
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of functionalized poly(1-butene) from 1,2-polybutadiene via sequential thiol-ene click reaction and ring-opening polymerization
Lin Tian a,
Jin Gub,
Hao Zhang*b and
Bo Dong*c
a,
Jin Gub,
Hao Zhang*b and
Bo Dong*c
aDepartment of Electrical Engineering, Jilin Technology College of Electronic Information, Jilin, 132021, PR China
bCollege of Food Science and Engineering, National Engineering Laboratory for Wheat and Corn Deep Processing, Jilin Agricultural University, Changchun 130118, PR China. E-mail: zhanghao3318@sina.com
cCollege of Materials Science and Engineering, Jilin University, Changchun 130022, PR China. E-mail: bdong0118@126.com
First published on 25th November 2020
Abstract
Poly(1-butene) is a kind of unique poly(α-olefin) material that displays exceptional creep resistance and environmental stress cracking resistance, and therefore currently finds wide application in the fields of packaging, films, pipes, etc. However, very few current researchers are paying attention to functional poly(1-butene) despite its great research significance, perhaps due to the general paucity of catalytic systems for synthesizing this material. Therefore, in the present study, we set out to develop an alternative method to prepare polar poly(1-butene)s. Specifically, 1,2-enriched poly(1,3-butadiene), used as a starting material, was partially hydrogenated to afford a quasi-poly(1-butene) polymer containing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds. These double bonds were further modified by subjecting the quasi-poly(1-butene) polymer to a thiol-ene reaction in the presence of thiol compounds, and a series of polar poly(1-butene)s that bore significantly improved surface properties were obtained. By using hydroxyl-containing thiol compounds, ring-opening polymerization (ROP) of ε-caprolactone was further implemented. Here, polar poly(ε-caprolactone) was incorporated as side chains, and we were able to control the chain length by adjusting the feeding ratio. The water contact angles of the resultant polymers, i.e., those containing the poly(ε-caprolactone) side chains, were as low as 59.4°, indicating a greater hydrophilicity resulting from the incorporation of these side chains.
C double bonds. These double bonds were further modified by subjecting the quasi-poly(1-butene) polymer to a thiol-ene reaction in the presence of thiol compounds, and a series of polar poly(1-butene)s that bore significantly improved surface properties were obtained. By using hydroxyl-containing thiol compounds, ring-opening polymerization (ROP) of ε-caprolactone was further implemented. Here, polar poly(ε-caprolactone) was incorporated as side chains, and we were able to control the chain length by adjusting the feeding ratio. The water contact angles of the resultant polymers, i.e., those containing the poly(ε-caprolactone) side chains, were as low as 59.4°, indicating a greater hydrophilicity resulting from the incorporation of these side chains.
Introduction
Poly(α-olefin)s (PAOs) such as poly(1-butene), poly(1-hexene), and poly(1-octene) currently find widespread applications in the fields of lubricants, impact modifiers, etc.1–7 However, as is the case for other petroleum-based polymer materials, PAOs display low surface energy levels and hence inferior properties with respect to adhesion, paintability, miscibility with inorganic fillers, etc.; and hence the goal of obtaining PAOs with improved hydrophilicity levels and developing functionalized PAOs is being pursued.8–10 Although copolymerization of α-olefin with polar monomers is a straightforward strategy to access a functionalized PAO, very few relevant catalytic systems are currently available due to the easy deactivation of active species by heteroatoms in polar comonomers.10–13 Therefore, post-functionalization has become considered to be a reliable and efficient alternative strategy.In the past few years, there have been extensive investigations on preparing functionalized poly(1-hexene)s and poly(1-octene)s, but very little attention has been paid to functionalized poly(1-butene), despite of its wide application in packaging materials, films, modifiers, etc., which is partially as result of, as stated above, the few available catalytic systems. 1,2-Enriched polybutadiene (PB) is structurally similar to poly(1-butene) when fully hydrogenated; and when it is partially hydrogenated, the remaining double bonds provide good reaction sites to be functionalized.14 So, in the present study, partially hydrogenated 1,2-PB was selected as a substrate to prepare functionalized poly(1-butene).
Compared with other carbon–carbon double bond modification methodologies such as oxidation,15–17 hydrozirconation,14,18 etc., the thiol-ene click reaction has attracted more attention because of its advantageous features, including (1) the ability to easily perform this reaction under mild conditions, (2) its broad substrate scopes, and (3) its good tolerance to diverse functionalities.19 These advantages facilitate the efficient incorporation of different functionalities into poly(1-butene) main chains. Moreover, in the past few decades, incorporation of the hydroxyl (–OH) group has garnered particular interest, not only because such use of –OH can increase the surface energy and simultaneously improve the adhesion properties of the polymer material, but also because –OH-functionalized polymer chains can be successfully employed as macroinitiators of ring-opening polymerizations (ROPs) of cyclic esters such as ε-caprolactone or lactides to prepare architecturally novel block,20–22 graft,23–25 and brush26,27 copolymers. So, herein, after hydroxyl-containing thiol compounds were subjected to the thiol-ene reaction to afford the –OH-functionalized quasi-poly(1-butene) chain structure, ROPs of ε-caprolactone from the –OH reaction site were subsequently carried out to eventually give poly(1-butene)-g-poly(ε-caprolactone) bearing polar poly(ε-caprolactone) groups as side chains (Scheme 1).
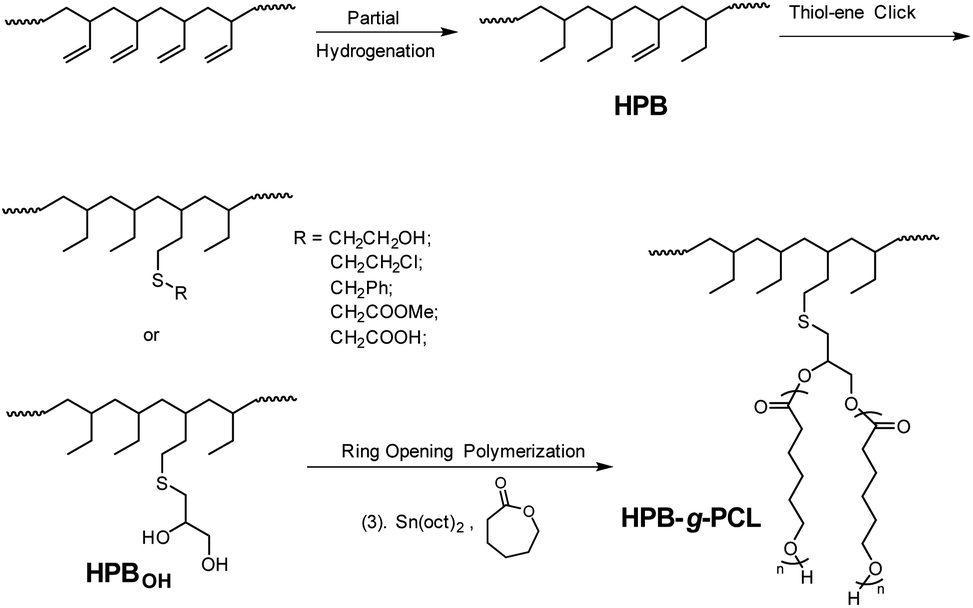 | ||
| Scheme 1 Functionalized poly(1-butene) accessed from 1,2-PB substrate, via sequential hydrogenation, thiol-ene click reaction, and ring-opening polymerization. | ||
Results and discussion
Atactic 1,2-PB with high vinyl content (1,2-content: 98.03%, Mn = 4.20 × 103, PDI = 1.09) for use as a substrate was prepared by performing anionic polymerization according to previous reports, specifically by using N,N-dipiperidine ethane (DPE) as a microstructure-regulating agent.14 Attempts to obtain higher-molecular-weight products by using reducing n-BuLi proved to be infeasible, probably due to side effects of DPE. Direct thiol-ene modification of 1,2-PB would give rise to intramolecular cyclization between the two neighboring vinyl groups;28 so, in order to obtain a structure similar to that of poly(1-butene), initial hydrogenations using the reducing agent p-toluenesulfohydrazide (TSH) was carried out to eliminate the adjacent vinyl structure. The hydrogenations were implemented under reflux conditions in the presence of TSH and simultaneously using tripropylamine (TPA) as a base to capture the resultant p-toluenesulfonic acid. In order to eventually obtain functionalized poly(1-butene) bearing different amounts of functionalities, hydrogenated 1,2-poly(butadiene)s (HPBs) showing different hydrogenation degrees (HDs), ranging from 6.83% to 100%, were obtained by adding different amounts of TSH under reflux conditions. The HDs were calculated according to eqn (1), and Fig. 1 shows the 1H NMR spectra for the resultant HPBs with different HDs. Here, for most of the samples, resonance signals at 4.93 and 2.12 ppm were observed and were assigned to the pendant H1 (CH2) and main-chain H2 (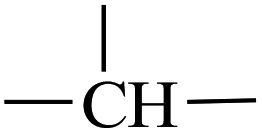 ) protons, respectively; these two signals monotonously decreased in peak intensity with increasing HD, and disappeared when 1,2-PB was completely hydrogenated. A resonance signal at 0.82 ppm was also observed for the hydrogenated samples and was assigned to the –CH3 protons of the ethyl group formed as a result of the hydrogenation; with increasing HD, this signal was observed to gradually increase in intensity.
) protons, respectively; these two signals monotonously decreased in peak intensity with increasing HD, and disappeared when 1,2-PB was completely hydrogenated. A resonance signal at 0.82 ppm was also observed for the hydrogenated samples and was assigned to the –CH3 protons of the ethyl group formed as a result of the hydrogenation; with increasing HD, this signal was observed to gradually increase in intensity.
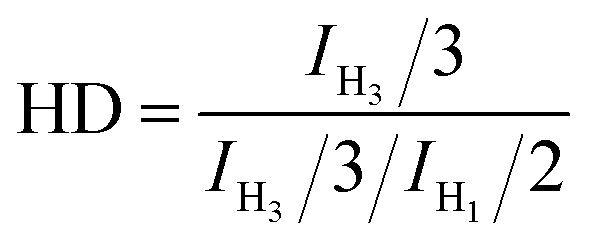 | (1) |
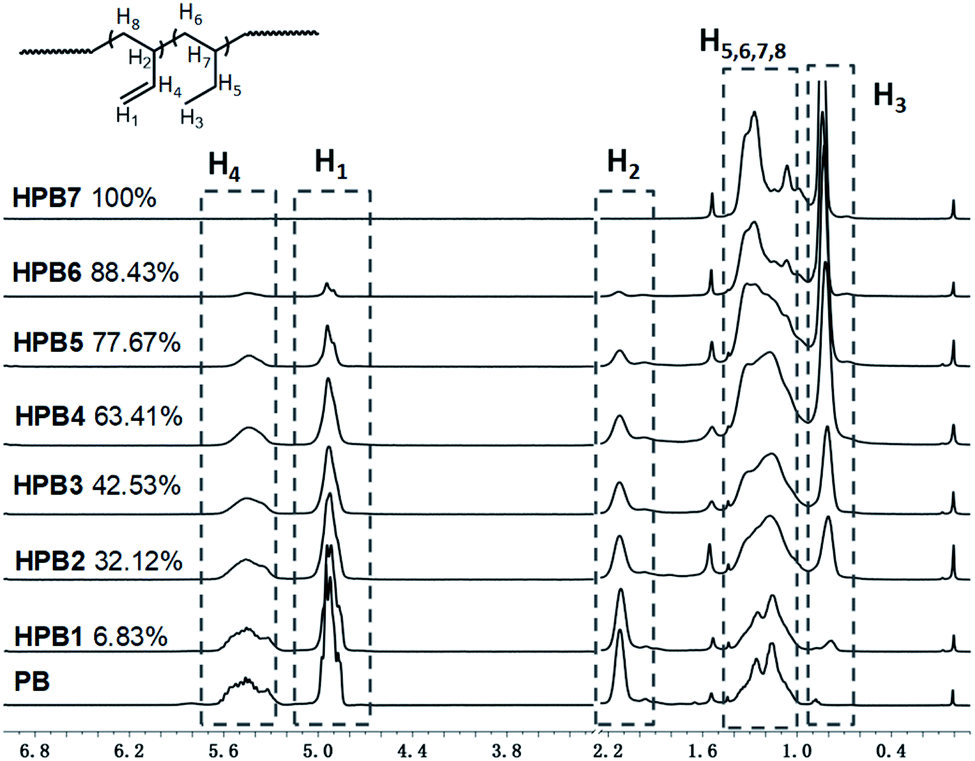 | ||
| Fig. 1 1H NMR spectra of HPBs with different degrees of hydrogenation (in CDCl3). | ||
With the HPBs bearing different amounts of remaining CC double bonds in hand, thiol-ene reactions in the presence of six different thiols were next carried out. The reactions were carried out for 2 h in THF solutions under inert argon atmosphere, 2,2′-azoisobutyronitrile (AIBN) was chosen as the radical source, and a 20-fold excess of thiol compounds with respect to the remaining CC double bonds in the HPB was applied. Using HPB6 with an HD of 88.43% as a representative example, the substrate reacted smoothly and efficiently with 2-mercapto-ethanol, 3-chloro-1-propanethiol, phenylmethanethiol, and methyl thioglycolate with functionalization degrees (FDs) of 90%, 92%, 96%, and 83%, respectively. Thiol-ene reactions between HPB6 and mercapto-1,2-propanediol or mercaptoacetic acid, however, were indicated to be less efficient under identical conditions, affording FDs of only 51% and 44%, respectively. Conducting the reactions with longer reaction times showed only little improvement of the FDs, which might have been due to the poor reactivities or poor solubilities of these two substrates in THF. All of these functionalized polymers were well identified by their 1H NMR spectra. For example, for mercapto-1,2-propanediol-functionalized HPB, resonance peaks were observed at 3.51–3.58 ppm and 3.68–3.83 ppm, and these peaks were assigned to methylene protons (a and c) and methyne proton (b), respectively (Fig. 2). Moreover, the product of the thiol-ene reaction yielded CC proton resonance peaks clearly weaker than those of the reactant. The thiol-ene reaction also showed little influence on the molecular weight of the polymer. As summarized in Table 1, the molecular weights of all of the obtained polymers were distributed around 4.6 × 103 Da.
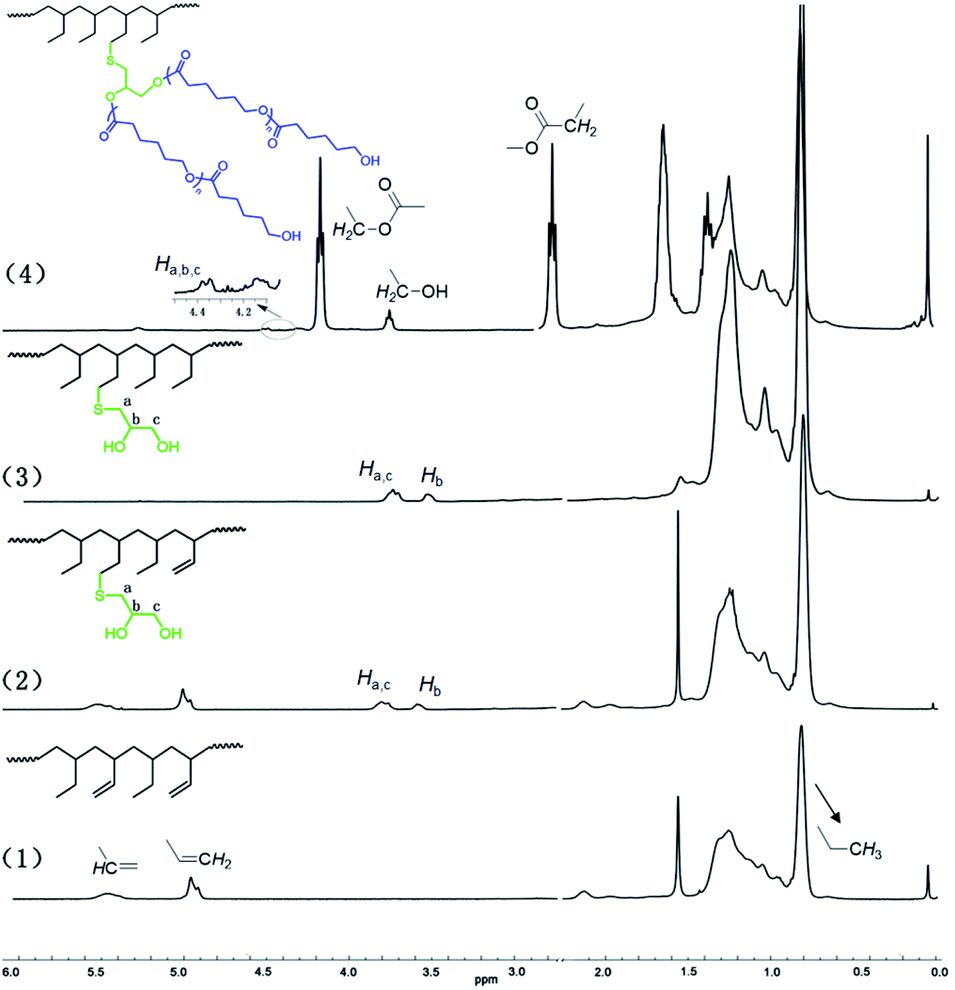 | ||
| Fig. 2 1H NMR spectra of (1) HPB6, (2) mercapto-1,2-propanediol-functionalized HPB6, (3) saturated mercapto-1,2-propanediol-functionalized HPB6, and (4) poly(1-butene)-g-poly(ε-caprolactone) in CDCl3. | ||
| Entry | Thiol | Mn (×103) | FD (%) | WCA (°) |
|---|---|---|---|---|
| 1 | 2-Mercapto-ethanol | 4.68 | 90 | 89.6 |
| 2 | 3-Chloro-1-propanethiol | 4.52 | 92 | 98.0 |
| 3 | Phenylmethanethiol | 4.48 | 96 | 103.8 |
| 4 | Methyl thioglycolate | 4.50 | 83 | 88.4 |
| 5 | Mercapto-1,2-propanediol | 4.69 | 51 | 81.2 |
| 6 | Mercaptoacetic acid | 4.81 | 44 | 70.2 |
| 7 | None | 4.58 | 0 | 108.1 |
We also attempted to carry out thiol-ene reactions on HPBs with greater vinyl contents. However, all of the resulting products were proved to be crosslinked polymers, even after our attempts to reduce the AIBN radical source, which hampered our efforts at further characterizing the obtained modified products.
Incorporation of the above polar thiol compounds can significantly improve the surface properties of the HPBs. The water contact angle (WCA) data are shown in Table 1. Compared to the non-polar HPB6 with a WCA of 108.1°, HPBs functionalized with 2-mercapto-ethanol, 3-chloro-1-propanethiol, methyl thioglycolate, and mercapto-1,2-propanediol showed lower WCA values of 89.6°, 98.0°, 88.4° and 81.2°, respectively. The greatest improvement was observed for the polymer incorporating mercaptoacetic acid; despite its lower functionalization degree, it still showed the smallest WCA, with a value of 70.2°.
For mercapto-1,2-propanediol-functionalized HPB, i.e., HPBOH, the pendant hydroxyl group can further serve as an initiation site for the ROP of cyclic esters. In the presence of 2 equivalents of stannous 2-ethyl-hexanoate (Sn(Oct)2), ε-caprolactone was successfully opened and grafted onto the mainchain of HPBOH. ε-Caprolactone monomer was, according to 1H NMR monitoring, completely consumed in the ROP reaction within 6 h. And due to the complete monomer conversions, we were able to regulate the poly(ε-caprolactone) side chain length by modulating the amount of ε-caprolactone. Fig. 3 shows the GPC curves of the resultant poly(1-butene)-g-poly(ε-caprolactone)s. Here, obviously different molecular weights of 4.69 × 103 to 5.23 × 103 and 6.94 × 103 Da were observed for different amounts of ε-caprolactone monomer added. Meanwhile, all of the GPC peaks were relatively narrow and unimodal, indicating relatively narrow molecular weight distributions (PDI values of 1.21 and 1.46) and indicating that all of the poly(ε-caprolactone)s were grafted onto the poly(1-butene) main chain rather than having formed ε-caprolactone homopolymers. Moreover, these narrow molecular weight distributions were also inconsistent with transesterifications having occurred, as such reactions would be expected to yield broad molecular weight distributions. Fig. 2 shows the NMR spectra acquired of the poly(1-butene)-g-poly(ε-caprolactone). Here resonance peaks corresponding to ε-caprolactone units can be clearly observed. For instance, resonance peaks can be observed at 4.02 and 2.26 ppm and they were assigned to the protons adjacent to the ester functionalities; and a triplet at 3.61 ppm was also observed, and was assigned to the terminal methylene protons in the terminally inserted ε-caprolactone. Besides, along with the incorporation of ε-caprolactone monomers, peaks corresponding to methylene protons (a and c) and the methyne proton (b) in the mercapto-1,2-propanediol units clearly downshifted from 3.51–3.58 ppm and 3.68–3.83 ppm to 4.12–4.44 ppm, with this downshift due to the strong electron-withdrawing effects of the carbonyl group, reflecting again the successful enchainment of ε-caprolactone monomers.
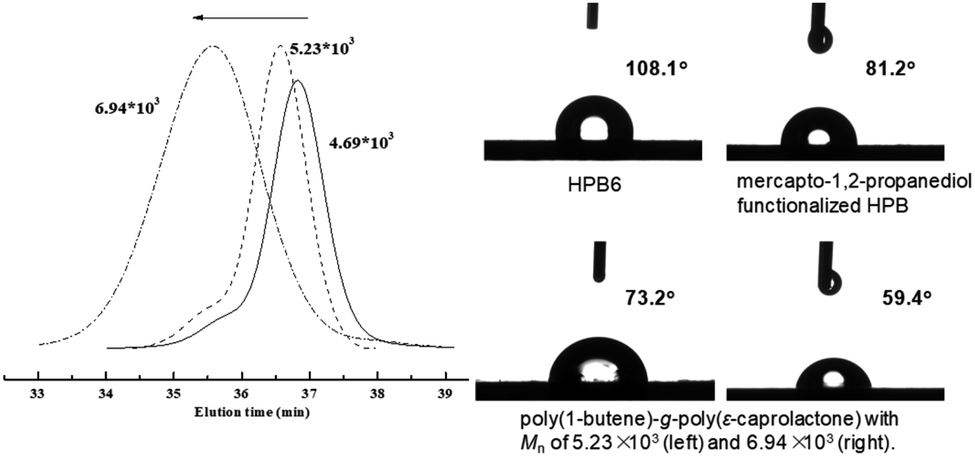 | ||
| Fig. 3 GPC curves of the poly(1-butene)-g-poly(ε-caprolactone)s and their water contact angles. | ||
These polymer products were also well characterized using FT-IR spectroscopy. With the incorporation of mercapto-1,2-propanediol, the vibration absorption signals that were originally located at 1637 cm−1 became obviously weakened (Fig. 4), and new stretching vibration signals of hydroxyl –OH at 3360 cm−1 appeared. After the ROP of ε-caprolactone, a characteristic ester group signal was also clearly observed at 1734 cm−1. Incorporation of poly(ε-caprolactone) was also found to significantly improve the hydrophilicity of poly(1-butene). As shown in Fig. 3, as the amount of poly(ε-caprolactone) was increased, the WCA dramatically decreased, from 108.1° to 73.2° and 59.4°, respectively.
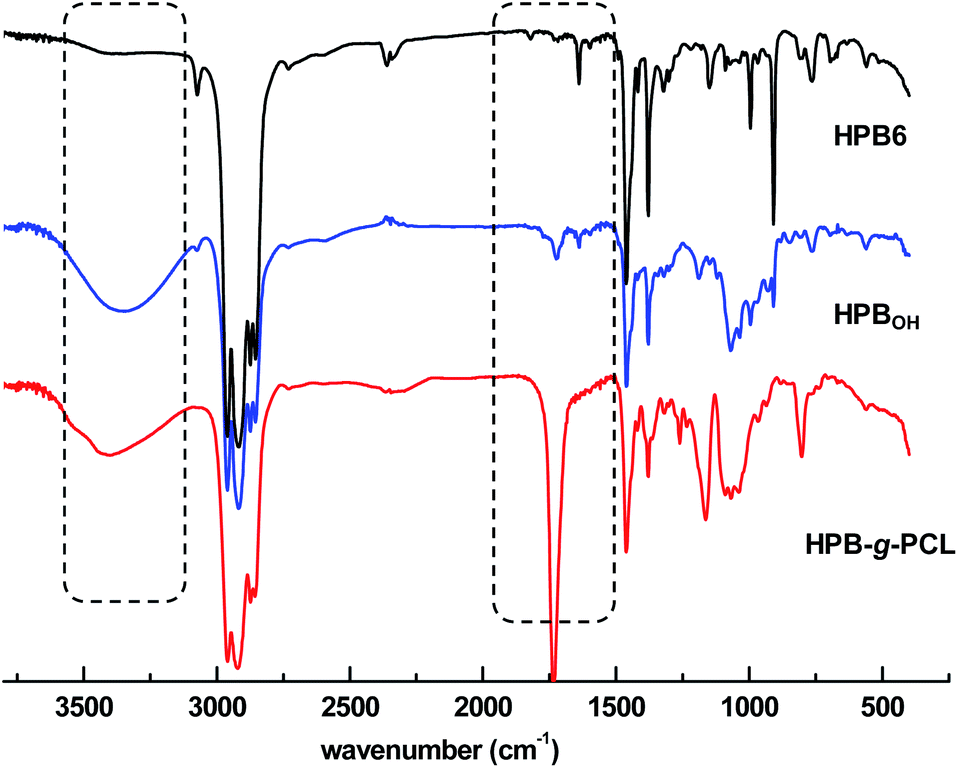 | ||
| Fig. 4 FT IR spectra of (1) HPB6, (2) mercapto-1,2-propanediol-functionalized HPB6, and (3) poly(1-butene)-g-poly(ε-caprolactone) in CDCl3. | ||
Conclusions
In summary, functional poly(1-butene) materials were prepared from 1,2-enriched poly(1,3-butadiene) by carrying out sequential partial hydrogenation and thiol-ene reactions. We were able to well regulate the functionalization degree by controlling the hydrogenation degrees or selecting an appropriate thiol compound. After functionalization, significantly improved surface properties, as evidenced from the water contact angles, were clearly observed. For hydroxyl-containing thiol compounds, after the thiol-ene reaction, the pendant hydroxyl groups were further used to initiate ring-opening polymerization of ε-caprolactone, to afford poly(1-butene)-g-poly(ε-caprolactone). We were also able to regulate the side chain length, i.e., the content of polar poly(ε-caprolactone), by choosing the feeding ratio. All of the obtained polar polymers were well identified using NMR and FT-IR spectroscopy.Experimental section
Materials and methods
All of the solvents (tetrahydrofuran (THF), hexane, toluene) were distilled from a sodium/potassium alloy with benzophenone. n-Butyllithium (1.6 M n-BuLi in hexane) was purchased from J&K Chemical. 1,3-Butadiene was obtained from Beifang Special Gas. 4-Methylbenzenesulfonhydrazide (TSH), tri(n-propyl)amine, 2-mercapto-ethanol, 3-chloro-1-propanethiol, phenylmethanethiol, methyl thioglycolate, mercapto-1,2-propanediol, tin(II) 2-ethylhexanoate (Sn(oct)2), and ε-caprolactone were purchased from Alfa Aesar and Sigma Aldrich. 1H NMR (400 MHz) spectra were recorded using a Varian Unity spectrometer in CDCl3 at ambient temperature using tetramethylsilane as an internal standard. FTIR spectra were recorded using a Bruker Vertex-70 FTIR spectrometer. The molecular weight (Mn) and molecular weight distribution (Mw/Mn) values of the polymers were determined at 30 °C by using a gel permeation chromatography (GPC) apparatus equipped with a Waters 515 HPLC pump, four columns (HMW 7 THF, HMW 6E THF × 2, HMW 2 THF), and a Waters 2414 refractive index detector. Tetrahydrofuran was used as the eluent at a flow rate of 1.0 ml min−1. The values of Mn and Mw/Mn were determined using polystyrene calibration. Contact angles were measured on a Ramé-hart 200-F1 standard goniometer at room temperature.C] = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and 10.9 g TPA ([TPA]/[CC] = 3:1); the resulting solution was refluxed for 48 h. After the reaction, the product was cooled to room temperature, and toluene was removed under reduced pressure until about 10 ml of solution remained; the resulting polymers were precipitated in ethanol and dried under vacuum, and further purification was performed by carrying out redissolution and reprecipitation three times.C]/[AIBN] ratio of 20/1/0.33. The resulting solution was degassed and heated to about 70 °C for 2 h under an argon atmosphere. After the reaction, THF was removed under reduced pressure, the resulting solution was poured into methanol, and the product was precipitated immediately. Further purification of the product was performed by carrying out redissolution and reprecipitation three times. After the thiol-ene reaction, not all of the double bonds were consumed; the remaining double bonds were further hydrogenated by performing a procedure similar to that described above.
:1) and 10.9 g TPA ([TPA]/[CC] = 3:1); the resulting solution was refluxed for 48 h. After the reaction, the product was cooled to room temperature, and toluene was removed under reduced pressure until about 10 ml of solution remained; the resulting polymers were precipitated in ethanol and dried under vacuum, and further purification was performed by carrying out redissolution and reprecipitation three times.C]/[AIBN] ratio of 20/1/0.33. The resulting solution was degassed and heated to about 70 °C for 2 h under an argon atmosphere. After the reaction, THF was removed under reduced pressure, the resulting solution was poured into methanol, and the product was precipitated immediately. Further purification of the product was performed by carrying out redissolution and reprecipitation three times. After the thiol-ene reaction, not all of the double bonds were consumed; the remaining double bonds were further hydrogenated by performing a procedure similar to that described above.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 32072169, 31801477).References
- T. C. M. Chung, Functionalization of Polyolefins, Academic Press, 1st edn, 2002 Search PubMed.
- R. G. Lopez, F. D'Agosto and C. Boisson, Prog. Polym. Sci., 2007, 32, 419–454 CrossRef.
- M. J. Yanjarappa and S. Sivaram, Prog. Polym. Sci., 2002, 27, 1347–1398 CrossRef CAS.
- J. Y. Dong and Y. L. Hu, Coord. Chem. Rev., 2006, 250, 47–65 CrossRef CAS.
- L. Guo, W. Liu and C. Chen, Mater. Chem. Front., 2017, 1, 2487–2494 RSC.
- W. Zhao, B. Dong, h. liu, Y. Hu and X. Zhang, Polym. Chem., 2020, 11, 2492–2501 RSC.
- Y. Chai, C. Wu, D. Liu, M. Run and D. Cui, Sci. China: Chem., 2019, 62, 761–766 CrossRef CAS.
- Y. Na and C. Chen, Angew. Chem., Int. Ed., 2020, 59, 7953–7959 CrossRef CAS.
- X. Song, L. Cao, R. Tanaka, T. Shiono and Z. Cai, ACS Macro Lett., 2019, 299–303, DOI:10.1021/acsmacrolett.9b00005.
- S. Liang, H. Zhang, R. Cong, H. Liu, F. Wang, Y. Hu and X. Zhang, RSC Adv., 2019, 9, 33465–33471 RSC.
- A. Hanifpour, N. Bahri-Laleh, M. Nekoomanesh-Haghighi and M. Karimi, J. Organomet. Chem., 2016, 819, 103–108 CrossRef CAS.
- K. Nomura, J. Liu, M. Fujiki and A. Takemoto, J. Am. Chem. Soc., 2007, 129, 14170–14171 CrossRef CAS.
- L. Fang, W.-P. Zhao, C. Han, Z. Chun-Yu, H. Liu, Y.-M. Hu and X.-Q. Zhang, Chin. J. Polym. Sci., 2019, 37, 462–470 CrossRef CAS.
- J. Zheng, F. Liu, Y. C. Lin, Z. J. Zhang, G. C. Zhang, L. Wang, Y. Liu and T. Tang, Macromolecules, 2012, 45, 1190–1197 CrossRef CAS.
- H. Zhang, Y. Li, C. Zhang, Z. Li, X. Li and Y. Wang, Macromolecules, 2009, 42, 5073–5079 CrossRef CAS.
- T. C. Chung, W. Janvikul, R. Bernard, R. Hu, C. L. Li, S. L. Liu and G. J. Jiang, Polymer, 1995, 36, 3565–3574 CrossRef CAS.
- Z. S. Yuan and M. Gauthier, Macromolecules, 2005, 38, 4124–4132 CrossRef CAS.
- Y. Lin, J. Zheng, F. Liu and T. Tang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1664–1671 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- C. J. Han, M. S. Lee, D. J. Byun and S. Y. Kim, Macromolecules, 2002, 35, 8923–8925 CrossRef CAS.
- J. O. Ring, R. Thomann, R. Mulhaupt, J. M. Raquez, P. Degee and P. Dubois, Macromol. Chem. Phys., 2007, 208, 896–902 CrossRef CAS.
- F. Wang, B. Dong, H. Liu, J. Guo, W. Zheng, C. Zhang, L. Zhao, C. Bai, Y. Hu and X. Zhang, Macromol. Chem. Phys., 2015, 216, 321–328 CrossRef CAS.
- N. Kashiwa, T. Matsugi, S. I. Kojoh, H. Kaneko, N. Kawahara, S. Matsuo, T. Nobori and J. I. Imuta, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3657–3666 CrossRef CAS.
- E. S. Park, M. N. Kim and J. S. Yoon, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 2561–2569 CrossRef CAS.
- M. A. Hong, L. Pan, B. X. Li and Y. S. Li, Polymer, 2010, 51, 3636–3643 CrossRef CAS.
- M. B. Runge, S. Dutta and N. B. Bowden, Macromolecules, 2005, 39, 498–508 CrossRef.
- W. Z. Yuan, J. Y. Yuan, F. B. Zhang, X. M. Xie and C. Y. Pan, Macromolecules, 2007, 40, 9094–9102 CrossRef CAS.
- N. ten Brummelhuis, C. Diehl and H. Schlaad, Macromolecules, 2008, 41, 9946–9947 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |