
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chromium-free Cu@Mg/γ-Al2O3 – an active catalyst for selective hydrogenation of furfural to furfuryl alcohol†
Racha Arundhathia,
Panyala Linga Reddyb,
Chanchal Samantaa and
Bharat L. Newalkar *a
*a
aCorporate Research & Development Centre, Bharat Petroleum Corporation Limited, Greater Noida, Uttar Pradesh-201306, India. E-mail: newalkarbl@bharatpetroleum.in
bDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai-400076, India
First published on 10th November 2020
Abstract
Development of a chromium (Cr)-free hydrogenation catalyst is very important to replace the existing hazardous Cr based catalyst used in the furfural hydrogenation to furfuryl alcohol. Herein, we report synthesis of well-dispersed copper nanoparticles supported on hydrothermally stable magnesium doped alumina (Cu@Mg/γ-Al2O3) for selective hydrogenation of furfural to furfuryl alcohol. The prepared catalyst was characterized by X-ray Photoelectron Spectroscopy (XPS), Auger Electron Spectroscopy (AES), Powder X-ray Diffraction (PXRD), Surface Area Analysis (SAA), High Resolution-Transmission Electron Microscopy (HR-TEM), Temperature Programmed Reduction/Desorption (TPR/TPD) and Inductively Coupled Plasma-Atomic Emission Spectroscopy (ICP-AES) to understand textural properties of the catalyst. The prepared catalyst was found to be highly active and selective with 99% conversion of furfural and 94% selectivity for furfuryl alcohol under solvent free conditions at 443.15 K and 2 MPa of hydrogen pressure. It was also observed that the Cu@Mg/γ-Al2O3 catalyst is reusable (up to six runs) while maintaining its high activity and selectivity (≥94%) in the hydrogenation of furfural to furfuryl alcohol.
1. Introduction
Furfural (FF) is considered as one of the most promising value-added chemicals that can be produced from lignocellulosic biomass and it is a useful raw material for C4 and C5 based chemical production.1,2 Furfural is industrially produced through the acid hydrolysis of agro-based biomass. Alternatively, it can be produced from woody biomass in an integrated forest biorefinery. Furfural is an important platform molecule and can be converted to other useful chemicals. Thus, many efforts have been devoted to the development of catalytic transformations of FF to other value-added chemicals such as furfuryl alcohol (FA), tetrahydro furfuryl alcohol (THFA) cyclopenatnone, 2-methyl furan (2-MF), 1,2 pentane diols, tetrahydrofuran (THF).3 Furfuryl alcohol (FA) is one of the most important derivative of furfural which has much wider applications than furfural. Around 62% of the globally produced furfural is converted to FA.4 FA is extensively used as a monomer for cross-linked polymers to produce furan resins, which are used in foundry binders. These resins have excellent chemical, thermal and mechanical properties along with the capability of resisting corrosion.5Commercially, FA is produced through catalytic hydrogenation of furfural using Cu–Cr based catalyst. Although Cu–Cr based catalyst is active and highly selective for furfural hydrogenation, negative environmental effect of Cr is a major concern for this catalyst. Despite the high yield associated with the traditional Cu–Cr catalyst, it is not ideal due to the presence of Cr2O3 (chromium oxide) which can cause severe environmental pollution.6 Therefore, development of Cr-free catalytic system with high activity, selectivity, recyclability and able to perform reaction under industrially relevant conditions is highly desirable for hydrogenation of FF to FA. Thus, industries are looking for Cr-free catalyst with similar performance in terms of selectivity and activity. In recent years, various Cr-free catalytic systems based on Pt, Pd, Ru, Rh, Ir and Au have been reported for hydrogenation of furfural and have shown good catalytic conversation under mild reaction conditions.7 However, undesired product formation because of high catalytic activity and cost factors of precious metals have limited the commercial applications of noble metal based catalysts. In comparison to this, development of non-precious catalyst system has attractive advantages to meet the challenges of cost effectiveness and sustainability for industrial usage. In search of this, supported and non-supported metal heterogeneous catalysts based on Fe, Co, Ni and Cu were explored.8 Along with attractive advantages, these catalysts have some disadvantages like low activity, need of higher reaction temperature and lower selectivity towards the desired products. Therefore, to enhance catalytic activity and selectivity, doping small amount of appropriate heteroatom have been explored.
Magnesium supported materials found to exert high catalytic activity in the furfural hydrogenation.9 Various Mg incorporated catalysts such as Cu–Mg/ZnO,10,11 Mg-doped Pt/Al2O3,12 Ni–Mg/SiO2,13 Mg-doped Co–Ni nanocatalyst,14 Mg–TiO2,15 Mg doped ZnO,16 graphene oxide/WS2/Mg-doped ZnO nanocomposite17 have been explored in hydrogenation, hydrogen storage and other type reactions.10–17 However, limited detailed studies are reported in the literature to demonstrate the performance of magnesium doped catalyst in FF hydrogenation to FA. Therefore, we have synthesized and studied performance of magnesium doped heterogeneous Cu@Mg/γ-Al2O3 catalyst for the selective hydrogenation of FF to FA under solvent free conditions.
2. Experimental
2.1 Materials and methods
All reagents were purchased from commercial suppliers and used without further purification. All hydrogenation experiments were carried out under hydrogen. Column chromatography was carried out with Merck silica gel 60–120 mesh and the products were characterized and quantified by GC detection. 1H NMR and 13C NMR (300 or 400 MHz and 75 or 100 MHz, respectively) spectra were recorded in CDCl3. Chemical shifts (δ) are reported in ppm using TMS as an internal standard, and spin–spin coupling constants (J) are given in Hz.2.2 Catalyst preparation
Cu@Mg/γ-Al2O3 catalyst was prepared in three steps as follows.Step 1: 200 mL of deionised water was taken in a 1 L four neck round bottom flask and equipped with an overhead mechanical stirrer. Mg(NO3)2 ·6H2O (6.63 g, 0.025 moles) was dissolved and 45 grams of γ-Al2O3 (45 g, 0.4413 moles) was added to Mg(NO3)2·6H2O dissolved solution. The resulting slurry was kept for stirring for 2 h and aged at 70 °C for 4 h. The solid product was isolated by filtration and dried at 110 °C for 12 h in an air oven. Mg doped alumina catalyst was then calcined at 350 °C in the presence of air for 5 h to obtain MgO/γ-Al2O3 and then cooled to room temperature.
Step 2: in a 250 mL round bottomed flask 100 mL of deionized water was taken and 22.11 g of Cu(NO3)2·3H2O (0.0915 moles) was added under stirring conditions at room temperature for complete dissolution of copper salt. To this solution, 20 g of calcined MgO/γ-Al2O3 catalyst added and stirred at room temperature for 2 h. The pH of the reaction mixture was maintained constantly (8 to 9) by the continuous addition of the base solution (30% NH4OH). The resulting slurry was aged at 70 °C for 2 h. The solid product was isolated by filtration, washed thoroughly with deionised water (to make the catalyst free from base) and dried at 110 °C for 12 h in oven.
Step 3: the copper supported on MgO/γ-Al2O3 was further calcined at 750 °C for 4 h to get CuO@Mg/γ-Al2O3. CuO@Mg/γ-Al2O3 was then reduced under 3 bar of H2 pressure at 350 °C to get the final desired reduced Cu(0)@Mg/γ-Al2O3 catalyst. Copper nanoparticles anchored on various supports like SiO2, TiO2, CeO2, MoO3 and γ-Al2O3 are also prepared (refer SI for catalyst preparation†) and their performance were checked for FF to FA conversion.
3. Results and discussions
3.1 Catalyst characterization
The catalyst did not show any phase change up to 200 °C and gradual conversion of Cu(0) to CuO is clearly observed after 200 °C. At 300 °C most of the Cu(0) is converted to CuO and the total conversion is observed at 400 °C (Fig. 1D). The XRD patterns of the calcined catalysts did not show any clear peaks corresponding to magnesium species. This could be due to the low concentration of Mg in the sample but it could also be a sign of high dispersion of Mg species, since no characteristics structures are observed.
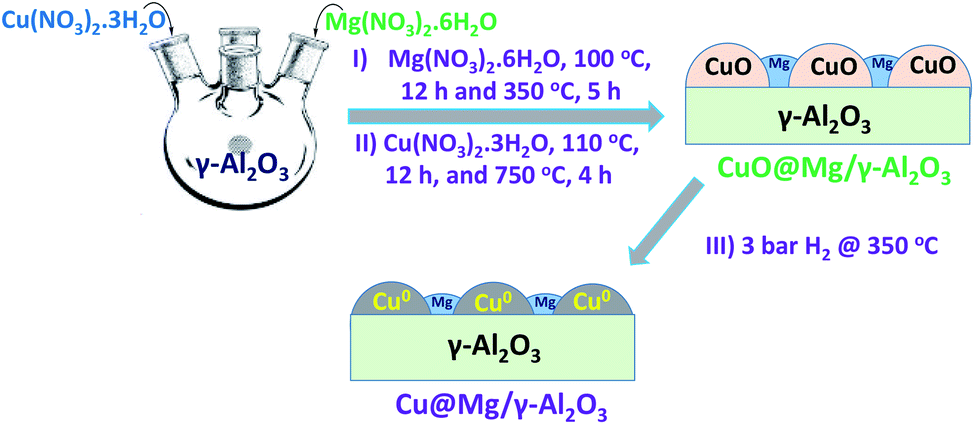 | ||
| Fig. 1 Schematic representation for synthesis of Cu@Mg/γ-Al2O3. | ||
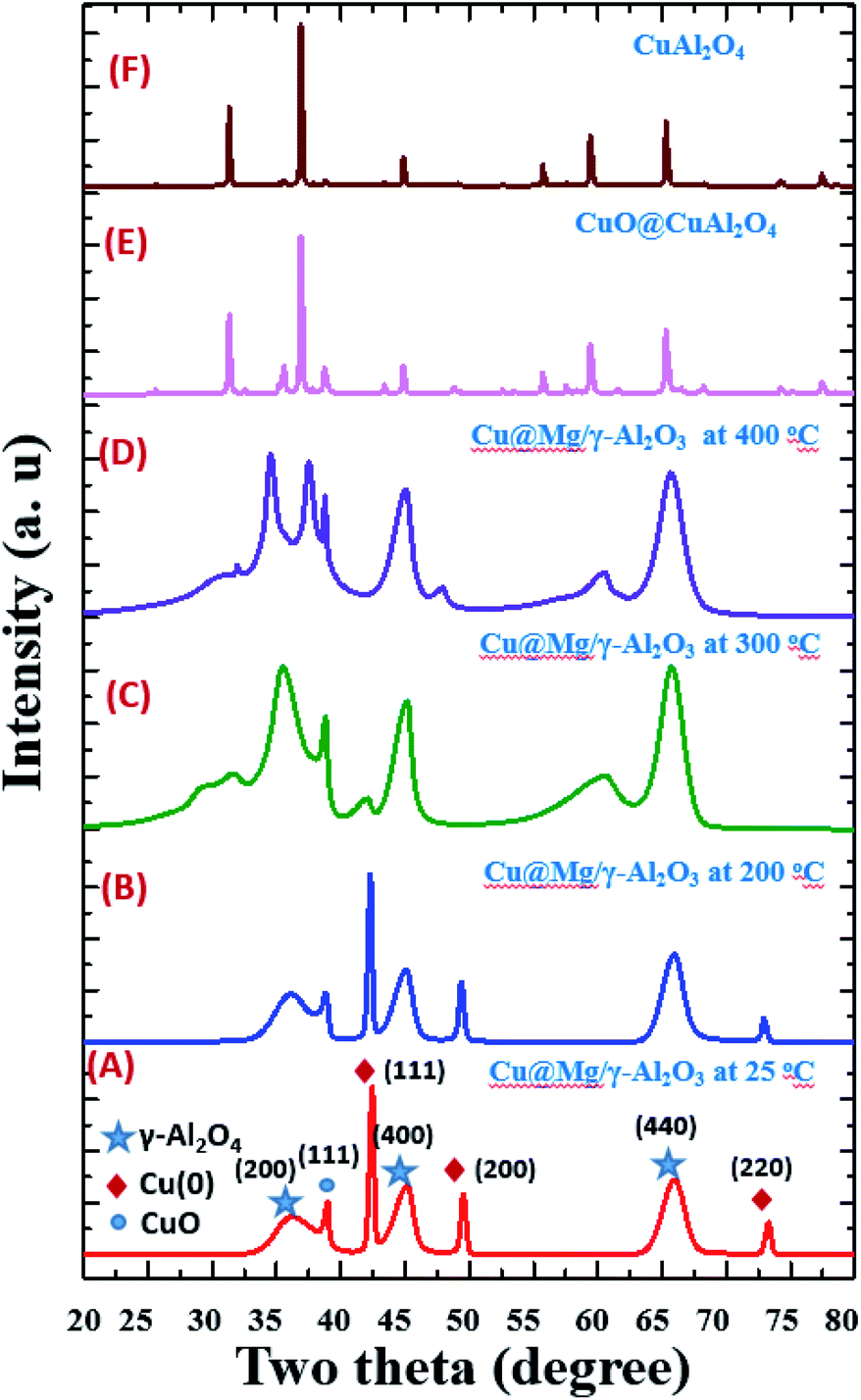 | ||
| Fig. 2 Temperature dependent XRD spectrum of Cu@Mg/γ-Al2O3 from 25 °C to 400 °C (images A–D), standard CuO@CuAl2O4 (E) and CuAl2O4 (F). | ||
 | ||
| Fig. 3 HR-TEM images [A–I] electron diffraction [SAED pattern, B] and elemental mapping [Cu, Mg and Al from J to L] of Cu@Mg/γ-Al2O3. | ||
To confirm the oxidation state and surface composition of the catalyst XPS experiments were recorded. As shown in Fig. 4, the binding energies of Cu@Mg/γ-Al2O3 was observed at 932.6 and 952.3 eV corresponds to the spin orbit coupling of Cu 2p3/2 and Cu 2p1/2 which confirmed the metallic nature Cu(0) of copper nanoparticles (Fig. 4B). The surface scanning of Cu@Mg/γ-Al2O3 by HR-TEM also confirms the uniform distribution of copper throughout the sample (Fig. 3E–H). The peak positions observed at 933.3 and 953.3 eV with the corresponding satellite peaks at 942.5 and 963.8 eV confirmed the Cu 2p core level in the +2 oxidation state (Fig. 4A) of CuO. In contrast, in the case of freshly reduced Cu(0)@Mg/γ-Al2O3, the binding energies were observed at 932.6 and 952.3 eV corresponding to the zero oxidation state of copper in the reduced catalyst and no satellite peaks at the corresponding positions were observed which strongly suggest the complete reduction of small amount of CuO present in Cu@Mg/γ-Al2O3 (Fig. 4A) to Cu(0)@Mg/γ-Al2O3(Fig. 4B). The very low shake satellite peak observed at 942.5 eV and 963.8 eV in Cu@Mg/γ-Al2O3 catalyst may be due to the small amount of copper oxide (CuO) formed due to copper propensity for aerial oxidation. The formation of Cu(0) nanoparticles by reduction was further confirmed with auger electron microscope (AES) analysis (refer ESI†). Cu@Mg/γ-Al2O3 (fresh) and reused Cu@Mg/γ-Al2O3 (recovered after 5th run) were characterized by Auger electron microscopic analysis to confirm the zero oxidation state of the copper, and no oxidation of metallic copper of Cu(0)@Mg/γ-Al2O3 was found even after successive runs. The kinetic energy for fresh and reused catalyst was found to be 919 eV and 918.7 eV.
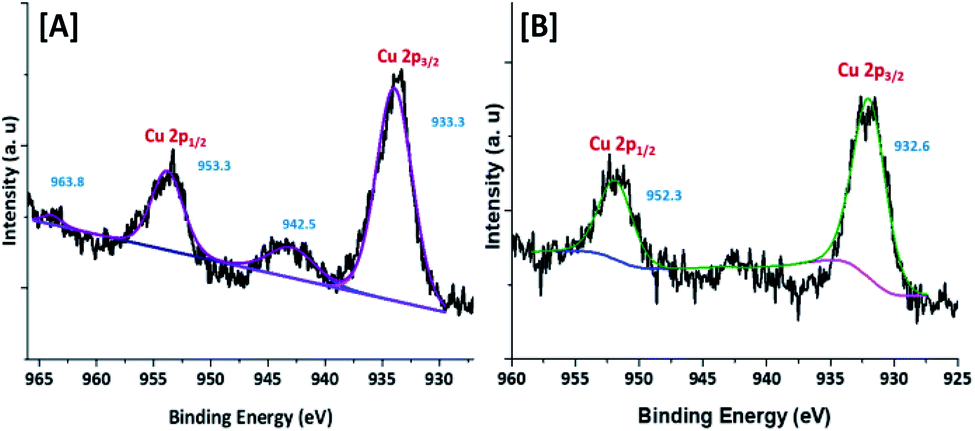 | ||
| Fig. 4 XPS spectrum of [A] Cu@Mg/γ-Al2O3 and [B] freshly reduced Cu(0)@Mg/γ-Al2O3. | ||
3.2 Catalytic activity: effect of support
As shown in the Table 1, for the catalytic hydrogenation of FF, copper nanoparticles anchored on various supports like SiO2, TiO2, CeO2, MoO3 and γ-Al2O3 were evaluated under solvent free reaction conditions at different temperatures. Among the screened catalysts, copper nanoparticles supported on γ-Al2O3 was found to give the best conversion of FF >99% with FA yield of 74% in 5 h, extending the reaction time to 12 h gave further improvement in reaction yield of FA to 83%. γ-Al2O3 is extensively used as support in various active catalysts because of its unique mechanical and chemical properties12–15 and high surface area. γ-Al2O3 supported catalysts show favourable activity in many reactions such CO2 hydrogenation,16 for hydrodesulfurization17 and reduction of NOx.18 However, the formation of side products could not lead the conversion to maximum yields (Table 1, entries 1 and 2). In case of SiO2, the deoxyhydrogenation product, 2-methyl furan (2-MF) is the major product with 40% yield along with only 10% yield of FA. On the other hand, other solid supports such as TiO2 resulted in the moderate conversion with less yield (Table 1, entry 4). Next, it was observed that CeO2 and MoO3 were less effective for the selective hydrogenation of FF to FA (Table 1, entries 5 and 6). Also, we have compared alumina supported precious metal catalysts like Pt, Rh, Ir and Pd with non-precious catalysts, among them Pt has shown good conversion towards deoxyhydrogenated furfural to 2-MF with 88% yield. Rh, Ir and Pd could give decarbonylated product furan as a major product with 87%, 96% and 99% of yields (Table 1, entries 7–10) respectively. Nevertheless, copper metal sites combined with an aluminium oxide site can play a major role in the activity, selectivity and stability of the samples. Thus, aluminium oxide acts mainly as structural and textural promoter, providing a larger surface area for solids containing copper oxide.18 The preparation of supported catalyst can be seen in ESI.†| Entry | Catalyst | Conv. [%] | Yield [%] | ||||
|---|---|---|---|---|---|---|---|
| FA | 2-MF | THFA | Furan | THF | |||
| a Reaction conditions: furfural (2.6 mol%); catalyst (0.1 g, noble metal 2 mol%); H2 (2 MPa); 443.15 K; 5 h.b Analyzed by GC using an toluene as internal standard.c Catalyst (0.1 g, Cu 0.4 mol%).d 12 h. | |||||||
| 1b | Cu/γ-Al2O3 | >99 | 74 | 16 | 3 | 2 | Trace |
| 2c,d | Cu/γ-Al2O3 | >99 | 83 | 0 | 1 | 4 | 0 |
| 3 | Cu/SiO2 | >60 | 10 | 40 | Trace | Trace | Trace |
| 4 | Cu/TiO2 | 20 | 15 | 2 | Trace | Trace | Trace |
| 5 | Cu/CeO2 | 45 | 23 | Trace | Trace | Trace | Trace |
| 6 | Cu/MoO3 | 20 | 8 | Trace | Trace | Trace | Trace |
| 7 | Pt/γ-Al2O3 | >90 | 12 | 88 | Trace | Trace | Trace |
| 8 | Rh/γ-Al2O3 | >99 | — | — | Trace | 87 | 5 |
| 9 | Ir/γ-Al2O3 | >99 | — | — | — | 96 | — |
| 10 | Pd/γ-Al2O3 | >99 | — | — | — | 99 | — |
Further, in order to design efficient catalytic system for FF hydrogenation copper nanoparticles supported on bimetallic supported system was examined. Recently, magnesium doped copper and iron based catalysts successfully employed for transfer hydrogenation of furfural using isopropanol as a hydrogen donor.19 The increase in activity can be attributed to the change in the electronic and physical structure of the catalyst and in-turn enhances increase in the absorption ability of the reacting molecules on the host material. The high catalytic activity with Mg doped supports can be attributed due to increasing adsorption sites of hydrogen on MgO surface with low coordination sites on the catalyst surface.20 Typically, MgO acts as a promoter to improve activity and/or selectivity acting as a basic site to polarise the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of furfural which in turn facilitate nucleophilic attack by hydrogen dissociatively adsorbed on adjacent Cu active sites. Besides this, the basic sites also help to reduce the concentration of Lewis acid centres on the support γ-Al2O3, which will decrease coke deriving from the acidic strength of γ-Al2O3. The same can be witnessed from CO2-TPD data wherein basic site concentration is found to enhance in presence of MgO (see ESI for CO2-TPD†). The effect of different metals as promoters such as: Co, Zn, Mg, Ga, Mn and Zr on catalytic activity of Cu/γ-Al2O3 in FA synthesis was also studied (Table 2). Based on the literature reports and in our present investigation to increase the catalytic activity of copper supported catalyst for hydrogenation of FF, magnesium has been doped in various molar ratios and their catalytic activity were investigated (Table 3). MgO increases the strength of the interaction between Cu and Al2O3 and induces a spillover effect between these phases. Synergistic catalytic effect between the catalytically active metallic copper species and the Lewis basic sites, which held the key to the hydrogenation reaction related to the hydrogen dissociation and the activation of the carbonyl groups attribute to its high catalytic efficiency. Another advantage of using MgO as a promoter for the preparation of Cu@Mg/γ-Al2O3 catalyst arises from the possibility of forming a CuO–MgO solid solution at any molar ratio due to close ionic radii of Mg2+ and Cu2+ cations (Mg2+ 0.65 Å and Cu2+ 0.73 Å) and the particular lattice parameters of this mixed metal oxide structure. The formation of this mixed oxide phase favors increased metal-support interaction, and thus prevents catalyst deactivation via sintering. On the other hand, the promotion of copper catalysts by magnesium oxides increases the amount of hydroxylalkyl intermediate group formed on the catalysts surface during the process compared to the reaction carried out with the unprompted catalysts. 1H NMR analysis of the reaction intermediate showed the formation of hydroxyalkyl intermediate in 9% yield, which transformed to FA with Cu–Mg/γ-Al2O3.
O bond of furfural which in turn facilitate nucleophilic attack by hydrogen dissociatively adsorbed on adjacent Cu active sites. Besides this, the basic sites also help to reduce the concentration of Lewis acid centres on the support γ-Al2O3, which will decrease coke deriving from the acidic strength of γ-Al2O3. The same can be witnessed from CO2-TPD data wherein basic site concentration is found to enhance in presence of MgO (see ESI for CO2-TPD†). The effect of different metals as promoters such as: Co, Zn, Mg, Ga, Mn and Zr on catalytic activity of Cu/γ-Al2O3 in FA synthesis was also studied (Table 2). Based on the literature reports and in our present investigation to increase the catalytic activity of copper supported catalyst for hydrogenation of FF, magnesium has been doped in various molar ratios and their catalytic activity were investigated (Table 3). MgO increases the strength of the interaction between Cu and Al2O3 and induces a spillover effect between these phases. Synergistic catalytic effect between the catalytically active metallic copper species and the Lewis basic sites, which held the key to the hydrogenation reaction related to the hydrogen dissociation and the activation of the carbonyl groups attribute to its high catalytic efficiency. Another advantage of using MgO as a promoter for the preparation of Cu@Mg/γ-Al2O3 catalyst arises from the possibility of forming a CuO–MgO solid solution at any molar ratio due to close ionic radii of Mg2+ and Cu2+ cations (Mg2+ 0.65 Å and Cu2+ 0.73 Å) and the particular lattice parameters of this mixed metal oxide structure. The formation of this mixed oxide phase favors increased metal-support interaction, and thus prevents catalyst deactivation via sintering. On the other hand, the promotion of copper catalysts by magnesium oxides increases the amount of hydroxylalkyl intermediate group formed on the catalysts surface during the process compared to the reaction carried out with the unprompted catalysts. 1H NMR analysis of the reaction intermediate showed the formation of hydroxyalkyl intermediate in 9% yield, which transformed to FA with Cu–Mg/γ-Al2O3.
| Sr. no. | Bimetallic catalyst | FF conv. [%] | FA yieldb [%] | ||||
|---|---|---|---|---|---|---|---|
| FA | 2-MF | Furan | THF | THFA | |||
| a Reaction conditions: furfural (2.6 mol%); catalyst (0.1 g, noble metal 2 mol%); H2 (2 MPa); 443.15 K; 5 h. In all the bimetallic catalysts, Cu/X metal ratios were maintained as 5 (10 wt% copper and 2 wt% X metal {X = Co, Zn, Mg, Ga, Mn and Zr}) on the support γ-Al2O3.b Analyzed by GC using toluene as an internal standard. | |||||||
| 1 | Cu–Co/γAl2O3 | 65 | 45 | 4 | 0 | 0 | 5 |
| 2 | Cu–Zn/γAl2O3 | 90 | 70 | 16 | 4 | 1 | 0 |
| 3 | Cu–Mg/γAl2O3 | >99 | 94 | 2 | 4 | 0 | 0 |
| 4 | Cu–Ga/γAl2O3 | 75 | 15 | 3 | 0 | 17 | |
| 5 | Cu–Mn/γAl2O3 | 80 | 32 | 8 | 5 | 3 | 0 |
| 6 | Cu–Zr/γAl2O3 | 80 | 40 | 0 | 0 | 0 | 0 |
| Sr. no. | Cu/Mg | FF conv. [%] | FA yieldb [%] | ||||
|---|---|---|---|---|---|---|---|
| FA | 2-MF | Furan | THF | THFA | |||
| a Reaction conditions: furfural (2.6 mol%); catalyst (0.1 g, Cu/Mg mol%); Cu/Mg = 4 (Cu: 0.2 mol% and Mg: 0.05 mol%); H2 (2 MPa); 443.15 K; 5 h.b Analyzed by GC using toluene as an internal standard. | |||||||
| 1 | 4 | >99 | 90 | 3 | 2 | 1 | 1 |
| 2 | 5 | >99 | 94 | 2 | 4 | 0 | 0 |
| 3 | 10 | >99 | 80 | 15 | 0 | 0 | 0 |
| 4 | 15 | >99 | 65 | 22 | 2 | 0 | 1 |
Among all, Cu–Mg supported catalysts showed the most superior activity performance in terms of FF conversion, FA formation rate, and yield. Therefore, the Cu–Mg was employed hereafter to compare with other support materials (Table 4). In the case of γ-Al2O3, FF conversion attributed 90% conversion with 85% of FA product yield, increase in reaction time to 5 h further increases the conversion to 99% with 94% of product selectivity (Table 4, entries 1 and 2). On the other hand, other solid supports such as SiO2, TiO2, CeO2 and MoO3 resulted in moderate yields with 2-MF and furan as a major side products (entries 8 to 11). The support γ-Al2O3 with high surface area than other supports (Table 2, entries 8 to 11) shows highly dispersed copper species achieved the highest catalytic activity compared to the sample containing copper on SiO2, TiO2, CeO2 and MoO3. The presence of γ-Al2O3 interacting with copper oxide provides a lower sintering under optimized reaction conditions.21 It is also clearly noticeable that the magnesium doped catalysts are comparatively higher in activity than magnesium free catalysts (Table 4, entries 1–5 vs. 6). These results clearly show that the combination of Cu(0) nanoparticles and Mg on γ-Al2O3 support is uniquely effective for the synthesis of FA from FF under solvent-free conditions. Furthermore, the addition of a second metal MgO as promoter has been found to improve activity and/or selectivity by acting as a basic site to polarise the ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CO bond facilitating nucleophilic attack by hydrogen dissociatively adsorbed on neighbouring Cu active sites. Moreover, with a capacity for oxygen storage, MgO can release oxygen to oxidize the carbon formed on the catalyst surface. Particularly, the presence of the basic centre will strengthen the chemisorption of carbonyl group. Next, to understand the importance of hydrogen pressure, the reaction was performed at 1 MPa, which took almost 10 h to complete the conversion of FF to FA with 93% selectivity of desired product (Table 4, entry 5). As evident from Tables 1 and 2 the optimum condition for hydrogenation of furfural is at 443.15 K for 5 h at 2 MPa to get maximum yield of FA.
CO bond facilitating nucleophilic attack by hydrogen dissociatively adsorbed on neighbouring Cu active sites. Moreover, with a capacity for oxygen storage, MgO can release oxygen to oxidize the carbon formed on the catalyst surface. Particularly, the presence of the basic centre will strengthen the chemisorption of carbonyl group. Next, to understand the importance of hydrogen pressure, the reaction was performed at 1 MPa, which took almost 10 h to complete the conversion of FF to FA with 93% selectivity of desired product (Table 4, entry 5). As evident from Tables 1 and 2 the optimum condition for hydrogenation of furfural is at 443.15 K for 5 h at 2 MPa to get maximum yield of FA.
| S. no. | Catalyst | H2 [MPa] | Time [h] | FF conv. [%] | Yield [%] | ||||
|---|---|---|---|---|---|---|---|---|---|
| FA | 2-MF | Furan | THF | THFA | |||||
| a Reaction conditions: FF (2.6 mol%); catalyst (2 g, Cu–Mg: Cu@Mg, Cu-0.2 mol%, Mg 0.05 mol%); H2 (2 MPa); 443.15 K.b Cu/Mg = 5.c Reuse 3. | |||||||||
| 1 | Cu–Mg/γ-Al2O3 | 2 | 3 | 90 | 85 | 0 | 3 | Trace | 0 |
| 2b | Cu–Mg/γ-Al2O3 | 2 | 5 | >99 | 94 | 2 | 4 | 0 | 0 |
| 3c | Cu–Mg/γ-Al2O3 | 2 | 5 | >99 | 94 | 2 | 4 | 0 | 0 |
| 4 | Cu–Mg/γ-Al2O3 | 2 | 4 | >99 | 90 | 4 | 4 | 1 | 0 |
| 5 | Cu–Mg/γ-Al2O3 | 1 | 10 | >99 | 93 | Trace | 5 | 2 | 0 |
| 6 | Cu/γ-Al2O3 | 2 | 10 | >99 | 74 | 16 | 3 | Trace | 2 |
| 7 | MgO/γ-Al2O3 | 2 | 10 | Trace | 0 | Trace | 0 | Trace | 0 |
| 8 | Cu–Mg/SiO2 | 2 | 5 | >99 | 44 | 0 | 26 | 9 | 17 |
| 9 | Cu–Mg/TiO2 | 2 | 5 | >99 | 39 | 49 | 8 | 4 | Trace |
| 10 | Cu–Mg/CeO2 | 2 | 5 | >99 | 2 | 53 | 0 | 0 | 0 |
| 11 | Cu–Mg/MoO3 | 2 | 5 | >99 | 32 | Trace | 0 | 0 | 0 |
3.3 Catalyst stability and reuse experiments
From an industrial perspective, one of the main advantages of using heterogeneous catalysts such as Cu@Mg/γ-Al2O3, is that they can be recovered and reused efficiently up to six consecutive runs (Fig. 5). The spent catalyst was recovered from the reaction mixture by simple centrifugation after the completion of the reaction and washed with EtOAc (3 × 10 mL) to remove all the organic substrates from the catalyst surface. No quantifiable amount of leached Cu was detected in the filtrate as determined by ICP (AES) studies of both fresh and spent catalyst. Furthermore, the HR-TEM images of the used catalyst did not show any significant change in the shape and size of the support as well as the particle size of the active species, Cu(0). This suggests that the morphology of the catalyst remains the same even after multiple reaction cycles Table 5.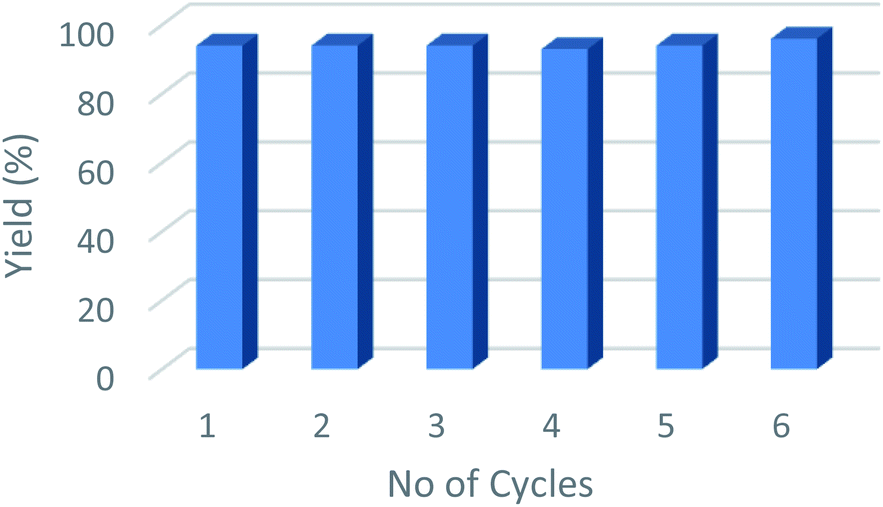 | ||
| Fig. 5 Recyclability of the catalyst for reduction of furfural up to six consecutive runs. | ||
| a Reaction conditions: FF (2.6 mol%); Cu@Mg/γ-Al2O3 (0.1 g); 443.15 K; H2 (2 MPa); 5 h; yields analyzed by GC using toluene as an internal standard. | ||||||
|---|---|---|---|---|---|---|
| Reaction run | Run 1 | Run 2 | Run 3 | Run 4 | Run 5 | Run 6 |
| FA yield (%) | 94 | 94 | 94 | 93 | 94 | 96 |
3.4 Representative reaction procedure
The hydrogenation of furfural was carried out in a 500 mL stainless steel autoclave PARR reactor. The vessel was charged with 2.6 mol of fufural, and 0.1 g of catalyst. The reactor was sealed, purged three times with H2 at 2 MPa, then pressurized to 2 MPa, heated to 443.15 K and stirred at 300 rpm for 5 h with continuous H2 flow (80 SLPH). Following the reaction, the autoclave was cooled to room temperature and the hydrogen gas was carefully released. The resulting reaction mixture was centrifuged and catalyst was separated from the reaction. The reaction mixture is then diluted with Toluene and analyzed by GC. Cu@Mg/γ-Al2O3 catalyst provides FA product in good to excellent yields over short reaction times compared with reported catalyst in the absence of solvents and strong bases (see ESI Table 1†). Furthermore, the catalyst was applicable to preparative scale reaction; 500 g of FF afforded 502 g of FA in 96% isolated yield (ESI†).4. Conclusions
The present study demonstrates high activity, selectivity and stability of chromium-free Cu-based catalyst supported on MgO-doped γ-Al2O3 in solvent free hydrogenation of furfural to furfuryl alcohol. The prepared catalyst is found to be selective in the hydrogenation of furfural to furfuryl alcohol with selectivity of 95% at 100% conversion of furfural. The remarkably high performance of this catalyst is attributed to the synergistic effect of copper and magnesium resulting in active Cu–Mg species on the surface of alumina.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank BPCL management for granting permission to publish the work and IIT-Bombay for providing necessary support in the catalyst characterizations.Notes and references
- H. Furkan, H. Isikgor and C. Remzi Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- K. Gupta, R. K. Rai and S. K. Singh, ChemCatChem, 2018, 10, 2326–2349 CrossRef CAS.
- X. Chen, L. Zhang, B. Zhang, X. Guo and X. Mu, Sci. Rep., 2016, 6, 28558–28570 CrossRef.
- A. Mandalika, L. Qin, T. K. Satob and T. Runge, Green Chem., 2014, 16, 2480–2489 RSC.
- J. B. Barr and S. B. Wallon, J. Appl. Polym. Sci., 1971, 15, 1079–1090 CrossRef CAS.
- (a) M. J. Taylor, L. J. Durndell, M. A. Isaacs, C. M. A. Parlett, K. Wilson, A. F. Lee and G. Kyriakou, Appl. Catal., B, 2016, 180, 580–585 CrossRef CAS; (b) M. M. Villaverde, N. M. Bertero, T. F. Garetto and A. Marchi, Catal. Today, 2013, 213, 87–92 CrossRef CAS.
- (a) M. J. Taylor, L. J. Durndell, M. A. Isaacs, C. M. A. Parlett, K. Wilson, A. F. Lee and G. Kyriakou, Appl. Catal., B, 2016, 180, 580–585 CrossRef CAS; (b) A. B. Merlo, V. Vetere, J. F. Ruggera and M. L. Casella, Catal. Commun., 2009, 10, 1665–1669 CrossRef CAS; (c) K. Fulajtarova, T. Sotak, M. Hronec, I. Vavra, E. Dobročka and M. Omastova, Appl. Catal., A, 2015, 502, 78–85 CrossRef CAS; (d) M. Lesiak, M. Binczarski, S. Karski, W. Maniukiewicz, J. Rogowski, E. Szubiakiewicz, J. Berlowska, P. Dziugan and I. Witońska, J. Mol. Catal. A: Chem., 2014, 395, 337–348 CrossRef CAS; (e) P. Reyes, D. Salinas, C. Campos, M. Oportus, J. Murcia, H. Rojas, G. Borda and J. L. Garcia Fierro, Quim. Nova, 2010, 33, 777–780 CrossRef CAS; (f) M. Tamura, K. Tokonami, Y. Nakagawa and K. Tomishige, Chem. Commun., 2013, 49, 7034–7036 RSC.
- (a) M. M. Villaverde, T. F. Garetto and A. J. Marchi, Catal. Commun., 2015, 58, 6–10 CrossRef CAS; (b) K. Xiong, W. Wan and J. G. Chen, Surf. Sci., 2016, 652, 91–97 CrossRef CAS; (c) R. V. Sharma, U. Das, R. Sammynaiken and A. K. Dalai, Appl. Catal., A, 2013, 454, 127–136 CrossRef CAS; (d) C. Xu, L. Zheng, J. Liu and Z. Huang, Chin. J. Chem., 2011, 29, 691–697 CrossRef CAS; (e) S. Srivastava, P. Mohanty, J. K. Parikh, A. K. Dalai, S. S. Amritphale and A. K. Khare, Chin. J. Catal., 2015, 36, 933–942 CrossRef CAS; (f) K. Yan and A. Chen, Fuel, 2014, 115, 101–108 CrossRef CAS; (g) J. Wu, G. Gao, J. Li, P. Sun, X. Long and F. Li, Appl. Catal., B, 2017, 203, 227–236 CrossRef CAS; (h) W. Yu, K. Xiong, N. Ji, M. D. Porosoff and J. G. Chen, J. Catal., 2014, 317, 253–262 CrossRef CAS; (i) H. Li, H. Luo, L. Zhuang, W. Dai and M. Qiao, J. Mol. Catal. A: Chem., 2003, 203, 267–275 CrossRef CAS; (j) H. Li, S. Zhang and H. Luo, Mater. Lett., 2004, 58, 2741–2746 CrossRef CAS; (k) W. Gong, C. Chen, H. Zhang, Y. Zhang, Y. Zhang, G. Wang and H. Zhao, J. Mol. Catal., 2017, 429, 51–59 CrossRef CAS; (l) B. J. Liu, L. H. Lu, B. C. Wang, T. X. Cai and K. Iwatani, Appl. Catal., A, 1998, 171, 117–122 CrossRef CAS; (m) T. V. Kotbagi, H. R. Gurav, A. S. Nagpure, S. V. Chilukuri and M. G. Bakker, RSC Adv., 2016, 6, 67662–67668 RSC; (n) S. Wei, H. Cui, J. Wang, S. Zhuo, W. Yi, L. Wang and Z. Li, Particuology, 2011, 9, 69–74 CrossRef CAS; (o) X. F. Chen, X. H. Li, H. S. Luo and M. H. Qiao, Appl. Catal., A, 2002, 233, 13–20 CrossRef CAS; (p) M. Audemar, C. Ciotonea, K. De Oliveira Vigier, S. Royer, A. Ungureanu, B. Dragoi, E. Dumitriu and F. Jerome, ChemSusChem, 2015, 8, 1885–1891 CrossRef CAS.
- (a) H. Chen, H. Ruan, X. Lu, J. Fu, T. Langrish and X. Lu, J. Mol. Catal., 2018, 445, 94–101 CrossRef CAS; (b) M. M. Villaverde, T. F. Garetto and A. J. Marchi, Catal. Commun., 2015, 58, 6–10 CrossRef CAS; (c) J. Wu, G. Gao, J. Li, P. Sun, X. Long and F. Li, Appl. Catal., B, 2017, 203, 227–236 CrossRef CAS.
- X. Kong, Z. Chen, Y. Wu, R. Wang, J. Chen and L. Ding, RSC Adv., 2017, 7, 49548–49561 RSC.
- Z. Lu, H. Yin, A. Wang, J. Hu, W. Xue, H. Yinand and S. Liu, J. Ind. Eng. Chem., 2016, 37, 208–215 CrossRef CAS.
- J. Escobara, M. C. Barrerab, V. Santesc and J. E. Terrazas, Catal. Today., 2017, 296, 197–204 CrossRef.
- A. Ramesh, P. Tamizhduraia, V. L. Mangesh, K. Palanichamy, S. Gopinatha, K. Sureshkumar and K. Shanthia, Int. J. Hydrogen Energy, 2019, 44(47), 25607–25620 CrossRef CAS.
- X. Ding, H. Ding, Y. Song, C. Xiang, Y. Li and Q. Zhang, Front. Chem., 2020, 7, 1–7 RSC.
- G. K. Meenashisundaram, M. H. Nai, A. Almajid and M. Gupta, Mater. Des., 2015, 65, 104–114 CrossRef CAS.
- E. Vinodkumar, R. Roshan and V. Kumar, ACS Appl. Mater. Interfaces, 2012, 4(5), 2717–2725 CrossRef.
- C. Chen, W. Yu, T. Liu, S. Cao and Y. Tsang, Sol. Energy Mater. Sol. Cells, 2017, 160, 43–53 CrossRef CAS.
- S. M. Jeong and S. D. Kim, Ind. Eng. Chem. Res., 2000, 39(6), 1911–1915 CrossRef CAS.
- J. Zhang, K. Dong, W. Luo and H. Guan, ACS Omega, 2018, 3, 6206–6216 CrossRef CAS.
- J. G. Skofronick, J. P. Toennies, F. Traeger and H. Weiss, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 035413 CrossRef.
- W. Hu, F. Donat, S. A. Scott and J. S. Dennis, RSC Adv., 2016, 6, 113016–113024 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08754h |
| This journal is © The Royal Society of Chemistry 2020 |