
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencecheML.io: an online database of ML-generated molecules†
Rustam Zhumagambetov a,
Daniyar Kazbek‡
a,
Mansur Shakipov‡a,
Daulet Maksuta,
Vsevolod A. Peshkov*b and
Siamac Fazli*a
a,
Daniyar Kazbek‡
a,
Mansur Shakipov‡a,
Daulet Maksuta,
Vsevolod A. Peshkov*b and
Siamac Fazli*a
aDepartment of Computer Science, School of Engineering and Digital Sciences, Nazarbayev University, Nur-Sultan, Kazakhstan. E-mail: siamac.fazli@nu.edu.kz
bDepartment of Chemistry, School of Sciences and Humanities, Nazarbayev University, Nur-Sultan, Kazakhstan. E-mail: vsevolod.peshkov@nu.edu.kz
First published on 22nd December 2020
Abstract
Several recent ML algorithms for de novo molecule generation have been utilized to create an open-access database of virtual molecules. The algorithms were trained on samples from ZINC, a free database of commercially available compounds. Generated molecules, stemming from 10 different ML frameworks, along with their calculated properties were merged into a database and coupled to a web interface, which allows users to browse the data in a user friendly and convenient manner. ML-generated molecules with desired structures and properties can be retrieved with the help of a drawing widget. For the case of a specific search leading to insufficient results, users are able to create new molecules on demand. These newly created molecules will be added to the existing database and as a result, the content as well as the diversity of the database keeps growing in line with the user's requirements.
1 Introduction
1.1 Motivation
In the 21st century, humankind will face many challenges in science and biomedicine that will require extensive discovery of new molecules and materials.1–5 When setting new goals associated with molecular design and synthesis, one should keep in mind that the fraction of “chemical space” explored to date is practically infinitesimal – less than one part of 105.6 At the same time, the existence of vast unexplored molecular horizons suggests that future major scientific tasks could be solved by finding optimal paths to the uncharted areas of chemical space and efficient ways to populate them.While the discovery of particular molecules, for example penicillin, can have a monumental impact on human society, unfortunately, the creation of novel drugs remains a very complex problem. Making and testing new compounds is a costly and time-consuming process since the number of potential candidates is overwhelming. A standard drug discovery campaign involves two very tedious processes that are high-throughput screening of available/accessible libraries of small molecules and subsequent hit-to-lead optimization. Considering that the “small molecule universe” (SMU), the set of all synthetically feasible organic molecules of 500 Da molecular weight or less, is estimated to contain over 1060 structures,6,7 an exhaustive search for active hits becomes fairly impractical.
Thus, it is desirable to develop appropriate computational tools to narrow down this enormous area of search. Therefore, virtual screening has become an increasingly popular strategy to mine for promising molecules among millions of existing and billions of virtual molecules.8–10 In the ideal case of computer-aided de novo drug design, the computer algorithms have to (i) create molecules, (ii) range them and assess their feasibility, (iii) perform conformational analysis, and (iv) identify the molecules, featuring specific structural properties that are responsible for the affinity towards certain biological receptors.11,12
Recently, computational methods have been well adopted by both chemical and pharmaceutical industries. The creation and development of mathematical tools, such as Machine Learning (ML) and in particular Deep Learning (DL) algorithms in combination with increased computational resources at a reduced price, contribute to further advances within the fields of materials design, drug discovery, and beyond.13–17
Therefore, we set a goal to implement, validate, and compare the molecular outputs of a number of recently established ML algorithms for de novo molecule generation. As a result, we created a unified database of virtual molecules in browse-able format – cheML.io.18 The resulting library has been incorporated into a web-page with a built-in drawing widget allowing to conduct substructure and similarity searches. In addition, a number of calculated molecular properties, such as molecular weight, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, number of H-acceptors/donors, and number of rotatable bonds, among others are accessible for each database member. Thus, researchers across all domains of chemical and biological sciences can witness how the rapidly growing field of ML technology can assist in the task of hit identification.19 Not only does our framework offer the possibility of retrieving the whole set of ML-generated molecules and employ them for virtual screening campaigns, but also new molecules can be generated on demand in a structural similarity driven fashion.
P, number of H-acceptors/donors, and number of rotatable bonds, among others are accessible for each database member. Thus, researchers across all domains of chemical and biological sciences can witness how the rapidly growing field of ML technology can assist in the task of hit identification.19 Not only does our framework offer the possibility of retrieving the whole set of ML-generated molecules and employ them for virtual screening campaigns, but also new molecules can be generated on demand in a structural similarity driven fashion.
1.2 A general introduction to machine learning
Generative machine learning frameworks for the creation of molecular libraries can be roughly classified into three categories and are based on autoencoders,20 recurrent neural networks (RNNs)21 and generative adversarial networks (GANs).22The idea of autoencoders can be traced back to the 80s.20,23 Autoencoders are neural networks that consist of an encoder and a decoder that tries to reconstruct the input. The encoder transforms the input into a compressed vector representation and the decoder attempts to recover the input from this compressed representation. A hidden layer with a limited number of nodes between the encoder and decoder represents the minimal amount of information that is needed to decode the original input. Such architectures can be used for denoising, dimensionality reduction and have more recently also been applied for drug discovery.24
Recurrent neural networks have been studied for more than 30 years.21 RNNs consist of several nodes that form a directed graph. In addition to processing input, they also receive their earlier outputs as an input. The output is, therefore, recurring as input in every time step. Applications of RNNs encompass data domains, where input data is “sequentially connected”, like natural language processing, music generation, text translation, automatic generation of image captions, among others.
Generative adversarial networks are a recently established framework for estimating generative models via an adversarial process.22 These types of models have been described as being “the most interesting idea in the last 10 years in Machine Learning” by Yann LeCun. Since their original publication in 2015, they have sparked a huge interest of the scientific community and led to a large number of interesting applications, such as image and video generation,25–27 music generation,28 tumor detection,29 generation and design of DNA30 as well as novel approaches for computational chemistry,31,32 namely the generation of new molecules with desired properties. GANs are designed as a zero sum game between two players (i.e. machine learning models): the discriminator D and the generator G. An artificial conflict is created between the two players that forces both players to get better throughout the game. D is a deep neural network that acts as a classifier. It looks at some input and will decide, whether this input is real or fake. The generator will take random noise and transforms it to produce outputs that resemble the training data. During the early stages of the game, the generator will not produce realistic outputs and the discriminator is trained to reject these kinds of images. However, G is trained to generate outputs that will fool D into believing his outputs are real. In terms of game theory, a Nash equilibrium is reached, when the generator has recovered the training distribution. At the Nash equilibrium, the best D can do is guess randomly and G can perfectly reproduce the training data. Since their original publication, there has been tremendous activity within this area of ML.
1.3 Digital representations of molecules
In order to apply machine learning tools for molecular generation, a means of encoding the molecules into a suitable digital representation is required.33 First, molecular structures need to be converted to line notations such as simplified molecular input line entry system (SMILES), Wiswesser Line Notation (WLN), or SYBYL Line Notation (SLN) formats.34,35 Then, these line notations are transformed into digital representations such as numerical vectors or graphs. Below we outline two common approaches for converting SMILES notations into the input for the machine learning algorithms.1.4 Overview of machine learning methods for molecular generation
To populate the database with ML-generated virtual molecules, we have implemented several machine learning frameworks. As we have mentioned previously, existing machine learning methods for molecular generation can be roughly divided into three major categories: GAN-based methods, autoencoder-based methods, and RNN-based methods. Excellent reviews providing comprehensive analysis of all available methods to date have recently appeared in the literature.14,15 Below we provide a brief description of several machine learning frameworks that we have utilized to populate our database of ML-generated molecules. A graphical representation of all considered methodologies, namely Objective Reinforced Generative Adversarial Network40 (ORGAN), Objective Reinforced Generative Adversarial Network for Inverse-Design Chemistry32 (ORGANIC), Conditional Diversity Network24 (CDN), Variational Autoencoder with Multilayer Perceptron41 (ChemVAE), Grammar Variational Autoencoder42 (GrammarVAE), Conditional Variational Autoencoder37 (CVAE), Recurrent Neural Networks43 (RNN), Junction Tree Variational Autoencoder39 (JT-VAE), a CycleGAN44 based model45 (MolCycleGAN) and Semi Supervised Variational Autoencoder46 (SSVAE), can be seen in Fig. 1.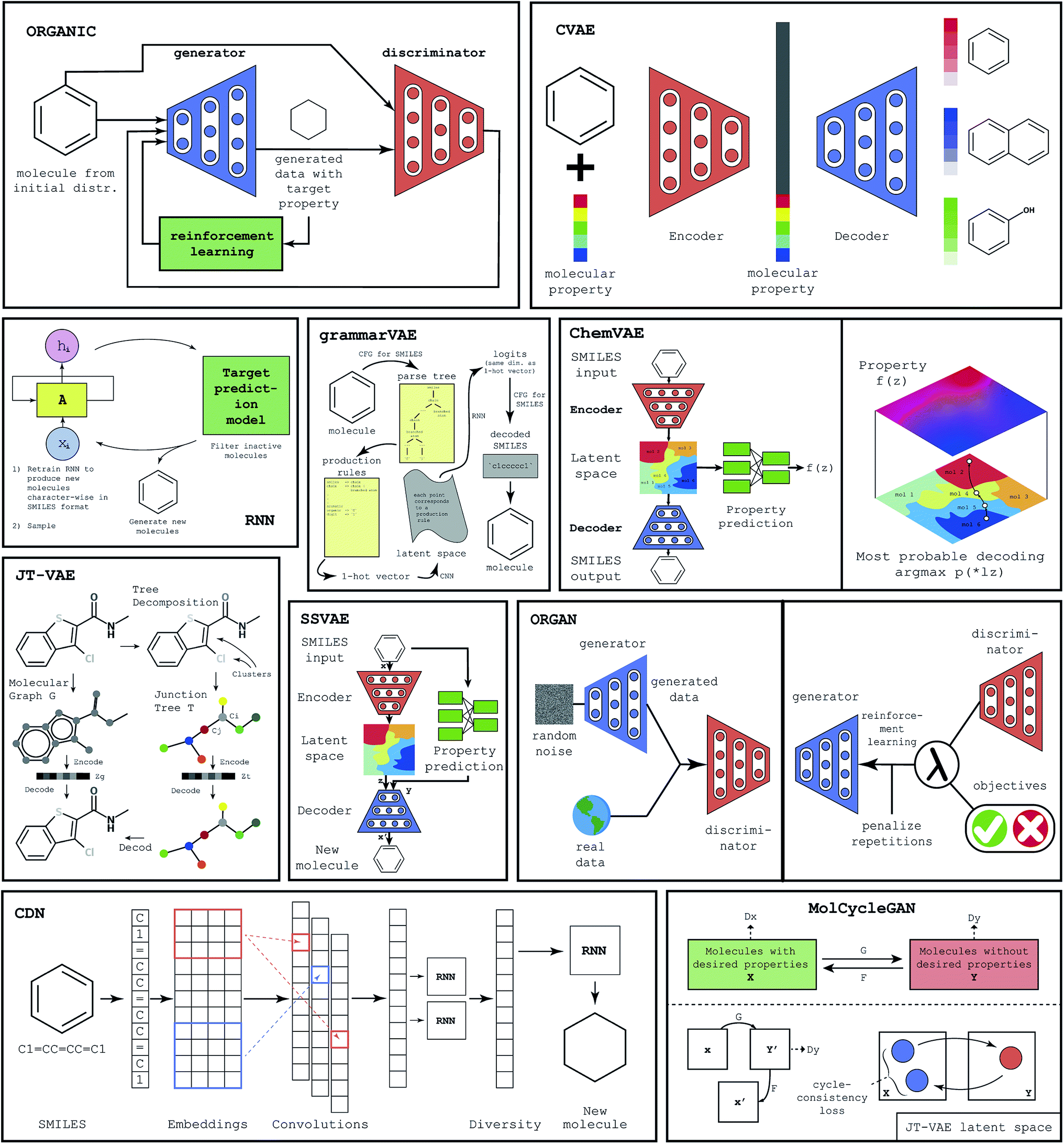 | ||
| Fig. 1 The graphical representations of machine learning algorithms used to create the database. | ||
ChemVAE is an autoencoder with a multilayer perceptron that is used for property prediction. While the autoencoder is employed to learn the latent space of valid molecules, the multilayer perceptron is used to generate molecules with desired properties. Trained jointly with the autoencoder, the perceptron organizes the latent space, grouping the molecules with similar property values in the same latent region. This allows the sampling of molecules with desired properties.
While methods like ORGAN or ORGANIC use a GAN and RNN to generate sequence data like the SMILES molecule representation, GrammarVAE attempts to avoid learning the syntax of the SMILES. Instead of having a GAN learn the syntax of the SMILES format, GrammarVAE uses the fact that SMILES can be represented by context-free grammar and learns the grammar rules, thus avoiding the generation of molecules that are syntactically invalid. To do this GrammarVAE calculates the parsing tree of a molecule, then converts it into one-hot-encoding, having the variational autoencoder learn the sequence of applied grammar rules, rather than individual characters.
JT-VAE deviates from the traditional approach to molecule generation. Instead of using the SMILES representation of the molecule, JT-VAE utilizes direct graph representation of the molecule. JT-VAE builds new molecules by using subgraphs of the old ones. While other methods often use atom combination approach, the JT-VAE uses component combination. Combination of the graph representation and the variational autoencoder almost always yields valid molecules. CVAE is aimed to generate molecules with several desired properties. It was observed that optimization for one property may unintentionally change other properties. In order to counter this effect, CVAE is designed as an artificial neural network that is suitable for optimization of multiple properties. To achieve this goal CVAE uses a conditional vector for both encoding and decoding. Such an architecture allows for the incorporation of properties into the latent space.
SSVAE is a semi supervised model that has advantages when dealing with datasets where only a part of the dataset is labeled with properties. It consists of 3 bi-directional RNNs, which are used for encoding, decoding, and predicting. In this model, the property prediction and molecule generation are combined in a single network. The novel molecules are decoded from the latent space, which is the product of the trained encoder.
ORGANIC is a framework for the generation of novel molecules, which is based on ORGAN. It is a more chemistry oriented version of ORGAN. The limitation of the ORGANIC is that it has an unstable output of molecules. The range of invalid molecules that are created deviates between 0.2 and 99 percent.
MolCycleGAN is based on the CycleGAN. To generate novel molecules with similar properties MolCycleGAN uses JT-VAE as a latent space producer, then utilizes GAN to produce the molecule. To feed the GAN a molecule with desired features is used and the resultant latent space embedding is then transformed back to the new molecule with a similar structure and desired properties.
2 System overview
2.1 Implementation of methods
Implementations of each method aside from RNN48 were mentioned in the original publications. All of the algorithms were written in Python with the help of either Pytorch49 or Tensorflow.50 Training of all the methods except for RNN was conducted using the samples of molecules from ZINC that were provided by the authors of the original implementations. For RNN we have used a ZINC-based training dataset, which was provided by the authors of JT-VAE.39 In addition, we ran implementations of CDN and RNN methods utilizing a 1.6 million molecules sample of ChEMBL51 as a training dataset. As a result, ca. 0.62 thousand out of the total 3.64 thousand molecules generated with CDN and ca. 0.65 million out of the total 0.96 million molecules generated with RNN were obtained based on the ChEMBL training data.2.2 Molecule storage and preprocessing
All generated molecules along with their properties are stored in PostgreSQL, a free and open-source relational database management system with a variety of modules that allows to use them in different contexts. For instance, RDKit Cartridge enables efficient search across molecules.Preprocessing is an important step in the management of a large molecular database, containing millions of members. Utilizing RDKit52 2.9 million molecules, produced by the above mentioned generative ML algorithms, were inserted into the database in canonical SMILES format. During the insertion a fraction of molecules were discarded: 174000 were invalid (according to RDKit) and for 633 RDKit was not able to construct canonical SMILES. In addition, all duplicates were removed. In total, 2.8 million molecules were inserted along with computed properties that are listed in Lipinski's rule of five.53 Moreover, we have computed other medicinal chemistry-relevant properties such as the number of rotatable bonds and the number of saturated rings.
The typical operations on databases composed of a large number of molecules involve searching by substructure and searching by similarity. To optimize such queries we have precomputed Morgan Fingerprints (Circular Fingerprints) as well as the RDKit implementation of Extended Connectivity Fingerprints.54
2.3 Searching molecules
Fig. 2 demonstrates two pages of the application: “draw molecule” and “query results”. The “draw molecule” page has a doodle widget that allows drawing molecules using common tools, such as bonds, atoms, ring tools, among others, and uses this information as an input for the query. This widget is adopted from an open-source javascript library called Kekule.js.55 The “query results” page lists molecules with some common properties that satisfy the given query. The resulting molecules can be sorted based on a range of molecular properties. In addition, the results can be downloaded as a csv file containing the molecules in SMILES format. Furthermore, the interface allows opting for the generation of additional molecules that are either similar to the original query or to any of the molecules retrieved from the database.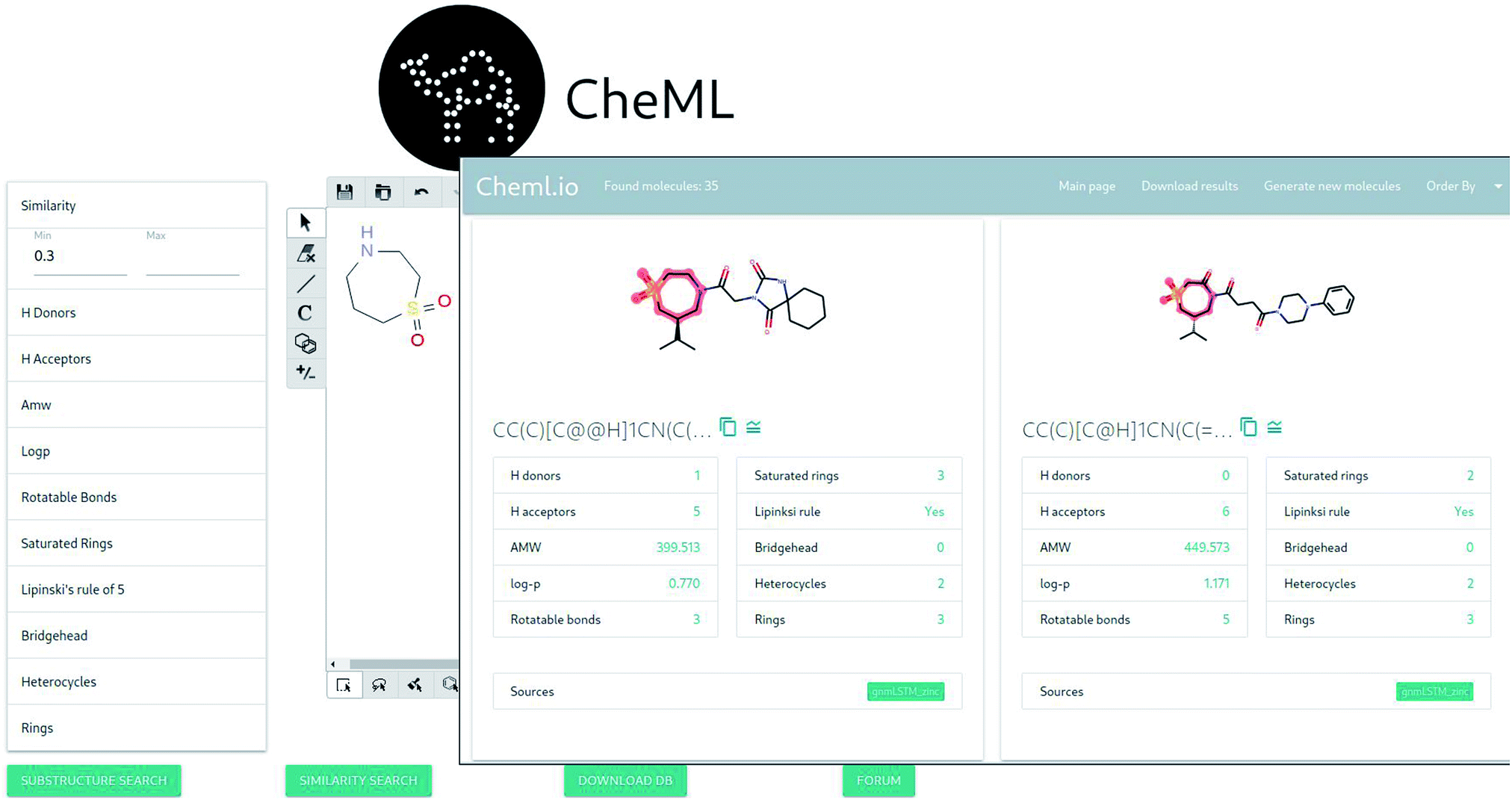 | ||
| Fig. 2 The screenshots of two main pages of the application. | ||
There are two types of queries: substructure search and similarity search. The substructure search performs the following operation:
Given a molecule X in SMILES format, find all molecules that contain X.
The similarity search performs the following operation:
Given a similarity threshold ε, find all molecules Y s.t. the dist(X,Y) < ε, where the dist(·,·) is the Tanimoto distance56.
By specifying the upper and lower bounds of molecular properties, it is possible to narrow down the search results.
2.4 Generation on demand
Currently, we employ Conditional Diversity Networks24 for the generation of new molecules on demand. Based on our observations, the algorithm achieves the best results when the training is performed for each request. Our current approach can be summarized as follows:1. Fetch molecules that are similar to the seed molecule from three databases: ZINC,57 ChEMBL51 and cheML.
2. Utilize these molecules as input data for the first training.
3. Fetch molecules from the above databases that contain the seed molecule as a substructure.
4. Utilize these molecules as input data for the second training.
5. Combine previous input data and use them as input for a third training.
6. Generate molecules using all three distinct models built by each of the above input data. Filter the resulting molecules based on their similarity score with the seed molecule. Exclude the molecules that are already present in the cheML.io database and those featuring the same structural backbone (i.e. different only by the stereochemical features) and send the outcome to the user by email.
7. Add novel molecules to the cheML.io database.
3 Results and discussion
3.1 Key characteristics of methods
To compare methods that were employed for the generation of molecules, we have looked into the several key characteristics of the machine learning algorithms: architecture, learning technique, molecular representations used, whether it can target desired property, computational resources needed to run it, and size of the training dataset. Please refer to the Table 1 for an overview.| Model | Architecture | Learning technique | Molecule representation | Property targeting | Computational costs | Training dataset size |
|---|---|---|---|---|---|---|
| JT-VAE | VAE | Autoencoder | Graph | Yes | Medium | 250k |
| RNN | RNN | Direct flow | SMILES | No | Low | 250k |
| GrammarVAE | VAE | Autoencoder | SMILES | No | High | 250k |
| ChemVAE | VAE | Autoencoder | SMILES | Yes | Medium | 250k |
| MolCycleGan | GAN | Direct flow | Latent vector | Yes | Medium | 250k |
| ORGAN | GAN | RL | SMILES | Yes | High | 1 million |
| ORGANIC | GAN | RL | SMILES | Yes | High | 250k |
| SSVAE | VAE | Autoencoder | SMILES | Yes | Medium | 310k |
| CDN | VAE | Autoencoder | SMILES | No | Low | 250k |
| CVAE | VAE | Autoencoder | SMILES | Yes | Medium | 500k |
3.2 Analysis across methods
As can be seen in Fig. 3, we did not aim to create a uniform number of molecules per method when implementing the studied algorithms. The main reason is due to the fact that some of the algorithms are more suitable for the generation of the bulk of molecules while others are more convenient for the targeted generation of specific molecules. For example, CVAE can be regarded as a specialized algorithm for the generation of molecules displaying specific properties while CDN is designed to generate similar yet diverse molecules when compared to a particular prototype. Thus, both CVAE and CDN were deployed by us for the generation of only a relatively small set of molecules ranging from several hundred to several thousand. On the other hand, considering its focus on structural similarity, CDN appeared to be the most suitable method for incorporating it into the generation on demand feature.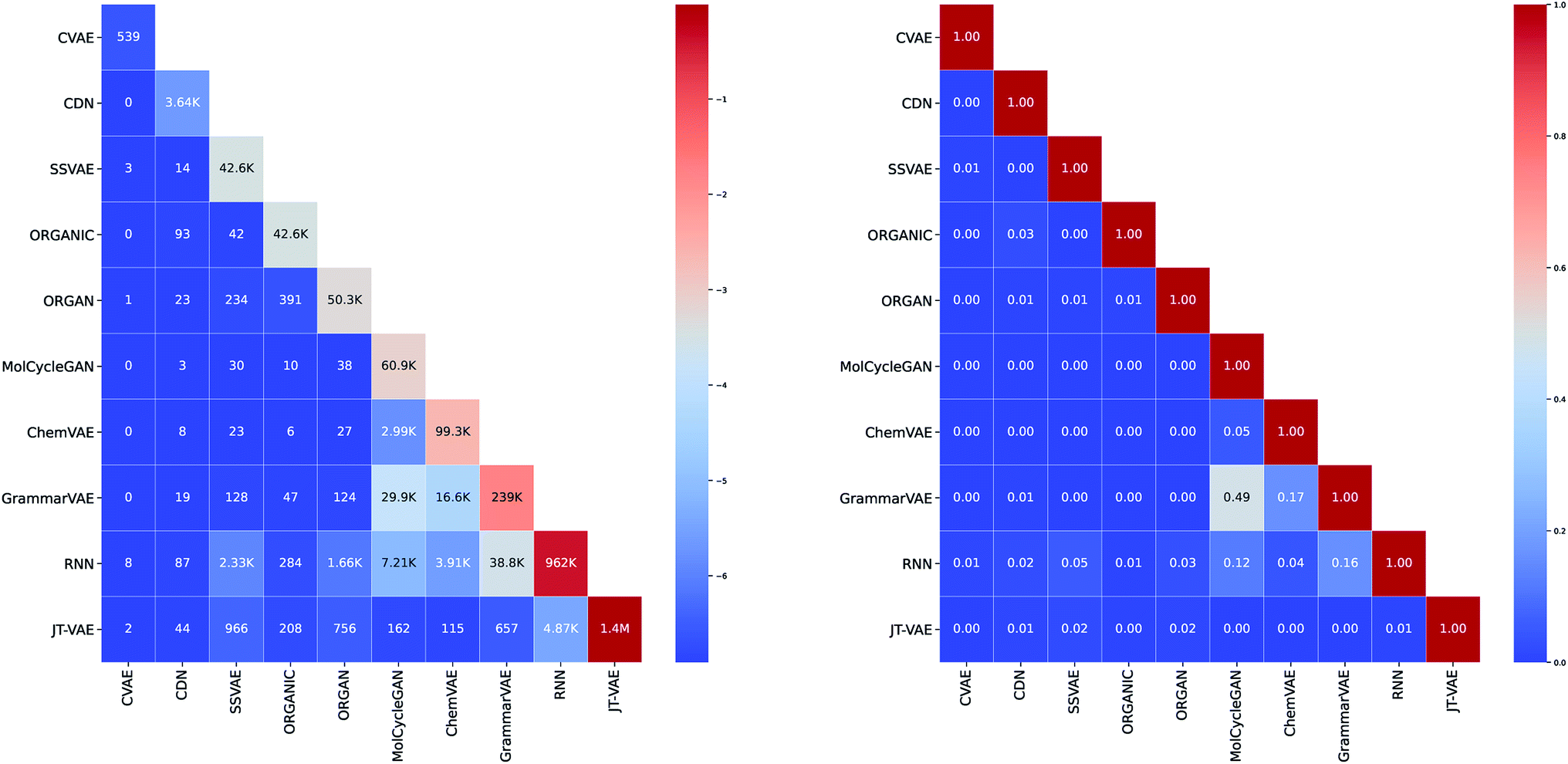 | ||
| Fig. 3 (Left) The diagonal of the matrix illustrates total number of molecules generated by each method. Intersections below the diagonal show number of same molecules that were generated by both methods. (Right) Each entry shows the proportion of shared molecules between each method. | ||
While the majority of generation algorithms shows a rather diverse output, when compared to the output of other algorithms (see Fig. 3), 49.07% of molecules generated by MolCycleGAN were also generated by GrammarVAE. This indicates that these methods may have a similar latent space despite that MolCycleGAN uses direct graph representation of the molecule while GrammarVAE uses the context-free grammar of the SMILES representation.
To further validate and compare the effectiveness of the implemented algorithms, metrics from the MOSES benchmarking framework58 were employed. Using the provided toolbox, we have calculated several representative metrics including novelty, internal diversity, and medical filters. Comparing the obtained results with the benchmark baselines indicates that almost all the methods demonstrated decent performance (see ESI for details†).
3.3 Diversity analysis
• Calculate a number of key molecular properties and structural parameters including molecular mass, number of atoms, number of chiral centers, number of rings, number of bridgehead atoms and number of heterocycles for the molecules from cheML, eMolecules and ZINC databases, as well as subsets of ML model outputs.
• Calculate the mean and standard deviation across above properties for each database, and subsets of cheML.
• Conduct statistical analyses to assess how the data fit the normal and uniform distributions property-wise; whether the variances across each property are equal between the databases; and whether the distributions of the data are somewhat similar between cheML.io and ZINC.
When comparing cheML.io with eMolecules and ZINC our database contains more diverse molecules across properties such as molecular mass and number of atoms, while the eMolecules database is the most diverse in terms of the number of rings and heterocycles. ZINC has the most variance for the number of chiral centers and bridgehead atoms (see Table 2).
| Database | Number of molecules | Exact molecular mass | Number of atoms | Number of chiral centers | Number of rings | Number of bridgehead atoms | Number of heterocycles |
|---|---|---|---|---|---|---|---|
| CheML | 2899276 |
373 ± 213 | 26.3 ± 15 | 0.23 ± 0.60 | 2.55 ± 1.02 | 0.023 ± 0.248 | 1.18 ± 0.93 |
| eMolecules | 26394586 |
331 ± 87.7 | 22.8 ± 6.46 | 0.0944 ± 0.489 | 2.63 ± 1.16 | 0.036 ± 0.319 | 1.35 ± 0.97 |
| ZINC | 30000000 |
321 ± 24.5 | 22.7 ± 2.0 | 1.399 ± 1.017 | 2.55 ± 0.90 | 0.076 ± 0.391 | 1.72 ± 0.95 |
| CheML JT-VAE | 1399265 |
323 ± 55 | 22.7 ± 3.9 | 0.0 ± 0.0 | 2.69 ± 0.93 | 0.024 ± 0.25 | 1.41 ± 0.89 |
| CheML RNN | 962245 |
475 ± 336 | 33.8 ± 23.8 | 0.30 ± 0.66 | 2.37 ± 1.12 | 0.0176 ± 0.23 | 0.80 ± 0.84 |
| CheML GrammarVAE | 239206 |
326 ± 59 | 22.7 ± 4.25 | 0.86 ± 0.90 | 2.63 ± 0.93 | 0.0238 ± 0.247 | 1.37 ± 0.92 |
| CheML ChemVAE | 99273 |
333 ± 63 | 22.9 ± 4.6 | 0.81 ± 0.9 | 2.72 ± 1.01 | 0.0540 ± 0.37 | 1.48 ± 0.95 |
| CheML MolCycleGAN | 60856 |
330 ± 61 | 23.0 ± 4.4 | 0.94 ± 1.00 | 2.75 ± 0.98 | 0.0400 ± 0.32 | 1.45 ± 0.96 |
| CheML ORGAN | 50262 |
273 ± 58 | 18.6 ± 4.1 | 0.28 ± 0.57 | 2.20 ± 0.75 | 0.029 ± 0.26 | 0.77 ± 0.72 |
| CheML ORGANIC | 42609 |
222 ± 60 | 15.8 ± 4.39 | 0.74 ± 0.82 | 1.39 ± 0.58 | 0.0022 ± 0.068 | 0.46 ± 0.55 |
| CheML SSVAE | 42606 |
355 ± 70 | 24.9 ± 4.8 | 0.0 ± 0.0 | 2.89 ± 0.92 | 0.034 ± 0.26 | 1.18 ± 0.87 |
| CheML CDN | 2415 | 385 ± 413 | 26.8 ± 27.8 | 0.44 ± 0.74 | 2.70 ± 1.28 | 0.167 ± 0.7 | 1.35 ± 1.07 |
| CheML CVAE | 539 | 304 ± 19 | 22.3 ± 1.48 | 0.87 ± 1.26 | 2.19 ± 0.52 | 0.0074 ± 0.122 | 0.69 ± 0.69 |
A test of normality was performed, which is based on D'Agostino and Pearson's test that combines skew and kurtosis to produce an omnibus test of normality.60,61 Our results indicate that all three databases' data are not normal for the considered properties. The implementation of the SciPy package was used for this and following statistical tests.62 Levene's test, which is optimal for highly non-normal data, showed that all property-wise differences in variances are statistically significant. This solidifies the claims stated in the previous paragraph. Lastly, a two-sample Kolmogorov–Smirnov test revealed that the distribution of values is not equal when comparing cheML.io with and the two other databases. This holds true for each property.
To compare ZINC and CheML side-by-side, normality tests were performed (based on D'Agostino and Pearson's test that combines skew and kurtosis to produce an omnibus test of normality) in order to determine the appropriate statistical tests for comparing variances and distributions. The normality tests indicated that most methods produce non-normal distributions with a significance level of α = 0.001. Please note that all p-values were corrected for multiple testing by application of Bonferroni correction.63 The only tests where the normality hypothesis could not be rejected were CheML CVAE for the properties: exact molecular weight, number of atoms, and number of rings.
The Kolmogorov–Smirnov test was used to deduce that the outputs of all models are statistically significantly different from uniform distributions along any property. Levene tests for equal variances yielded that variances vary statistically significantly when comparing the ML model outputs with the ZINC sample for all 6 molecular properties, which can be seen in Table 2. The only exception, where the null hypothesis of equal variance could not be rejected was for CheML CVAE vs. ZINC for the number of chiral centers. Two-sample Kolmogorov–Smirnov test indicates that the difference between ML model outputs and ZINC sample is statistically significant across all properties, with only two exceptions being CheML CVAE vs. ZINC for the number of bridgehead atoms and CheMl CVAE vs. ZINC for the same property.
According to the vast majority of tests, variances and distributions are not equal when comparing ML model outputs to the ZINC sample. Out of 120 statistical tests between CheML and ZINC in total, 117 yielded statistical significance. It is important to note that the CheML CVAE sub-dataset which constituted 2 out of 3 tests where the null hypothesis could not be rejected, is also the smallest set among the outputs, comprising only 539 molecules.
In addition, further statistical tests were performed, where each of the molecular properties' variances were compared utilizing Levene's test across all considered methods. Also, Kolmogorov–Smirnov tests were applied for testing equal distributions. These results can be obtained from Tables S2 and S3 in ESI.†
3.4 Performance of generation on demand
As was mentioned in the system overview section, we use the CDN as an algorithm for the generation of molecules on demand. To improve the proportion of correctly generated molecules we have substituted the SMILES character parser to a SMILES grammar parser. The original method of converting SMILES strings to number vectors involved assigning a number to each character of the SMILES string. For example, the atom H would be codified as 23, atom S as 24 and atom Si as a combination of two numbers 24 and 25 that should be placed consecutively.Thus, if number 25 would appear as standalone in the resulting vector, the whole vector would be discarded because the corresponding string and associated molecule would be invalid. To eliminate such cases we have used the SMILES grammar parser that breaks the SMILES string into morphemes, i.e. atoms and supporting elements, like stereoisomers. While the grammar parser does not eliminate syntactic errors it helps with standalone atom parts.
Utilizing the uniform training dataset for every generation request mainly resulted in a production of completely irrelevant molecules. However, when we have introduced the application of case specific training datasets described in the previous section the reliability of generation on demand feature has greatly improved.
During testing, we have observed that all the inputs for the generation on demand could be roughly divided into three categories: small molecules representing common structural motifs widely found in more complex molecules, medium-sized molecules that are not so widespread as subunits for other molecules, and large complex molecules that cannot be identified as substructures of any molecules from ZINC, ChEMBL or cheML.io. Owing to these differences, inputs from each of the above categories might require their own approach for assembling the training datasets. For example, the similarity-based training dataset for small molecules could be readily assembled from any database. However, due to the small size of the input molecule, the resulting training dataset might include molecules that are rather different from the initial one in the sense that they would not contain it as a substructure. Thus, adding a substructure-based training dataset and blending it with a similarity-based training dataset has generally led to a more balanced outcome for the generation requests featuring small and medium-sized molecules as inputs. On the other hand, for large and complex molecules that can not be found as substructures of other molecules, the only option is to use a similarity-based training dataset. Therefore, we have designed a 3-stage process for assembling the training datasets that accounts for the above mentioned variations and provides optimal results for any type of input structure without the need for manual categorizing.
4 Conclusions
In summary, we have surveyed and tested a number of ML algorithms for the de novo generation of organic molecules. A database of 2.8 million molecules originating from these efforts has been integrated into a user-friendly webpage framework that allows to perform substructure and similarity searches and browse the results in an interactive fashion. To facilitate and structurize the browsing process all the molecules in the database have been supplemented with a range of calculated molecular properties and structural features. Thus, the outcome of every search can be ordered, based on the chosen property or structural parameter. In addition, the molecules resulting from each specific search request as well as the overall database can be readily retrieved in a csv file, where molecules appear in SMILES format. When a specific search leads to insufficient results, users are able to request the generation of new molecules with the aid of the Conditional Diversity Network (CDN) algorithm. The user can opt to generate the molecules that are either similar to the original input structure or to any of the molecules retrieved from the database. All the newly generated molecules will be directly incorporated into the database assuring its continuous broadening and improvement. The generation on demand feature will also undergo further tuning and enhancement. In order to facilitate this process, the users will be provided with the possibility to leave their feedback and suggestions.We hope this dynamic online database may eventually provide assistance to researchers that are interested in the biological validation of ML-generated molecules and thereby justify the means of creating them.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Nazarbayev University Faculty Development Grant (240919FD3926). The authors would also like to acknowledge the support of NPO Young Researchers Alliance and Nazarbayev University Corporate Fund “Social Development Fund” for grant under their Fostering Research and Innovation Potential Program.Notes and references
- N. R. Council, Beyond the Molecular Frontier: Challenges for Chemistry and Chemical Engineering, The National Academies Press, Washington, DC, 2003 Search PubMed
.
- W. H. Sauer and M. K. Schwarz, J. Chem. Inf. Comput. Sci., 2003, 43, 987 CrossRef CAS
- S. L. Schreiber, Nature, 2009, 457, 153 CrossRef CAS
- S. Dandapani and L. A. Marcaurelle, Nat. Chem. Biol., 2010, 6, 861 CrossRef CAS
- S. Wu, Y. Kondo, M.-a. Kakimoto, B. Yang, H. Yamada, I. Kuwajima, G. Lambard, K. Hongo, Y. Xu, J. Shiomi, C. Schick, J. Morikawa and R. Yoshida, npj Comput. Mater., 2019, 5, 66 CrossRef
- R. S. Bohacek, C. McMartin and W. C. Guida, Med. Res. Rev., 1996, 16, 3 CrossRef CAS
- P. G. Polishchuk, T. I. Madzhidov and A. Varnek, J. Comput.-Aided Mol. Des., 2013, 27, 675 CrossRef CAS
- B. K. Shoichet, Nature, 2004, 432, 862 CrossRef CAS
- S. Ghosh, A. Nie, J. An and Z. Huang, Curr. Opin. Chem. Biol., 2006, 10, 194 CrossRef CAS
- D. Stumpfe and J. Bajorath, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 260 CAS
- G. Schneider and U. Fechner, Nat. Rev. Drug Discovery, 2005, 4, 649 CrossRef CAS
- A. M. Virshup, J. Contreras-García, P. Wipf, W. Yang and D. N. Beratan, J. Am. Chem. Soc., 2013, 135, 7296 CrossRef CAS
- A. Kadurin, A. Aliper, A. Kazennov, P. Mamoshina, Q. Vanhaelen, K. Khrabrov and A. Zhavoronkov, Oncotarget, 2017, 8, 10883 CrossRef
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Science, 2018, 361, 360 CrossRef CAS
- D. C. Elton, Z. Boukouvalas, M. D. Fuge and P. W. Chung, Mol. Syst. Des. Eng., 2019, 4, 828 RSC
- G. Chen, Z. Shen, A. Iyer, U. F. Ghumman, S. Tang, J. Bi, W. Chen and Y. Li, Polymers, 2020, 12, 163 CrossRef CAS
- N. Brown, P. Ertl, R. Lewis, T. Luksch, D. Reker and N. Schneider, J. Comput.-Aided Mol. Des., 2020, 34, 709 CrossRef CAS
- Cheml.io, accessed September 2020, https://cheml.io Search PubMed.
- A. Zhavoronkov, Y. A. Ivanenkov, A. Aliper, M. S. Veselov, V. A. Aladinskiy, A. V. Aladinskaya, V. A. Terentiev, D. A. Polykovskiy, M. D. Kuznetsov, A. Asadulaev, Y. Volkov, A. Zholus, R. R. Shayakhmetov, A. Zhebrak, L. I. Minaeva, B. A. Zagribelnyy, L. H. Lee, R. Soll, D. Madge, L. Xing, T. Guo and A. Aspuru-Guzik, Nat. Biotechnol., 2019, 37, 1038 CrossRef CAS
- Y. Le Cun and F. Fogelman-Soulié, Intellectica, 1987, 1, 114 Search PubMed
- D. E. Rumelhart, G. E. Hinton and R. J. Williams, Nature, 1986, 323, 533 CrossRef
- I. J. Goodfellow, J. Pouget-Abadie, M. Mirza, B. Xu, D. Warde-Farley, S. Ozair, A. Courville and Y. Bengio, Proceedings of the 27th International Conference on Neural Information Processing Systems - Volume 2, ed. Z. Ghahramani, M. Welling, C. Cortes, N. D. Lawrence and K. Q. Weinberger, MIT Press, Cambridge, MA, USA, 2014, p. 2672 Search PubMed
- I. Goodfellow, Y. Bengio and A. Courville, Deep learning, The MIT Press, Cambridge, Massachusetts, 2016 Search PubMed
- S. Harel and K. Radinsky, Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining - KDD ’18, London, United Kingdom, 2018, p. 331 Search PubMed
- H. Zhang, T. Xu, H. Li, S. Zhang, X. Wang, X. Huang and D. Metaxas, 2017 IEEE International Conference on Computer Vision (ICCV), 2017, p. 5908 Search PubMed
- C. Ledig, L. Theis, F. Huszár, J. Caballero, A. Cunningham, A. Acosta, A. Aitken, A. Tejani, J. Totz, Z. Wang and W. Shi, 2017 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), 2017, p. 105 Search PubMed
- C. Vondrick, H. Pirsiavash and A. Torralba, Proceedings of the 30th International Conference on Neural Information Processing Systems, Red Hook, NY, USA, 2016, p. 613 Search PubMed
- L.-C. Yang, S.-Y. Chou and Y.-H. Yang, Proceedings of the 18th International Society for Music Information Retrieval Conference, ISMIR 2017, Suzhou, China, October 23-27, 2017, p. 324 Search PubMed
- T. Schlegl, P. Seeböck, S. M. Waldstein, U. Schmidt-Erfurth and G. Langs, Information Processing in Medical Imaging, ed. M. Niethammer, M. Styner, S. Aylward, H. Zhu, I. Oguz, P.- T. Yap and D. Shen, Springer International Publishing, Cham, 2017, vol. 10265, p. 146 Search PubMed
- N. Killoran, L. J. Lee, A. Delong, D. Duvenaud and B. J. Frey, arXiv, 2017, preprint, arXiv:1712.06148v1, https://arxiv.org/abs/1712.06148v1.
- A. Kadurin, S. Nikolenko, K. Khrabrov, A. Aliper and A. Zhavoronkov, Mol. Pharm., 2017, 14, 3098 CrossRef CAS
- B. Sanchez-Lengeling, C. Outeiral, G. L. Guimaraes and A. Aspuru-Guzik, ChemRxiv, 2017 DOI:10.26434/chemrxiv.5309668.v3
- L. David, A. Thakkar, R. Mercado and O. Engkvist, J. Cheminf., 2020, 12, 56 CAS
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31 CrossRef CAS
- J. W. Raymond and P. Willett, J. Comput.-Aided Mol. Des., 2002, 16, 521 CrossRef CAS
- A. Cadeddu, E. K. Wylie, J. Jurczak, M. Wampler-Doty and B. A. Grzybowski, Angew. Chem., Int. Ed., 2014, 53, 8108 CrossRef CAS
- J. Lim, S. Ryu, J. W. Kim and W. Y. Kim, J. Cheminf., 2018, 10, 31 Search PubMed
- T. Blaschke, O. Engkvist, J. Bajorath and H. Chen, J. Cheminf., 2020, 12, 68 CAS
- W. Jin, R. Barzilay and T. Jaakkola, Proceedings of the 35th International Conference on Machine Learning, Stockholmsmässan, Stockholm Sweden, 2018, p. 2323 Search PubMed
- G. L. Guimaraes, B. Sanchez-Lengeling, C. Outeiral, P. L. C. Farias and A. Aspuru-Guzik, arXiv, 2018, preprint, arXiv:1705.10843v3, https://arxiv.org/abs/1705.10843v3
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268 CrossRef
- M. J. Kusner, B. Paige and J. M. Hernández-Lobato, Proceedings of the 34th International Conference on Machine Learning-Volume 70, 2017, p. 1945 Search PubMed
- M. H. S. Segler, T. Kogej, C. Tyrchan and M. P. Waller, ACS Cent. Sci., 2018, 4, 120 CrossRef CAS
- J.-Y. Zhu, T. Park, P. Isola and A. A. Efros, 2017 IEEE International Conference On Computer Vision (ICCV), 2017, p. 2242 Search PubMed
- Ł. Maziarka, A. Pocha, J. Kaczmarczyk, K. Rataj and M. Warchoł, Artificial Neural Networks and Machine Learning – ICANN 2019, Workshop and Special Sessions, Cham, 2019, p. 810 Search PubMed
- S. Kang and K. Cho, J. Chem. Inf. Model., 2019, 59, 43 CrossRef CAS
- L. Yu, W. Zhang, J. Wang and Y. Yu, Proceedings of the Thirty-First AAAI Conference on Artificial Intelligence, San Francisco, California, USA, 2017, p. 2852 Search PubMed
- Implementation of RNN from https://github.com/LamUong/Generate-novel-molecules-with-LSTM was used.
- A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein, L. Antiga, A. Desmaison, A. Kopf, E. Yang, Z. DeVito, M. Raison, A. Tejani, S. Chilamkurthy, B. Steiner, L. Fang, J. Bai and S. Chintala, Advances in Neural Information Processing Systems 32, ed. H. Wallach, H. Larochelle, A. Beygelzimer, F. dAlché-Buc, E. Fox and R. Garnett, Curran Associates, Inc., Red Hook, NY, USA, 2019, p. 8024 Search PubMed
- M. Abadi, P. Barham, J. Chen, Z. Chen, A. Davis, J. Dean, M. Devin, S. Ghemawat, G. Irving, M. Isard, M. Kudlur, J. Levenberg, R. Monga, S. Moore, D. G. Murray, B. Steiner, P. Tucker, V. Vasudevan, P. Warden, M. Wicke, Y. Yu and X. Zheng, Proceedings of the 12th USENIX Conference on Operating Systems Design and Implementation, USA, 2016 Search PubMed
- D. Mendez, A. Gaulton, A. P. Bento, J. Chambers, M. De Veij, E. Félix, M. P. Magariños, J. F. Mosquera, P. Mutowo, M. Nowotka, M. Gordillo-Marañón, F. Hunter, L. Junco, G. Mugumbate, M. Rodriguez-Lopez, F. Atkinson, N. Bosc, C. J. Radoux, A. Segura-Cabrera, A. Hersey and A. R. Leach, Nucleic Acids Res., 2018, 47, D930 CrossRef
- RDKit: Open-source cheminformatics, accessed September 2020, http://www.rdkit.org Search PubMed
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3 CrossRef CAS
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742 CrossRef CAS
- C. Jiang, X. Jin, Y. Dong and M. Chen, J. Chem. Inf. Model., 2016, 56, 1132 CrossRef CAS
- G. Maggiora, M. Vogt, D. Stumpfe and J. Bajorath, J. Med. Chem., 2014, 57, 3186 CrossRef CAS
- T. Sterling and J. J. Irwin, J. Chem. Inf. Model., 2015, 55, 2324 CrossRef CAS
- D. Polykovskiy, A. Zhebrak, B. Sanchez-Lengeling, S. Golovanov, O. Tatanov, S. Belyaev, R. Kurbanov, A. Artamonov, V. Aladinskiy, M. Veselov, A. Kadurin, S. Johansson, H. Chen, S. Nikolenko, A. Aspuru-Guzik and A. Zhavoronkov, arXiv, 2020, preprint, arXiv:1811.12823v5, https://arxiv.org/abs/1811.12823v5.
- eMolecules, https://www.emolecules.com/info/products-data-downloads.html, accessed September 2020 Search PubMed.
- R. d'Agostino, Biometrika, 1971, 58, 341 CrossRef
- R. d'Agostino and E. S. Pearson, Biometrika, 1973, 60, 613 Search PubMed
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S. J. van der Walt, M. Brett, J. Wilson, K. Jarrod Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. Carey, İ. Polat, Y. Feng, E. W. Moore, J. Vand erPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E. A. Quintero, C. R. Harris, A. M. Archibald, A. H. Ribeiro, F. Pedregosa, P. van Mulbregt and SciPy 1.0 Contributors, Nat. Methods, 2020, 17, 261 CrossRef CAS
- C. Bonferroni, Pubblicazioni del R Istituto Superiore di Scienze Economiche e Commericiali di Firenze, 1936, 8, p. 3 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07820d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |