
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen sulfide sensing using an aurone-based fluorescent probe†
Arjun Kaflea,
Shrijana Bhattaraia,
Justin M. Miller *ab and
Scott T. Handy*ab
*ab and
Scott T. Handy*ab
aMolecular Bioscience Program, Middle Tennessee State University, Murfreesboro, TN 37132, USA. E-mail: justin.miller@mtsu.edu; scott.handy@mtsu.edu
bDepartment of Chemistry, Middle Tennessee State University, Murfreesboro, TN 37132, USA
First published on 22nd December 2020
Abstract
Hydrogen sulfide detection and sensing is an area of interest from both an environmental and a biological perspective. While many methods are currently available, the most sensitive and biologically applicable ones are fluorescence based. In general, these fluorescent probes are based upon large, high-molecular weight, well-characterized fluorescent scaffolds that are synthetically demanding to prepare and difficult to tune and modify. In this study, we have reported a new reduction-based, rationally designed and synthesized turn-on fluorescent probe (Z)-2-(4′-azidobenzylidene)-5-fluorobenzofuran-3(2H)-one (6g) utilizing a low molecular weight aurone fluorophore. During these studies, the modular nature of the synthesis was used to quickly overcome problems with solubility, overlap of excitation of the probe and reduced product, and rate of reaction, resulting in a final compound that is efficient and sensitive for the detection of hydrogen sulfide. The limitation of slow reaction and the reduced fluorescence in a biologically relevent medium was solved by employing cationic surfactant cetyltrimethyl ammonium bromide (CTAB). The probe features a high fluorescence enhancement, fast response (10–30 min), and good sensitivity (1 μm) and selectivity for hydrogen sulfide.
Introduction
The detection of hydrogen sulfide has been an important goal for some time.1–5 From an environmental perspective, hydrogen sulfide is a noxious and toxic by-product of various industrial processes, particularly the Kraft pulping process.6 From a biological perspective, hydrogen sulfide has increasingly been recognized as a critical very small signaling molecule akin to nitric oxide and carbon monoxide that can be diagnostic for a range of pathological conditions7 including kidney disease,8 Down's syndrome,9 and cirrhosis of the liver.10 This recognition, combined with the desire to better understand the role and localization/transport of hydrogen sulfide, has resulted in the development of a number of probes for live cell imaging.11Thanks to its high sensitivity, rapid response, and generally simple sample preparation, fluorescence spectroscopy has become the most common approach for the design of new hydrogen sulfide sensors. Within these sensors, several different approaches have been developed, particularly the reduction of nitro and azido groups,12–22 the chelation with copper (II) complexes,23–25 nucleophilic thiolysis,26–40 and conjugate additions and cyclizations.41–45 For all of these approaches, one key consideration is avoiding reaction with other common sulfur-containing compounds, particularly glutathione and cysteine. A concern with existing probes is that they are mostly based upon common bulky scaffolds including fluorescein, xanthene, and rhodamine which have inherent limitations in terms of cell permeability and solubility in the cellular environment. Additionally, these molecules often require more lengthy synthetic sequences for their preparation and significant effort is required in order to modify their intrinsic spectral range.
To address this situation, we envisioned using the aurone system as an alternative fluorescent scaffold. Aurones are a small family of natural products primarily responsible for the golden pigmentation present in plants with yellow flowers such as dahlias, snapdragons, and cosmos.46–48 Due to their unusual exocyclic alkene, they absorb at longer wavelengths compared to the more common flavones. At the same time, this ring system is easy to synthesize in a variety of ways, particularly the acid or base catalyzed condensation of an aldehyde and a benzofuranone.49–52 Indeed, this condensation can even be performed under effectively neutral conditions, rendering it highly functional group compatible as well as generally high yielding.
Aurones have been reported to be fluorescent, with the first significant study reported by Bane and co-workers.53 Since this report on the fluorescent properties of a series of amino-substituted aurones, a few other studies have explored variations of the benzofuranone portion either experimentally or computationally.54–56
Interestingly, application of aurone fluorescence has been virtually unexplored. To date, only two reports have appeared in the literature (Fig. 1). In the first, hydroxyaurone 1 was reported as a selective sensor for mercury (II) in aqueous solution as well as living cells.57 It exhibited high selectivity and a linear decrease in fluorescence intensity in the presence of low micromolar concentrations of mercury (II) salts. An earlier report noted that 4-hydroxyaurones 2 exhibited changes in their UV and fluorescence spectra in the presence of cyanide anion.58 p-Bromo-substituted compound 2b was particularly interesting as it exhibited a dramatic increase in emission upon irradiation at 469 nm in acetonitrile in the presence of cyanide anion as well as a visual color change. Other anions did not induce a similar change. As a result, aurones certainly have the potential to be fluorescent sensors.
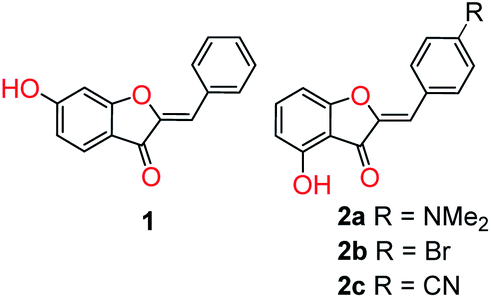 | ||
| Fig. 1 Aurone-based fluorescent probes. | ||
With this in mind, we envisioned utilizing an aurone scaffold to develop an azide-reduction based turn-on fluorescent probe which in the presence of H2S undergoes reduction to the corresponding amine accompanied by a significant shift in the absorption and emission spectra. For this purpose, we explored 4′-azidoaurone 6a as a simple model probe which ultimately on appropriate optimization led us to the rationally designed H2S selective probe 6g.
Experimental section
Materials and methods
ACS grade chemicals and solvents were used as received without further purification. All reactions were conducted under an air atmosphere. 1H and 13C NMR spectra were recorded on a JEOL AS (500 or 300 MHz) NMR instrument and chemical shifts were recorded in ppm with reference to tetramethyl silane (TMS), chloroform-d or DMSO-d6.59 The following conventions are used for multiplicities: s, singlet; d, doublet; t, triplet; m, multiplet; dd, doublet of doublet; dt, doublet of triplet; ddd, doublet of doublet of doublet; br, broad. A Cary 630 FT-IR (Agilent Technologies) was used to collect IR spectra. Solid samples were used for collecting the IR-spectra. The high-resolution mass spectra (HRMS) were acquired on a Waters Synapt HDMS QToF as solutions in MeOH containing 1% trifluoroacetic acid. Fluorescence spectra were collected using an F-4500 fluorescence spectrophotometer (Hitachi, Japan) with the slit width for excitation and emission set at 5 nm and photomultiplier voltage set at 700v. Excitation wavelength details are provided in-text as relevant. An HP-8452A Diode Array Spectrophotometer (Agilent Technologies Inc.) was used to record UV-visible absorption spectra. Reactions were monitored by Thin Layer Chromatography (TLC). All extracts were concentrated under reduced pressure using a Buchi Rotary Evaporator. Products were purified by flash silica gel (32-63u) column chromatography.General procedure for the synthesis of 4′-azido benzaldehyde derivatives
In a 3 dram glass vial containing a magnetic stir bar, 1.0 mmol of fluorinated aromatic aldehyde and 1.5 mmol (1.5 equivalent) of sodium azide (NaN3) were dissolved in 2 mL of dimethylsulfoxide (DMSO) and heated for 2–3 h at 70–90 °C (70 °C for multi-fluorosubstituted) in a sand bath. After the completion of the reaction, it was diluted with DI water and extracted with ethylacetate (×4). To remove residual DMSO the organic fraction was further washed with DI water and brine followed by drying over anhydrous MgSO4. The organic layer was concentrated in vacuo and purified by flash column chromatography using 2–10% ethyl acetate: hexane to obtain the desired 4′-azido benzaldehydes.General procedure for the synthesis of aurones (6a–6g)
All the aurones 6a–6g were synthesized based on the literature.52 In a 3-dram glass vial, benzofuranone (1 equivalent) was dissolved in 5 mL of glacial acetic acid containing a magnetic stir bar. To this solution 4′-azidobenzaldehyde (1.5 equivalent) and 2–3 drops (0.2 mL) of concentrated HCl were added and stirred at room temperature for up to 3 hours. In most cases the reaction afforded a precipitate, normally after 30 min, indicating the completion of the reaction. After the reaction was complete, it was poured into ice-cold DI water. The precipitate obtained was filtered and the residue was washed multiple times with water and allowed to air dry. No further purification was required. For some of the aurones, which did not precipitate out efficiently, the water diluted reaction mixture was neutralized with saturated NaHCO3 and extracted with ethyl acetate, concentrated under reduced pressure using a Rotary Evaporator and purified by flash column chromatography. The characterization data for 6a–6f is provided in ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane) yield: 50% (70 mg); yellow solid, mp 137–138 °C. 1H NMR (500 MHz, CDCl3) δ 7.89 (d, J = 8.5 Hz, 1H), 7.45 (dd, J = 6.5, 3.0 Hz, 1H), 7.38 (td, J = 8.5, 3.0 Hz, 1H), 7.29 (dd, J = 8.5, 3.5 Hz, 1H), 7.10 (d, J = 8.5 Hz, 1H), 6.85 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 183.9, 162.1, 159.0 (d, 1JC–F = 244.7 Hz), 147.4, 141.9, 133.3, 129.0, 124.4 (d, 2JC–F = 26.1 Hz), 122.5 (d, 3JC–F = 8.0 Hz), 119.7, 114.2 (d, 3JC–F = 7.8 Hz), 113.1, 110.36 (d, 2JC–F = 24.3 Hz). IR (neat) 2128, 1707, 1658, 1598, 1484, 1263, 1190, 832 cm−1.
:hexane) yield: 50% (70 mg); yellow solid, mp 137–138 °C. 1H NMR (500 MHz, CDCl3) δ 7.89 (d, J = 8.5 Hz, 1H), 7.45 (dd, J = 6.5, 3.0 Hz, 1H), 7.38 (td, J = 8.5, 3.0 Hz, 1H), 7.29 (dd, J = 8.5, 3.5 Hz, 1H), 7.10 (d, J = 8.5 Hz, 1H), 6.85 (s, 1H). 13C NMR (125 MHz, CDCl3) δ 183.9, 162.1, 159.0 (d, 1JC–F = 244.7 Hz), 147.4, 141.9, 133.3, 129.0, 124.4 (d, 2JC–F = 26.1 Hz), 122.5 (d, 3JC–F = 8.0 Hz), 119.7, 114.2 (d, 3JC–F = 7.8 Hz), 113.1, 110.36 (d, 2JC–F = 24.3 Hz). IR (neat) 2128, 1707, 1658, 1598, 1484, 1263, 1190, 832 cm−1.General procedure for the synthesis of amino derivatives (7a–7g)
For the synthesis of the corresponding amines (7a–7g) of the probe candidates, one equivalent of the aurone (6a–6g) was mixed with an excess of NaHS (4 equivalents) in a 1-dram glass vial containing a magnetic stir bar. To the mixture, 1.5 mL of MeCN with a few drops of DI H2O was added and stirred for 30 minutes (acetone can also be used as a solvent). After a few minutes, the solution turned bright red. Once the reaction was complete, based on the TLC, the mixture was concentrated under vacuum and subjected to flash column chromatography (eluted with 5% MeOH:DCM). The characterization data for 7a–7f is provided in the ESI.†
:DCM) yield: 85% (19.5 mg); dark red solid, mp 178–180 °C. 1H NMR (500 MHz, DMSO-D6) δ 7.74 (d, J = 8.5 Hz, 2H), 7.65–7.58 (m, 2H), 7.57 (dd, J = 7.5, 2.5 Hz, 1H), 6.88 (s, 1H), 6.66 (d, J = 8.5 Hz, 2H), 6.16 (s, 2H). 13C NMR (75 MHz, DMSO-d6) δ 181.6, 160.5, 158.2 (d, 1JC–F = 240.9 Hz), 152.1, 144.3, 134.2, 123.7 (d, 2JC–F = 26.0 Hz), 122.6 (d, 3JC–F = 8.1 Hz), 118.7, 116.2, 114.7 (d, 3JC–F = 8.1 Hz), 113.9, 109.3 (d, 2JC–F = 24.2 Hz). IR (neat) 3420, 3320, 3216, 1689, 1633, 1562, 1514, 1480, 1272, 1156, 817, 762 cm−1. HRMS calcd for C15H11FNO2 256.0774, observed 256.0071.Solution preparation method for spectroscopic measurement
Stock solutions (10, 5, and 1 mM) of the probe candidates (6a–6g) and their respective amines were prepared in DMSO. Similarly, the stock solutions of NaHS (10, 5 and 1 mM), cetyl trimethylammonium bromide (CTAB) (5 and 1 mM) and various biologically relevant sulfide sources L-cysteine, reduced glutathione, and 2-mercaptoethanol (5 and 1 mM) were prepared in phosphate buffered saline (PBS) (1X, pH = 7.4, without Ca and Mg). The detailed sample preparation procedures for the experiments can be found in the ESI.† All UV-visible absorption and emission spectra were collected in various concentrations of DMSO (0.5, 5, 50, and 100%) and/or MeCN in PBS-buffer (1X, pH = 7.4, without Ca and Mg) in 3 mL and 1.5 mL cuvettes respectively, in the presence and absence of 1 mM CTAB. The detection limit was determined using different concentrations of 7g (0.1–50 μM) in different solvent systems (biological and non-aqueous). Limits of detection were defined based on linearity and signal-to-noise ratios for the upper and lower limits, respectively. All emission intensities used to determine detection limits were derived from triplicate experimental measurements. Rate profiles of the reduction reaction as well as the selectivity study of the probe were performed by the incubation of 20 μM 6g with 100 μM H2S source (NaHS) in either 0.5% v/v DMSO in PBS/1 mM CTAB (biological) or 90% v/v DMSO/10% v/v PBS (non-aqueous) solvent systems. The reactions were excited at 465 nm and emission spectra spanning from 480 to 700 nm were collected at 2 minute intervals. All nonlinear least squares (NLLS) analyses were performed using a single-exponential function given as:| Y = A(1 − e−kapp×t) + b |
Results and discussion
Probe design
All of the fluorescent probe candidates 6a–6g were synthesized as outlined in Scheme 1. The reaction involved the Knoevenagel-type condensation of a benzofuranone 5 with a 4-azidobenzaldehyde derivative 4 under acidic reaction conditions.52 Since the reaction product generally precipitates out in cold water, the products were separated by filtration. As depicted in Scheme 1, the 4-azidoaldehydes were prepared by direct azidation of the corresponding 4-fluorobenzaldehydes with NaN3 in DMSO.60 The 4′-azidoaurone derivatives 6(a–g) were further reduced by NaHS to give the corresponding amines 7.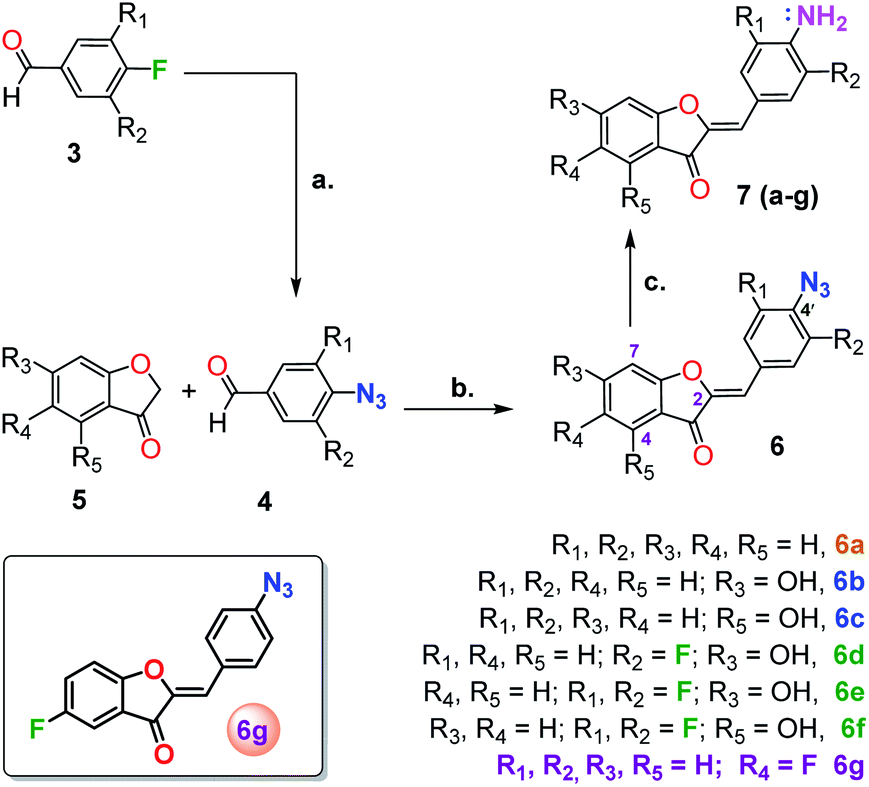 | ||
| Scheme 1 Aurone-based probe synthesis. (a) 1.5 equiv. NaN3, DMSO, 70–90 °C, 2–5 h (b) 1.1 equiv. 4, glacial acetic acid, 3 drops conc. HCl, rt, 3 h (c) 4 equiv. NaHS, 2 mL MeCN or acetone, few drops DI water, 30 min. | ||
Based on the fact that highly water soluble H2S predominantly (>75%) exists as HS− under physiological conditions, NaHS was utilized as the source of H2S for this study.61 Although the model probe 6a (100 μM) was found to react efficiently (∼7 min) with 5 equivalent of NaHS in a 1:1 or higher ratio of DMSO in phosphate buffer saline (PBS) medium, its utility was greatly limited in highly aqueous medium (<1% DMSO) as the probe precipitates out below 20% DMSO or MeCN in PBS. Additionally, increasing the aqueous fraction of the solvent was accompanied by a significant decrease in the fluorescence intensity of the corresponding amine. Nevertheless, this observation provided the proof of concept for the utility of an aurone scaffold for hydrogen sulfide detection.
To improve solubility with increasing ratios of water to DMSO, we introduced a hydroxyl (–OH) group at C-6 or C-4 of the benzofuranone moiety of the model probe 6a to give aurones 6b and 6c, respectively. As expected, this rendered them highly soluble in largely aqueous media (as low as 5% DMSO in PBS). Unfortunately, it also resulted in significant spectral overlap, limiting the selective excitation of the starting azides and/or the corresponding amine products (Fig. 2 and S1†).
Next, leaving the hydroxyl group intact on the benzofuranone moiety, the effects of substitution in the phenyl ring on the reactivity and electronic properties of the probe molecule were explored. As suggested by Henthorn and Pluth in their mechanistic study of H2S-mediated reduction of aryl azides, the introduction of fluorine was reported to result in roughly a 2.2-fold rate increase for each fluorine substitution.62–65 Utilizing this same strategy, we synthesized both mono- and di-fluoro substituted hydroxylated candidates 6d–6f. Despite their outstanding aqueous solubility, all of them resulted in highly overlapped absorption spectra with their corresponding amines 7d–7f similar to that observed with 6b and 6c. The result was that there was still no excitation window available for the selective excitation of reactant or product to monitor the reaction (Fig. 2). This situation can be attributed to the electron donating effect of the hydroxyl group making the carbonyl group less electron deficient which results in a reduction in the resonance mediated electron donation from the amine.
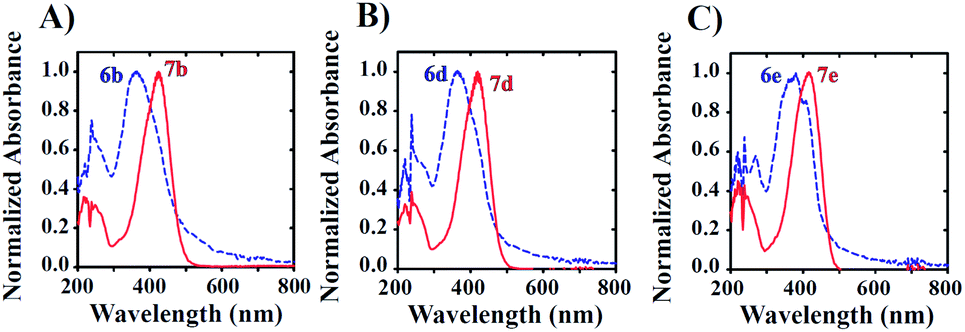 | ||
| Fig. 2 UV-visible spectra illustrating spectral overlap. Samples (50 μM) were prepared in 5% DMSO in PBS (v/v) for the study. | ||
Since, phenyl ring functionalization of aurones did not resolve the issue of spectral overlap of the azido and amino compounds, we decided to introduce fluorine in the benzofuranone moiety. This addition has the potential to increase the electrophilicity of the carbonyl carbon, which in turn will increase the resonance meditated electron donation from amino group. This increase in polarization should impart significant differences in the electronic properties of the two compounds, thereby improving their spectral separation. The impact of fluorine in the benzofuranone moiety on the rate of azide reduction was less clear, but was expected to be modest. As an initial test, 6g and its corresponding amine 7g were synthesized. Interestingly, the introduction of fluorine at C-5 of benzofuranone moiety not only resolved the problem with spectral overlap, but also afforded considerable solubility to the probe in largely aqueous media (as low as 0.5% DMSO in PBS buffer) without the need for any hydroxyl group. This ultimately provided us with the desired turn-on fluorescent probe 6g for the detection of H2S.
Spectral characterization of 6g and 7g
To evaluate its utility as a fluorescent probe, the excitation and emission properties of probe 6g (reactant) and corresponding amine 7g (product) were characterized under varied solvent conditions. Fig. 3 presents excitation and emission spectra collected with either molecule in the presence of 0.5, 50, 90, or 100% v/v DMSO and phosphate buffered saline (PBS) solution. All 6g excitation spectra recorded in DMSO/PBS mixtures exhibit two distinct excitation modes that are not significantly dependent on DMSO/PBS ratio (Fig. 3A). Excitation maxima are observed at 346/394 nm, 362/414 nm, 346/402 nm, and 350/406 nm for conditions of 0.5, 50, 90, or 100% v/v DMSO, respectively (dashed lines in Fig. 3A).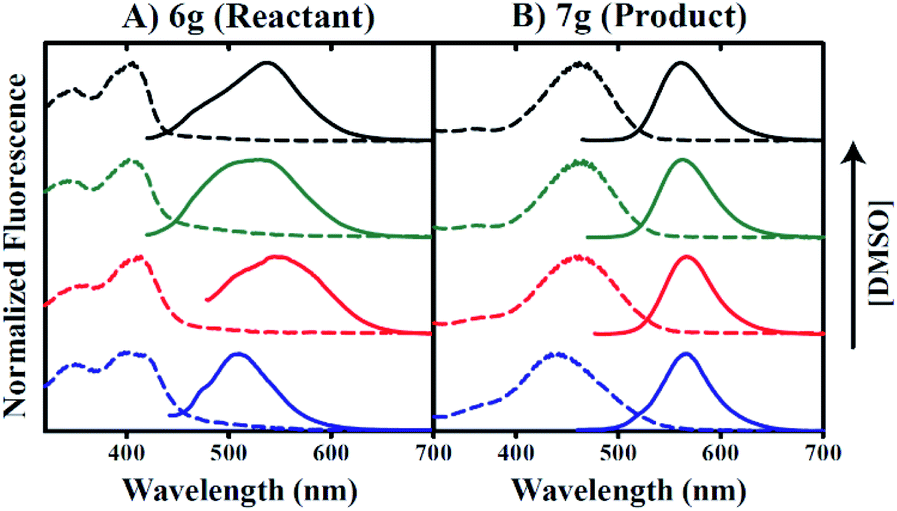 | ||
| Fig. 3 Evaluation of chromophore DMSO-sensitivity. (A and B) Normalized excitation and emission spectra are represented from bottom to top of frame in order of ascending [DMSO]. All spectra are colored blue, red, green, or black to indicate 0.5% v/v, 50% v/v, 90% v/v, or 100% v/v DMSO, respectively. Excitation and emissions spectra are represented as either dashed or solid lines, respectively. Peak maxima are discussed in-text. Panel (A) and (B) represent spectral data corresponding to reactant 6g or product 7g, respectively. | ||
Emission spectra of 6g indicate solvent sensitivity when excited at 405 nm where maximum fluorescence is observed for 0.5% DMSO/99.5% PBS conditions at 510 nm (solid lines in Fig. 3A). This contrasts with the red-shifted 6g emission maxima of 545, 531, and 539 nm for 50%, 90%, and 100% v/v DMSO conditions, respectively. In comparison, maximal 7g absorption occurs at 442 nm in the presence of 0.5% DMSO, whereas excitation maxima equal to 458, 462, and 462 nm are observed for conditions including 50, 90, and 100% v/v DMSO, respectively (Fig. 3B). However, maximal emissions occur at 566, 566, 563, and 561 nm for 0.5, 50, 90, and 100% v/v DMSO, respectively, thereby indicating minimal solvent sensitivity for product species fluorescence. Comparison of the spectra in Fig. 3 indicates that excitation of 6g/7g mixtures using wavelengths in the 440–460 nm range should allow for selective observation of product emission.
Inspection of raw 7g fluorescence intensities as a function of increasing DMSO concentration suggests that an aqueous environment may contribute to quenched product emission. Our data indicate a 33-fold increase in fluorescence intensity for conditions including 90% versus 0.5% v/v DMSO. Thus, the use of 6g as a fluorescent sulfide probe may be limited in biological applications under these conditions. In this context, cationic surfactant cetyltrimethylammonium bromide (CTAB) has been utilized in the literature to enhance both the rate of the reaction and the intensity of the fluorescence.66,67 Based on this precedent, we successfully employed CTAB to enhance the reactivity of probe 6g in highly aqueous medium (0.5% DMSO in PBS).
Fig. 4 indicates that the inclusion of 1 mM CTAB in 0.5% v/v DMSO does not impact 6g excitation properties (dashed lines in Fig. 4A), but instead promotes a red-shift in the apparent emission maximum from 510 nm to 550 nm in the absence versus presence of CTAB respectively. In contrast, 7g excitation properties in 0.5% v/v DMSO are perturbed by the inclusion of CTAB such that maximal absorption shifts from 442 nm to 462 nm in the absence versus presence of surfactant, respectively (Fig. 4B). However, Fig. 4B reveals no impact of CTAB on the wavelength of maximum emission.
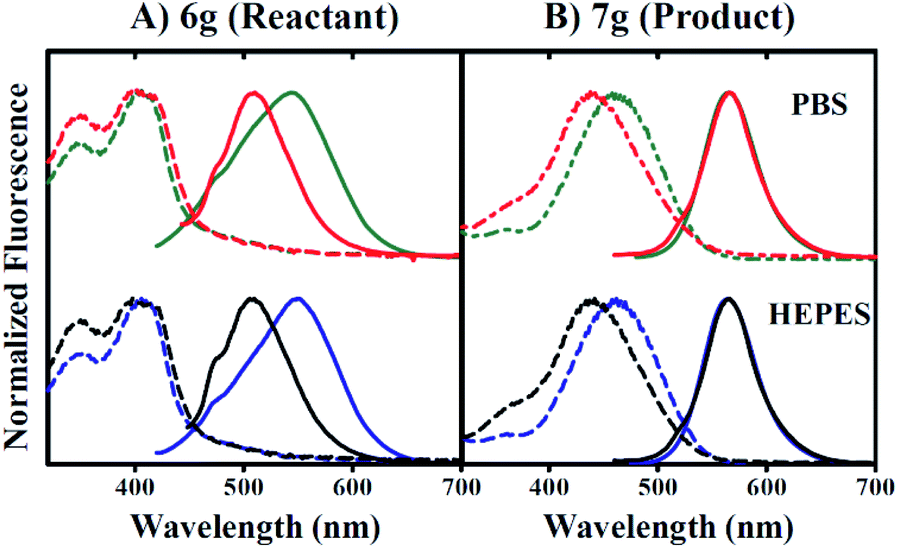 | ||
| Fig. 4 CTAB influences product emission properties. Normalized excitation/emission spectra are presented in the presence of 0.5% v/v DMSO and varied buffer components. Excitation and emission spectra are represented by dashed or solid lines, respectively. Spectra corresponding to conditions including PBS buffer without or with 1 mM CTAB are colored red or green, respectively (top panel). Spectra corresponding to conditions including HEPES buffer without or with 1 mM CTAB are colored black or blue, respectively (bottom panel). Peak maxima are discussed in-text. Panel (A) and (B) represent spectral data corresponding to reactant 6g or product 7g respectively. | ||
Furthermore, raw 7g fluorescence intensities are observed to increase 42-fold for 0.5% v/v DMSO conditions including CTAB versus conditions that do not. Our data also indicate that these trends occur independent of buffer components included in the aqueous fraction since the inclusion of PBS or HEPES buffers yielded identical results. The data presented in Fig. 4A and B collectively suggest that 7g may be utilized in aqueous solution as a fluorescent probe under conditions that include CTAB.
Effect of pH on spectral properties of 6g and 7g
The effect of pH on 6g and 7g spectral properties was also examined. Excitation and emission spectra were collected in PBS buffer previously pH adjusted to pH = 3.0, 4.8, 7.5, 9.2, and 11 in the absence and presence of CTAB as described above. Fig. S2A and B† illustrate that 6g experiences an altered Stokes shift that depends on whether CTAB is included. In the absence of CTAB, we note excitation and emission maxima occur at 424 and 500 nm, respectively, at pH = 3.0. A similar observation is made when the pH is adjusted to 4.8, where excitation and emission maxima occur at 426 and 502 nm, respectively. At pH = 3.0 and 4.8, the Stokes shift is calculated as 76 nm. However, at pH 7.5 and above, we observe a blue-shift, where excitation maxima are observed at 400 nm when determined at pH = 7.5, 9.2, and 11.0. Corresponding emission maxima were observed at 527, 528, and 528 nm for pH = 7.5, 9.2, and 11.0, respectively. The calculated Stokes shift for pH ≥ 7.5 is ∼128 nm. In contrast, when CTAB is included, no effect of pH is observed such that maximal excitation is observed at 404 nm independent of pH. Emission maxima are observed at wavelengths that increase with increasing pH such that maxima are observed at 511 (Stokes shift = 107 nm), 527 (Stokes shift = 123 nm), 547 (Stokes shift = 143 nm), 547, and 550 (Stokes shift = 146 nm) nm for pH = 3.0, 4.8, 7.5, 9.2, and 11.0, respectively. In contrast 7g exhibits pH-independent excitation maxima at 440 and 460 nm when CTAB is absent or present, respectively. The emission maximum for 7g is observed at 565 nm independent of pH, yielding a Stokes shift equal to 125 or 105 depending on the absence or presence of CTAB, respectively. Taken together, we note 7g spectral properties to be largely independent of solution pH. However, we do note an increase in raw emission intensities for 7g when CTAB is included, where mild fluorophore quenching is observed at pH below 4.8. At pH ≥ 4.8, emission intensities are observed to be independent of pH.Dynamic range study
The use of any fluorescent probe requires knowledge of the dynamic range under specific working conditions. As such, we next determined the useful working range of 7g in conditions including either increased DMSO concentrations or 0.5% v/v DMSO/1 mM CTAB. Knowledge of the dynamic range under these separate conditions allows for both biological and non-aqueous applications. Fig. 5A demonstrates that the titration of 7g in 0.5% v/v DMSO, 99.5% v/v PBS, and 1 mM CTAB with excitation at 465 nm yields emission spectra with maxima observed at ∼565 nm. A secondary plot of maximum fluorescence intensity versus [7g] clearly reveals non-linear behavior for concentrations exceeding 20 μM (Fig. 5B). Our data also indicate that fluorescence intensities can be reliably estimated for [7g] as low as 1 μM based on spectra signal-to-noise ratio and deviation from linearity. Thus, the useful dynamic range for 7g in the presence of 0.5% v/v DMSO, 99.5% PBS, and 1 mM CTAB spans from 1 μM to 20 μM.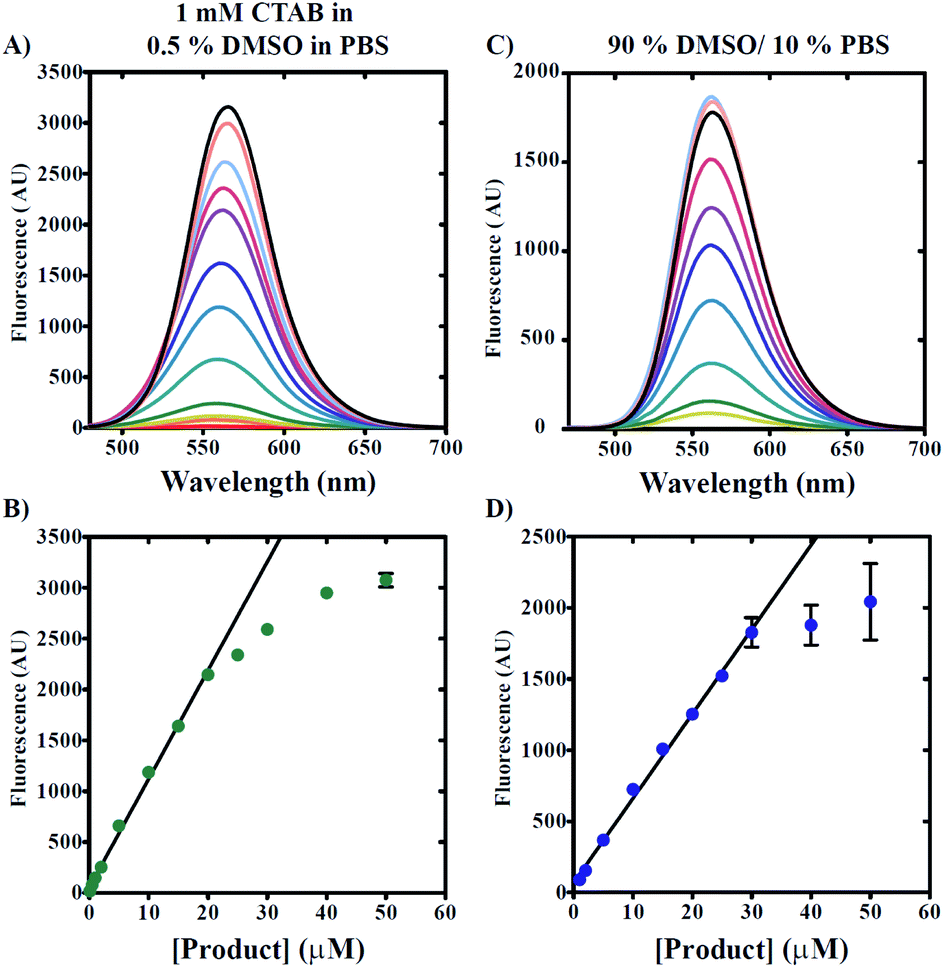 | ||
| Fig. 5 Determination of product 7g range of detection. Product 7g was titrated separately into conditions including either 1 mM CTAB and 0.5% v/v DMSO in PBS (A and B) or 90% v/v DMSO/10% v/v PBS buffer (C and D) with excitation at 465 nm. Emission spectra are represented as solid lines (A and C). Signal intensity observed at 560 nm plotted as a function of [7g] reveals linear range of detection (B and D). All data points represent average values determined from at least three independent experiments. Error bars indicate ± standard deviation. Solid lines are the result of Linear Least Squares (LLS) fits. For conditions with 1 mM CTAB and 0.5% v/v DMSO in PBS (B), LLS fit yields estimates of slope and y-intercept equal to 50 ± 24 (AU) and 107 ± 3 AU μM, respectively. For conditions with 90% v/v DMSO/10% v/v PBS buffer (D), LLS fit yields estimates of slope and y-intercept equal to 68 ± 24 (AU) and 60 ± 1 AU μM, respectively. | ||
Fig. 5C and D reveal a similar trend when measurements are made under conditions including 90% v/v DMSO and 10% v/v PBS solution. However, elevated DMSO concentrations appear to increase the useful linear concentration range such that non-linearity is observed for [7g] greater than 30 μM. We note the observation of decreased reproducibility when 7g concentrations are utilized that exceed 25 μM in 90% v/v DMSO/10% v/v PBS as indicated by the increased standard deviation of mean fluorescence intensities presented in Fig. 5D.
Reaction kinetics and solvent studies
Quantification of sulfide concentration in any sample using “turn-on” fluorescent sensing requires knowledge of kinetic rates of reaction under varied solution conditions. Rate profiles describing the reduction of azide reactant to amine product were performed by the incubation of 20 μM 6g with 100 μM NaHS in either 0.5% v/v DMSO/99.5% PBS/1 mM CTAB or 90% v/v DMSO/10% v/v PBS solvent systems at 25 °C. Samples were excited at 465 nm and emission spectra spanning from 480 to 700 nm were collected at 2 minute intervals. Fig. 6 demonstrates a time-dependent increase in fluorescence intensity for both conditions.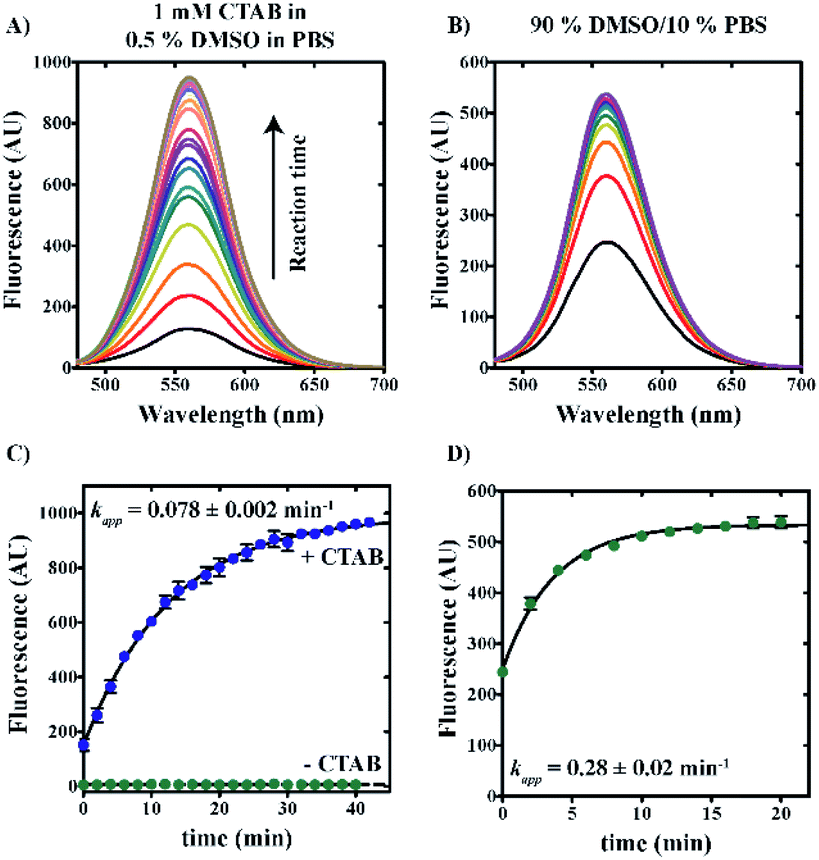 | ||
| Fig. 6 Compound 6g reaction rate constant is dependent on solvent condition. Reaction profiles describing the reduction of azide reactant to amine product by the incubation of compound 6g with 100 μM NaHS in the presence of either 0.5% v/v DMSO in PBS ±1 mM CTAB (A) or 90% v/v DMSO/10% v/v PBS buffer (B). Emission spectra are represented as solid lines and reveal increasing signal intensity as a function of reaction time (A and B). All data points represent average values determined from at least three independent experiments (C and D). Error bars indicate ± standard deviation. Solid lines are the result of nonlinear least squares (NLLS) fits with rate constants indicated. | ||
Fig. 6C shows a plot of maximum fluorescence intensity as a function of time for aqueous reaction conditions including 0.5% v/v DMSO/99.5% v/v PBS in the presence or absence of 1 mM CTAB. In the presence of 1 mM CTAB, reaction completion is achieved within approximately one hour (blue spheres, Fig. 6C). In contrast, no reaction is observed in the absence of CTAB (green spheres, Fig. 6C). By comparison, reactions performed in 90% v/v DMSO/10% v/v PBS are observed to reach completion within 20 minutes (green spheres, Fig. 6D).
Nonlinear least squares (NLLS) analysis of the data shown in Fig. 6C estimates the apparent rate constant, amplitude, and y-intercept as 0.078 ± 0.002 min−1, 842 ± 8 AU, and 149 ± 7 AU, respectively. In comparison, NLLS analysis of reaction data collected in 90% v/v DMSO/10% v/v PBS (Fig. 6D) estimates the apparent rate constant, amplitude, and y-intercept as 0.28 ± 0.02 min−1, 284 ± 7 AU, and 249 ± 6 AU, respectively. These data collectively indicate that reactions performed in a polar aprotic environment occur with increased reaction rates relative to those performed under aqueous conditions.
NLLS analyses of data presented in Fig. 6C and D yield estimates of the y-intercept as nonzero values. Reference to Fig. 4 illustrates that excitation at 465 nm of a mixture of 6g and 7g may yield an apparent fluorescence intensity that contains contributions from both reactant and product. Moreover, no product could be present at time zero. Therefore, we interpret the observation of a nonzero y-intercept to reflect the contribution of reactant emission to the overall observed signal.
To determine whether contaminating reactant emission leads to errors in our estimation of reaction rate, we performed a series of reactions using independent excitation wavelengths wherein 20 μM 6g was incubated with 100 μM NaHS in 0.5% v/v DMSO/99.5% PBS/1 mM CTAB. Samples were excited at 465, 480, and 500 nm with spectra collected at 2 minute intervals as described above. Fluorescence time courses are presented in Fig. 7A, which qualitatively demonstrate a decrease in apparent amplitude and y-intercept with increasing excitation wavelength. Fig. 7B predicts no significant dependence in the apparent rate constant describing product formation on excitation wavelength. NLLS analyses of the data presented in Fig. 7A yield estimates of the rate constant that fluctuate about a mean value equal to 0.075 ± 0.006 min−1.
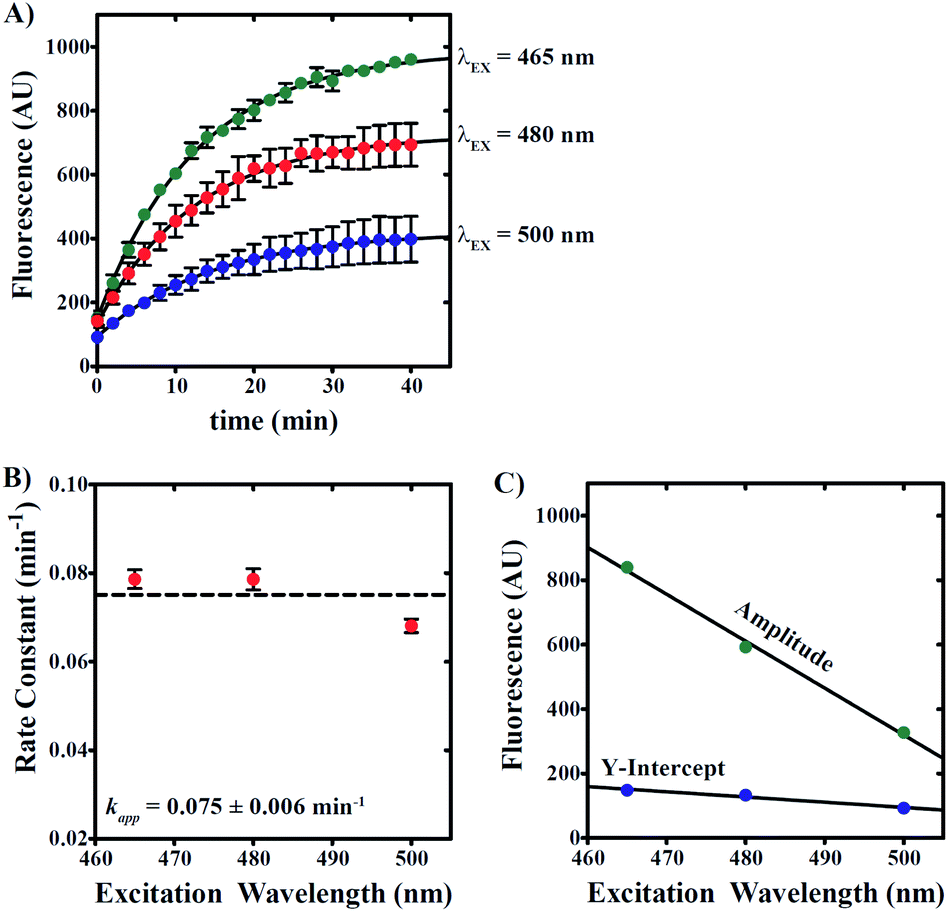 | ||
| Fig. 7 Compound 6g reaction rate constant is independent of excitation wavelength. (A) Kinetic time courses were collected using excitation wavelengths equal to 465 (green spheres), 480 (red spheres), and 500 nm (blue spheres). Reactions were performed by incubating 20 μM 6g with 100 μM NaHS in 1 mM CTAB and 0.5% v/v DMSO in PBS. Reaction progress was monitored by fluorescence intensity observed at 560 nm. Solid lines are the result of nonlinear least squares (NLLS) fits to a single-exponential function. (B) First-order rate constants plotted as a function of excitation wavelength with average apparent rate constant indicated. (C) estimates of amplitude (green spheres) and y-intercept (blue spheres) describing time courses in (A) obtained from NLLS analyses. Solid lines are the result of LLS fits with slopes equal to −14.6 ± 0.9 and −1.6 ± 0.3 AU nm−1, respectively. All data points represent average values determined from at least three independent experiments. Error bars indicate ± standard deviation. | ||
In addition, the apparent amplitude and y-intercept are observed to linearly decrease as excitation wavelength is increased, which is an observation consistent with the excitation of sample at non-maximal excitation wavelengths for both reactant and product (Fig. 7C). Given that the y-intercept represents signal derived at time zero prior to any product formation, we interpret the observation of a y-intercept that decreases with increasing excitation wavelength to suggest that longer excitation wavelengths favor decreased background signal associated with reactant emission. Taken together, the data presented in Fig. 7 collectively indicates that reactant contribution to overall apparent fluorescence signal does not interfere in the estimation of reaction rate constants.
Moreover, knowledge of the dependence of time course y-intercept on excitation wavelength allows for baseline correction to remove reactant signal contributions, thereby allowing for conversion of fluorescence intensity values into concentration units. By subtracting baseline emissions from subsequent time points, the resulting emission values can be readily converted into concentration units if a standard curve relating product concentration to emission value is available. Fig. 8 highlights this ability for time courses collected in the presence of 1 mM CTAB in 0.5% DMSO in PBS or 90% DMSO/10% PBS. Though the estimated rate constants are unaltered, Fig. 8 now readily allows for estimation of hydrogen sulphide concentration in real-time.
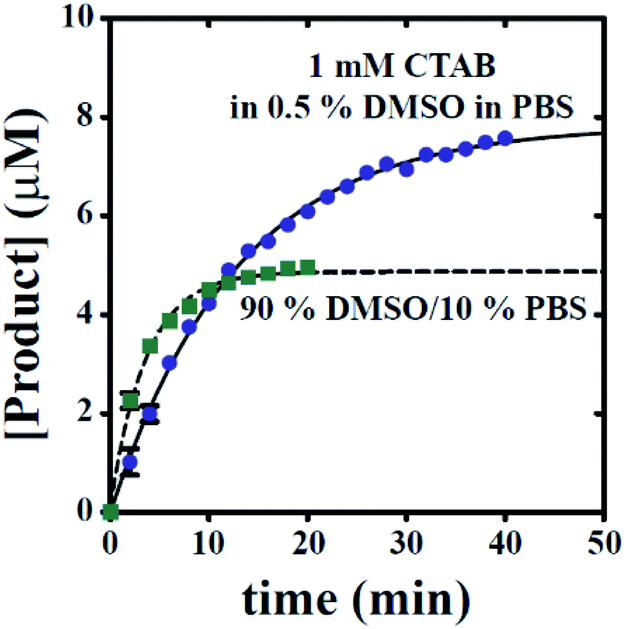 | ||
| Fig. 8 All time courses can be converted into plots of [Product] versus time. Blue spheres represent conditions including 1 mM CTAB, 0.5% DMSO in PBS (condition A). Green squares represent conditions including 90% DMSO and 10% PBS (condition B). The solid line represents the results of a NLLS fit with A, kapp, and b estimated as 7.9 ± 0.1, 0.08 ± 0.002, and −0.03 ± 0.07, respectively. The dashed line represents the results of a NLLS fit with A, kapp, and b estimated as 4.8 ± 0.1, 0.28 ± 0.02, and 0.1 ± 0.1, respectively. Error bars indicate ± standard deviation. | ||
Selectivity for sulfide studies
Any use of 6g as a molecular probe will require knowledge of its reaction specificity. To evaluate reaction specificity, a series of fluorescence time courses were collected using the same methodology as described above. In place of NaHS, three potential sulfide-donors were employed: L-cysteine, reduced glutathione, and 2-mercaptoethanol. All emission spectra reflect the reaction of 100 μM sulfide-donor with 20 μM 6g in 0.5% v/v DMSO, 99.5% v/v PBS, and 1 mM CTAB. As expected, NaHS yields the observation of significant product emission at λEM = 560 nm (Fig. 9). In contrast, the inclusion of either L-cysteine or reduced glutathione yielded no observed product formation. Fig. 9 illustrates that a mild increase in emission was observed when 2-mercaptoethanol was utilized as a sulfide-donor. However, 2-mercaptoethanol reactivity with 6g is much slower on the timescale associated with NaHS:6g reaction. Taken together, we conclude that 6g chemical reactivity is selective for free sulfide.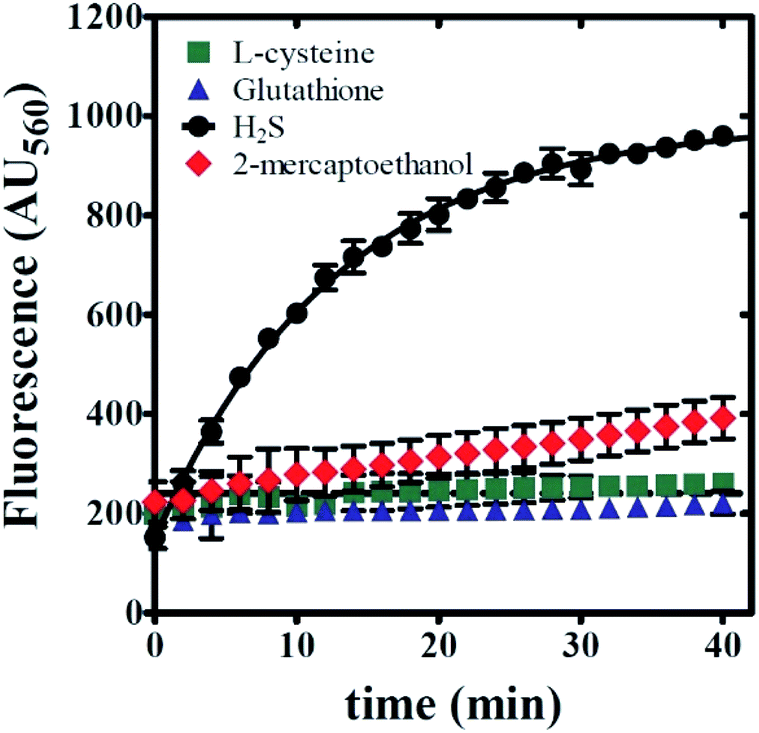 | ||
| Fig. 9 Compound 6g displays chemical reactivity that is selective for sulfide. Kinetic time courses were collected using an excitation wavelength equal to 465 nm. Reactions were performed by incubating 20 μM 6g with 100 μM sulfide-donor in 1 mM CTAB and 0.5% v/v DMSO in PBS. Sulfide-donors examined include L-cysteine (green squares), glutathione (blue triangles), NaHS (black circles), and 2-mercaptoethanol (red diamonds). Reaction progress was monitored by fluorescence intensity observed at 560 nm. Solid lines are the result of nonlinear least squares (NLLS) fits to a single-exponential function. All data points represent average values determined from at least three independent experiments. Error bars indicate ± standard deviation. | ||
Sensing mechanism
In this study the turn-on probe 6g, in the presence of HS− undergoes an irreversible chemical transformation to amine 7g thereby transducing signals by converting the molecular recognition phenomenon into an intense fluorescence signal.Based on the mechanistic study by Henthorn and Pluth, this reaction is suggested to proceed via nucleophilic attack of hydrogen sulfide anion (HS−) on the terminal electrophilic nitrogen of azido group forming an azidothiol intermediate species which on subsequent HS− intermolecular attack is converted to the respective amine 7g.64 The amine subsequently undergoes a spontaneous internal charge transfer (ICT) via resonance to its corresponding highly fluorescent zwitterionic form as depicted in Fig. 10A. The presence of CTAB may further stabilize the charged species via electrostatic interactions. This consequently leads to the stabilization of the high quantum yield state resulting in an enhanced emission intensity as evident in Fig. 6A, C, and S3.† It has been suggested that CTAB works by changing the local concentration of the reactants and the pH value of the reaction mixture. The CTAB micelles increase the local concentration of anions including OH− and HS− around it (Fig. 10B), which increases the local pH of the solution. This in turn decreases the [H2S]/[HS−] ratio thereby contributing an overall higher HS− concentration around the micelles.66 The effect of pH on dissociation of H2S in an aqueous medium is summarized in Fig. 10C.61 At lower pH (∼3) the fluorescence intensity of the product is expected to decrease due to protonation of the amine which shuts down the ICT process (Fig. S2E and S3†). This is apparent from Fig. S3† which clearly demonstrates how the emission intensity of the product 7g is affected by the pH although UV-absorption remains unaffected. In this overall phenomenon, CTAB micelles serve as a scaffold to recruit HS− and probe 6g in the vicinity of each other, thereby significantly increasing the reaction rate (Fig. 6A and C) along with fluorescent intensities.
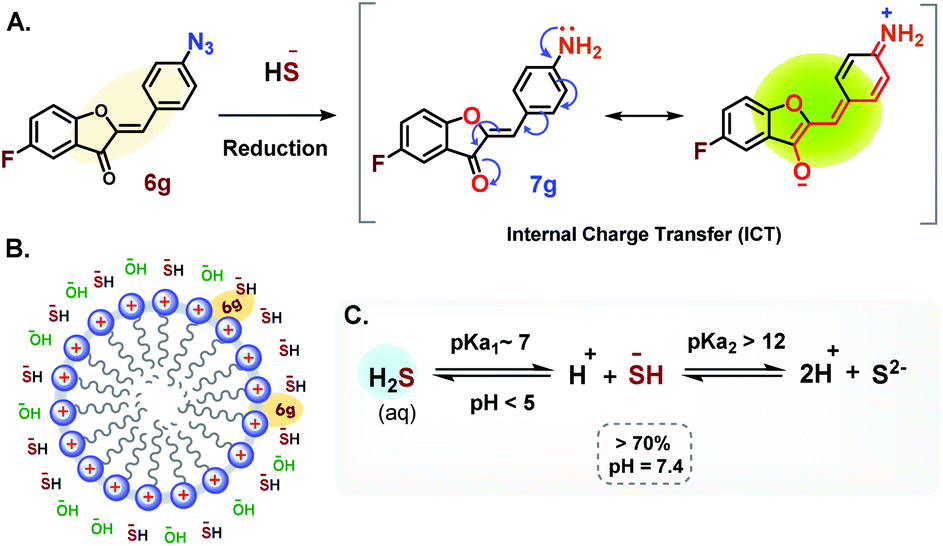 | ||
| Fig. 10 Sensing mechanism of 6g. (A) Reaction scheme showing the reduction of azide (–N3) to the corresponding amine. The resonance responsible for the fluorescence is depicted in parenthesis. (B) Schematic representation of CTAB micelle and its role in recruiting and changing the local concentration of 6g and HS−. (C) Reaction scheme representing the dissociation of H2S in aqueous medium. | ||
Conclusions
In short, an aurone-based sensor for hydrogen sulphide has been developed that exhibits good sensitivity and selectivity. Further, the development of this probe molecule has demonstrated the strength of the aurone framework in terms of the ease with which new compounds can be prepared to modify the reactivity and physical properties (solubility in this case) to fit the demands of various sensing applications. Based upon these results, future optimized compounds can be designed and prepared for a wide range of environmental as well as biological applications. Given the ease and flexibility in the synthesis of the aurone framework, many additional sensing applications can be envisioned, particularly applying the lessons learned in this project. Efforts to this end are underway and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the Molecular Biosciences program at Middle Tennessee State University.Notes and references
- Z. Guo, G. Chen, G. Zeng, Z. Li, A. Chen, J. Wang and L. Jiang, Analyst, 2015, 140, 1772–1786 RSC.
- J. Li, C. Yin and F. Huo, RSC Adv., 2015, 5, 2191–2206 RSC.
- F. Yu, X. Han and L. Chen, Chem. Commun., 2014, 50, 12234–12249 RSC.
- V. S. Lin, A. R. Lippert and C. J. Chang, Methods Enzymol., 2015, 554, 63–80 CAS.
- B. Peng and M. Xian, Methods Enzymol., 2015, 554, 47–62 CAS.
- S. L. M. Rubright, L. L. Pearce and J. Peterson, Nitric Oxide, 2017, 71, 1–13 CrossRef.
- G. K. Kolluru, X. Shen, S. C. Bir and C. G. Kevil, Nitric Oxide, 2013, 35, 5–20 CrossRef CAS.
- A. F. Perna and D. Ingrosso, Nephrol. Dial. Transplant., 2012, 27, 486–493 CrossRef CAS.
- P. Kamoun, M.-C. Belardinelli, A. Chabli, K. Lallouchi and B. Chadefaux-Vekemans, Am. J. Med. Genet., Part A, 2003, 116, 310–311 CrossRef.
- S. Fiorucci, E. Antonelli, A. Mencarelli, S. Orlandi, B. Renga, G. Rizzo, E. Distrutti, V. Shah and A. Morelli, Heptaology, 2005, 42, 539–548 CrossRef CAS.
- For a recent example, see: Y. Chen, X. Shang, C. Li, Z. Xue, H. Chen, H. Wu and T. Wang, Sci. Rep., 2018, 8, 16159 CrossRef.
- S. K. Bae, H. Heo, D. J. Choi, D. Sen, E.-H. Joe, B. R. Cho and H. M. Kim, J. Am. Chem. Soc., 2013, 135, 9915–9923 CrossRef CAS.
- T. S. Bailey and M. D. Pluth, J. Am. Chem. Soc., 2013, 135, 16697–16704 CrossRef CAS.
- J. Katla and S. Kanvah, Photochem. Photobiol. Sci., 2018, 17, 42–50 RSC.
- Y.-S. Zeng, R.-C. Gao, T.-W. Wu, C. Cho and K.-T. Tan, Bioconjugate Chem., 2016, 27, 1872–1879 CrossRef CAS.
- S.-A. Choi, C. S. Park, O. S. Kwon, H.-K. Giong, J.-S. Lee, T. H. Ha and C.-S. Lee, Sci. Rep., 2016, 6, 26203 CrossRef CAS.
- Y. Guo, T. Zeng, G. Shi, Y. Cai and R. Xie, RSC Adv., 2014, 4, 33626–33628 RSC.
- K. Xiang, Y. Liu, C. Li, B. Tian, T. Tong and J. Zhang, Dyes Pigm., 2015, 123, 78–84 CrossRef CAS.
- Y. Jiang, Q. Wu and X. Chang, Talanta, 2014, 121, 122–126 CrossRef CAS.
- R. Wang, F. Yu, L. Chen, H. Chen, L. Wang and W. Zhang, Chem. Commun., 2012, 48, 11757–11759 RSC.
- H. Peng, Y. Cheng, C. Dai, A. L. King, B. L. Predmore, D. J. Lefer and B. Wang, Angew. Chem., Int. Ed., 2011, 50, 9672–9675 CrossRef CAS.
- T. Ozdemir, F. Sozmen, S. Mamur, T. Tekinay and E. U. Akkaya, Chem. Commun., 2014, 50, 5455–5457 RSC.
- X. Qu, C. Li, H. Chen, J. Mack, Z. Guo and Z. Shen, Chem. Commun., 2013, 49, 7510–7512 RSC.
- F. Hou, L. Huang, P. Xi, J. Cheng, X. Zhao, G. Xie, Y. Shi, G. Cheng, X. Yao, D. Bai and Z. Zeng, Inorg. Chem., 2012, 51, 2425–2460 CrossRef.
- A. A. Abd-Elaal, S. M. Tawfik and Y.-I. Lee, J. Mol. Liq., 2017, 247, 35–42 CrossRef CAS.
- J. Lv, F. Wang, J. Qiang, X. Ren, Y. Chen, Z. Zhang, Y. Wang, W. Zhang and X. Chen, Biosens. Bioelectron., 2017, 48, 96–100 CrossRef.
- X. Cao, W. Lin, K. Zheng and L. He, Chem. Commun., 2012, 48, 10529–10531 RSC.
- D. Maity, A. Raj, P. K. Samanta, D. Karthigeyan, T. K. Mundu, S. K. Pati and T. Govindaraju, RSC Adv., 2014, 4, 11147–11151 RSC.
- S. Das and P. Sahoo, Sens. Actuators, B, 2019, 291, 287–292 CrossRef CAS.
- Y. Chen, X. Shang, C. Li, Z. Xue, H. Chen, H. Wu and T. Wang, Sci. Rep., 2018, 8, 1–9 CrossRef.
- Y. Wang, L. Yang, X.-R. Wei, R. Sun, Y.-J. Xu and J.-F. Ge, Anal. Methods, 2018, 10, 5291–5296 RSC.
- G. Asaithambi and V. Periasamy, J. Photochem. Photobiol., A, 2019, 369, 97–105 CrossRef CAS.
- H. Wang, X. Wu, S. Yang, H. Tian, Y. Liu and B. Sun, Dyes Pigm., 2019, 160, 757–764 CrossRef CAS.
- H. Wang, J. Wang, S. Yang, H. Tian, Y. Liu and B. Sun, Food Chem., 2018, 257, 150–154 CrossRef CAS.
- J. Cui, T. Zhang, Y.-Q. Sun, D.-P. Li, J.-T. Liu and B.-X. Zhao, Sens. Actuators, B, 2016, 232, 705–711 CrossRef CAS.
- B. Peng, W. Chen, C. Liu, E. W. Rosser, A. Pacheco, Y. Zhao, H. C. Aguilar and M. Xian, Chem.–Eur. J., 2014, 20, 1010–1016 CrossRef CAS.
- D. Maity, A. Raj, P. K. Samanta, D. Karthigeyan, T. K. Kundu, S. K. Pati and T. Govindaraju, RSC Adv., 2014, 4, 11147–11151 RSC.
- J. Chao, M. Xu, Y. Zhang, F. Huo, Y. Liu, X. Wang and C. Yin, Spectrochim. Acta, Part A, 2019, 214, 227–232 CrossRef CAS.
- H. Zhang, J. Chen, H. Xiong, Y. Zhang, W. Chen, J. Sheng and X. Song, Org. Biomol. Chem., 2019, 17, 1436–1441 RSC.
- J.-B. Li, Q. Wang, H.-W. Liu, L. Yuan and X.-B. Zhang, Chem. Commun., 2019, 55, 4487–4490 RSC.
- F. Chen, D. Han, H. Liu, S. Wang, K.-B. Li, S. Zhang and W. Shi, Analyst, 2018, 143, 440–448 RSC.
- X. Shang, J. Li, Y. Feng, H. Chen, W. Guo, J. Zhang, T. Wang and X. Xu, Front. Chem., 2018, 6, 202 CrossRef.
- S. K. Patra, S. K. Sheet, B. Sen, K. Aguan, D. R. Roy and S. Khatua, J. Org. Chem., 2017, 82, 10234–10246 CrossRef CAS.
- Y. Qian, B. Yang, Y. Shen, Q. Du, L. Lin, J. Lin and H. Zhu, Sens. Actuators, B, 2013, 182, 498–503 CrossRef CAS.
- X. Hou, X. Guo, Z. Luo, H. Zhao, B. Chen, J. Zhao and J. Wang, Anal. Methods, 2014, 6, 3223–3226 RSC.
- R. Haudecoeur and A. Boumendjel, Curr. Med. Chem., 2012, 19, 2861–2875 CrossRef CAS.
- C. Zwergel, F. Gaascht, S. Valente, M. Diederich, D. Bagrel and G. Kirsch, Nat. Prod. Commun., 2012, 7, 389–394 CrossRef CAS.
- A. V. Popova, S. P. Bondarenko and M. S. Frasinyuk, Chem. Heterocycl. Compd., 2019, 55, 285–299 CrossRef CAS.
- C. Lee, E. Chew and M. Go, Eur. J. Med. Chem., 2010, 45, 2957–2971 CrossRef CAS.
- R. S. Varma and M. Varma, Tetrahedron Lett., 1992, 33, 5937–5940 CrossRef CAS.
- I. Hawkins and S. T. Handy, Tetrahedron, 2013, 69, 9200–9204 CrossRef CAS.
- A. T. Geissman and J. B. Harborne, J. Am. Chem. Soc., 1955, 77, 4622–4624 CrossRef.
- N. Shanker, O. Dilek, M. Mukherjee, D. W. McGee and S. L. Bane, J. Fluoresc., 2011, 21, 2173–2184 CrossRef CAS.
- Y. Xue, Y. Dou, L. An, Y. Zheng and Y. Liu, RSC Adv., 2016, 6, 7002–7010 RSC.
- C. Espinosa-Bustos, D. Coretes-Arriagada, M. A. Soto-Arriaza, J. Robinson-Duggon, N. Pizarro, A. R. Cabrera, D. Ruentealba and C. O. Salas, Photochem. Photobiol. Sci., 2017, 16, 1268–1276 RSC.
- K. Munoz-Becerra, N. Villegas-Escobar, C. Zuniga-Loyola, D. Cortes-Arriagada and A. Toro-Labbe, Mol. Phys., 2019, 117, 1451–1458 CrossRef CAS.
- M. Zhang, Y.-T. Bao, W. Yang, H.-F. Xiao, Z.-X. Han, X. Wu and L. Yang, J. Heterocycl. Chem., 2018, 55, 1130–1135 CrossRef CAS.
- H. Chen, Y. Sun, C. Zhou, D. Cao, Z. Liu and L. Ma, Spectrochim. Acta, Part A, 2013, 116, 389–393 CrossRef CAS.
- H. E. Gottlieb and V. Lotlyr, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS.
- A. Kafle, S. Yossef and S. T. Handy, Tetrahedron Lett., 2020, 61, 151899 CrossRef CAS.
- Q. Li and J. R. Lancaster Jr, Nitric Oxide, 2013, 35, 21–34 CrossRef CAS.
- Z. Zhu, Y. Li, C. Wei, X. Wen and Z. Xi, Chem.–Asian J., 2016, 11, 68–71 CrossRef CAS.
- C. Wei, R. Wang, L. Wei, Z. Li, Z. Xi and L. Yi, Chem.–Asian J., 2014, 9, 3586–3592 CrossRef CAS.
- J. Zhang, Y. Gao, X. Kang, Z. Zhu, Z. Wang, Z. Xi and L. Yi, Org. Biomol. Chem., 2017, 15, 4212–4217 RSC.
- H. A. Henthorn and M. D. Pluth, J. Am. Chem. Soc., 2015, 137, 15330–15336 CrossRef CAS.
- H. Tian, J. Qian, H. Bai, Q. Sun, L. Zhang and W. Zhang, Anal. Chim. Acta, 2013, 768, 136–142 CrossRef CAS.
- J. Zhang and W. Guo, Chem. Commun., 2014, 50, 4214–4217 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08802a |
| This journal is © The Royal Society of Chemistry 2020 |