (DSC) using a TA Instruments Model 2000, measured in a temperature range of −120 °C to 25 °C under a nitrogen atmosphere and a heating rate of 5 °C min−1. The compositions of the copolymers, as well as their microstructures, were calculated utilizing nuclear magnetic resonance in ASCED model equipment with a power of 400 MHz trademark Bruker using deuterated chloroform (CDCl3) as solvent at room temperature. The isomer 3,4 in relation with the isomer 1,4 was determined by 1H Nuclear Magnetic Resonance (NMR) using 16 scans for the measurements. Cis/trans ratios were calculated by 13C NMR by integrating signals of the olefinic groups. The reactivity ratios of the monomers were calculated using the Fineman–Ross and Kelen–Tödüs methods using the instant compositions obtained from the samples at conversions less than 10%. Where r1 and r2 are the reactivity ratios of 1,3-butadiene monomers and terpenic monomers respectively; F is the ratio of the instantaneous concentrations of the grafted monomers in the chain and f is the ratio of the monomers in the feed.where:
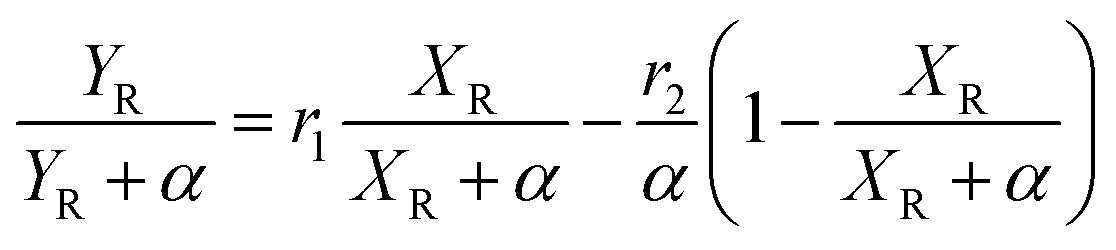
XRM being the highest and lowest XRm obtained from the Fineman–Ross method.
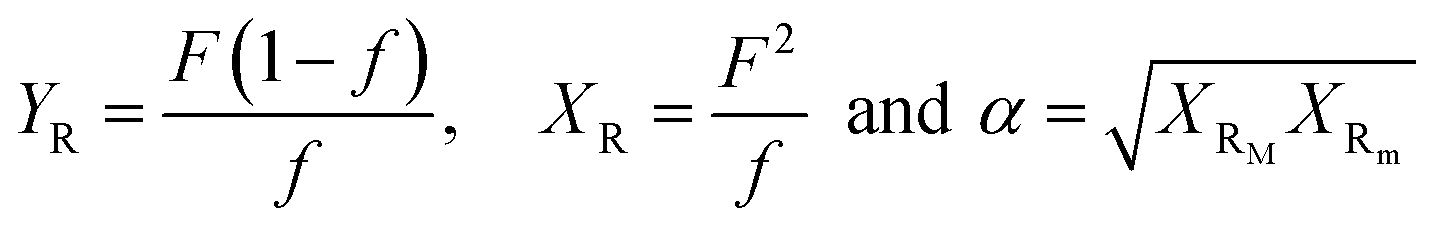
Results and discussion
With the aim of making “more sustainable” elastomers, (with a significant content of a renewable component), with high stereoselectivity and molecular weights (which is generally reflected in good mechanical properties of the materials), we herein report the copolymerization of butadiene with the bio-based terpenes myrcene and farnesene, conceived by CCTP using a ternary Nd-based catalytic system, comprising neodymium versatate/diisobutylaluminum hydride/dimethyldichlorosilane (NdV3/DIBAH/DMDCS, see Fig. 1). Similar catalytic systems have been previously demonstrated to stereospecifically polymerize butadiene.14,27,28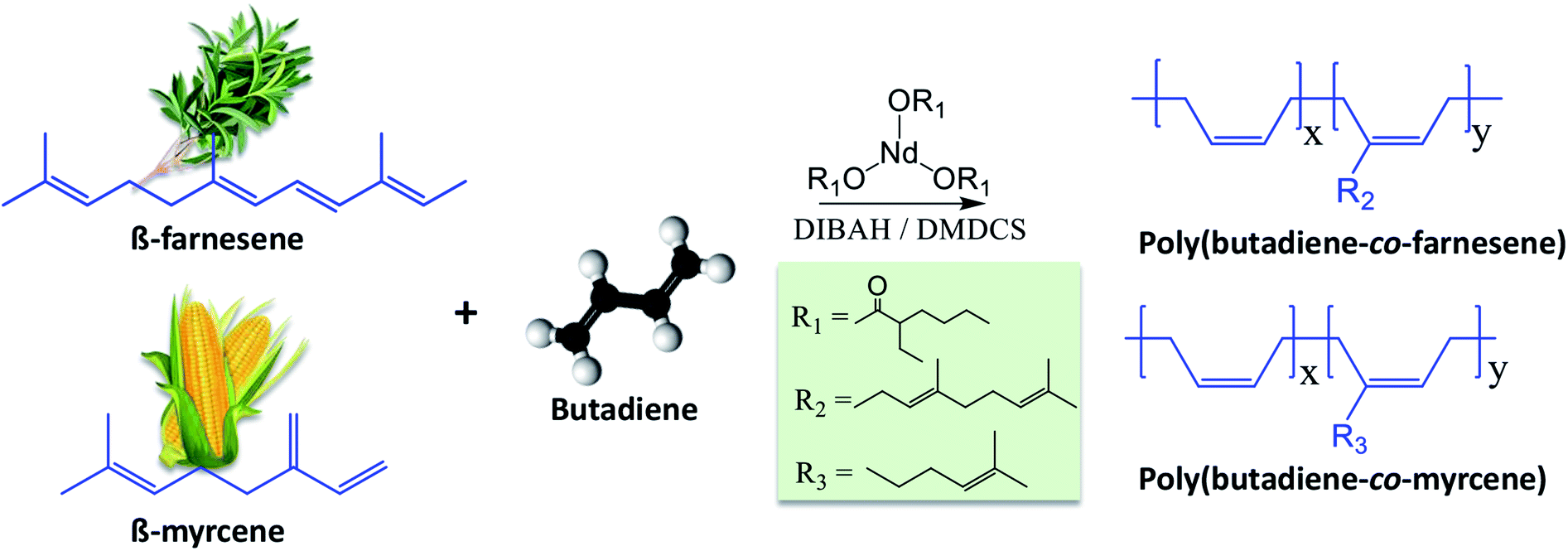 | ||
| Fig. 1 Schematic representation of the copolymerization of β-myrcene and trans-β-farnesene with 1,3-butadiene using a ternary catalytic system comprising of NdV3, DIBAH, and DMDCS. | ||
NdV3 is used in combination with an alkylaluminum (here DIBAH), which acts as a co-catalyst by behaving as a Lewis acid, in a way that it creates free-coordination sites in the metal complex and thus generating the active catalytic species that initiate the polymerization. Halide donors (here DMDCS), on the other hand, are used to enhance the microstructure-control, increase the catalytic activity, and promoting the polymerization via coordinating the Nd atoms and thus promoting the cis coordination of the monomer molecules. The molar ratios used in this work Al/Nd = 30 and Cl/Nd = 1, lead to narrow molecular weight distributions because they allow polymerizing Bd in the CCTP regime. For this reason, narrow distributions could be expected in the copolymers. We employed this catalytic system to copolymerize Bd with both Fa and My, and the most relevant results are summarized in Table 1.
| Runa | Theoretical terpeneb content-% | (Co)polymer compositions (experimental)c | Terpene weight fraction | Ad | kappe | Mw (kg mol−1) | Đf | Tg g(°C) | |
|---|---|---|---|---|---|---|---|---|---|
| Bd-% | Terpene-% | ||||||||
| a Synthesis of PBMy and PBFa carried out by an isothermal process, with temperatures of 70 °C and 60 °C respectively, maintaining constant stirring of 100 rpm. n.d. = not determined.b Theoretical molar (%) terpene content used for the synthesized of the copolymers.c Molar (%) compositions calculated via 1H NMR.d Catalytic activity (kgpolymer/mol−1 h−1). Calculated after 2 h of reaction for poly(butadiene-co-myrcene) and after 30 min for poly(butadiene-co-farnesene).e Apparent first-order rate constant (L mol−1 min−1) calculated considering the kinetic law d[M]/dt = ka[Nd][M], where kapp = k[Nd], and from the plots ln(1 − x)−1 = f(t), where x is the conversion.f Dispersity index (Mw/Mn) calculated by SEC.g Determined by DSC. | |||||||||
| PB1 | 0 | 100 | 0 | 0 | 364.4 | 165.5 | 85 | 1.9 | −107 |
| PBMy1 | 10 | 94.7 | 5.3 | 0.12 | 409.3 | 33.6 | 119 | 2.0 | −103 |
| PBMy2 | 25 | 87.9 | 12.1 | 0.26 | 114.5 | 30.9 | 164 | 2.5 | −99 |
| PBMy3 | 50 | 72.1 | 27.9 | 0.49 | 103.0 | 14.7 | 297 | 3.4 | −87 |
| PBMy4 | 75 | 47.9 | 52.1 | 0.74 | 83.8 | 12.6 | 560 | 3.7 | −79 |
| PMy | 100 | 0 | 100 | 1 | 109.7 | 5.1 | 1259 | 3.7 | −64 |
| PB2 | 0 | 100 | 0 | 0 | 242.9 | 165.4 | 53 | 2.8 | −106 |
| PBFa1 | 5 | 97.3 | 2.7 | 0.07 | 299.8 | 219.2 | n.d. | n.d. | −103 |
| PBFa2 | 10 | 94.9 | 5.1 | 0.12 | 228.0 | 140.6 | 59 | 1.9 | −101 |
| PBFa3 | 30 | 84.3 | 15.7 | 0.32 | 200.4 | 99.0 | 65 | 1.7 | −97 |
| PBFa4 | 50 | 63.1 | 36.9 | 0.53 | 291.3 | 89.4 | 115 | 2.1 | −88 |
| PBFa5 | 75 | 47.4 | 52.6 | 0.74 | 267.9 | 67.5 | 145 | 2.5 | −82 |
| PFa | 100 | 0 | 100 | 1 | n.d. | n.d. | 375 | 3.6 | −75 |
The kinetics of the copolymerizations were greatly influenced by the terpene incorporation, as can be observed by tracking the time-conversion relations (Fig. 2). PB homopolymer reaches full conversion after 1 h, while the PMy homopolymer (Fig. 2a) reaches only 80% of conversion after 8 h. This could be due to the My monomer purity which can have a strong effect on the polymerization rate, presumably because the present isomers have a slower insertion rate than the β-myrcene monomer to the active centers, as it's the case of limonene,29 this is then reflected in the catalytic activity (see Table 1), where a gradual drop was observed upon terpene units incorporation. A similar effect was observed in the copolymerizations of Bd and Fa (Fig. 2b), where a decrease in the copolymerization rate is also observed as a function of terpenic content. Another factor that could also influence the catalytic performance is the viscosity increase in the reaction (not studied here), which was significantly increased when feeding a large quantity of My or Fa. Shorter reaction times were required to synthesize poly(butadiene-co-myrcene) (PBMy) copolymers. However, in these reactions, we utilized a monomer/Nd ratio of 2500:1, compared to 4000:1 which was used for poly(butadiene-co-farnesene) (PBFa); whereas decreasing the monomer/catalyst ratio increases the interaction probability between the monomer and an active site, which consequently increases the ratio of monomer binding to the polymer chain. The use of higher monomer/Nd ratios in the synthesis of PBFa copolymers was aimed to synthesize copolymers of comparable molecular weight in relation to the PBMy copolymers, following previous works carried out in our research group.
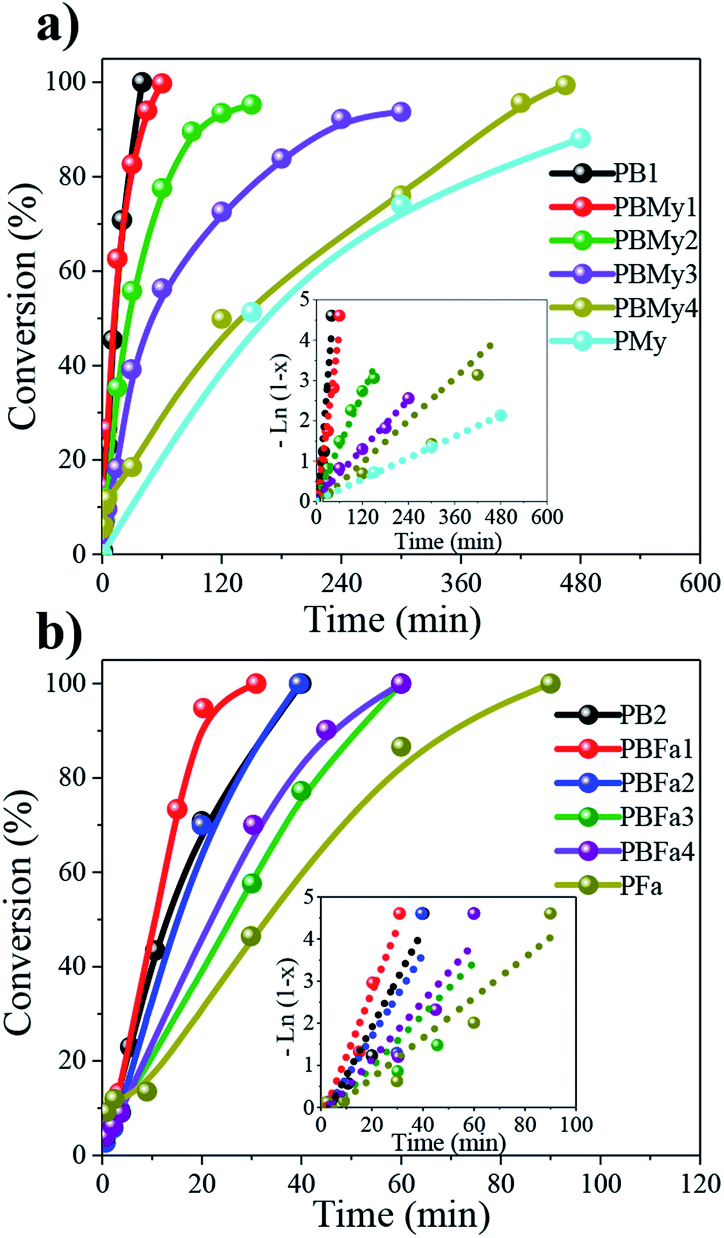 | ||
| Fig. 2 Rate of conversion in the synthesis of different (a) poly(butadiene-co-myrcene) and (b) poly(butadiene-co-farnesene) copolymers. Inlet figures show the first-order kinetics of the polymerizations. | ||
Both polymerizations revealed pseudo-first-order polymerization kinetics (inlet Fig. 2a and b) with good linearity between monomer –Ln(1 − x) vs. time, indicating a constant propagation of the growing polymeric chains, therefore being living polymerization processes. The calculated kinetic constant (kapp) values are shown in Table 1.
We furthermore demonstrated the successful copolymerization by obtaining the 1H NMR spectra of the produced copolymers (Fig. 3). The spectra display the appearance of the peaks at 5.1 ppm, which grows as a function of polyterpene content. The appearance of peaks at the range of 1.6 to 2.1 ppm in both systems is unequivocal evidence of the presence of the terpenic component in the synthesized copolymers. Moreover, the composition of the copolymers was calculated by the integral area relation of the peaks at δ = 5.1 and 5.5 ppm, corresponding to the double bond within the polymer backbone of the polyterpenes and PB, respectively. The compositions are summarized in Table 1.
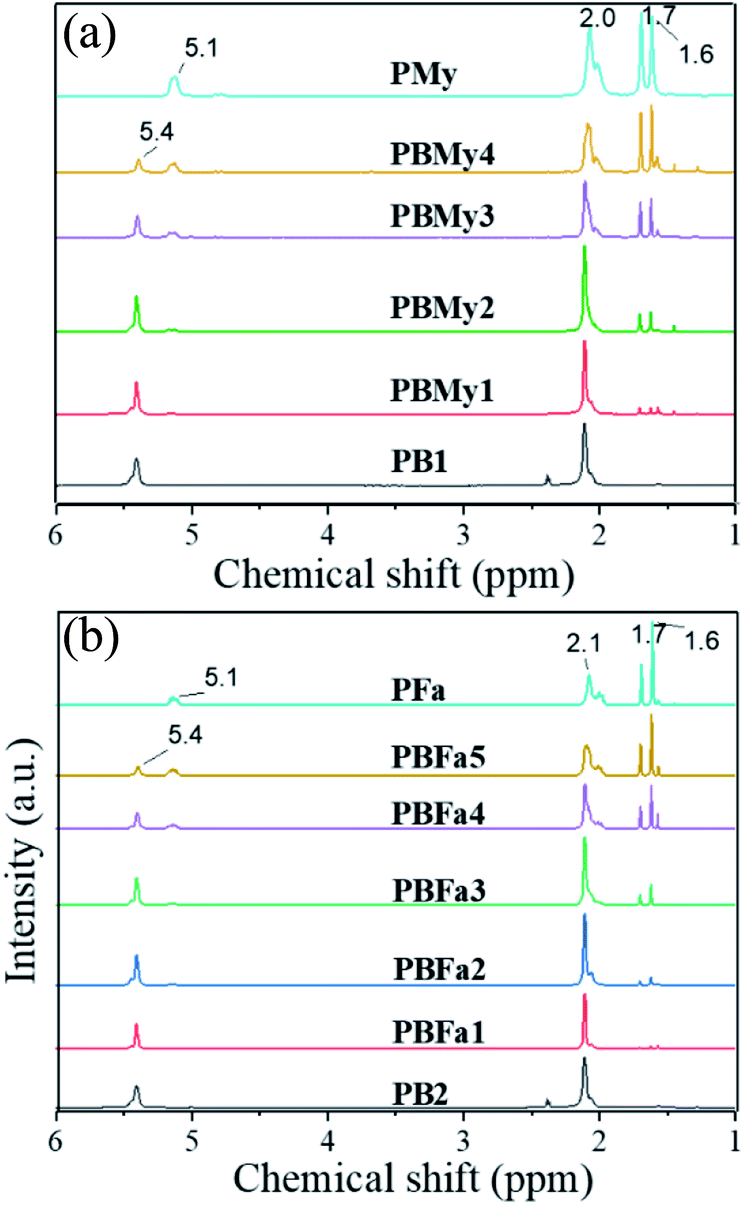 | ||
| Fig. 3 1H NMR spectra of different (a) PBMy and (b) PBFa copolymers. | ||
Microstructure of the homopolymers and copolymers
The microstructure of the synthesized (co)polymers was further examined by NMR and the results are summarized in Table 2. We estimated the isomer 1,4- in relation to the vinyl-1,2 of PB, PBMy3 and PBFa4. As well as the relation of 1,4 towards 3,4 in the case of PMy and PFa homopolymers. The calculations were done by integrating the signals in the olefinic region. The results are shown in Table 2.| Run | 1,4-cis (%) | 1,4-trans (%) | Vinyl-1,2 (%) | 3,4 (%) |
|---|---|---|---|---|
| PB1 | 95.7 | 3.8 | 0.5 | — |
| PMy | 90.3 | 3.4 | — | 6.3 |
| PFa | 91.8 | 4.8 | — | 3.4 |
| PBMy3 | 95.2 | 2.6 | 2.2 | — |
| PBFa4 | 95.7 | 2.3 | 2.0 | — |
On the other hand, the 1,4-cis content was estimated via 13C NMR (spectra are shown in ESI†), and the results show that PB homopolymer (PB1) is 95.7 %-cis while PMy and PFa have 90.3% and 91.8% of 1,4-cis, respectively. Moreover, the terpene insertion does not seem to alter the microstructure control (of the PB segment), as the 1,4-cis content of PBMy3 and PBFa4 (50% theoretical terpene content) was found to be 95.2% and 95.7, respectively (spectra shown in ESI†). These results were expectable, considering that these types of Nd-based catalytic systems are known to polymerize dienes in a stereocontrolled way, leading to high-cis PB. As well as have good control of the molecular weight characteristics of the polymers.24–26
Thermal behavior of the homopolymers and copolymers
The high cis structure of the polymers was further corroborated from the Tg values of the PB homopolymers which were −107 °C and −106 °C (Table 1), which are the values reported for a high cis-1,4 PB.30,31 The Tg corresponding to the different homopolymers (PB, PMy, and PFa) and copolymers (PBMy and PBFa) was characterized by DSC and the values are reported in Table 1. As shown in Table 1, the Tg of PB1 and PMy were −107 and −64 °C, and PB2 and PFa were −106 and −75 °C respectively. While all copolymers exhibited a single Tg situated between the glass transition temperature of the two homopolymers in each copolymer system, which indicated that the copolymers were random. It can be seen in Fig. 4 that the Tg of the different kinds of copolymers depends on the monomer ratio or copolymer composition, where the Tg gradually increases as a function of My and Fa content in the copolymers. The shift of the Tg towards higher temperatures can be attributed to the lateral or alkyl group of both myrcene and farnesene, which tend to decrease the mobility of the copolymer chain.32 On the other hand, a quasi-linear relationship of the Tg as a function of the terpene-weight fraction was observed (see Fig. 4). Those gradual movements of Tg indicate that the two monomers are randomly copolymerized, i.e., a random distribution of Bd and terpene sequences in the copolymer.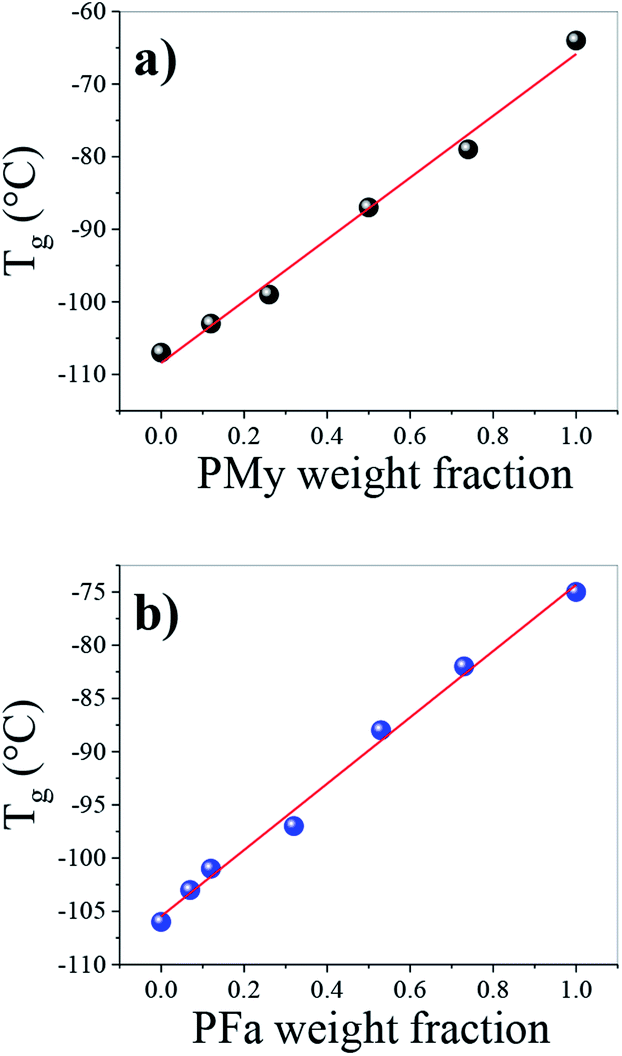 | ||
| Fig. 4 Influence of terpene content over the glass transition temperature of the synthesized (a) PBMy and (b) PBFa copolymers. | ||
Determination of reactivity ratios
To further understand the chemical composition of the copolymers, the reactivity constants of the different monomers during the copolymerizations were studied by two linear methods, the Fineman–Ross and the Kelen Tüdös. The graphs of the Fineman–Ross and Kelen–Tüdös methods for calculating the reactivity relationships of Bd and My monomers are shown in Fig. 5a and b. The obtained values of the reactivity constants of rBd and rMy from the Fineman–Ross method are 0.44 and 0.35, respectively, and from the Kelen–Tüdös method 0.66 and 0.63, respectively. In both cases, the reactivity of the Bd monomer was slightly higher than that of My monomer; however, the reactivity constants of both monomers can be considered similar. These reactivity constant values indicate that the polymer chains ending on butadiene or myrcene prefer to react with the comonomer before reacting with a monomer of the same species. Additionally, the product rBdrMyr < 1 indicates that the synthesized copolymers possess an alternated distribution of the comonomers along the chains; this behaviour should be more evident in the copolymers with composition proximately to 50/50 %-mol. Fig. 5c and d shows the graphs of the Fineman–Ross and Kelen–Tüdös methods for calculating the reactivity constants of Bd and Fa monomers. The values of reactivity constants were calculated as 0.15 and 2.37 employing the Fineman–Ross method, and 0.21 and 1.57 through the Kelen–Tüdös method, for rBd and rFa respectively. As can be observed from these values, unlike the PBMy copolymers, in the PBFa, the terpenic monomer is more reactive than butadiene towards the homo-propagation processes; however, as the product of the reactivity constants (rBdrFa) is less to 1, the resulting composition should be alternating with small blocks of farnesene at the beginning of the copolymerization reaction without forming a tapered microstructure. This behaviour should be more evident in copolymers with composition proximately to 50/50 %-mol.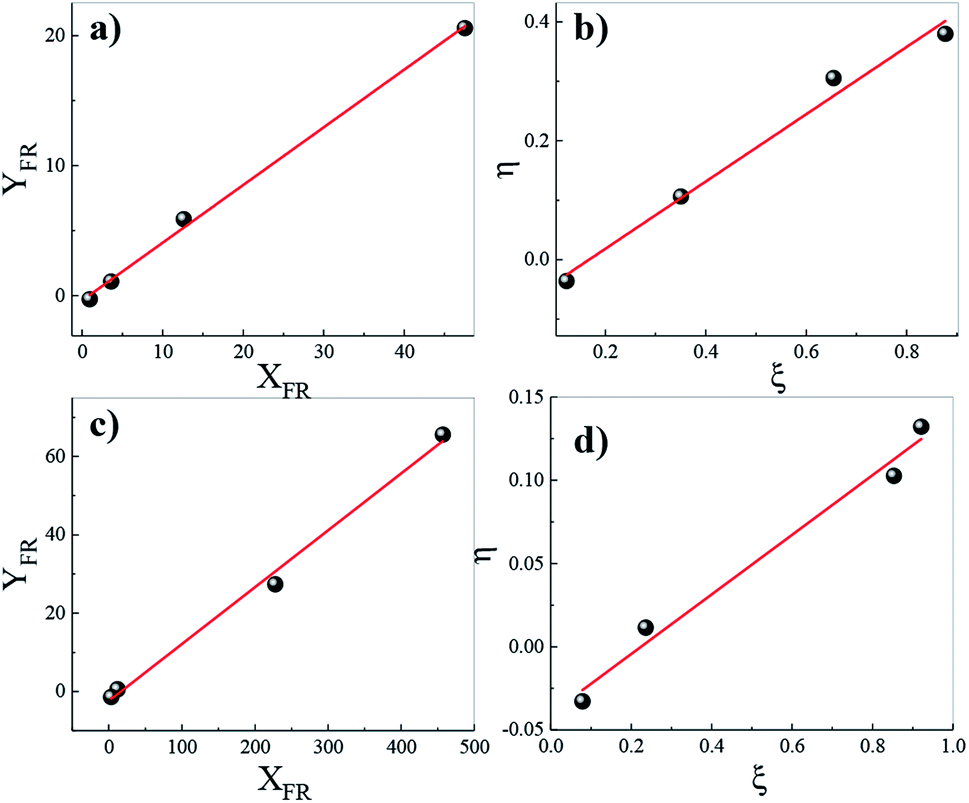 | ||
| Fig. 5 Determination of reactivity ratios by the Fineman–Ross (a and c) and Kelen–Tüdös (b and d) methods of (a and b) PBMy, (b–d) PBFa. | ||
Molecular weight characteristics of the homopolymers and copolymers
The successful insertion of the terpenic segments was also demonstrated by studying the molecular weight characteristics of the polymers, shown in Fig. 6 and Table 1. As can be observed, the molecular weight increases gradually as a function of terpenic content in the copolymers. Furthermore, the presence of one curve in the molecular weight distribution (MWD) indicates the formation of the copolymer. The main reason for this increase in molecular weight could be attributed to the incorporation of monomers (My and Fa) of higher molecular weight in the copolymer chain. Lastly, at low content of the terpenic monomer, the dispersity index (Ð) was found to be rather low, however, it tends to increase upon the incorporation of the terpenic segments, as can be seen in Table 1. This behavior can be explained because large amounts of terpene must disturb the CCTP regime by the differences in their propagation rates. Moreover, Ð also depends on chain transfer reactions, which are responsible for partially deactivating the catalyst, but also generating multiple catalytic active sites with different kinetic behaviors (therefore leading to polymer chains of variable length). This was reflected in the MWD, see Fig. 6, as one can see that the obtained copolymers with higher terpene content have broader distributions.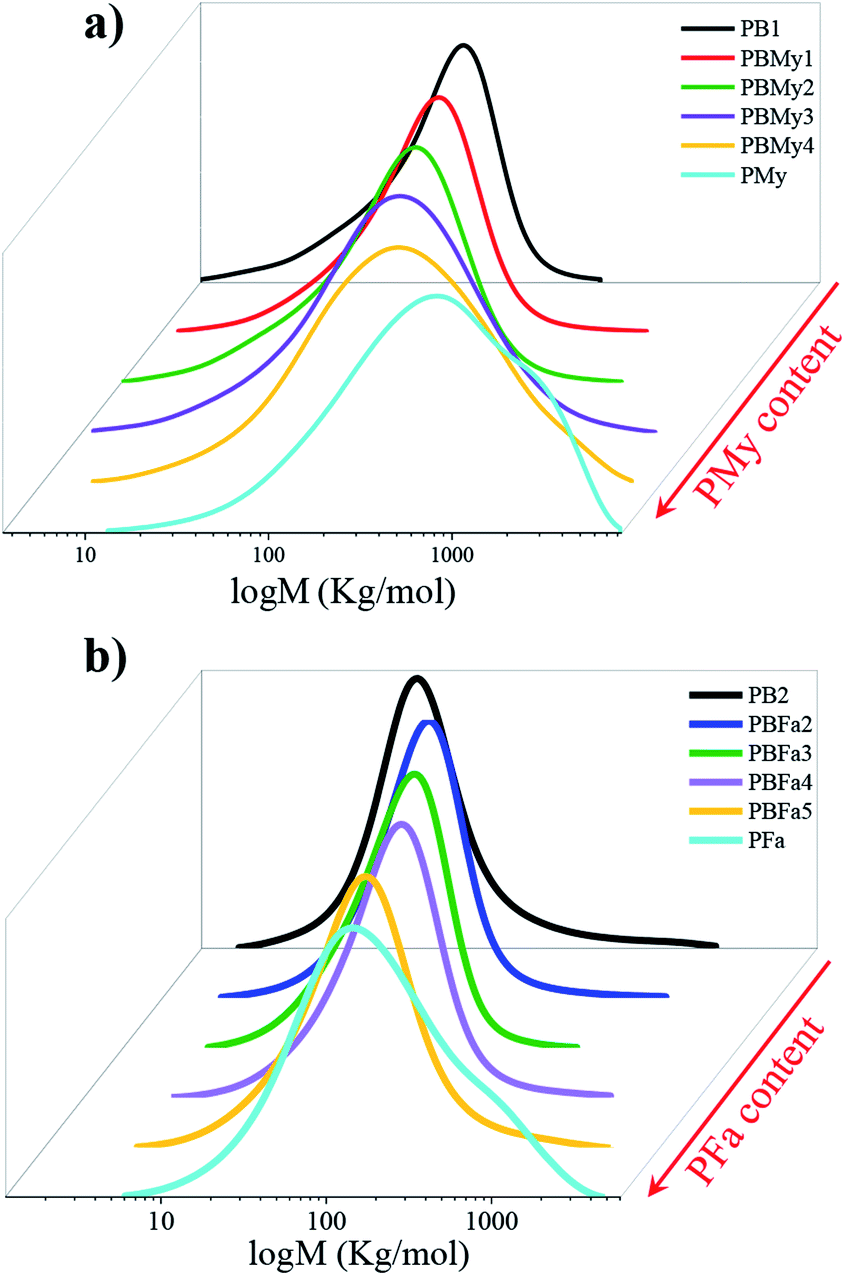 | ||
| Fig. 6 Evolution of the MWD as a function of terpene content of (a) poly(butadiene-co-myrcene) and (b) poly(butadiene-co-farnesene). | ||
A rheological characterization of the copolymers was performed in order to elucidate the influence of the terpene incorporation, using solely PBFa copolymers as model system. The flow curves, shown in the ESI (Fig. S6†), show a non-monotonic increase in the apparent viscosity of PB upon incorporation of farnesene segments. This behavior is presumably promoted by both, the inherent higher viscosity of the polyterpenes, but also the higher molecular weight of the copolymers that enhances entanglement.
Conclusions
In this work, we demonstrate the statistical copolymerization of 1,3-butadiene with two different terpenes, β-myrcene and trans-β-farnesene. The polymerizations were carried out via coordination polymerization using a ternary catalytic system based on neodymium. The results indicate that the catalytic system allows us to obtain PB with 95% 1,4-cis content, which is furthermore preserved upon copolymerization with the terpenes. Moreover, by tuning the terpenic content, we can obtain copolymers with variable molecular weight characteristics, as well as glass transition temperature, which goes from −106 to −75 °C, or −107 to −64 °C upon the incorporation of farnesene and myrcene units, respectively. Additionally, the reactivity constants were determined by the Fineman–Ross and Kelen–Tüdös methods from which we established that, in both copolymers, there is a random arrangement of the monomers in the copolymer chains. We believe that our results provide relevant insights into the synthesis of more sustainable elastomers and establish the basis for the gradual substitution of monomers from fossil sources by biobased monomers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of the Mexican National Council of Science and Technology (CONACyT) through the Basic Science project 258278, and the FORDECYT project 296356. The authors thank José Díaz Elizondo, Judith Cabello Romero, and Jesús Alfonso Mercado for their technical support in the characterization of samples.References
- Z. Wang, M. S. Ganewatta and C. Tang, Prog. Polym. Sci., 2020, 101, 101197 Search PubMed.
- A. J. Marzocca, S. Goyanes and A. L. R. Garraza, Rev. Latinoam. Metal. Mater., 2010, 30, 67 Search PubMed.
- L. Friebe, O. Nuyken, H. Windisch and W. Obrecht, Macromol. Chem. Phys., 2002, 203, 1055 Search PubMed.
- S. K.-H. Thiele and D. R. Wilson, J. Macromol. Sci., 2003, 43, 581 Search PubMed.
- V. K. Srivastava, M. Maiti, G. C. Basak and R. V. Jasra, J. Chem. Sci., 2014, 126, 415 Search PubMed.
- P. Zinck, Polym. Int., 2012, 61, 2 Search PubMed.
- P. Sahu and A. K. Bhowmick, Ind. Eng. Chem. Res., 2019, 58, 20946 Search PubMed.
- P. Sahu, P. Sarkar and A. K. Bhowmick, ACS Sustainable Chem. Eng., 2017, 5, 7659 Search PubMed.
- P. Sahu, A. K. Bhowmick and G. Kali, Processes, 2020, 8, 1 Search PubMed.
- P. Sarkar and A. K. Bhowmick, RSC Adv., 2014, 4, 61343 Search PubMed.
- F. Della Monica and A. W. Kleij, Polym. Chem., 2020, 11, 5109 Search PubMed.
- D. H. Lamparelli, V. Paradiso, F. Della Monica, A. Proto, S. Guerra, L. Giannini and C. Capacchione, Macromolecules, 2020, 53, 1665 Search PubMed.
- L. Valencia, F. J. Enríquez-Medrano, H. R. López González, R. Handa, H. S. Caballero, R. M. Carrizales, J. L. Olivares-Romero and R. E. Díaz de León Gómez, RSC Adv., 2020, 10, 36539 Search PubMed.
- A. Bahena, I. Magaña, H. R. López González, R. Handa, F. J. Enríquez-Medrano, S. Kumar, R. M. Carrizales, S. Fernandez, L. Valencia and R. E. Díaz de León Gómez, RSC Adv., 2020, 10, 36531 Search PubMed.
- P. M. Dewick, Medicinal Natural Products: A Biosynthetic Approach, 3rd edn, 2009 Search PubMed.
- A. Behr and L. Johnen, ChemSusChem, 2009, 2, 1072 Search PubMed.
- G. W. Dawson, D. C. Griffiths, J. A. Pickett, M. C. Smith and C. M. Woodcock, J. Chem. Ecol., 1982, 8, 1111 Search PubMed.
- K. R. Benjamin, I. R. Silva, J. P. Cherubim, D. McPhee and C. J. Paddon, J. Braz. Chem. Soc., 2016, 27, 1339 Search PubMed.
- J. Raynaud, J. Y. Wu and T. Ritter, Angew. Chem., Int. Ed., 2012, 51, 11805 Search PubMed.
- X. Yu, M. Li, J. Hong, X. Zhou and L. Zhang, Chem.–Eur. J., 2019, 25, 2569 Search PubMed.
- W. Li, J. Zhao, X. Zhang and D. Gong, Ind. Eng. Chem. Res., 2019, 58, 2792 Search PubMed.
- B. Liu, L. Li, G. Sun, D. Liu, S. Li and D. Cui, Chem. Commun., 2015, 51, 1039 Search PubMed.
- B. Liu, D. Tao Liu, S. Hui Li, G. Ping Sun and D. Mei Cui, Chin. J. Polym. Sci., 2016, 34, 104 Search PubMed.
- D. Leon and Y. A. De Santiago-rodriguez, Macromol. Symp., 2013, 325, 125 Search PubMed.
- F. J. Enr, R. M. Carrizales, K. R. Acosta and H. R. López, Can. J. Chem. Eng., 2016, 94, 823 Search PubMed.
- R. D. De León, R. López, L. Valencia, R. Mendoza, J. Cabello and J. Enríquez, Key Eng. Mater., 2018, 779, 115 Search PubMed.
- R. P. Quirk and A. M. Kells, Polym. Int., 2000, 49, 751 Search PubMed.
- R. P. Quirk, A. M. Kells, K. Yunlu and J.-P. Cuif, Polymer, 2000, 41, 5903 Search PubMed.
- K. Yao and C. Tang, Macromolecules, 2013, 46, 1689 Search PubMed.
- L. A. Valencia López, F. J. Enríquez-Medrano, H. M. Textle, F. S. Corral, H. R. L. González, C. S. Thomas, F. H. Gámez, J. L. O. Romero and R. E. D. De León Gómez, J. Mex. Chem. Soc., 2016, 60, 141 Search PubMed.
- F. J. Enríquez-Medrano, L. A. V. López, Y. A. de Santiago-Rodríguez, F. S. Corral, H. S. Caballero, M. L. L. Quintanilla and R. D. de León-Gómez, J. Polym. Eng., 2015, 35, 1 Search PubMed.
- Q. Gan, Y. Xu, W. Huang, W. Luo, Z. Hu, F. Tang, X. Jia and D. Gong, Polym. Int., 2020, 69, 763 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 13C NMR spectra of some of the synthesized polymers. See DOI: 10.1039/d0ra09280k |
| This journal is © The Royal Society of Chemistry 2020 |