
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom “S” to “O”: experimental and theoretical insights into the atmospheric degradation mechanism of dithiophosphinic acids†
Zhipeng Wang‡
a,
Yixiang Zhang‡b,
Jingjing Liub,
Lianjun Songa,
Xueyu Wanga,
Xiuying Yanga,
Chao Xu c,
Jun Li*b and
Songdong Ding*a
c,
Jun Li*b and
Songdong Ding*a
aCollege of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: dsd68@163.com
bDepartment of Chemistry, Key Laboratory of Organic Optoelectronics, Molecular Engineering of the Ministry of Education, Tsinghua University, Beijing 100084, China
cCollaborative Innovation Center of Advanced Nuclear Energy Technology, Institute of Nuclear and New Energy Technology, Tsinghua University, Beijing 100084, China
First published on 4th November 2020
Abstract
Dithiophosphinic acids (DPAHs, expressed as R1R2PSSH) are a type of sulfur-donor ligand that have been vastly applied in hydrometallurgy. In particular, DPAHs have shown great potential in highly efficient trivalent actinide/lanthanide separation, which is one of the most challenging tasks in separation science and is of great importance for the development of an advanced fuel cycle in nuclear industry. However, DPAHs have been found liable to undergo oxidative degradation in the air, leading to significant reduction in the selectivity of actinide/lanthanide separation. In this work, the atmospheric degradation of five representative DPAH ligands was investigated for the first time over a sufficiently long period (180 days). The oxidative degradation process of DPAHs elucidated by ESI-MS, 31P NMR, and FT-IR analyses is R1R2PSSH → R1R2PSOH → R1R2POOH → R1R2POO–OOPR1R2, R1R2PSSH → R1R2PSS–SSPR1R2, and R1R2PSSH → R1R2PSOH → R1R2POS–SOPR1R2. Meanwhile, the determination of pKa values through pH titration and oxidation product by PXRD further confirms the S → O transformation in the process of DPAH deterioration. DFT calculations suggest that the hydroxyl radical plays the dominant role in the oxidation process of DPAHs and the order in which the oxidation products formed is closely related to the reaction energy barrier. Moreover, nickel salts of DPAHs have shown much higher chemical stability than DPAHs, which was also elaborated through molecular orbital (MO) and adaptive natural density portioning (AdNDP) analyses. This work unambiguously reveals the atmospheric degradation mechanism of DPAHs through both experimental and theoretical approaches. At the application level, the results not only provide an effective way to preserve DPAHs but could also guide the design of more stable sulfur-donor ligands in the future.
Introduction
Separation of trivalent actinides (An3+) over trivalent lanthanides (Ln3+) is one of the most critical and challenging steps in spent fuel reprocessing owing to their very similar chemical and physical properties.1,2 Previous reports indicate that An3+ and Ln3+ could be effectively separated using soft N-donor or S-donor ligands.3–5 In comparison with N-donor ligands, the S-donor ligands have shown much higher selectivity for An3+/Ln3+ separation.4 For example, the purified CYANEX® 301, which is a typical aliphatic substituted dithiophosphinic acid (DPAH, expressed as R1R2PSSH) ligand, could selectively extract Am3+ over Eu3+ with high separation factors (SFAm/Eu) up to 5900 in HNO3 solution at pH 3.5–4.0.5 However, when CYANEX® 301 was exposed to oxidation environment such as atmosphere or HNO3 for a certain time, significant decreases of its selectivity to Am3+ over Eu3+ could be observed, which is expected to limit its use in real applications.6,7To uncover the underlying chemical mechanism accounted for the decrease of An3+/Ln3+ selectivity by DPAHs and help improve the stability of dithiophosphinic acids, a few investigations have been carried out on the oxidation behavior of DPAHs in aqueous solution. It has been shown that a high concentration of non-oxidizing mineral acid such as H2SO4 and HCl hardly had obvious detrimental influence on the structure of CYANEX® 301, whereas the oxidizing HNO3 could destruct the ligand rapidly even at low acidity and for a short testing time.8,9 With respect to the oxidation products and the possible oxidation pathways of the aliphatic substituted DPAH, there are still different understandings and insights. Wang et al. discovered the formation of monothiophosphinic acid (RRPOSH) as the degradation product of (n-pentyl)2PSSH and (n-butyl)2PSSH in the case of contacting with 1.0 mol L−1 or 3.0 mol L−1 HNO3 for 10 hours via 31P NMR and FT-IR analyses.10 However, Menoyo et al. proposed that CYANEX® 301 could be oxidized to RRPSOH, RRPS, RRPOOH and RRPO after contacting with 5.0 mol L−1 HNO3 for 10 days using FT-IR, FT-Raman and GC-MS monitorings.11 The oxidation pathway was afforded as follow: RRPSSH → RRPSOH → RRPOOH. Nevertheless, a further study by Marc et al. indicated the formation of dimer product of CYANEX® 301 in contact with 1.0 mol L−1 HNO3 for 18 hours by means of 31P NMR spectra and single crystal analyses,12 which was also supported by Groenewold et al. via ESI-MS spectra and collision-induced dissociation spectra tests.13 The assessing oxidation pathway was presented as RRPSSH → RRPSS–SSPRR → RRPOS–SOPRR → RRPOO–OOPRR → RRPO–O–OPRR. So far, there has been no scientific consensus about the oxidation mechanism of DPAH.
The stability properties of aromatic group substituted DPAHs have also been studied. (ClPh)2PSSH is one of the most representative structures of aromatic DPAHs. Modolo et al. performed the hydrolysis experiments of (ClPh)2PSSH in 0.5–3.0 mol L−1 HNO3, HCl, or H2SO4.14 It was found that (ClPh)2PSSH decomposed completely after contacting with 3.0 mol L−1 HNO3 for more than 20 days. To avoid this adverse impact, reducing agents or radical scavengers such as amidosulfuric acid, urea, or hydrazine must be added into the biphasic extraction system.15,16 However, this stabilizing method has led to a more complex extraction system and, unfortunately, did not completely solve the problem of oxidative degradation of DPAHs. Moreover, it was also found DPAH could be oxidized in ambient environment.17,18 For example, CYANEX® 301 could degrade obviously in 3 hours when exposed to the air. In contrast, this ligand would be very stable in an inert atmosphere, suggesting that DPAH are very sensitive to the oxidizing atmosphere. This oxidation issue in ambient environment not only deteriorate the extraction performances of DPAHs, but also requires special protection measures during their transportation and storage, which in turn limits their applications in advanced fuel cycle. Therefore, investigation on DPAH oxidation behavior at ambient environment is of great significance. Unfortunately, to the best of our knowledge, data are very scarce on the oxidation stability of DPAHs at ambient environment. As a result, the understanding on the oxidation processes of DPAHs at ambient environment is superficial due to the lack of sufficient experimental evidences and theoretical supports.
In the present work, by taking consideration of the impact of substituent on the oxidation, five representative DPAHs (Fig. S1†) bearing different alkyl and/or aryl substituent groups were intentionally synthesized and their oxidation processes were systematically monitored at ambient environment for 180 days using ESI-MS, 31PNMR, FT-IR, PXRD, and pH titration methods. Meanwhile, along with the oxidation of DPAHs, the extraction behavior of Am3+ and Eu3+ by these ligands was also investigated. Moreover, a detailed oxidation mechanism of DPAHs was proposed through the help of density functional theory and ab initio study.
Experimental section
General
The radioactive trace solution of 241Am3+ with radionuclidic purity was supplied by China Institute of Atomic Energy. The HNO3 solution containing Eu3+ was prepared from Eu2O3·6H2O (99.99%, Aldrich, USA). All the other reagents used in this work were of AR grade and used as received. 85% aqueous H3PO4 was utilized as external standard for 31P NMR. The investigated ligands of this work were prepared by referring to previous literature.19The ESI-MS spectra were collected on Bruker Amazon SL spectrometer (Bruker Inc., Switzerland). 31P NMR spectra were recorded by Varian Inova NMR spectrometer (240 MHz) (Bruker Inc., Switzerland). FT-IR spectra were recorded on Nicolet 6700 Fourier transform infrared spectrometer (Thermo Fisher Scientific Inc., USA). Powder X-ray diffraction (PXRD, Shimadzu 6100, Japan) patterns were obtained using Cu Kα radiation (0.1542 nm) at 40 kV and 30 mA. The Lei Ci PHS-3C type pH meter (Shanghai Precision & Scientific Instrument Co. Ltd., China) was used for determining pKa values of DPAH compounds as well as pH values of aqueous phase. The NaI(Tl) scintillation counter (China National Nuclear Inc., China) was used for detecting the radioactivity of 241Am3+ in each phase. The inductively coupled plasma atomic emission spectrometer (ICP-AES, IRIS Advantage, Thermo Elemental Inc., USA) was applied to determining the concentration of Eu3+ in aqueous phase.
Oxidation procedures
Adequate amount of each DPAH sample was separately placed in Φ = 3.5 cm open beaker and exposed to the ambient environment. 20 μL aliquots were withdrawn from each sample for ESI-MS, 31P NMR, FT-IR characterizations and pKa values determination with appropriate time intervals. The solid oxidation product was successively filtered from the sample, washed with ethanol and dried in oven prior to the PXRD test.pH titration
0.75 mmol DPAH was dissolved in EtOH/H2O (99/1%, v/v) mixture (50 mL) and titrated by 0.05 mol L−1 NaOH aqueous solution at 25.0 ± 0.5 °C. The pH values were recorded with pH meter. Each ligand was titrated in triplicates. The pKa values were determined through the stepwise linear regression method20,21 and the purity of examined compounds was also calculated.Extraction
The DPAH ligands were dissolved in toluene to obtain the organic phase with a concentration of 0.5 mol L−1 DPAH. The aqueous phase with different concentrations of HNO3 contains trace amount of 241Am3+, 200 ppm Eu3+, and 1.0 mol L−1 NaNO3. Equal volumes of the two phases were stirred thoroughly at 25.0 ± 0.5 °C for 30 min to ensure phase transfer equilibrium. After phase separation by centrifugation, 0.5 mL specimens of each phase was pipetted into test tubes. The radiocounts of 241Am3+ in both organic phase and aqueous phase were measured separately. The concentration of Eu3+ in aqueous phase was analyzed by ICP-AES. The distribution ratio (D) for 241Am3+ and Eu3+ was defined as the ratio of metal ion concentration in the organic phase ([M]org.) to that in the aqueous phase ([M]aq.), DM = [M]org./[M]aq.. The separation factor (SFAm/Eu) was expressed as the ratio of DAm and DEu, SFAm/Eu = DAm/DEu.Theoretical calculations
The substituent groups of all the reactants in the experiment do not participate in the reactions, so methyl was used to replace the alkyl or aryl groups in order to improve the calculation efficiency. The simplified dithiophosphonic acids were labeled as MeDPAH in the following discussions. The structures of reactants, intermediates, transition states and products were optimized by B3LYP functions (density functional theory) combining with the 6-311++G(d,p) basis set using Gaussian 09 Program.22 The harmonic vibrational analyses were conducted to confirm the minima character of the obtained geometries at the same level of theory (i.e. the local minimal with positive frequencies and the saddle points with only one imaginary frequency). The intrinsic reaction coordinates (IRC)23 calculations were performed to confirm that the transition states connect the designated reactants and products. In order to obtain the highly accurate relative energies, single-point energies of all the optimized stationary point optimized at the B3LYP/6-311++G(d,p) level of theory were refined using CCSD(T) method24 in combination with 6-311++G(d,p) basis set. If it is not otherwise specified, all the relative energies mentioned below are calculated from CCSD(T)/6-311++g(d,p) level of theory. Gibbs free energy was calculated with single point energies calculated from CCSD(T)/6-311++g(d,p) level and the sum of zero-point energy corrections and entropy corrections. Translational and rotational entropies are neglected for solid state substance. The software of CYLview25 was used to draw the geometric structures of the stationary points on the potential energy surface (PES).The hydroxyl radical has been recognized as the most important oxidant in the atmosphere, which always acts as initiator of oxidation and degradation of organic compound.26–32 As a result, the hydroxyl radical was also considered as the main oxidant for the oxidation of these dithiophosphonic acids investigated in this work. It is noted that, the effects of oxygen and water molecules in the atmospheric degradation of MeDPAH were also considered. However, these reactions were not competitive with respect to the reactions with hydroxyl radical. Therefore, we only detailed the reaction mechanism of hydroxyl radical with MeDPAH below, and the other reactions were presented in the ESI.†
The Ni(MeDPA)2 complex was fully optimized using the Amsterdam Density Functional program (ADF2016.106).33 The generalized gradient approximation (GGA) with the PBE functional and the TZ2P Slater type basis set34 were used for all atoms. The scalar-relativistic ZORA formalism was adopted to account for the relativistic effects.35,36 In order to gain further insights into the chemical bonds of Ni(MeDPA)2, we performed AdNDP analysis to generate the multi-centered localized orbitals using Multiwfn codes.37
Results and discussion
Identification of oxidation products
The oxidation products of DPAHs were identified with the help of spectroscopic techniques including ESI-MS, 31P-NMR, FT-IR, etc. For clarity, the following discussions mainly focus on the results of one representative DPAH L1, which shows the most obvious changes during the oxidation experiment.The oxidative degradation process of DPAHs was strictly monitored by ESI-MS. The ESI-MS spectra of L1 with oxidation time of 0 d and 180 d are shown in Fig. 1. The numbers in brackets are the generation order of species. Other mass spectra of L1 with oxidation time of 5 d, 10 d, 20 d, 30 d, 60 d, 90 d, 120 d, and 150 d are summarized in ESI as Fig. S2.† As we can see, the pure ligand L1 with molecule formula of R1R2PSSH can be observed at m/z = 287.1057. After 180 days oxidation, several oxidation products successively appeared at m/z = 271.1285, 571.1880, 255.1514, 539.2336, and 507.2793, which can be attributed to R1R2PSOH, R1R2PSS–SSPR1R2, R1R2POOH, R1R2POS–SOPR1R2, and R1R2POO–OOPR1R2, respectively.12,13,16,17 The emergence of different types of impurities means that DPAH can degrade not only through the replacement of sulphur atom by oxygen atom, but also via coupling reaction to form dimers. This result indicates that the appearance of both oxo-substitute monomer and coupling-dimer of the previous works is possible even the oxidation environment is not exactly the same. Regularly, all of the products were observed step by step, in the sequence of R1R2PSSH → R1R2PSOH → R1R2POOH → R1R2POO–OOPR1R2, R1R2PSSH → R1R2PSS–SSPR1R2, and R1R2PSSH → R1R2PSOH → R1R2POS–SOPR1R2, severally. Meanwhile, ESI-MS spectra of L2–L5 (Fig. S3–S6 in ESI†) show the same variation process as L1, revealing the common degradation regularities of DPAH ligands in air atmosphere. Even so, it should be noted that the occurrence time of new decomposition species is different for each ligand. Specifically, the first oxidative product R1R2PSOH appeared at 5 d for L1, whereas that of L2, L3, L4, and L5 is 20 d, 10 d, 10 d, and 20 d, separately. In addition, the appearance time of the last oxidative species is 90 d for L1, 150 d for L2, 120 d for L3, 120 d for L4, and 180 d for L5. Herein, the stability of five ligands can be preliminarily arranged as follows: L5 > L2 > L4 ≈ L3 > L1.
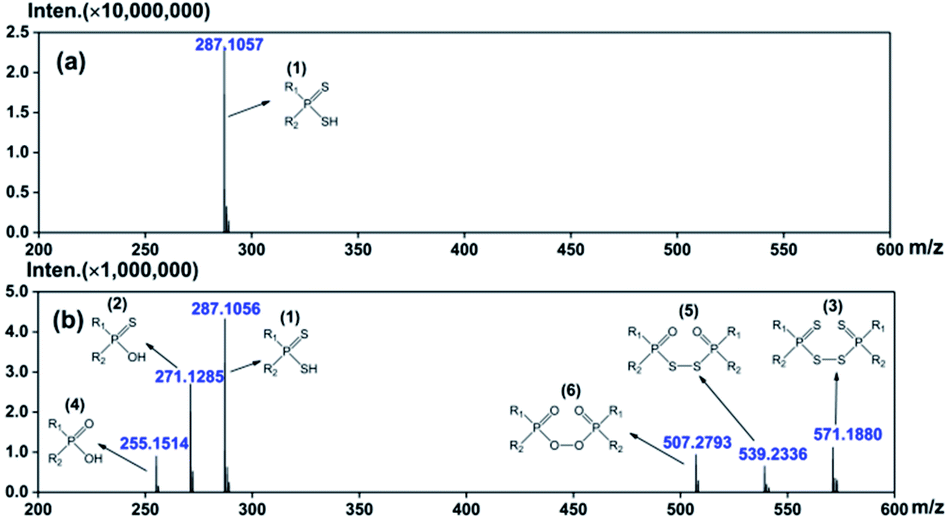 | ||
| Fig. 1 ESI-MS spectra of L1 at ambient environment for 0 d (a) and 180 d (b). | ||
The 31P NMR spectra of L1 with different oxidation degrees are shown in Fig. 2(a). It is clear that the decomposition species increase with the extension of testing time. The signal peaks at 62.12, 86.32, 71.95, 58.26, 72.71, and 73.85 ppm are assigned to R1R2PSSH, R1R2PSOH, R1R2PSS–SSPR1R2, R1R2POOH, R1R2POS–SOPR1R2, and R1R2POO–OOPR1R2, respectively,10–12,14,16,17 which coincided with the occurrence order of mass spectra. Further, the ratios of each product can be roughly calculated through the integrals of corresponding peaks and shown in Fig. 2(b). It can be seen that proportions of the impurities grow with the increasing of oxidation time. After degradation of 180 days, only 31% of the pure L1 is left. Whilst the residual ratios of pure L2, L3, L4, and L5 are 49%, 38%, 41%, and 64%, severally (Fig. S7–S10 in ESI†). The above intercomparison reveals that the stabilities of ligands are correlated with the variation in substituents. The DPAH molecules attached with o-trifluoromethyl phenyl (L5, L2, and L4) show good resistance to the air, indicating that the introduction of o-trifluoromethyl phenyl group is conducive to the air oxidation resistance. On the basis of ESI-MS and 31P NMR analyses, L5 is considered as the most stable ligand.
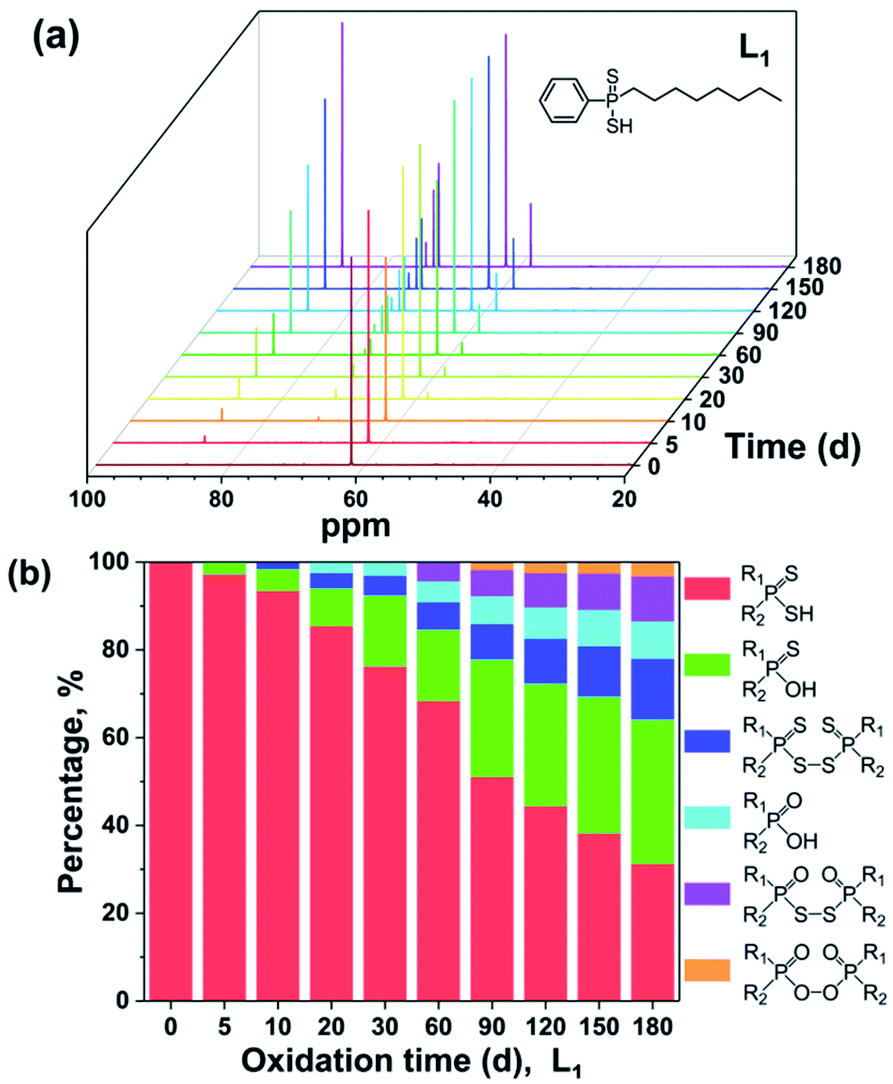 | ||
| Fig. 2 31P NMR spectra (a) and percentages of oxidation products (b) of L1 at ambient environment for different oxidation time. | ||
To further determine the molecular structure of the oxidation products, we examined the ligands by FT-IR spectroscopy in succession. Spectra of L1–L5 are presented as Fig. S11 in ESI.† The variations in the IR spectra are rather complicated. Nonetheless, some regularities on the functional groups evolution can be summarized. The characteristic peaks of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S at 631 cm−1 and 501 cm−1 gradually flatted with the extension of exposure time in the air.9,11,38 At the same time, the peaks distributed from 1000 cm−1 to 1280 cm−1 become more obvious, which can be attributed to the formation of PO and P–OH functional groups. Beyond the general phenomena of L1–L5, some detailed distinctions have also been captured. In the spectra of L1, L3, L4, and L5, peaks ascribed to PS ranging from 750 cm−1 to 830 cm−1 disappear gradually, indicating the reduction of R1R2PSSH molecules. Besides, the P–OH absorptions in the spectra of L2, L3, and L4, distributed in the range of 870 cm−1 to 985 cm−1, become stronger over the examined time, which further reveals the replacement of sulfur by oxygen during oxidation. Moreover, an unusual band belonging to S–S group vibration was observed near 480 cm−1 in the graphs of L3 and L4, indicating the occurrence of coupling reaction during degradation.
S at 631 cm−1 and 501 cm−1 gradually flatted with the extension of exposure time in the air.9,11,38 At the same time, the peaks distributed from 1000 cm−1 to 1280 cm−1 become more obvious, which can be attributed to the formation of PO and P–OH functional groups. Beyond the general phenomena of L1–L5, some detailed distinctions have also been captured. In the spectra of L1, L3, L4, and L5, peaks ascribed to PS ranging from 750 cm−1 to 830 cm−1 disappear gradually, indicating the reduction of R1R2PSSH molecules. Besides, the P–OH absorptions in the spectra of L2, L3, and L4, distributed in the range of 870 cm−1 to 985 cm−1, become stronger over the examined time, which further reveals the replacement of sulfur by oxygen during oxidation. Moreover, an unusual band belonging to S–S group vibration was observed near 480 cm−1 in the graphs of L3 and L4, indicating the occurrence of coupling reaction during degradation.
In the process of the experiment, slight rotten egg smell diffused from the samples, which may demonstrate the generation of hydrothion (H2S). In addition, the color of L1 and L3 changed from dark green to light yellow, whereas L2, L4, and L5 with the color of orange get darker over the measurement period. Interestingly, yellow solid separated out from the samples (Fig. S12†), which is speculated to be sulfur. For further analysis of the precipitate composition, the yellow solid was characterized by PXRD and compared with the diffraction standard spectrogram of sulfur. Corresponding characterization results are shown in Fig. S13.† The diffraction peaks of the test sample match with those of the simulated rhombic sulfur39 very well, proving the formation of S powder during the degradation of DPAHs.
As a phosphonic acid ligand, structural changes may have effects on its properties, such as the degree of H+ dissociation. Thus, the variation on the pKa values of five ligands was also recorded in the examined 180 d. As can be seen from Fig. 3, all the pKa values of the five ligands increase with the extension of monitoring time, which further indicates the replacement of S atom by O atom during the oxidation.40 Through a comparison, the ΔpKa values between 0 d and 180 d of five ligands are sequenced as L5 > L2 > L4 ≈ L3 > L1, which is in accordance with the foregoing experimental results.
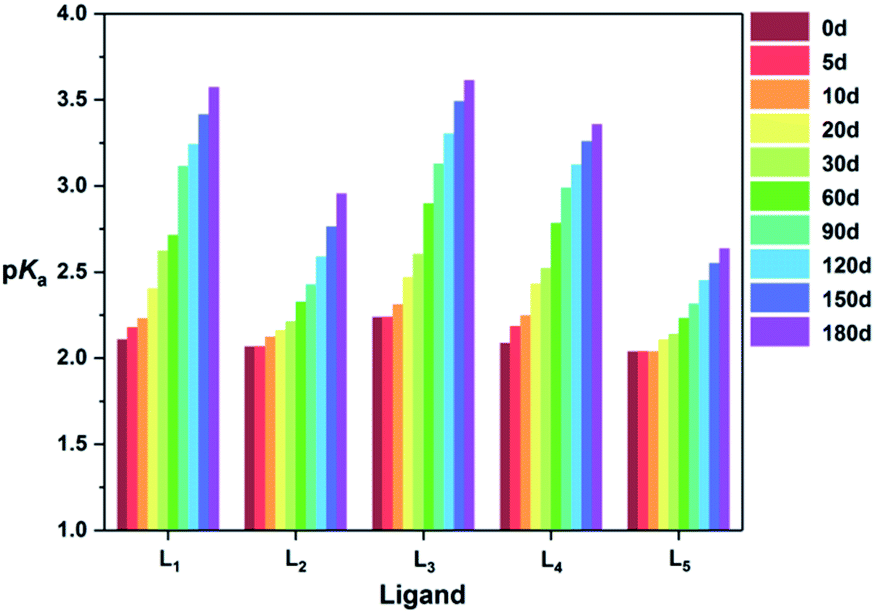 | ||
| Fig. 3 pKa values of L1–L5 at ambient environment for different oxidation time. | ||
Influence of DPAH oxidation on extraction
The degradation of DPAH extractants may have effect on their extraction behavior. Thus, the influence of oxidation time on the extraction of Am3+ and Eu3+ by the five DPAHs was investigated. The D values and SFAm/Eu values of L1–L5 at different time are shown in Fig. S14,† respectively. Both DAm and DEu increase more than one magnitude after 180 days degradation, which originate from the strong extractability of coordinated oxygen atom transformed from sulfur atom.6,7 Besides, the increase in DEu is more obvious than DAm, leading to the decrease of separability. After 180 days oxidation, the SFAm/Eu values of L1–L5 decrease from 9.5 to 2.9, 150 to 23, 1060 to 16, 17 to 1.6, and 1800 to 150, respectively. The changes on the SFAm/Eu values of DPAH ligands imply that air oxidation does have great influence on the extraction of Am3+ and Eu3+. In the meantime, five DPAHs show different resistance abilities to the air. Among the investigated ligands, L5 still holds a high SFAm/Eu values over 100 even oxidized by air for 180 days, exhibiting a good potential application prospects in the future.Preservation of DPAHs
During the synthesis of DPAH ligands, the crude product was converted to nickel salt form for recrystallization. Then the Ni-salt was acidized by HCl to obtain the target ligand with satisfying purity.19 Intriguingly, we found that all the Ni-salts of the investigated ligands were still very pure after placing in the ambient environment for 180 days (Fig. S15†). It means that the transformation of DPAH ligand to Ni-salt can reduce the reactiveness of ligand oxidation effectively. More importantly, it will provide a convenient preservation method of DPAH ligand with great significance. We consider that the formation of coordination bond S⋯Ni⋯S can stabilize the compound, preventing degradation of ligand even in contact with the air directly. Actually, this view point can be accepted due to the fact that sulphur acts as a proton acceptor and tends to form a more stable complex with divalent 3 d transition metal ions such as nickel or cobalt.41,42 The detailed analysis by theoretical calculation has been performed in the latter section.Oxidation mechanism
To understand and explain the details of the oxidation reaction in a molecular level, the reactions of MeDPAH with hydroxyl radical, oxygen and water were calculated theoretically, respectively. Here, the Gibbs free energy surface for the reaction between MeDPAH and hydroxyl radical were displayed (Fig. 4, S16 and S17†) owing to its competitive advantage in reaction kinetics.26 The profiles for the other reactions were presented in ESI as Fig. S19–S21.†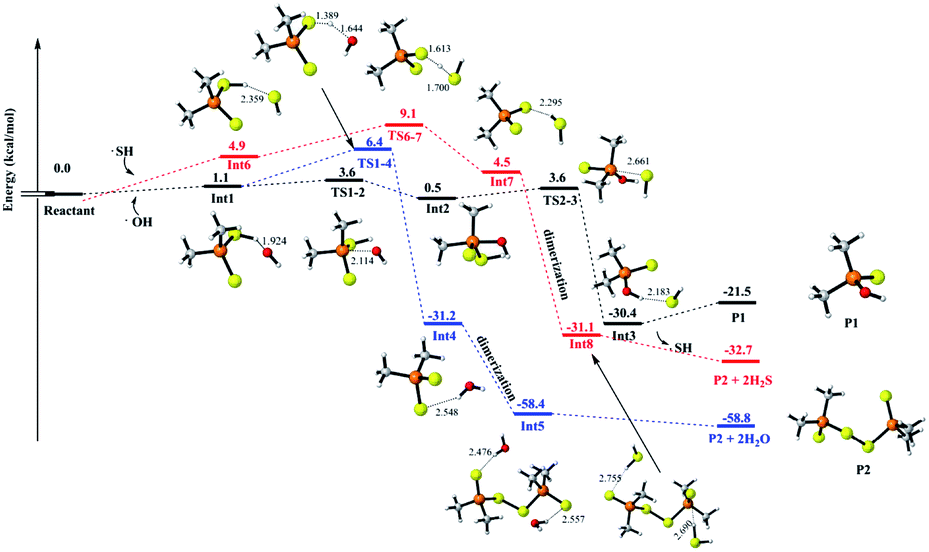 | ||
| Fig. 4 Gibbs free energy profile from MeDAPH to generate P1 (R1R2PSOH) and P2 (R1R2PSS–SSPR1R2) at CCSD(T)/6-311++G(d,p)//B3LYP/6-311++G(d,p) level (in kcal mol−1). The name of species is labeled below and the corresponding energy is labeled above. The unit of bond length is angstrom. | ||
According to the order of intermediates generation in the whole oxidation process, the entire reaction mechanism is divided into three stages. In each stage, three different reaction pathways have been considered, which are filled in black, red, and blue for visual differentiation. As indicated by the black path in Fig. 4, the reaction initiates from hydrogen bonded complex Int1, which locates 1.1 kcal mol−1 higher than the total energy of the initial reactant. As the reaction proceeds, Int1 connects to intermediate Int2 via transition state TS1-2, where the hydroxyl radical adds to the phosphorous atom with an energy barrier of 2.4 kcal mol−1. As Int2 forms, the reaction proceeds to pass through an energy barrier of 3.2 kcal mol−1 to produce a hydrogen-bonded complex Int3 via TS2-3. The SH radical leaves via the cleavage of P–SH bond in TS2-3 to generate Int3, followed by the production of P1 (R1R2PSOH) which is the first oxidation product detected in experiment. The first oxidation stage is accompanied by the substitution of SH in MeDPAH with OH to produce P1, where the cleavage of P–SH bond with 3.2 kcal mol−1 energy barrier is the rate-determined step.
Alternatively, the reaction can proceed via hydrogen abstraction reaction to produce Int4 (the blue path). In TS1-4, the hydrogen transfers from sulfur atom to oxygen atom with an energy barrier of 5.3 kcal mol−1. Once Int4 forms, it will get together immediately with the existing hydrated (CH3)2PS2 radical to produce Int5 followed by leaving of H2O to produce P2 (R1R2PSS–SSPR1R2). P2 is the second oxidation intermediate detected in experiment with the total energy barrier of 6.4 kcal mol−1.
As we all know, most radicals have very high reaction activity, so the byproduct SH radical43,44 produced in the reaction path labeled in black was also considered as one of the oxidants in the whole reaction pathways, which has been depicted in Fig. 4, S16 and S17† in red path. As shown in Fig. 4, the SH radical approaches the initial reactant to form a hydrogen bond complex Int6 with a Gibbs free energy of 4.8 kcal mol−1 because of entropy decrease. Then the reaction proceeds via the similar reaction pathways as that marked in blue. However, this path is different in that the H2S acts as a kind of byproduct (Fig. 4) instead of H2O. The generation of H2S here further confirmed the component of the rotten egg smell gas captured in the experiment.
By comparing the three different ways in Fig. 4, it can be seen that the whole energy barrier for P1 production is 3.6 kcal mol−1 (labeled black), while the energy barrier for P2 production is 6.4 kcal mol−1 (labeled blue). The lower energy barrier for P1 production is consistent with the appearance of P1 before P2 in the experiment. It is worth noting that, although the energy barrier for P2 production with the path labeled in red is only 9.0 kcal mol−1, the SH radical is one of the byproducts of the pathway for P1 production. Therefore, the reaction pathway labeled as red will take place after the blue one.
Remarkably, the geometries of P1 are very similar to the reactant, with only one alteration that the thiol was substituted by the hydroxyl. Therefore, as shown in Fig. S16,† the three reaction pathways of forming P3 and P4 from P1 are similar to those shown in Fig. 4. It is worth noting that, for the (CH3)2PS2 radical in Int12, the calculated spin density indicates that the single electron is mainly distributed over the sulfur atom (Fig. S18†), so the dimerization of Int12 takes place between two sulfur atom with an barrierless process. This could also explain the detected species R1R2POS–SOPR1R2 instead of R1R2PSO–OSPR1R2 in the experiment. Analogously, the similar three pathways of generating P5 from P4 are depicted in Fig. S17.† All energy barriers of these reaction pathways shown in Fig. 4, S16, and S17† are summarized in Table 1. Coincidentally, it can be seen from Table 1, the black reaction pathways are always more favorable than the red ones in Fig. 4 and S16,† which is consistent with the appearance order of the oxidation products.
| Pathway | Energy barriers, (kcal mol−1) | ||
|---|---|---|---|
| In Fig. 4 | In Fig. S16 | In Fig. S17 | |
| Black | 8.9 | 9.2 | 20.7 |
| Blue | 5.3 | 1.2 | 20.3 |
| Red | 9.1 | 11.3 | 28.9 |
Apart from hydroxyl radical, the oxygen and water molecules in the atmosphere might also degrade DPAH ligands to some extent. Thus, the effects of these species were considered, respectively. The oxidation pathways of oxygen are shown in Fig. S19.† The initiate complex Int24, locating at 1.5 kcal mol−1 relative to the total energy of DPAH and oxygen, reacts to generate P2 and P5 successively through a series of intermediate products and transition states. Meanwhile, oxysulfide will release in the process of Int27 → Int17, which may be one of the compositions of the pungent gas. In this profile, the whole energy barriers of P2 and P5 are 28.4 kcal mol−1 and 65.7 kcal mol−1, respectively, which are much higher than those of hydroxyl radical oxidation route. Therefore, the oxygen oxidation is not competitive. It is noteworthy that the byproduct hydroperoxyl radical is also considered to be an important oxidant in the atmosphere. Thus, the possible oxidation reactions involving hydroperoxyl radical was further analyzed as Fig. S20.† From the calculations, the whole energy barrier of P1 formation is 25.7 kcal mol−1, which is much higher than that of hydroxyl radical oxidation processes. As a result, the oxidation by hydroperoxyl radical is negligible.
In addition to the oxygen, the effect of water molecule in degrade process was described in Fig. S21.† The whole energy barrier of the generation of P1 is 42.9 kcal mol−1, which is higher than that of hydroxyl radical oxidation obviously. It is worth noting that the concentration of hydroxyl radical in the troposphere is approximately 10.9 × 105 molecules cm−3,45 which is rather low in comparison with that of oxygen and water molecules. However, the generation rate of ˙OH in the atmosphere includes two parts, formation of transient O atom from O3 by photodissociation and reaction between water vapor and transient O atom.46 In other words, ˙OH and O3 are dynamic equilibrated. In this work, the reaction between hydroxyl radical and dithiophosphinic acid molecules can be regarded as an initiation step in the entire oxidation process. With the consumption of hydroxyl radical, new ˙OH will be generated continuously through the ozone-hydroxyl radical dynamic equilibrium. Essentially, the O3 molecules are actually consumed and hydroxyl radical act as “catalyst” in this process. Besides, the concentration of O3 is 50–85 ppb by volume in air in Beijing,47 which is comparable with the content of O2 and H2O. Herein, the oxidation by oxygen and water molecules is confirmed as uncompetitive kinetically in comparison with hydroxyl radical.
The stability of Ni(MeDPA)2
The canonical molecular orbital (MO) diagram can give in-depth bonding information which is related to the stability of Ni(MeDPA)2 complexes.48,49 Fig. 5 presents the MO energy levels calculated at the PBE/TZ2P level of theory. The doubly frontier occupied orbitals 8 b3g, 4 b2g, 19 ag, and 18 ag are mainly non-bonding Ni-based 3 d orbitals. Clearly, the occupied bonding MOs 9 b1g, 8 b1g, and 17 ag show the interaction between Ni and S atoms on both sides. Upon the electron donation from metal to ligand orbitals, the virtual 9 b1g orbital of (MeDPA)2 was significantly stabilized by 3 dx2−y2 orbital of the central Ni atom to form σ-type bonding. The rest MOs mainly come from (MeDPA)2, little contributed from Ni 3 d orbitals. The 3 d orbital from Ni overlaps with 8 b1g orbital to form the Ni–S σ bond (8 b1g MO). Further, the 14 ag orbital in (MeDPA)2 is significantly stabilized by interacting with 4 s orbital of Ni to form 17 ag MO (Ni–S σ bond). These three Ni–S σ-type bonds are deeply buried beneath and strongly stabilized with respect to individual MeDPAH molecule (the MO energy levels of MeDPAH are shown in Fig. S22†). Overall, the inherent chemical bonding between Ni and the surrounding ligand is dominantly derived from the 9 b1g orbital, with 10 b1g as its corresponding antibonding counterpart. In addition, each pair of two adjacent sulfurs atoms can form π-type bonds via MO 11 b3u and 16 b2u, whereas these bonds are absent in MeDPAH case. These π-type interactions give rise to the considerable decrease of the S⋯S distance from 3.496 in MeDPAH to 3.147 Å in Ni(MeDPA)2.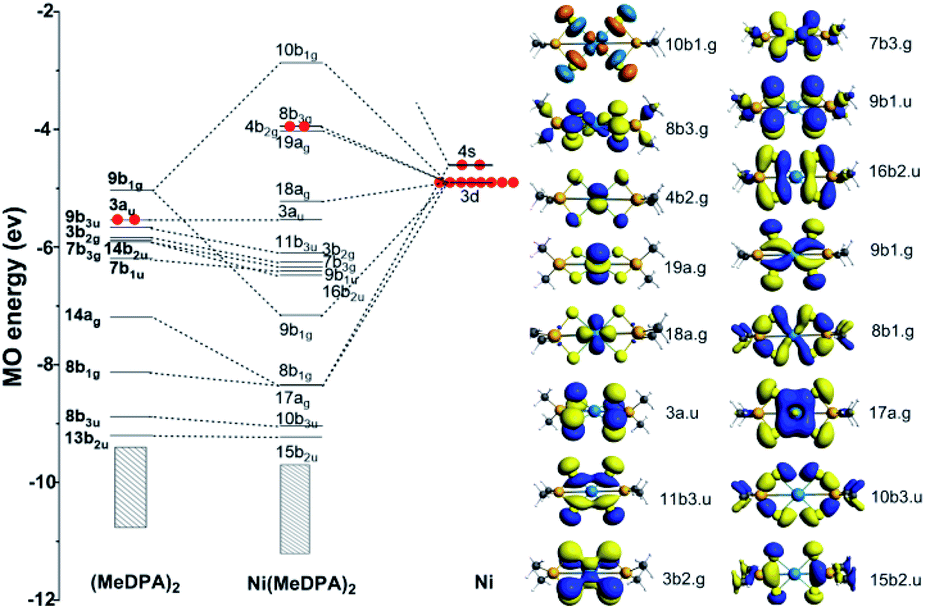 | ||
| Fig. 5 The MO bonding scheme of D2h Ni(MeDPA)2 at the level of PBE/TZ2P (isovalue = 0.03), illustrating the bonding interactions between (MeDPA)2 and Ni fragments. | ||
The chemical bonding in Ni(MeDPA)2 can be further understood by AdNDP analysis.50,51 As the AdNDP results (the 1c–2e and 2c–2e bonds are not listed) shown in Fig. S23,† there are two 4c–2c π bonds contributed by the S 3p orbitals, and these delocalized bonds strengthen the interactions between the two isolated MeDPA. The remaining delocalized bonds are all in-plane σ bonds. The interactions between Ni and (MeDPA)2 are mainly achieved by 5c–2e σ bonds constituted by S 3p orbitals as well as Ni 4 s and 3 d orbitals, respectively, which hold the whole complex together steadily. Meanwhile, the three 6c–2e σ bonds strengthen the S–P bonds. Overall, because of these delocalized bonds in compound Ni(MeDPA)2, it is difficult to break the S–P or S–Ni bond in the air, determining the high stability of Ni-salts.
Conclusions
For the first time, the oxidation behavior of five DPAH ligands with typical structures at ambient environment was systematically investigated through ESI-MS, 31P NMR, FT-IR, and pH titration. It has been shown that DPAH molecules were oxidized in the following pathways: R1R2PSSH → R1R2PSOH → R1R2POOH → R1R2POO–OOPR1R2, R1R2PSSH → R1R2PSS–SSPR1R2, and R1R2PSSH → R1R2PSOH → R1R2POS–SOPR1R2. The introduction of o-trifluoromethyl phenyl group was helpful to improve the oxidation resistance of DPAHs. Along with the oxidation of DPAHs, both DAm and DEu increased, whereas SFAm/Eu decreased dramatically. Remarkably, the ligand L5 bearing two o-CF3Ph substituent groups could still effectively separate Am3+ over Eu3+ after 180 days oxidation, suggesting a good application prospect. Theoretical calculations revealed that the oxidation process of DPAHs was mainly governed by a hydroxyl radical oxidation mechanism. Meanwhile, the order in which the oxidation products formed was closely related to the reaction energy barrier. Due to the stabilization effect of the delocalized bonds, the nickel salt of DPAH showed much better stability as compared to the pure DPAH. In conclusion, the results of this work not only give a rational oxidation mechanism and an effective preservation method for DPAHs at ambient environment, but also provide important guidance to design more stable S-donor ligands for actinide/lanthanide separation in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are very grateful for the financial support by the National Science Foundation of China (Grant No. 11675115, 11975161, and 91426302). Comprehensive training platform of specialized laboratory (College of chemistry, Sichuan University) is acknowledged for ESI-MS, 31P NMR, FT-IR, PXRD, and ICP-AES analyses. The calculations were performed using supercomputers at Tsinghua National Laboratory for Information Science and Technology.References
- D. Lundberg and I. Persson, Coord. Chem. Rev., 2016, 318, 131–134 CrossRef CAS.
- G. T. Seaborg, Radiochim. Acta, 1993, 61, 115–122 CAS.
- C. L. Xiao, C. Z. Wang, L. Y. Yuan, B. Li, H. He, S. A. Wang, Y. L. Zhao, Z. F. Chai and W. Q. Shi, Inorg. Chem., 2014, 53, 1712–1720 CrossRef CAS.
- F. W. Lewis, M. J. Hudson and L. M. Harwood, Synlett, 2011, 18, 2609–2632 Search PubMed.
- Y. J. Zhu, J. Chen and R. Z. Jiao, Solvent Extr. Ion Exch., 1996, 14, 61–68 CrossRef CAS.
- Y. J. Zhu, J. Chen and G. R. Choppin, Solvent Extr. Ion Exch., 1996, 14, 543–553 CrossRef CAS.
- C. Hill, C. Madic, P. Baron, M. Ozawa and Y. Tanaka, J. Alloys Compd., 1998, 271, 159–162 CrossRef.
- K. C. Sole, J. Brent Hiskey and T. L. Ferguson, Solvent Extr. Ion Exch., 1993, 11, 783–796 CrossRef CAS.
- J. Chen, R. Z. Jiao and Y. J. Zhu, Solvent Extr. Ion Exch., 1996, 14, 555–565 CrossRef CAS.
- F. Wang, C. Jia, D. F. Pan and J. Chen, Ind. Eng. Chem. Res., 2013, 52, 18373–18378 CrossRef CAS.
- B. Menoyo, M. P. Elizalde and A. Almela, Anal. Sci., 2002, 18, 799–804 CrossRef CAS.
- P. Marc, R. Custelcean, G. S. Groenewold, J. R. Klaehn, D. R. Peterman and L. H. Delmau, Ind. Eng. Chem. Res., 2012, 51, 13238–13244 CrossRef CAS.
- G. S. Groenewold, D. R. Peterman, J. R. Klaehn, L. H. Delmau, P. Marc and R. Custelcean, Rapid Commun. Mass Spectrom., 2012, 26, 2195–2203 CrossRef CAS.
- G. Modolo and S. Seekamp, Solvent Extr. Ion Exch., 2002, 20, 195–210 CrossRef CAS.
- C. Madic, F. Testard, M. J. Hudson, J. Liljenzin, B. Christansen, M. Ferrando, A. Facchini, A. Geist, G. Modolo, G. Espartero and J. Mendoza, CEA-Report 6066, 2004 Search PubMed.
- M. E. Freiderich, D. R. Peterman, J. R. Klaehn, P. Marc and L. H. Delmau, Ind. Eng. Chem. Res., 2014, 53, 3606–3611 CrossRef CAS.
- P. Marc, Doctoral dissertation, Oak Ridge National Laboratory, 2011 Search PubMed.
- H. Ma, O. Kökkılıç, C. M. Marion, R. S. Multani and K. E. Waters, Can. J. Chem. Eng., 2018, 96, 1585–1596 CrossRef CAS.
- Z. P. Wang, N. Pu, Y. Tian, C. Xu, F. Wang, Y. Liu, L. R. Zhang, J. Chen and S. D. Ding, Inorg. Chem., 2019, 58, 5457–5467 CrossRef CAS.
- C. X. Zeng and Q. Hu, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2008, 47, 71–74 Search PubMed.
- G. P. Gillman, Soil Res., 1985, 23, 643–646 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision C.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- K. Ishida, K. Morokuma and A. Komornicki, J. Chem. Phys., 1977, 66, 2153–2156 CrossRef CAS.
- R. J. Bartlett, J. Chem. Phys., 1989, 93, 1697–1708 CrossRef CAS.
- C. Y. Legault, CYLVIEW, 1.0 b, Université de Sherbrooke, 2009 Search PubMed.
- R. Atkinson, Atmos. Environ., 2000, 34, 2063–2101 CrossRef CAS.
- R. Atkinson and J. Arey, Chem. Rev., 2003, 103, 4605–4638 CrossRef CAS.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- M. Gnanaprakasam, L. Sandhiya and K. A. Senthilkumar, Theor. Chem. Acc., 2017, 136, 131–147 Search PubMed.
- M. Ng, D. K. Mok, E. P. Lee and J. M. Dyke, J. Phys. Chem. A, 2017, 121, 6554–6567 CrossRef CAS.
- B. Wei, J. Sun, Q. Mei and M. He, Comput. Theor. Chem., 2018, 1129, 1–8 CrossRef CAS.
- P. O. Wennberg, K. H. Bates, J. D. Crounse, L. G. Dodson, R. C. McVay, L. A. Mertens, T. B. Nguyen, E. Praske, R. H. Schwantes, M. D. Smarte, J. M. S. Clair, A. P. Teng, X. Zhang and J. H. Seinfeld, Chem. Rev., 2018, 118, 3337–3390 CrossRef CAS.
- G. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS.
- E. Van Lenthe, A. Ehlers and E. J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- E. Van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- G. X. Tian, Y. J. Zhu, J. M. Xu, P. Zhang, T. D. Hu, Y. N. Xie and J. Zhang, Inorg. Chem., 2003, 42, 735–741 CrossRef CAS.
- K. Huang, X. Feng, X. M. Zhang, Y. T. Wu and X. B. Hu, Green Chem., 2016, 18, 1859–1863 RSC.
- K. C. Sole and J. B. Hiskey, Hydrometallurgy, 1992, 30, 345–365 CrossRef CAS.
- B. K. Tait, Hydrometallurgy, 1993, 32, 365–372 CrossRef CAS.
- C. Bourget, B. Jakovljevic and D. Nucciarone, Hydrometallurgy, 2005, 77, 203–218 CrossRef CAS.
- S. C. Herndon and A. R. Ravishankara, J. Phys. Chem. A, 2006, 110, 106–113 CrossRef CAS.
- G. S. Tyndall and A. R. Ravishankara, Int. J. Chem. Kinet., 1991, 23, 483–527 CrossRef CAS.
- M. Z. Li, E. Karu, C. Brenninkmeijer, H. Fischer, J. Lelieveld and J. Williams, npj Clim. Atmos. Sci., 2018, 1, 1–7 CrossRef CAS.
- A. M. Thompson, Science, 1992, 256, 1157–1165 CrossRef CAS.
- Y. X. Wang, J. M. Hao, M. B. Mcelroy, J. W. Munger, H. Ma, C. P. Nielsen and Y. Q. Zhang, Tellus, 2010, 62, 228–241 CrossRef.
- S. X. Hu, J. J. Liu, J. K. Gibson and J. Li, Inorg. Chem., 2018, 57, 2899–2907 CrossRef CAS.
- J. Q. Wang, C. X. Chi, H. S. Hu, L. Y. Meng, M. B. Luo, J. Li and M. F. Zhou, Angew. Chem., Int. Ed., 2018, 57, 542–546 CrossRef CAS.
- W. L. Li, T. T. Chen, D. H. Xing, X. Chen, J. Li and L. S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E6972–E6977 CrossRef CAS.
- D. Y. Zubarev and A. I. Boldyrev, J. Org. Chem., 2008, 73, 9251–9258 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization data of all compounds; data of extraction and theoretical calculations. See DOI: 10.1039/d0ra08841b |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2020 |