
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeveloping non-isocyanate urethane-methacrylate photo-monomers for 3D printing application†
Neelima Singh,
Hadi Bakhshi * and
Wolfdietrich Meyer
* and
Wolfdietrich Meyer
Department of Functional Polymer Systems, Fraunhofer Institute for Applied Polymer Research IAP, Geiselbergstraße 69, 14476 Potsdam, Germany. E-mail: hadi.bakhshi@iap.fraunhofer.de; Tel: +49-331-568-1425
First published on 15th December 2020
Abstract
Urethane-methacrylate photo-monomers were prepared via a non-isocyanate route for the 3D printing application. The monomers were synthesized through reacting aliphatic amines, i.e. 1,6-hexanediamine, 1,4-butanediol bis(3-aminopropyl) ether, or n-butylamine, with cyclic carbonates, i.e. ethylene carbonate or propylene carbonate, followed by the methacrylation of the generated hydroxylurethanes. The effects of the chemical structure of monomers on their photo-reactivity and physicomechanical properties of the cured samples were studied. Propylene carbonate generated side methyl groups within the urethane block, which significantly limited the crystallization of the monomers resulting in high photo-reactivity (Rp,max = 6.59 × 10−2 s−1) and conversion (DBCtotal = 85%). The ether bonds of 1,4-butanediol bis(3-aminopropyl) ether decreased the intermolecular hydrogen bonding between urethane blocks, which not only improved the photo-reactivity (Rp,max = 8.18 × 10−2 s−1) and conversion (DBCtotal = 86%) of the monomer but led to a high crosslinking density (νc = 5140 mol m−3) and more flexibility for the cured sample. An ink was developed based on the monomers and successfully 3D printed on a digital light processing machine. In the absence of toxic isocyanates and tin compounds, the non-isocyanate route can be employed to develop urethane-methacrylates with desirable photo-reactivity and physicomechanical properties as good candidates to formulate inks for 3D printing of biomedical materials.
Introduction
Urethane-methacrylates are interesting photo-monomers to generate networks with high mechanical stability. These monomers are used in the formulation of UV-curable coatings1 and adhesives,2 dental restorative materials,3 and stereolithography.4,5 The intermolecular hydrogen bonding between the urethane blocks of monomers grants the pre-association of molecules and thus 3–6 times faster photo-curing compared to their corresponding non-hydrogen bonding methacrylates.6–8 Furthermore, the hydrogen bonding causes the superior mechanical strength for the cured samples.9,10 Therefore, urethane-methacrylates are ideal candidates for developing fast-curable formulations, where high mechanical properties such as tensile and flexural strengths for the final material are expected.Urethane-methacrylates are usually synthesized from the reaction of polyisocyanates and hydroxyalkyl methacrylates.10–12 The principal limitation of this route is the high toxicity of isocyanates to the environment and humans.13–15 Isocyanates are one of the frequently reported sources for chemical-induced occupational asthma16 since they can react with hydroxyl, amine, and carboxylic acid functions of the human body's proteins in terms of inhalation or skin/eye contact.13–15 Although it is assumed that isocyanates are completely converting during the synthesis, traces of unreacted isocyanate residues were detected in the final polyurethane materials,17,18 which are toxic for the users.19,20 Tin-based catalysts, e.g. dibutyl-tin-dilaurate (DBTDL), which is not removed after the reaction led to the toxicity for urethane-methacrylates even after photo-curing.21,22 Therefore, synthesis of urethane-methacrylates excluding isocyanates and tin-based catalysts is valuable, especially for the fabrication of biomedical, food packaging, and children products.
One non-isocyanate route to synthesize urethane-methacrylates is through the ring-opening reaction of cyclic carbonates with primary amines, which generates hydroxylurethanes.7,23–26 In contrast to isocyanates, cyclic carbonates are not toxic or moisture-sensitive to require special safety care during storage and handling.27 The pendant hydroxyl groups allow further functionalization reactions, e.g. methacrylation.7,23,24,28,29 On this subject, Wang et al.28 synthesized urethane-methylates through the mentioned non-isocyanate route as reactive diluents for UV-curable polyurethane coatings. Meng et al.23,24 prepared also urethane-methylates via the same route for emulsion polymerization.
Recently, photo-curing 3D printing technology is vastly utilizing for medical applications.30,31 It can be used to rapidly manufacture personalized implants and organs, which perfectly match the patient's damaged tissue.31 The suitable mechanical strength, bioactivity, biodegradability/biostability, and particularly biocompatibility of the employed materials are effective for the proper function of the 3D printed implant in the body and tissue repairing process. Regarding biocompatibility, the majority of photo-curable inks are cytotoxic, due to the unreacted monomers, photoinitiator resides, and toxic impurities. Therefore, developing photo-curable 3D printing inks for direct and long-term implantation in the body is still in research. As mentioned before, the synthesis of urethane-methacrylates through a non-isocyanate route is valuable for developing a biocompatible photo-curable ink. On this subject, Warner et al.32 synthesized a non-isocyanate urethane-ally compound to develop thiol–ene inks for digital light processing (DLP) printing, which did not expose any toxic effect against murine myoblasts.
In this work, we prepared urethane-methacrylate photo-monomers to develop 3D printable inks employing non-isocyanate urethane chemistry without using toxic isocyanates and tin compounds. For this purpose, different aliphatic primary diamines or amine, i.e. 1,6-hexanediamine (1,6-HDA), 1,4-butanediol bis(3-aminopropyl)ether (1,4-BBE), or n-butylamine (n-BA), were reacted with cyclic carbonates, i.e. ethylene carbonate (EC) or propylene carbonate (PC), to generate hydroxylurethanes. Later, the hydroxyl groups of hydroxylurethanes reacted with methacrylate anhydride (MAAn) to synthesize urethane-methacrylates. The effects of the chemical structure of monomers on their photo-reactivity and physicomechanical properties of the cured samples were studied. The 3D printability of a formulation based on the monomers was tested on a DLP printer.
Experimental
All experimental details including materials, synthesis procedures, instruments, methods are provided in the ESI.†Results and discussion
Synthesis of urethane-methacrylates
Non-isocyanate urethane chemistry was employed to develop urethane-methacrylate photo-monomers in two steps. In the first step, aliphatic primary diamines (1,6-HDA or 1,4-BBE) or amine (n-BA) were reacted with cyclic carbonates (EC or PC) without using any catalyst to generate hydroxylurethanes (Fig. 1).7,23,24,28,33 FTIR spectra obtained from the reaction mixtures showed the disappearance of the peak at 1795 cm−1 corresponding to the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bond of the cyclic carbonate moiety and the appearance of three new peaks at 1525, 1710, and 3320 cm−1 attributing to the generated urethane and hydroxyl groups (Fig. S1 in ESI†).34,35 The aminolysis of EC with the primary amines was fast without using any catalyst. For example, the reaction of EC with 1,6-HDA was completed within 1 h at 25 °C. However, the reaction of PC with 1,6-HDA was very slow requiring a high reaction time (48 h) and temperature (60 °C, Fig. S1 in ESI†). The low reactivity of PC against the primary amines has been previously reported.36 The presence of the electron-releasing methyl group within PC decreases the partial polarity of the carbonyl bond and consequently reduces its reactivity for the nucleophilic reactions.37,38
O) bond of the cyclic carbonate moiety and the appearance of three new peaks at 1525, 1710, and 3320 cm−1 attributing to the generated urethane and hydroxyl groups (Fig. S1 in ESI†).34,35 The aminolysis of EC with the primary amines was fast without using any catalyst. For example, the reaction of EC with 1,6-HDA was completed within 1 h at 25 °C. However, the reaction of PC with 1,6-HDA was very slow requiring a high reaction time (48 h) and temperature (60 °C, Fig. S1 in ESI†). The low reactivity of PC against the primary amines has been previously reported.36 The presence of the electron-releasing methyl group within PC decreases the partial polarity of the carbonyl bond and consequently reduces its reactivity for the nucleophilic reactions.37,38
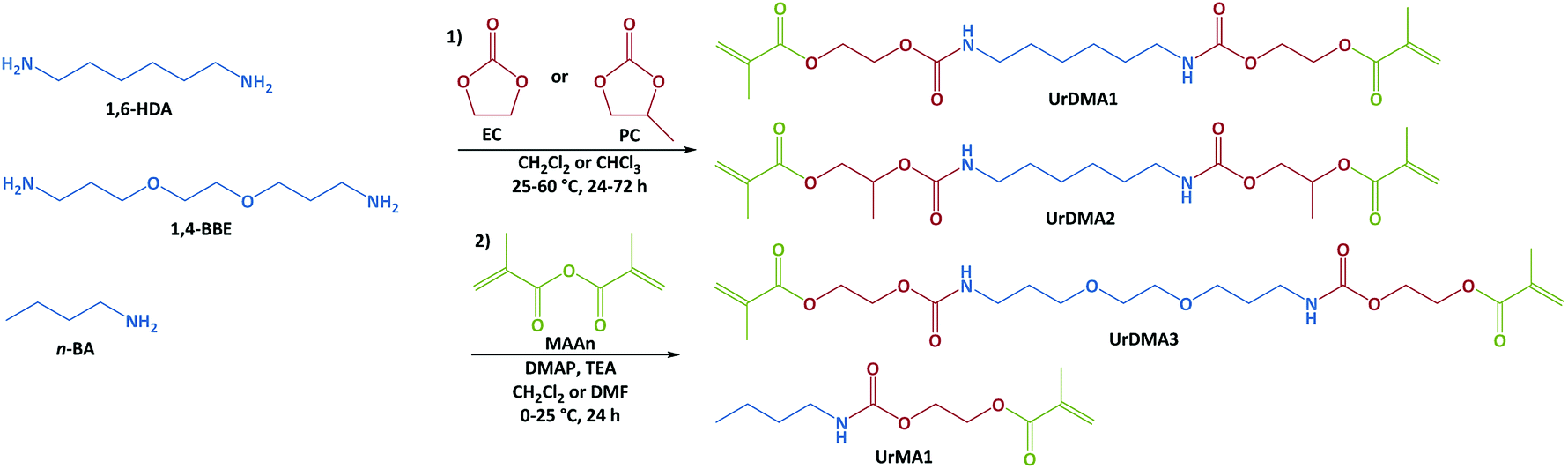 | ||
| Fig. 1 Two-step non-isocyanate route for the synthesis of urethane-methacrylate monomers. | ||
The methyl group of PC can result in constitutional isomerization during the ring–opening reaction. Therefore, the reaction of PC and 1,6-HAD led to three products; one with two primary hydroxyl groups, one with two secondary hydroxyl groups, and one with a primary and a secondary hydroxyl groups (synthesis of UrDMA2 in ESI†). According to the 1H-NMR integration values (Fig. S2 in ESI†), the ring–opening reaction of PC from γ-position of the methyl group (59%) resulting in the primary hydroxyl group was more possible than the β-position (41%) leading to the secondary hydroxyl group, which was in agreement with previous reports.36,39 It can be attributed to the steric hindrance effect of the methyl moiety during the attack of nucleophiles (primary amine groups). Increasing the reaction temperature from 25 °C to 60 °C did not change the ratio of primary to secondary hydroxyl groups within the final product (Urdiol2). In the case of EC, only one hydroxylurethane, with the primary hydroxyl groups was obtained (synthesis of UrDMA1, UrDMA3, and UrMA1 in ESI†). The chemical structure of all synthesized hydroxylurethanes was studied by FTIR and NMR spectroscopies. The results were in agreement with their expected molecular structures (Fig. S2 and S3 in ESI†). According to the 1H-NMR integration values, the yield of the aminolysis reaction was 100% for all synthesized hydroxylurethanes.
In the second step, the hydroxyl groups of hydroxylurethanes were reacted with MAAn using DMAP as a catalyst and TEA as an acid scavenger at room temperature (Fig. 1).7,23,24,28 Due to the exothermic nature of the methacrylation reaction, the hydroxylurethane solutions were initially cooled down to 0 °C. The chemical structure of all synthesized monomers was studied by FTIR and NMR spectroscopies (ESI†). FTIR spectra confirmed the successful methacrylation, while the sharpness of the peak at 3320 cm−1 relating to the hydroxyl groups of hydroxylurethanes was decreased and a shoulder peak at 1640 cm−1 regarding the double bonds (CC) of methacrylate moieties appeared. The NMR spectra for UrDMA1 are presented in Fig. 2. All peaks are assigned with the corresponding protons or carbons in the embedded molecular structure of UrDMA1. The urethane protons yielded two signals at 6.87 and 7.21 ppm regarding the pseudo E and Z conformations.33,40 The protons of the methacrylate moieties appeared at 1.88, 5.69, and 6.03 ppm.7 Meanwhile, the carbons of carbonyl bonds within the urethane and methacrylate moieties led to signals at 156.39 and 166.87 ppm.7,33,40 According to the 1H-NMR integration values, the yield of the methacrylation reaction was 100% for all synthesized monomers.
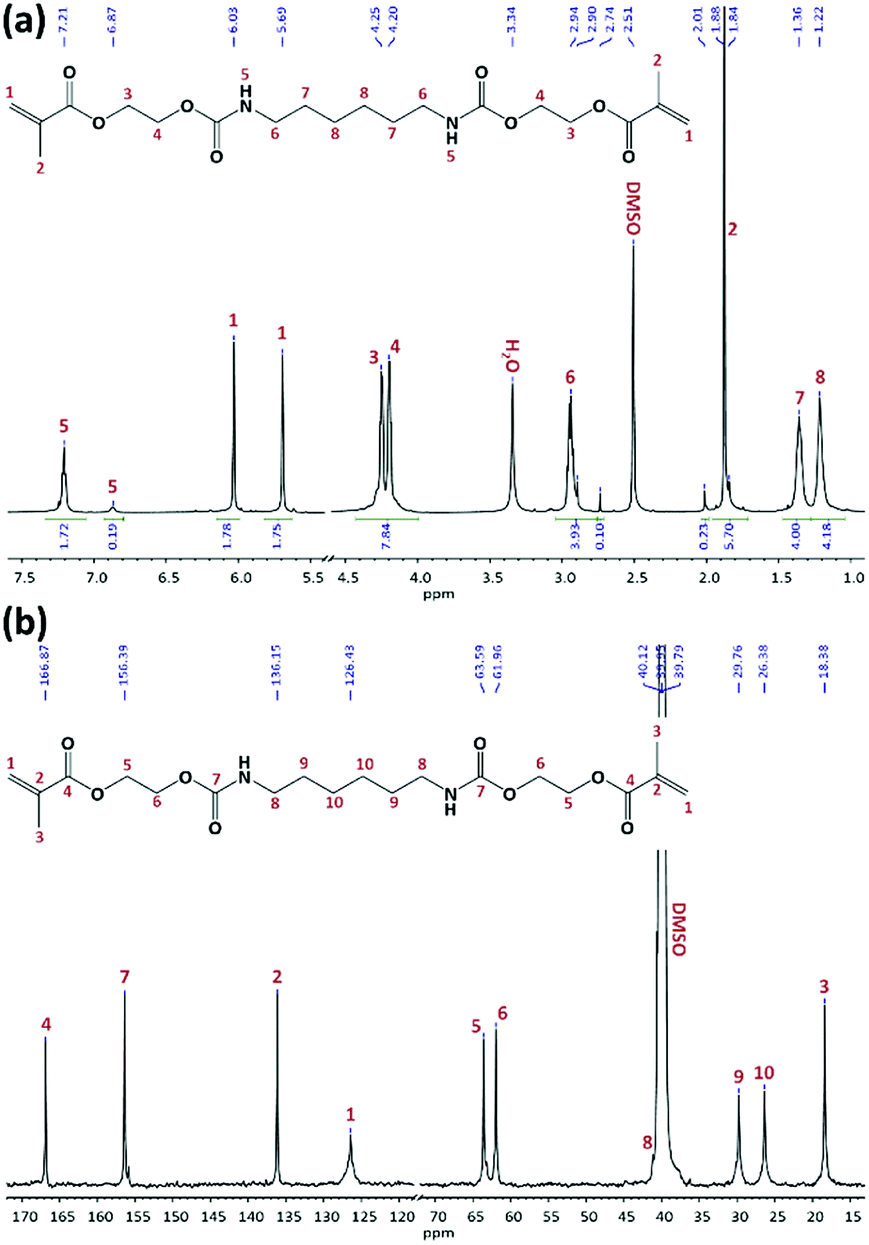 | ||
| Fig. 2 1H-NMR (a) and 13C-NMR (b) spectra for UrDMA1 in DMSO-d6. | ||
The physical state of the photo-monomers is important for the ease of formulating photo-curable inks. Therefore, the melting points (Tm) of the monomers were evaluated by DSC (Table 1 and Fig. S4 in ESI†). Due to the thermal-sensitivity of the monomers, the first heating cycle was used for the extraction of Tm values (peak maximum). UrDMA1 was a white powder with a Tm of 76 °C, thus it needs to be melted or dissolved in a reactive dilute before photo-curing. The Tm of UrDMA1 was depended on its purity. Thus, the Tm of UrDMA1 synthesized here with high purity was 20 °C higher than the reported value (56–57 °C (ref. 7)). UrDMA2 was a colorless low-viscose liquid that did not show any Tm by cooling, while it underwent a glass transition (Tg) at −57 °C (Fig. S4 in ESI†). The UrDMA2 molecules as a mixture of three constitutional isomers were sterically hindered by two methyl groups to form intermolecular hydrogen bonding, pack in and make a crystalline domain.7 UrDMA3 was a waxy solid with a Tm of 31 °C. The lower Tm and crystallinity (ΔHm = 5.0 J g−1) of UrDMA3 comparing to UrDMA1 (ΔHm = 109.9 J g−1) is attributed to the presence of two ether bonds within its molecular structure, which can make hydrogen bonds with the urethane moieties and consequently increases the ratio of intramolecular to intermolecular hydrogen bonding. Monofunctional UrMA1 was a colorless liquid with a Tm of 3 °C. Due to low viscosity (35 mPa s), it can be used as a reactive diluent for other difunctional urethane-methacrylates.
| Monomer | DSC | Photo-DSC | ||||||
|---|---|---|---|---|---|---|---|---|
| Tm (°C) | ΔHm (J g−1) | Tp (°C) | Rp,max (s−1) | tmax (s) | ΔHp,total (J g−1) | DBCtotal (%) | t95% (s) | |
| a Tp: temperature of photo-curing, Rp,max: maximum photo-curing rate, tmax: time to reach Rp,max, ΔHp,total: total generated photo-curing heat, DBCtotal: total double bond conversion, t95%: time to reach 95% of DBCtotal. | ||||||||
| UrDMA1 | 76 | 109.9 | 25 | 4.72 × 10−3 | 61.4 | 5.8 | 2 | 99.3 |
| 80 | 2.68 × 10−2 | 13.3 | 121.2 | 47 | 49.6 | |||
| UrDMA2 | — | — | 25 | 6.59 × 10−2 | 13.4 | 254.8 | 85 | 33.6 |
| UrDMA3 | 31 | 5.0 | 25 | 8.18 × 10−2 | 10.9 | 182.9 | 86 | 29.1 |
| UrMA1 | 3 | 34.1 | 25 | 6.61 × 10−2 | 19.5 | 247.5 | 98 | 32.9 |
Tp: temperature of photo-curing, Rp,max: maximum photo-curing rate, tmax: time to reach Rp,max, ΔHp,total: total generated photo-curing heat, DBCtotal: total double bond conversion, t95%: time to reach 95% of DBCtotal.
Photo-reactivity of urethane-methacrylates
The photo-reactivity of the monomers was studied using photo-DSC in isothermal mode. Each monomer was mixed with ethyl phenyl(2,4,6-trimethylbenzoyl)phosphinate (TPO-L, 3 wt%) as a photoinitiator before analysis at 25 °C with a UV intensity of 1 W m−2. UrDMA1 was warmed up to melt before mixing with TPO-L. The photo-curing rate (Rp) and double bond conversion (DBC) values as a function of time were calculated from photo-DSC data (Fig. S5 in ESI†) and presented in Fig. 3 and Table 1. UrDMA1 containing TPO-L, which was solid at room temperature showed very low photo-reactivity (DBCtotal = 2%) at 25 °C. Therefore, photo-DSC was repeated for this sample at a higher temperature (80 °C). All samples exhibited the auto-acceleration and auto-deceleration phenomenon, i.e. an initial increase and a later decrease in Rp, respectively (Fig. 3a), as well as a maximum limiting conversion during the photo-curing process (Fig. 3b). This complex behavior is because the mobility of the methacrylate groups gradually decreases over the photo-curing time. Initially, the Rp of liquid monomers, an overall of the propagation and termination rates, is constant and chemical-controlled. Later, due to an increase in the viscosity of the photo-curing mixture the coupling of macroradicals for the termination is diffusion-limited, while the monomers are still mobile for the propagation, thus Rp increases (auto-acceleration). Finally, the reaction mixture is transformed from a liquid to a rubbery or glassy network, which significantly restricts the diffusion of monomers to reach the macroradicals, therefore Rp decreases (auto-deceleration). Due to the gelation or vitrification, all methacrylate groups can not react and the final conversion is less than unity.11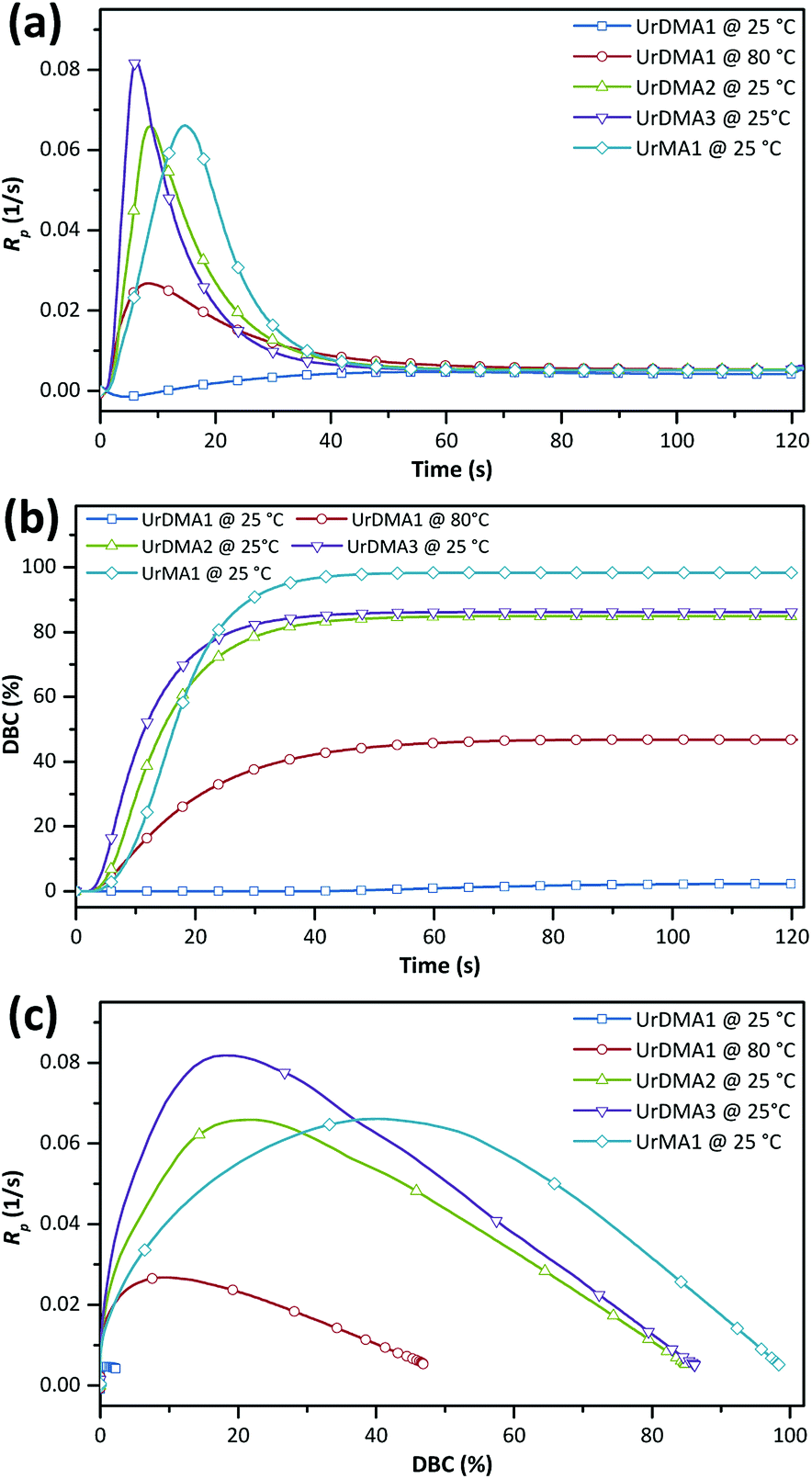 | ||
| Fig. 3 Photo-curing rate (a and c) and double bond conversion (b) for urethane-methacrylates obtained from photo-DSC data. | ||
UrDMA1 cured even at 80 °C showed a lower photo-reactivity (Rp,max = 2.68 × 10−2 s−1) and conversion (DBCtotal = 47%) comparing to UrDMA2 (Rp,max = 6.59 × 10−2 s−1, DBCtotal = 85%). Although UrDMA1 molecules have higher potency to pre-associate via intermolecular hydrogen bonding comparing to the amorph UrDMA2 molecules, the reaction of methacrylate groups within this high viscosity melted mixture (at 80 °C) was diffusion-limited. The photo-reactivity of the synthesized UrDMA1 was lower than the reported values (Rp,max = 9 × 10−2 s−1, DBCtotal = 78% (ref. 7)), which can be related to the differences in the purity of monomer and experiment parameters. UrDMA3 with the balanced intermolecular hydrogen bonding (UrDMA1 > UrDMA3 > UrDMA2) and viscosity (UrDMA1 > UrDMA3 > UrDMA2) displayed the highest photo-reactivity (tmax = 10.9 s, Rp,max = 8.18 × 10−3 s−1) and conversion (DBCtotal = 86%) compared to UrDMA1 and UrDMA3. Higher conversion of UrDMA3 is also related to the higher molecular flexibility of the corresponding photo-curing mixture (UrDMA3 > UrDMA2 > UrDMA1)11 arising from two flexible ether bonds within its molecular structure. Plotting Rp versus DBC presented interesting facts (Fig. 3c). Reaching high DBCtotal values is important since the mechanical strength of the urethane-methacrylates is dependent on their conversion during photo-curing.12 UrDMA1 with higher viscosity and lower molecular flexibility reached Rp,max at lower conversion (9.2%), while Rp,max for UrDMA2 and UrDMA3 observed at higher conversions (21.3% and 18.0%, respectively) due to lower viscosity and higher molecular flexibility of the corresponding photo-curing mixtures.
Monofunctional UrMA1 showed a slower photo-curing (tmax = 19.5 s) but higher conversion (DBCtotal = 98%) comparing to the difunctional urethane-methacrylates. Due to the mono-functionality, the viscosity of the photo-curing mixture did not increase sharply leading to a delayed gelation point, where the whole methacrylate groups remained mobile and active until the end of the polymerization. Due to the same reason, UrMA1 reached Rp,max at much higher conversion (40.3%).
Thermal, physical, and mechanical properties of cured samples
Mixtures of each monomer and TPO-L (3 wt%) were poured into Teflon or silicone molds (depth of 0.5 mm) and cured with a UV lamp under an Ar atmosphere. All the cured samples were denoted with a prefix “X” following the name of the corresponding monomer. The thermal transitions of the cured sample were determined using DSC. The first and second heating cycles were used for the extraction of Tg values (middle point of base-line change, Table 2 and Fig. S6 in ESI†). For all difunctional urethane-methacrylates, the Tg values of the cured samples were significantly increased in the second heating cycle comparing to the first cycle, probably due to the thermally-initiated polymerization of the unreacted methacrylate groups within the samples at elevated temperatures (120–200 °C). The Tg values for the cured samples were also determined via DMA (peak of the tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ curve, Fig. 4 and Table 2). As expected, the Tg values obtained from DMA were higher than the values determined by DSC, due to differences in the analysis principles and parameters,41,42 but represented the same trend. XUrDMA3 showed the lowest Tg values compared to XUrDMA1 and XUrDMA2 due to the existence of flexible ether bonds within its structure, which can confirm the high flexibility for the photo-curing UrDMA3 mixture as a reason for its high conversion. Meanwhile, XUrDMA2 had lower Tg values than XUrDMA1 due to the sterical hindrance effect of two methyl groups increasing the free volume between the crosslinked urethane-methacrylate backbones.
δ curve, Fig. 4 and Table 2). As expected, the Tg values obtained from DMA were higher than the values determined by DSC, due to differences in the analysis principles and parameters,41,42 but represented the same trend. XUrDMA3 showed the lowest Tg values compared to XUrDMA1 and XUrDMA2 due to the existence of flexible ether bonds within its structure, which can confirm the high flexibility for the photo-curing UrDMA3 mixture as a reason for its high conversion. Meanwhile, XUrDMA2 had lower Tg values than XUrDMA1 due to the sterical hindrance effect of two methyl groups increasing the free volume between the crosslinked urethane-methacrylate backbones.
| Sample | DSC | DMA | ||||||
|---|---|---|---|---|---|---|---|---|
| Tg (°C) | Tg (°C) | tanδmax |
|
|
Trubbery (K) | νc (mol m−3) | ||
| 1st cycle | 2nd cycle | |||||||
a tanδmax: loss coefficient (loss modulus/storage modulus) at Tg, 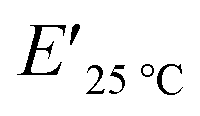 : storage modulus at 25 °C, : storage modulus at 25 °C, 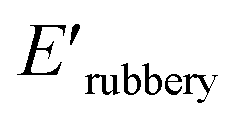 : storage modulus in the rubbery region, T: the absolute temperature at the beginning of the rubbery plateau. : storage modulus in the rubbery region, T: the absolute temperature at the beginning of the rubbery plateau. |
||||||||
| XUrDMA1 | 50 | 100 | 126 | 0.41 | 2100 | 24 | 140 | 2360 |
| XUrDMA2 | 45 | 81 | 85 | 0.32 | 1720 | 105 | 127 | 10550 |
| XUrDMA3 | 45 | 57 | 80 | 0.40 | 1670 | 49 | 108 | 5140 |
| XUrMA1 | 25 | 24 | 51 | 1.50 | 1070 | 20 | 55 | 2500 |
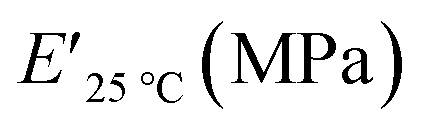
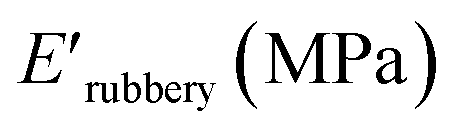
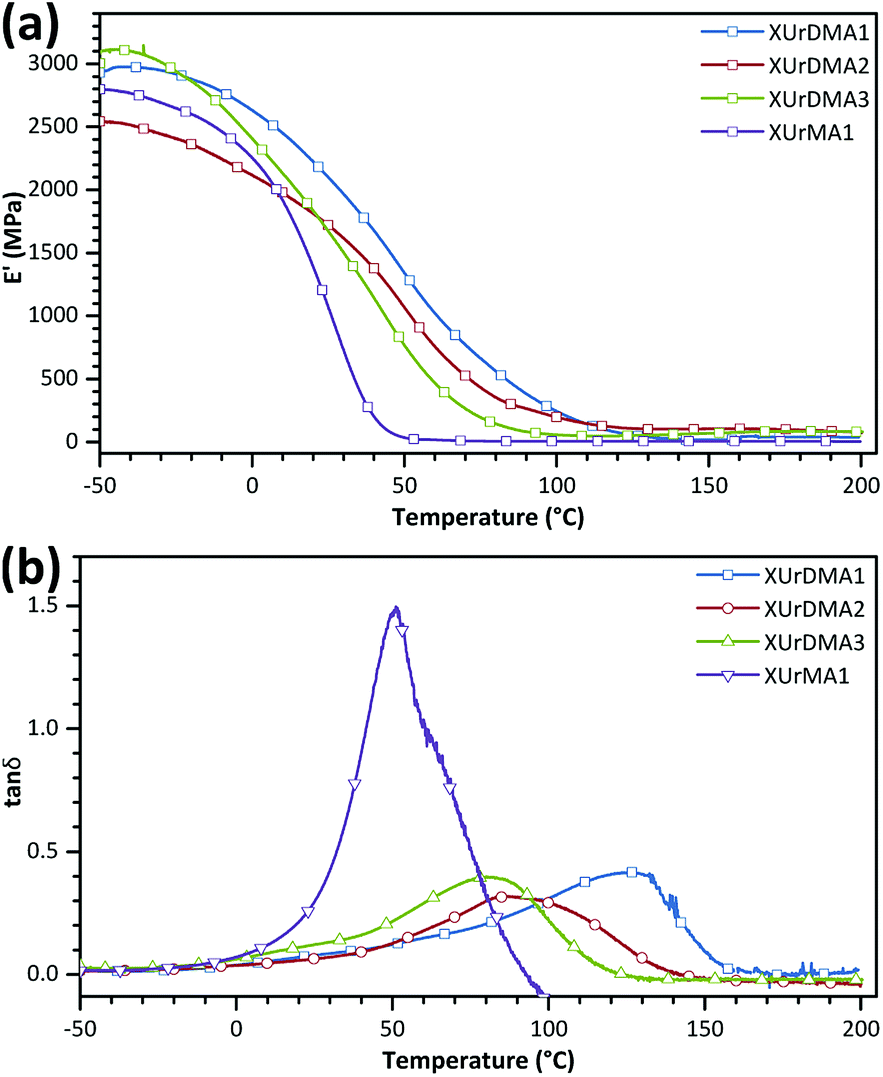 | ||
| Fig. 4 Storage modules and tanδ curves for cured samples. | ||
The XUrMA1 supposed to be a thermoplastic displayed an exothermic melting peak at 188 °C (ΔHm = 3.6 J g−1). Due to the mono-functionality of UrMA1, i.e. less possibility for chemical crosslinking, XUrMA1 showed lower Tg values and a higher loss coefficient (tanδmax = 1.50) comparing to other cured samples based on the difunctional monomers (Fig. 4b). Due to the complete conversion of the methacrylate group of UrMA1 during the photo-curing process (DBCtotal = 98%), the Tg value obtained from DSC for XUrMA1 did not change in the second heating cycle compared to the first cycle (Fig. S6 in ESI†).
The viscoelastic data obtained from DMA (Fig. 4a and Table 2) showed higher storage modulus (E′) at room temperature for XUrDMA1 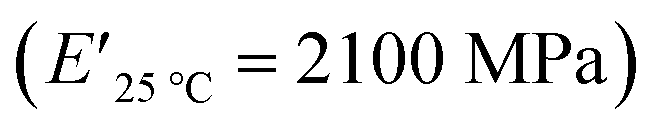 compared to XUrDMA2
compared to XUrDMA2 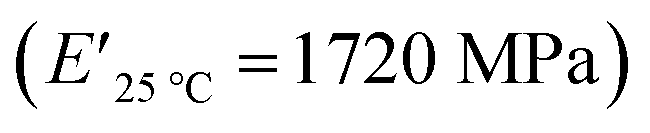 and XUrDMA3
and XUrDMA3 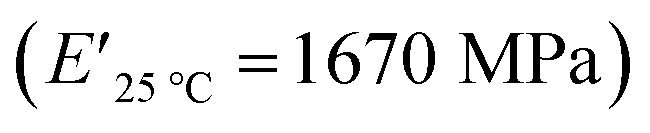 , which is attributed to the higher ability of its urethane-methacrylate backbone for hydrogen bonding resulting in the physical crosslinking. In contract, at the elevated temperatures (beginning of the rubbery, 108–140 °C), where no hydrogen bonding occurs, XUrDMA1 displayed a lower E′ value
, which is attributed to the higher ability of its urethane-methacrylate backbone for hydrogen bonding resulting in the physical crosslinking. In contract, at the elevated temperatures (beginning of the rubbery, 108–140 °C), where no hydrogen bonding occurs, XUrDMA1 displayed a lower E′ value 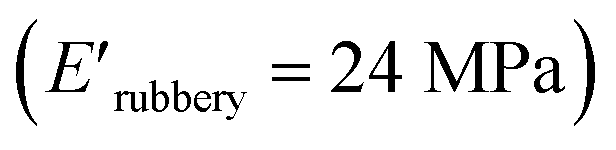 compared to XUrDMA2
compared to XUrDMA2 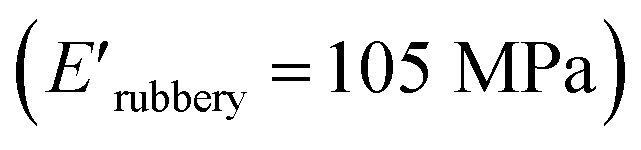 and XUrDMA3
and XUrDMA3 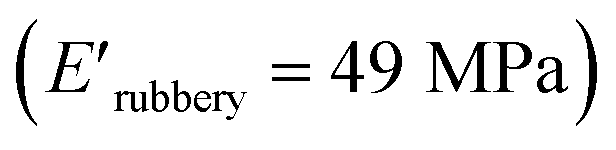 , which can be related to the lower content of crosslinking points within its structure. Therefore, the crosslink density (νc, mole number of network chains per unit volume) for all cured samples was calculated based on DMA data.43 As expected, XUrDMA1 had the lowest νc value (2360 mol m−3) due to the low conversion of methacrylate groups (47%). The higher νc value for XUrDMA2 (10550 mol m−3) compared to XUrDMA3 (5140 mol m−3), with the similar DBCtotal values, can be explained based on the lower molecular weight of its urethane-methacrylate backbone (457 g mol−1 versus 517 g mol−1).
, which can be related to the lower content of crosslinking points within its structure. Therefore, the crosslink density (νc, mole number of network chains per unit volume) for all cured samples was calculated based on DMA data.43 As expected, XUrDMA1 had the lowest νc value (2360 mol m−3) due to the low conversion of methacrylate groups (47%). The higher νc value for XUrDMA2 (10550 mol m−3) compared to XUrDMA3 (5140 mol m−3), with the similar DBCtotal values, can be explained based on the lower molecular weight of its urethane-methacrylate backbone (457 g mol−1 versus 517 g mol−1).
The gel content of the cured samples was determined through extraction with acetone (Table 3). A conversion of 47% during the photo-curing UrDMA1 was enough to provide a gel content of 100% for XUrDMA1, similar to XUrDMA2 and XUrDMA3. Surprisingly, XUrMA1 based on the monofunctional monomer exposed a gel content of 89%, which can be attributed to the backbiting effect, i.e. radical abstraction of tertiary hydrogens for butyl moiety,44,45 during the photo-curing process, which led to partial crosslinking of the poly(urethane-methacrylate) chains. Checking the gel content of XUrMA1 using more polar solvents, i.e. THF and DMSO, resulted in the same values. It is worth to mention that the XUrMA1 sample was unstable in acetone and broken into small pieces, which demonstrated the low level of chemical crosslinking within its structure.
| Sample | Gel content (%) | Water absorption (%) in PBS after 7 d | TGA | ||
|---|---|---|---|---|---|
| T5% (°C) | T50% (°C) | T90% (°C) | |||
| a T5%: temperature with weight loss of 5%, T50%: temperature with weight loss of 50%, T90%: temperature with weight loss of 90%. | |||||
| XUrDMA1 | 100 ± 1 | 4 ± 1 | 290 | 429 | 479 |
| XUrDMA2 | 99 ± 1 | 4 ± 1 | 212 | 402 | 472 |
| XUrDMA3 | 99 ± 1 | 4 ± 1 | 217 | 405 | 451 |
| XUrMA1 | 89 ± 4 | 2 ± 1 | 215 | 357 | 436 |
The bulk hydrophilicity of the cured samples was evaluated by measuring the water absorption in phosphate-buffered saline (PBS, pH = 7.4) at 37 °C (Table 3 and Fig. S7 in ESI†). All cured samples showed a water absorption of less than 4% in PBS. XUrMA1 displayed a lower water absorption (2%) comparing to other cured samples based on the difunctional monomers (4%), which can be attributed to the higher hydrophobicity of its urethane-methacrylate backbone as well as the presence of crystalline domains resisting against the penetration of water molecules inside the samples. The presence of hydrophilic ether bonds within the XUrDMA3 backbone facilitated the absorption of water molecules, reaching 4% in 1 d (Fig. S7 in ESI†), but did not increase the overall equilibrium water absorption after 7 d compared to XUrDMA1 (Table 3). It is worth to mention that the absorbed water molecules could effectively plasticize the urethane-methacrylate backbone of the cured samples. For example, the hard and brittle XUrDMA1 sample turned flexible after immersing in PBS (Fig. 5).
 | ||
| Fig. 5 Pictures for XUrDMA1 before (left) and after (right) immersion in PBS. | ||
The thermal stability of the cured samples was evaluated by TGA (Table 3 and Fig. S8 in ESI†). Results revealed that all samples were thermally stable at least up to 212 °C (T5%, the temperature at which 5% weight loss took place) under an N2 atmosphere. The cured samples underwent a two-step thermal degradation (Fig. S8 in ESI†) starting with a weight loss at 210–350 °C regarding the thermal degradation of urethane bonds.34,43 Urethane bonds are known to be relatively thermally unstable generating primary amine/olefin or secondary amine/carbon dioxide upon degradation.34,43 The second weight loss at 350–480 °C is attributed to the degradation of their uncrosslinked/crosslinked aliphatic backbones.34,43,46 Surprisingly, XUrDMA1 with lower DBCtotal and νc values exposed higher thermal stability (T5% = 290 °C) than XUrDMA2 (T5% = 212 °C) and XUrDMA3 (T5% = 217 °C). One explanation could be the thermally-initiated polymerization of the unreacted methacrylate groups within samples at elevated temperatures, which is in agreement with the increasing of Tg values in the second heating cycle of DSC (Table 2 and Fig. S6 in ESI†). As expected, XUrMA1 with a partially crosslinked structure demonstrated a weight loss profile at lower temperatures compared to other cured samples based on the difunctional monomers (Fig. S8 in ESI†).
3D printing of urethane-methacrylates
The 3D printability of the monomers was evaluated on a commercial DLP printer operating at 365 nm. For this purpose, a mixture of monomers containing TPO-L was used as an ink (the formulation is not reported). The ink was very reactive and successfully 3D printed to a complex object with a curing time of 3 s for one layer of 100 μm (Fig. 6). After 3D printing, the objects were washed thoroughly with isopropanol to remove the unreacted ink and post-cured with a UV lamp. The printed objects displayed high accuracy and precision as the designed model (Fig. S9 in ESI†). | ||
| Fig. 6 Pictures for 3D printed test object using an ink based on the monomers. | ||
Conclusions
Urethane-methacrylate monomers were synthesized via a safe and environmentally friendly route without using any toxic isocyanates or tin-based catalysts. The chemical structure of the urethane block was effective on the photo-reactivity (Rp and DBCtotal) of the monomers and the physicomechanical properties of the cured samples. The incorporation of side methyl groups or ether bonds within the urethane block can be employed to change the intermolecular hydrogen bonding for improving the photo-reactivity of the monomer as well as the flexibility and crosslinking density of the cured samples. An ink was developed based on the monomers and successfully 3D printed using a DLP machine. Through the non-isocyanate route, a wide range of starting materials, i.e. amines and cyclic carbonates, can be used to synthesis new urethane-methacrylates with desirable photo-reactivity and physicomechanical properties for the 3D printed samples. We are now using these monomers for 3D printing of flexible biomedical materials, which will be published in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support by the Federal Ministry of Education and Research of Germany in the framework of “ProMatLeben – Polymere” (project number 13XP5087E, PolyKARD).References
- J. W. Hong, H. K. Cheon, S. H. Kim, K. H. Hwang and H. K. Kim, Prog. Org. Coat., 2017, 110, 122–127 Search PubMed.
- M. Wrosch, G. Xian and V. M. Karbhari, J. Appl. Polym. Sci., 2008, 107, 3654–3662 Search PubMed.
- M. Yin, F. Liu and J. He, J. Mech. Behav. Biomed. Mater., 2016, 57, 157–163 Search PubMed.
- J. Seo, D. I. Kushner and M. A. Hickner, ACS Appl. Mater. Interfaces, 2016, 8, 16656–16663 Search PubMed.
- D. K. Patel, A. H. Sakhaei, M. Layani, B. Zhang, Q. Ge and S. Magdassi, Adv. Mater., 2017, 29, 1606000 Search PubMed.
- J. F. G. A. Jansen, A. A. Dias, M. Dorschu and B. Coussens, Macromolecules, 2003, 36, 3861–3873 Search PubMed.
- H. J. Assumption and L. J. Mathias, Polymer, 2003, 44, 5131–5136 Search PubMed.
- T. Y. Lee, T. M. Roper, E. S. Jönsson, C. A. Guymon and C. E. Hoyle, Macromolecules, 2004, 37, 3659–3665 Search PubMed.
- M. T. Lemon, M. S. Jones and J. W. Stansbury, J. Biomed. Mater. Res., Part A, 2007, 83, 734–746 Search PubMed.
- I. M. Barszczewska-Rybarek, Dent. Mater., 2009, 25, 1082–1089 Search PubMed.
- I. Sideridou, V. Tserki and G. Papanastasiou, Biomaterials, 2002, 23, 1819–1829 Search PubMed.
- I. Barszczewska-Rybarek, Polym. Bull., 2017, 74, 4023–4040 Search PubMed.
- MDI and TDI: a safety, health and the environment: a source book and practical guide, ed. D. C. Allport, D. S. Gilbert and S. M. Outterside, J. Wiley, New York, 2003 Search PubMed.
- C. A. Redlich, D. Bello and A. V. Wisnewski, in Environmental and occupational medicine, Lippincott Williams & Wilkins, Philadelphia, 4th edn, 2007, pp. 502–516 Search PubMed.
- J. E. Lockey, C. A. Redlich, R. Streicher, A. Pfahles-Hutchens, P. Bert, J. Hakkinen, G. L. Ellison, P. Harber, M. Utell, J. Holland, A. Comai and M. White, J. Occup. Environ. Med., 2015, 57, 44–51 Search PubMed.
- C. A. Redlich and M. H. Karol, Int. Immunopharmacol., 2002, 2, 213–224 Search PubMed.
- S. Gagné, J. Lesage, C. Ostiguy and H. Van Tra, Analyst, 2003, 128, 1447–1451 Search PubMed.
- C. A. Krone, J. T. A. Ely, T. Klingner and R. J. Rando, Bull. Environ. Contam. Toxicol., 2003, 70, 328–335 Search PubMed.
- C. A. Krone and T. D. Klingner, Pediatr. Allergy Immunol., 2005, 16, 368–379 Search PubMed.
- D. Bello, C. A. Herrick, T. J. Smith, S. R. Woskie, R. P. Streicher, M. R. Cullen, Y. Liu and C. A. Redlich, Environ. Health Perspect., 2007, 115, 328–335 Search PubMed.
- M. C. Tanzi, P. Verderio, M. G. Lampugnani, M. Resnati, E. Dejana and E. Sturani, J. Mater. Sci.: Mater. Med., 1994, 5, 393–396 Search PubMed.
- M. Nath, Appl. Organomet. Chem., 2008, 22, 598–612 Search PubMed.
- L. Meng, X. Wang, M. Ocepek and M. D. Soucek, Polymer, 2017, 109, 146–159 Search PubMed.
- L. Meng, M. D. Soucek, Z. Li and T. Miyoshi, Polymer, 2017, 119, 83–97 Search PubMed.
- Y. Deng, S. Li, J. Zhao, Z. Zhang, J. Zhang and W. Yang, RSC Adv., 2014, 4, 43406–43414 Search PubMed.
- S. Li, J. Zhao, Z. Zhang, J. Zhang and W. Yang, RSC Adv., 2015, 5, 6843–6852 Search PubMed.
- M. S. Kathalewar, P. B. Joshi, A. S. Sabnis and V. C. Malshe, RSC Adv., 2013, 3, 4110 Search PubMed.
- X. Wang and M. D. Soucek, Prog. Org. Coat., 2013, 76, 1057–1067 Search PubMed.
- M. Decostanzi, C. Bonneaud and S. Caillol, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1224–1232 Search PubMed.
- H. Quan, T. Zhang, H. Xu, S. Luo, J. Nie and X. Zhu, Bioact. Mater., 2020, 5, 110–115 Search PubMed.
- Q. Yan, H. Dong, J. Su, J. Han, B. Song, Q. Wei and Y. Shi, Engineering, 2018, 4, 729–742 Search PubMed.
- J. J. Warner, P. Wang, W. M. Mellor, H. H. Hwang, J. H. Park, S.-H. Pyo and S. Chen, Polym. Chem., 2019, 10, 4665–4674 Search PubMed.
- G. Rokicki and A. Piotrowska, Polymer, 2002, 43, 2927–2935 Search PubMed.
- H. Bakhshi, H. Yeganeh, S. Mehdipour-Ataei, A. Solouk and S. Irani, Macromolecules, 2013, 46, 7777–7788 Search PubMed.
- H. Bakhshi and S. Agarwal, Polym. Chem., 2016, 7, 5322–5330 Search PubMed.
- M. Blain, L. Jean-Gérard, R. Auvergne, D. Benazet, S. Caillol and B. Andrioletti, Green Chem., 2014, 16, 4286–4291 Search PubMed.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3678–3685 Search PubMed.
- R. M. Garipov, V. A. Sysoev, V. V. Mikheev, A. I. Zagidullin, R. Ya. Deberdeev, V. I. Irzhak and Al. Al. Berlin, Dokl. Phys. Chem., 2003, 393, 289–292 Search PubMed.
- F. Camara, S. Benyahya, V. Besse, G. Boutevin, R. Auvergne, B. Boutevin and S. Caillol, Eur. Polym. J., 2014, 55, 17–26 Search PubMed.
- C. Li, S. Li, J. Zhao, Z. Zhang, J. Zhang and W. Yang, J. Polym. Res., 2014, 21, 498 Search PubMed.
- S. Kasapis, I. M. Al-Marhoobi and J. R. Mitchell, Carbohydr. Res., 2003, 338, 787–794 Search PubMed.
- C. A. Gracia-Fernández, S. Gómez-Barreiro, J. López-Beceiro, J. Tarrío Saavedra, S. Naya and R. Artiaga, Polym. Test., 2010, 29, 1002–1006 Search PubMed.
- H. Bakhshi, H. Yeganeh, A. Yari and S. K. Nezhad, J. Mater. Sci., 2014, 49, 5365–5377 Search PubMed.
- H. Bakhshi, H. Bouhendi, M. J. Zohuriaan-Mehr and K. Kabiri, J. Appl. Polym. Sci., 2010, 117, 2771–2780 Search PubMed.
- H. Bakhshi, M. J. Zohuriaan-Mehr, H. Bouhendi and K. Kabiri, J. Mater. Sci., 2011, 46, 2771–2777 Search PubMed.
- H. Bakhshi and S. Agarwal, J. Mater. Chem. B, 2017, 5, 6827–6834 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and extra results. See DOI: 10.1039/d0ra06388f |
| This journal is © The Royal Society of Chemistry 2020 |