
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRelationship between surface hydrophobicity and flux for membrane separation†
Gil-Seon Kanga,
Youngbin Baek*b and
Ji-Beom Yoo *c
*c
aSKKU Advanced Institute of Nanotechnology (SAINT), Sungkyunkwan University, 2066 Seobu-ro, Jangan-gu, Suwon 16419, Republic of Korea
bDepartment of Biotechnology, Sungshin Women's University, Dobongro 76gagil, Gangbuk-gu, Seoul 01133, Republic of Korea
cSchool of Advanced Materials Science and Engineering (BK21), Sungkyunkwan University, 2066 Seobu-ro, Jangan-gu, Suwon 16419, Republic of Korea
First published on 4th November 2020
Abstract
Surface hydrophobicity of anodic aluminum oxide (AAO) membranes was controlled via carbon coating using the CVD method or O2 plasma treatment with insignificant changes of pore diameter. This study first demonstrated that a larger hydrophobic pore surface and hydrophilic membrane surface are favorable for developing high performance membranes.
Membrane technology is widely used for liquid separation and purification, such as in various water treatments, protein purification, virus removal, and artificial organs.1 Despite great progress in membrane development, it is still challenging to increase both flux and rejection, which overcome the ‘trade-off’ line,2 and to prevent membrane fouling.3 Although strategies for antifouling membranes, including the role of surface chemistry, such as hydrophobicity, charge, and roughness, have been well established,4,5 it is still challenging to develop membranes that have faster water permeability with reasonable rejection.
Because conventional polymers rely on the ‘trade-off’ phenomenon, carbon nanomaterials (CNMs), such as carbon nanotubes (CNTs) and graphene oxide (GO)/reduced GO (rGO), have been attractive for overcoming the limitation of polymer-based membranes.6,7 The strategies for CNT-based membranes and GO/rGO-based membranes to achieve high flux are using fast transport of water molecules through the pore's hydrophobic surface and/or a very thin active layer for GO/rGO membranes.8–11 For example, GO membranes also showed increased flow allowed by a thin active layer (i.e., flux is inversely proportional to the length of the pore) by applying various membranes,12 whereas rGO membranes have been applied only to a water vapor passing-through process (i.e., membrane distillation).13 Additionally, the water flow through the inner wall of CNTs is several hundreds to thousands times faster than the theoretical flow because of the low interaction force between water molecules and CNT's hydrophobic wall.14,15 However, the energetic requirements, found by using molecular dynamic simulation, needed to pass water molecules through the hydrophobic CNT end were several hundred atm of applied pressure.16 Lee et al.17 reported that CNT wall membranes showed approximately 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 L m−2 h−1 bar of water permeability using O2 plasma etching (to use the inner pores of CNT by removing the its fullerene cap and possibly to introduce the hydrophilic surface) and densification technique (to increase pore density and to use the outer wall as pores). Given the results from previous studies, it is still unclear how the hydrophobicity of the pore surface affects both flux and rejection behaviors.
000 L m−2 h−1 bar of water permeability using O2 plasma etching (to use the inner pores of CNT by removing the its fullerene cap and possibly to introduce the hydrophilic surface) and densification technique (to increase pore density and to use the outer wall as pores). Given the results from previous studies, it is still unclear how the hydrophobicity of the pore surface affects both flux and rejection behaviors.
In this study, we experimentally demonstrate that the surface hydrophobicity of an AAO membrane affects the flow behavior, distinguished to the membrane surface and pore surface; it is more favorable to transport water through the membranes having a hydrophilic membrane surface and a hydrophobic pore surface.
We fabricated anodized alumina oxide (AAO) membranes and controlled their hydrophobicity by using carbon coating via a CVD method (more details are described in the ESI; † digital pictures of AAO and C-AAO membranes are provided in Fig. S1†). Fig. 1 shows the hydrophobicity and surface chemistry of the AAO membrane and carbon-coated AAO (C-AAO) membrane with 60 nm of pore diameter. As shown in Fig, 1a and b, the contact angle of the AAO membrane of 19° ± 2°, is in good agreement with the values in the literature for non-porous alumina (∼12°).18 The membrane surface became more hydrophobic, to 65° ± 3° in the C-AAO membrane (Fig. 1b).
 | ||
| Fig. 1 The surface analysis of AAO and carbon-coated AAO membranes. (a)The image of a pristine AAO membrane and (b) a C-AAO membrane by contact angle. (c) C 1s and (d) O 1s XPS spectrum of C-AAO. (e) Optical image of C-AAO cross-section, and (f) Raman microscopy (λ = 514 nm) of each position. | ||
XPS spectra of carbon (Fig. 1c) and oxygen (Fig. 1d) obtained from a C-AAO membrane indicate successful carbon deposition on the AAO membrane. According to the XPS spectrum of C 1s (Fig. 1c), the graphitic sp2 carbon shows C–C bonding (284.6 eV), C–O bonding (286.5 eV), a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group (288.0 eV), and an O–CO group (289.4 eV).19–22
O group (288.0 eV), and an O–CO group (289.4 eV).19–22
The O 1s spectrum of C-AAO in Fig. 1d shows Al–O bonding (531.6 eV) and C–O bonding (533.2 eV),22,23 whereas that of AAO had only Al–O bonding (Fig. S2†). These Al–OH groups chemically reacted with carbon in a high-temperature CVD process, eventually forming the thin carbon layer.24 Additionally, we used cross-sectional Raman spectroscopy to evaluate carbon coating on the pore surfaces, where the positions are marked in Fig. 1e. We observed typical Raman spectra of an amorphous carbon through each point, shown in Fig. 1f. The D-band at 1350 cm−1 and the G-band at 1580 cm−1 represent the sp3 bonds and sp2 bond, respectively. The ratio of ID/IG at five points was approximately 1.3 (Fig. 1e and f), consistent with graphene oxide.19,25,26 These results indicate that C-AAO membranes were more hydrophobic, not only on the top surface (i.e., membrane surface), but also on the pore surface of the membranes.
We evaluated the water flow velocities of AAO and C-AAO membranes with 40–80 nm of pore diameter using a dead-end filtration system (Fig. S3†), with results shown in Fig. 2. The flow velocity increased with increasing pore diameter of both AAO and C-AAO membranes, as expected. However, the flow velocity of C-AAO membranes was faster in all ranges of pore diameter than that of AAO membranes, almost consistent with the theoretical flow velocity calculated from the Hagen–Poiseuille (H–P) equation in the no-slip condition (eqn (1) in ESI†); the water flow velocity of C-AAO membranes increases from 0.7 × 10−4 m s−1 to 3.5 × 10−4 m s−1, whereas that of AAO membranes increases from 0.3 × 10−4 m s−1 to 2.9 × 10−4 m s−1 as pore diameter varies from 40 nm to 80 nm. Note that the changes of pore diameter and membrane thickness were negligible after carbon coating and an average pore diameter and pore density, used as 1.02 × 1010 cm−2 of all membranes, were measured from SEM images using a ‘Measure IT’ program (Fig. S4†). Since the physical properties (e.g. pore diameter, pore density, and membrane thickness) of both AAO and C-AAO membranes were the same, the faster flow in C-AAO membranes can be explained by the hydrophobic surface, consistent with previous studies which showed that water molecules rapidly pass through the hydrophobic wall surface of carbon nanotubes.17,27,28 It is well known that hydrophobic pore surfaces lead to a frictionless flow and weaken interfacial force between water molecules and carbon-coated surface, resulting in faster flow.19,29,30 Note that the adhesion energy of water on alumina is ∼800 mJ m−2,31 whereas that of water on graphite is about 100 mJ m−2.32
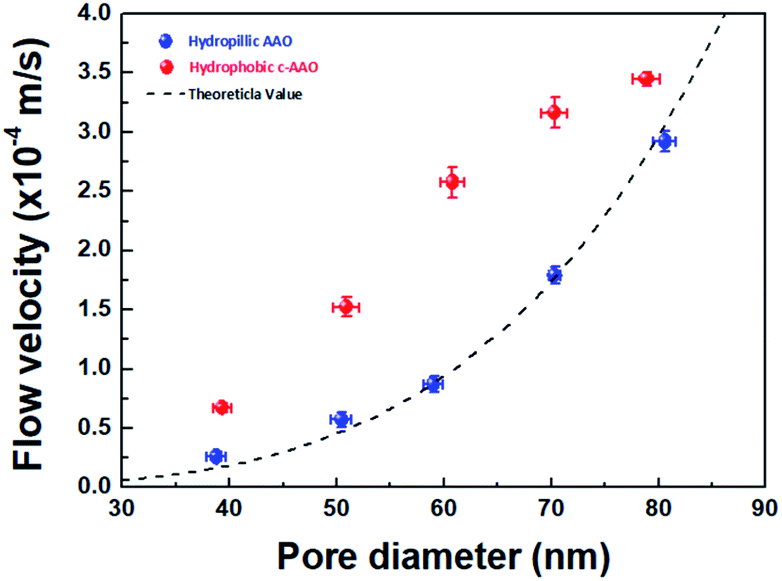 | ||
| Fig. 2 Flow velocities with various pore diameters depending on the surface chemistry. The theoretical flow velocity (black line) as a function of pore diameter. The blue dot is the flow velocity of a hydrophilic surface (AAO membrane), and the red dot is that of a hydrophobic surface (C-AAO membrane) by experimental water velocity. | ||
To find the extent of faster flow for C-AAO membranes, enhancement factor (ε) and slip length (Ls) along with the experimental flux (Qexp) with a unit of L m−2 h−1, and theoretical flux (QHP, calculated from H–P eqn) are represented in Table 1. Enhancement factor was calculated from Qexp divided by QHP, and slip length was calculated using the H–P eqn in the slip condition (eqn (2) in ESI†); more details are described in the ESI.† Enhancement factor increased from 1.2 to 3.6, which also affected the increased slip length, from 3.1 nm to 15.2 nm when the pore diameter decreased from 80 nm to 40 nm. These results indicate that water flow velocity increases with smaller pore diameter because of the larger hydrophobic effects.
| Diametera (nm) | Theoretical flux (QHP: L m−2 h−1) | Experimental flux (Qexp: L m−2 h−1) | Enhancement factor (ε) | Slip length (Ls: nm) |
|---|---|---|---|---|
| a Pore diameter were measured from SEM image (see ESI Fig. S4) using Measure IT program, standard deviation for pore diameter was ±2. | ||||
| 40 | 66.8 | 243 ± 13 | 3.6 | 15.2 ± 2.2 |
| 50 | 163.0 | 518 ± 29 | 3.2 | 13.4 ± 1.5 |
| 60 | 338.1 | 928 ± 46 | 2.7 | 12.0 ± 0.8 |
| 70 | 626.3 | 1118 ± 46 | 1.8 | 6.4 ± 1.2 |
| 80 | 1068.4 | 1242 ± 20 | 1.2 | 3.1 ± 1.0 |
We did another experiments to evaluate the flux behavior according to the solvent's viscosity (shown in Fig. S5†). We obtained data using acetone (0.32 cP), methanol (0.6 cP), water (1.0 cP), ethanol (1.1 cP), and IPA (2.3 cP) at ambient temperature. The results also showed that the flow of C-AAO membranes was much faster than that of AAO membranes, in a good agreement with theoretical flux calculations using viscosity and density of all solvents.33–39
The negative effects of a hydrophobic surface can limit how the water molecules enter the gate of pore; high pressure (e.g., a few hundred atm) is needed to pass water molecules through a theoretically hydrophobic surface composed with CC bonds with a few nm of pore diameter. Therefore, it is favorable that membranes have a relatively hydrophilic membrane surface with a hydrophobic pore surface; this scheme is presented in Fig. 3a.
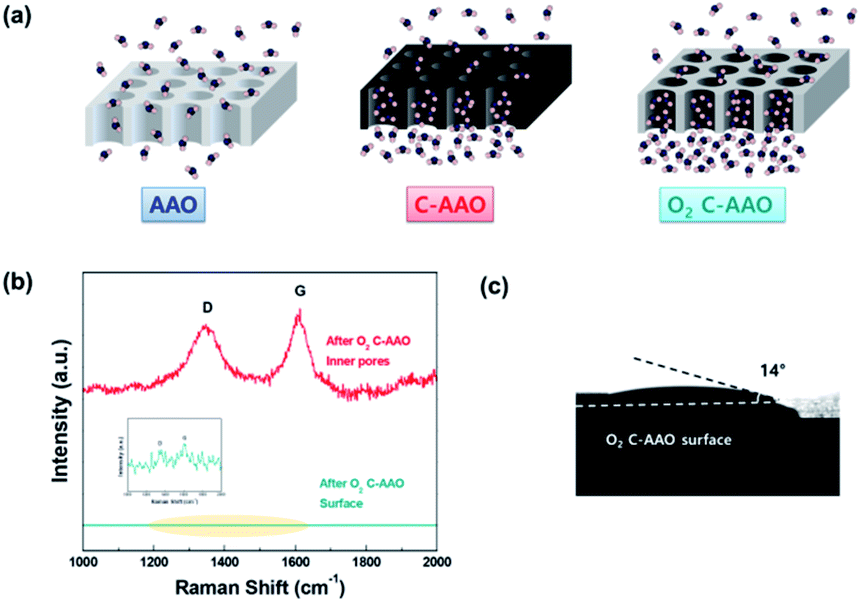 | ||
| Fig. 3 (a) Schematic of AAO (left), C-AAO (middle) and O2 C-AAO membranes (right). (b) Raman spectra of an O2 C-AAO membrane; upper red line, representing the inner pore surface indicates the presence of D and G bands, while the lower green line, representing the membrane surface, indicates there is no carbon on the membrane surface. (c) Contact angle image of the O2 C-AAO membrane. | ||
In order to control the hydrophobicity of the membrane surface, we treated 60 nm of C-AAO membrane with O2 plasma. Fig. 3a shows the Raman spectra of the pore surface and membrane surface of the O2 plasma-treated C-AAO membrane (referred as an O2 C-AAO membrane), indicating that coated carbon maintained in pores (ratio of ID/IG maintained as 1.3) while no carbon existed on the membrane surface. This change made the membrane surface more hydrophilic again (14° of contact angle), below the value of C-AAO and comparable with AAO membranes (Fig. 3b; the dramatically changed surface hydrophobicity can also be found in the movie file of the ESI†). These carbons are etched on the C-AAO membrane surface, where the carbon atoms break and remove oxygen bonds and the oxygen atoms have functional groups. The hydrogen atoms of water weakly bond with the surface oxygen negative charges of the O2 C-AAO membrane. For this reason, water molecules spread faster than the AAO membrane surface, as confirmed by the green dot in the water-flux graph shown in Fig. 4a. The water flux of the C-AAO membrane was about 928 L m−2 h−1, which was increased to 1160 L m−2 h−1 when only the membrane surface became more hydrophilic.
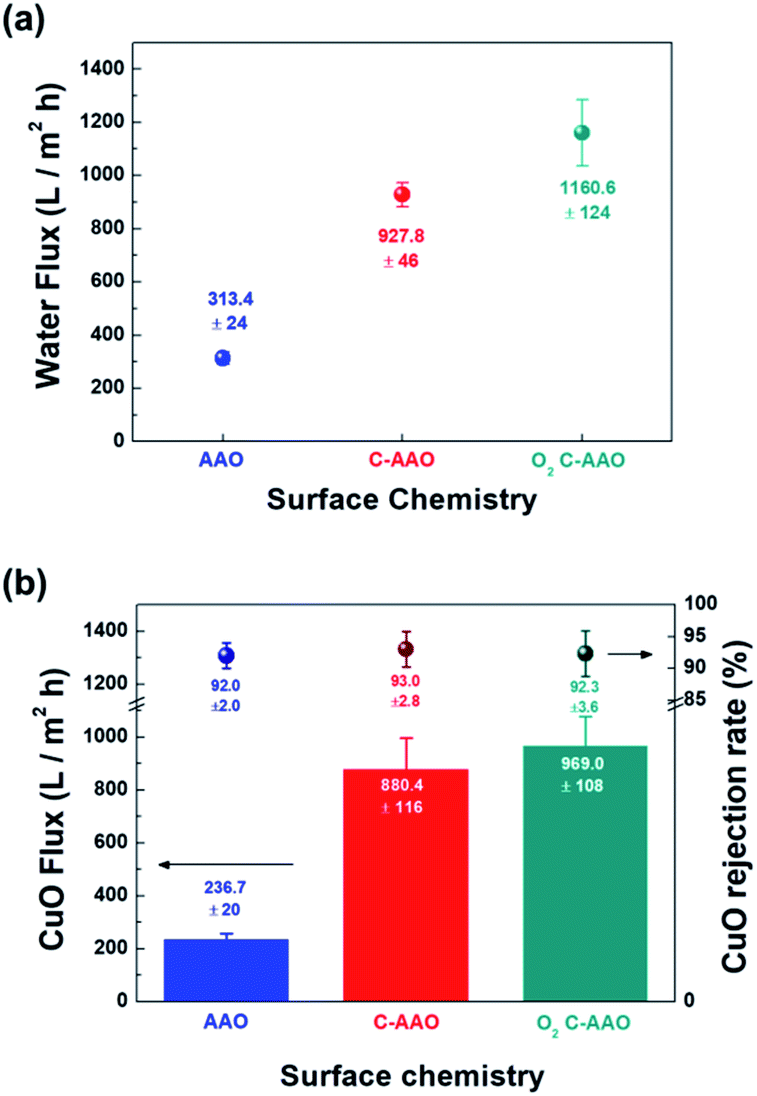 | ||
| Fig. 4 (a) Water fluxes of each membrane depending on the external and inner surface. (b) CuO molecules (about 64 nm) rejection rate depending on the surface chemistry of 60 nm AAO membranes as UF. The blue dot is the rejection rate of the AAO membrane, and the red dot and green dot that of the C-AAO membrane and O2-treated C-AAO membrane, respectively. The blue, red, and green bars are CuO Flux with each CuO molecule removal of the AAO membrane, C-AAO membrane, and surface-treated C-AAO membrane. | ||
We did another experiment to evaluate the rejection property using 16 mg L−1 of CuO solution, shown in Fig. 4b. The hydrodynamic diameter of the synthesized CuO nanoparticles40 was approximately 64 nm, measured using dynamic light scattering (Fig. S7†). Flux behaviors of AAO, C-AAO, and O2 C-AAO membranes were similar to that using DI water, and the rejection properties of all three membranes were similar to 92%, consistent with the calculated value from the Ferry–Renkin eqn (see ESI†).
Conclusions
We first demonstrated that the hydrophilic entrance surface and hydrophobic pore surface are favorable for membrane used for liquid separation. Carbon coating by the CVD method on AAO membranes changed the hydrophobicity of both surfaces by changing the pore size insignificantly. Explaining that two different membranes have the same physical properties, but different hydrophobicities on both entrance and pore surfaces (i.e. C-AAO, and O2 C-AAO membranes in this study). This hydrophilic entrance membrane surface implies that the hydrophobicity of a pore surface would play a critical role in flux behavior at few tens of pore diameter, providing an important insight in to develop high-performance membranes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Industrial Strategic technology development program (10085617, Measurement of Mechanical Properties of 2D materials using Bulge test) funded By the Ministry of Trade, industry & Energy (MOTIE, Korea) and by the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science, ICT and Future Planning (NRF-2018R1C1B5086300).Notes and references
- R. W. Baker, Membrane technology and applications, 3rd edition, 2012 Search PubMed.
- H. B. Park, J. Kamcev, L. M. Robeson, E. Menachem and B. D. Freeman, Science, 2017, 6343 Search PubMed.
- W. Guo, N. Huu-Hao and J. Li, Bioresour. Technol., 2012, 122, 27–34 CrossRef CAS.
- D. Rana and T. Matsuura, Chem. Rev., 2010, 110, 2448–2471 CrossRef CAS.
- J. R. Werber, C. O. Osuji and E. Menachem, Nat. Rev. Mater., 2016, 1, 1–15 Search PubMed.
- S. Daer, J. Kharraz, A. Giwa and S. W. Hasan, Desalination, 2015, 367, 37–48 CrossRef CAS.
- Y. H. Teow and A. W. Mohammad, Desalination, 2019, 451, 2–17 CrossRef CAS.
- B. J Hinds, N. Chopra, T. Rantell, R. Andrews, V. Gavalas and L. G. Bachas, Science, 2004, 303, 62–65 CrossRef.
- J. K. Holt, H. G. Park, Y. Wang, M. Stadermann, A. B. Artyukhin, C. P. Grigoropoulos, A. Noy and O. Bakajin, Science, 2006, 312, 1034–1037 CrossRef CAS.
- D. Cohen-Tanugi and J. C. Grossman, Nano Lett., 2012, 12, 3602–3608 CrossRef CAS.
- J. Abraham, K. S. Vasu, C. D. Williams, K. Gopinadhan, Y. Su, C. T Cherian, J. Dix, E. Prestat, S. J. Haigh, I. V. Grigorieva, P. Carbone, A. K Geim and R. R. Nair, Nat. Nanotechnol., 2017, 12, 546 CrossRef CAS.
- Y. Wei, Y. Zhang, X. Gao, Z. Ma, X. Wang and C. Gao, Carbon, 2018, 139, 964–981 CrossRef CAS.
- A. Abdel-Karim, J. M. Luque-Alled, S. Leaper, M. Alberto, X. Fan, A. Vijayaraghavan, T. A. Gad-Allah, A. S. El-Kalliny, G. Szekely, S. I. A. Ahmed, S. M. Holmes and P. Gorgojo, Desalination, 2019, 452, 196–207 CrossRef CAS.
- G. Hummer, J. C. Rasaiah and J. P. Noworyta, Nature, 2001, 414, 188–190 CrossRef CAS.
- S. Krishnan and F. A. Armstrong, Chem. Sci., 2012, 3, 1015–1023 RSC.
- J. H. Walther, K. Ritos, E. R. Cruz-Chu, C. M Megaridis and P. Koumoutsakos, Nano Lett., 2013, 13, 1910–1914 CrossRef CAS.
- B. H. Lee, Y. Baek, M. Lee, D. Jeong, H. H. Lee, J. Yoon and Y. H. Kim, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- D. Megias-Alguacil, E. Tervoort, C. Cattin and L. J. Gauckler, J. Colloid Interface Sci., 2010, 353, 512–518 CrossRef.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463–470 CrossRef CAS.
- S. Stankovich, R. D. Piner, X. Q. Chen, N. Q. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155–158 RSC.
- J. Haeberle, K. Henkel, H. Gargouri, F. Naumann, B. Gruska, M. Arens, M. Tallarida and D. Schmeiber, J. Nanotechnol., 2013, 4, 732–742 CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray photoelectron spectroscopy, Physical Electronics, Inc, 1995 Search PubMed.
- J. Joubert, A. Salameh, V. Krakoviack, F. Delbecq, P. Sautet, C. Coperet and J. M. Basset, J. Phys. Chem. B, 2006, 110, 23944–23950 CrossRef CAS.
- F. Tuinstra and J. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- S. Reich and C. Thomsen, Philos. Trans. R. Soc. London, Ser. A, 2004, 362, 2271–2288 CrossRef CAS.
- Y. Baek, C. Kim, D. K. Seo, T. Kim, J. S. Lee Jeo, Y. H. Kim, K. H. Ahn, S. S. Bae, S. C. Lee, J. Lim, K. Lee and J. Yoon, J. Membr. Sci., 2014, 460, 171–177 CrossRef CAS.
- H. G. Park and Y. Jung, Chem. Soc. Rev., 2014, 43, 565–576 RSC.
- M. Whitby and N. Quirke, Nat. Nanotechnol., 2007, 2, 87–94 CrossRef CAS.
- C. H. Ahn, Y. Baek, C. Lee, S. O. Kim, S. Kim, S. Lee, S. H. Kim, S. S. Bae, J. Park and J. Yoon, J. Ind. Eng. Chem., 2012, 18, 1551–1559 CrossRef CAS.
- P. M. Hobbs and A. J. Kinloch, J. Adhes., 1998, 66, 203–228 CrossRef CAS.
- F. M. Fowkes, in Chemistry and Physics of Interfaces, American Chemical Society, Washington D.C, 1971, p. 154 Search PubMed.
- W. Fu, T. Pei, Y. Mao, G. Li, Y. Zhao and L. Chen, J. Membr. Sci., 2019, 572, 453–463 CrossRef CAS.
- X. Yang, L. Yan, J. Ma, Y. Bai and L. Shao, J. Membr. Sci., 2019, 591, 117353 CrossRef.
- H. Sun, Y. Zhang, H. Sadam, J. Ma, Y. Bai, X. Shen, J. Kim and L. Shao, J. Membr. Sci., 2019, 582, 1–8 CrossRef CAS.
- H. Jiang, Q. Zhao, P. Wang and X. Zhai, J. Membr. Sci., 2019, 588, 117148 CrossRef CAS.
- C. Lei, Z. Li, Q. Gao, R. Fu, W. Wang, Q. Li and Z. Liu, J. Membr. Sci., 2020, 608, 118192 CrossRef CAS.
- J. Huang, X. Cheng, Y. Zhang, K. Wang, H. Liang, P. Wang, J. Ma and L. Shao, Cell Reports Physical Science, 2020, 1, 100034 CrossRef.
- X. Cheng, Z. Sun, X. Yang, Z. Li, Y. Zhang, P. Wang, H. Liang, J. Ma and L. Xhao, J. Mater. Chem. A, 2020, 8, 16933–16942 RSC.
- Y. Cao, Y. Xu, H. Hao and G. Zhang, Mater. Lett., 2014, 114, 88–91 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07262a |
| This journal is © The Royal Society of Chemistry 2020 |