
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy on the performance of NiO/ZnxZr1−x catalysts for CO2 hydrogenation
Shuai Chang†
 ad,
Wenshuo Mao†cd,
Wei Naab,
Wengui Gao*ab,
Gaocheng Qucd and
Hua Wangabd
ad,
Wenshuo Mao†cd,
Wei Naab,
Wengui Gao*ab,
Gaocheng Qucd and
Hua Wangabd
aFaculty of Metallurgy and Energy Engineering, Kunming University of Science and Technology, Kunming 650093, China. E-mail: gao_wengui@126.com
bState Key Laboratory of Complex Nonferrous Metal Resources Clean Utilization, Kunming University of Science and Technology, Kunming 650093, China
cFaculty of Chemical Engineering, Kunming University of Science and Technology, Kunming 650093, China
dEngineering Research Center of Metallurgical Energy Conservation and Emission Reduction, Ministry of Education, Kunming University of Science and Technology, Kunming 650093, China
First published on 25th November 2020
Abstract
The NiO/ZnxZr1−x (x represents the molar mass of Zn) catalyst was prepared by the impregnation method and tested in CO2 methanation. The activity results show that NiO/Zn0.3Zr0.7 has a higher CO2 conversion rate and methane selectivity than NiO/ZnO and NiO/ZnO–ZrO2. Combined with N2 adsorption–desorption, H2-TPR, CO2-TPD, H2-TPD, XRD, TEM, XPS and FTIR and other characterization methods, the physical and chemical properties of NiO/ZnO–ZrO2 were studied. The incorporation of ZnO into NiO/ZrO2 forms a ZnO–ZrO2 solid solution, and the combination of the solid solution weakens the interaction between NiO and the oxide support, thereby promoting the reduction and dispersion of NiO. The H2-TPR experiment results show that, because ZnO–ZrO2 forms a solid solution, NiO is better dispersed on the surface, resulting in a significant reduction in the reduction temperature of NiO. Using FTIR to conduct CO2 adsorption and methanation experiments on NiO/ZnxZr1−x to determine the adsorbed species and intermediates, the results show that CO2 methanation follows the formate pathway.
1. Introduction
Carbon dioxide is the main gas causing the greenhouse effect. The greenhouse effect has led to global warming, melting of Antarctic glaciers, and frequent disastrous climates, causing a series of environmental problems. Therefore, there should be some strategies to reduce the accumulation of this gas in the atmosphere. Dorner et al.1 reviewed the modified Fischer–Tropsch catalysts in converting CO2 to value-added hydrocarbons. Porosoff et al.2 provided an overview of the various catalysts used for CO2 conversion to CO, CH3OH, and light alkanes and olefins. Converting carbon dioxide into methane is also a method of resource utilization of carbon dioxide while reducing carbon dioxide in the air.3,4 Since methane is not only an excellent fuel, it can also be converted into olefins and other fossil fuels.Methane is the main component of natural gas, a clean, efficient fossil fuel and energy carrier with low carbon emissions, high heat output and easy complete combustion compared to oil and coal, and is safe to use and transport.5–7 Methanation of carbon dioxide is also called the Sabatier reaction, which was first reported by Paul Sabatier in 1902.8
Since the reaction was first developed, it has attracted widespread attention. In this reaction, the main catalyst systems studied include noble metal catalysts and other transition metal catalysts,9–12 among which Ni, Rh and Ru are considered to have the best catalytic performance for CO2 hydromethanation.13,14 Although Rh and Ru have higher activity and methane selectivity, their high price limits their industrial use.15,16 Nickel-based supported catalysts have attracted great interest in the hydrogenation of CO2 because of its high activity and low price, which have been extensively studied in this reaction system.17 Al2O3, TiO2, SiO2, ZrO2, and CeO2 have been used as supports.12,18–21 Among these oxide support materials, ZrO2 has excellent properties, including high thermal stability, abundant surface active sites, basic sites and oxygen vacancies, so ZrO2 has been widely studied as a catalytic support.22–24 At present, the CO2 conversion rate of NiO/ZrO2 catalyst is still not ideal.
ZnO is a good additive in CO2 hydrogenation, but there is no conclusion about adding ZnO as a promoter to NiO/ZrO2 catalyst system for CO2 hydromethanation. There are also few reports on the reaction path of NiO/ZnxZr1−x catalytic system to CO2 methanation and the species formed on the catalyst surface. This article performed performance tests on NiO/ZrO2, NiO/Zn0.3Zr0.7 and NiO/ZnO catalysts. Fourier-transform infrared spectroscopy (FTIR) studies further provide insights into the methane formation pathways and surface intermediates of NiO/ZnxZr1−x catalysts.
2. Experimental
2.1 Preparation of catalysts
NiO/ZrO2, NiO/Zn0.3Zr0.7 and NiO/ZnO catalysts were prepared by co-precipitation and impregnation methods. Taking NiO/Zn0.3Zr0.7 three-component catalyst as an example, the first step is the preparation of the support. A certain amount of Zn(NO3)2·6H2O and Zr(NO3)4·5H2O was dissolved in deionized water according to the substance ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 to prepare a nitrate mixed solution with a metal ion concentration of 1 mol L−1, denoted as A; in addition, weigh a certain amount of Na2CO3 and dissolve in deionized water to prepare a solution of equal concentration, denoted as B. Under stirring at 80 °C, add A and B dropwise to the beaker at the same time, adjust the dropping speed of A and B to control the pH of the solution to about 7. The mixture was stirred continuously in an 80 °C water bath for 3 h, then removed and cooled at room temperature for 2 h. Then it was washed thoroughly with deionized water. Finally, the new composite supports were obtained by drying at 110 °C for 12 h and then calcining at 500 °C for 3 h.
:7 to prepare a nitrate mixed solution with a metal ion concentration of 1 mol L−1, denoted as A; in addition, weigh a certain amount of Na2CO3 and dissolve in deionized water to prepare a solution of equal concentration, denoted as B. Under stirring at 80 °C, add A and B dropwise to the beaker at the same time, adjust the dropping speed of A and B to control the pH of the solution to about 7. The mixture was stirred continuously in an 80 °C water bath for 3 h, then removed and cooled at room temperature for 2 h. Then it was washed thoroughly with deionized water. Finally, the new composite supports were obtained by drying at 110 °C for 12 h and then calcining at 500 °C for 3 h.
The impregnation method synthesizes nickel catalyst supported on metal oxide. Ni(NO3)2·6H2O is dissolved in deionized water at room temperature. The support material is added to the aqueous solution, and the resulting suspension is heated to 80 °C and stirred until all the water has evaporated. Finally, it was dried at 110 °C for 12 h and calcined at 500 °C in air for 3 h. The Ni load is fixed at 10% by weight.
2.2 Catalyst characterizations
X-ray diffraction (XRD) method was used to characterize the bulk phase and structure of the catalyst. XRD measurements were performed on a desktop X-ray diffractometer (Rigaku D/max-Rc) equipped with a CuKα radiation source, at 40 kV and 100 mA, in the scanning angle (2θ) range of 10–90° at scanning speed of 2° min−1. Specimen was prepared by packing around 0.2 g sample powder in a glass holder. The X-ray photoelectron spectroscopy (XPS) spectrum is given on K-Alpha+.An Autosorb-iQ-C type physical adsorption instrument purchased from Kantar was used to measure the nitrogen adsorption and desorption curve of the catalyst. Before the measurement, the sample was degassed at 300 °C for 3 h to remove physically absorbed water and impurities on the surface. The specific surface area was calculated by the Brunauer–Emmett–Teller (BET) method, and the pore size distribution was analyzed by the Barrett–Joyner–Halenda (BJH) method.
Temperature-programmed H2 reduction (H2-TPR), temperature-programmed CO2 desorption (CO2-TPD) and temperature-programmed H2 desorption (H2-TPD) were carried out on a Quantachrome automated chemisorption analyzer (chemBET pulsar TPR/TPD). For H2-TPR, 0.1 g catalyst of pellets (20–40 mesh) of the catalyst were loaded in a quartz U-tube and heated from room temperature to 450 °C at the heating rate of 10 °C min−1 and maintained for 30 min in He flow. Then, the samples were cooled down to room temperature, followed by heating to 800 °C in a 10% H2/Ar flow at the flow rate of 30 ml min−1. The H2 consumption in H2-TPR process was continuously monitored as a function of increasing temperature using a thermal conductivity detector (TCD). The catalyst was firstly reduced, and then pre-treated in He at 300 °C for 40 min. Next, the samples were saturated with 10% CO2/Ar at around 30 °C for 60 min and then flushed with He to evacuate the surface physisorbed CO2. The CO2-TPD experiment is carried out at a temperature of 30 to 500 °C. The procedure of H2-TPD is the same as CO2-TPD.
The materials formed on the catalyst surface were analyzed by FTIR. For CO2 methanation, first measure the catalyst at 450 °C with 10% H2/Ar flow rate at 30 ml min−1 for 1 h, and then purge with Ar for 30 min at 450 °C. Cool to the required temperature, obtain the background spectrum in the Ar atmosphere, and finally switch Ar to CO2/H2 (1:4) mixed gas and send it to the system. For CO2 adsorption, under the conditions of a temperature of 200–500 °C and a heating rate of 10 °C min−1, a 10% CO2/Ar gas with a flow rate of 30 ml min−1 was added, and the spectrum was collected. The wave number range is 800–4000 cm−1. The spectrum recorded 64 scans at a resolution of 4 cm−1.
The TEM of the catalyst was performed on a Tecnai G2 TF30 S-Twin high-resolution transmission electron microscope of FEI Company in the Netherlands.
2.3 Catalytic activity measurement
The catalytic performance test is carried out under atmospheric pressure. The catalyst powder was tableted and crushed in to uniform particles of 20–40 mesh, and 500 mg of the catalyst was put into a quartz tube with an inner diameter of 8 mm. The test temperature of 400 to 500 °C is strictly determined by a thermocouple inserted inside the catalytic bed. Before starting the evaluation, the sample was reduced at 450 °C for 2 h at a H2 flow rate of 45 ml min−1 to obtain an activated catalyst. After activating the catalyst, the temperature drops to the desired temperature. The feed gas was then flowed at a rate of 100 ml min−1 to reach a GHSV of 12000 h−1. The composition of the feed gas is as follows: CO2/H2 (1:4) as the balance gas. The gas products were analyzed by a gas chromatograph (Agilent Technologies 6890 USA) equipped with TCD detectors for H2, CO and CO2, and then FID detectors for CH4, CH3OH and other hydrocarbons. The catalytic tests were carried out at different temperatures, ranging from 400 to 500 °C. CO2 conversion and CH4 selectivity were calculated as follows: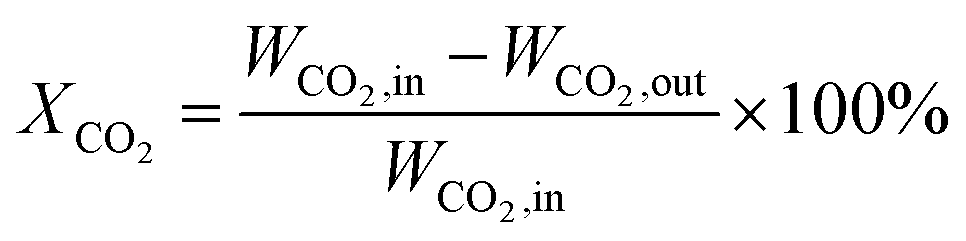
where WCO2, in and WCO2, out are the relative volume percentages of CO2 in the feed and exhaust gas, and WCH4, out and WCO, out are the relative volume percentages of CH4 and CO in the exhaust gas.
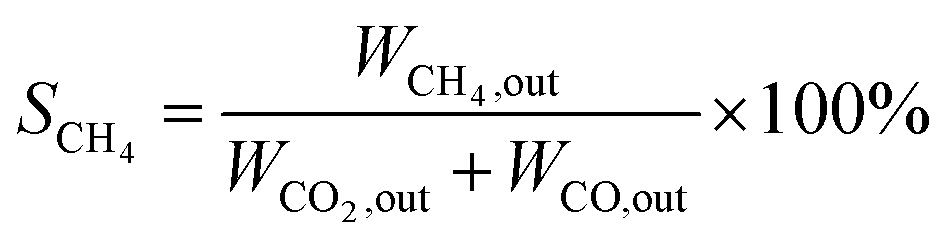
3. Results and discussion
3.1 Structural and textural properties
Fig. 1 is the XRD pattern of the NiO/ZnxZr1−x catalyst. X-ray diffraction (XRD) patterns show that ZrO2 prepared by co-precipitation is mainly a mixture of monoclinic and tetragonal phases. The diffraction peaks located at 30.52°, 35.38°, 50.88°, and 60.5° are related to the existence of the tetragonal t-ZrO2. The characteristic diffraction peaks of monoclinic phase m-ZrO2 can be observed at 2θ for 24.16°, 28.24°, 31.5°, 34.12° and 49.34°. The characteristic diffraction peaks of NiO were observed at 37.16° and 43.16°. The characteristic diffraction peaks of ZnO can be observed at 2θ, 31.28°, 34.44°, 36.26°, 47.56° and 56.52°. The addition of ZnO (30%) to ZrO2 causes ZrO2 to change from monoclinic to tetragonal. This indicates that ZnO–ZrO2 solid solution is formed when the ZnO content is 30%.25 It can be seen in the enlarged image that when the Zn concentration is 30%, the XRD of ZrO2 shifts to a higher angle. These facts further confirm the conclusion that ZnO–ZrO2 is in a solid solution state.26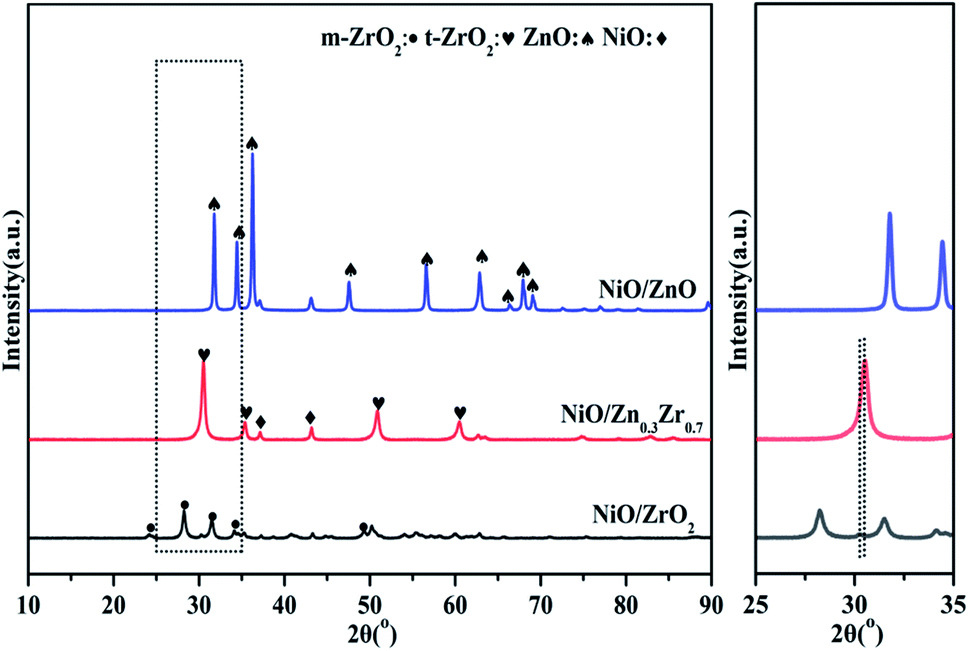 | ||
| Fig. 1 XRD curves of NiO/ZnxZr1−x catalysts. | ||
Fig. 2 is a TEM image of NiO/ZnxZr1−x after reduction. Considering that the ionic radius of Zn2+ (0.74 Å) is smaller than that of Zr4+ (0.82 Å).27 When ZnO is doped into the lattice of ZrO2, the (011) crystal surface pitch of ZrO2 changes from 0.295 nm to 0.264 nm and the XRD of the (011) pitch of ZrO2 moves to a larger angle, this information further establishes that ZnO–ZrO2 exists in a solid solution form.25 These facts further confirm the conclusion that ZnO–ZrO2 is in a solid solution sate. Fig. 2b–d are the distribution of Ni on ZnO–ZrO2, ZrO2 and ZnO respectively. It can be seen from the figure that the dispersion of Ni on the composite carrier ZnO–ZrO2 is the best, and the particle size of Ni is the smallest, with an average particle size of about 5 nm. Ni has good dispersibility on the ZrO2 carrier, and the average particle size is about 10 nm. On ZnO carriers, Ni has the largest particle size and poor dispersion, and the size of Ni particles directly affects the catalytic activity.
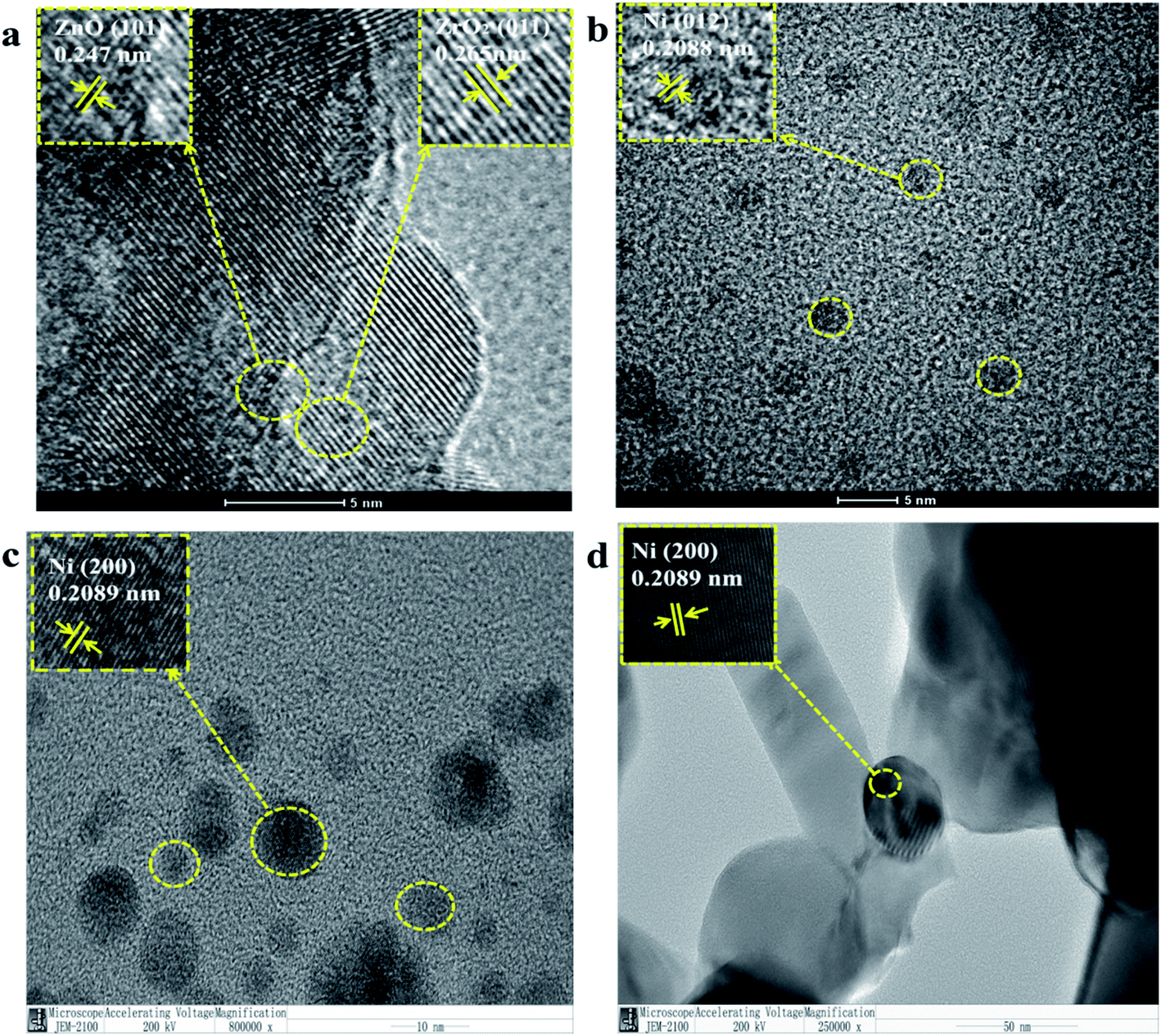 | ||
| Fig. 2 TEM image of NiO/ZnxZr1−x after reduction, (a and b) NiO/Zn0.3Zr0.7; (c) NiO/ZrO2; (d) NiO/ZnO. | ||
The N2 adsorption–desorption isotherm curve of the NiO/ZnxZr1−x catalyst is shown in Fig. 3. The detailed textural parameters were listed in Table 1. The isotherms of all the catalysts exhibited typical IV type isotherms with a H2-type hysteresis loop according to the IUPAC classification. As shown in Fig. 3, when Zn is added to the NiO/ZrO2 catalyst, the area of the hysteresis loop tends to increase, which means that the volume of the mesoporous structure increases. As shown in Table 1, the BET surface area (14.136 m2 g−1) and average pore volume (0.06 cm3 g−1) of NiO/Zn0.3Zr0.7 are larger than NiO/ZrO2 and NiO/ZnO. Using ZnO–ZrO2 as the carrier can provide a high specific surface area and ensure a high degree of dispersion of NiO on the carrier, so that the Ni/Zn0.3Zr0.7 catalyst has excellent catalytic performance.
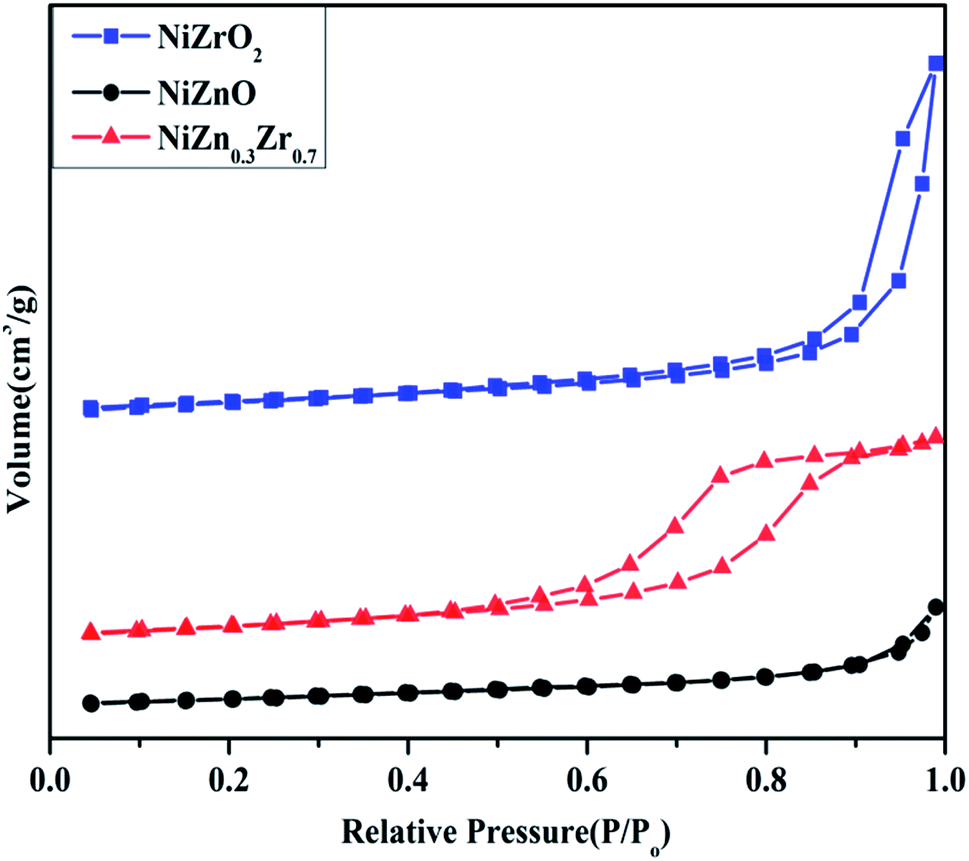 | ||
| Fig. 3 NiO/ZnxZr1−x catalyst adsorption and desorption curve of nitrogen. | ||
| Sample | SBET (m2 g−1) | Dp (nm) | Vp (cm3 g−1) |
|---|---|---|---|
| Ni/ZrO2 | 11.18 | 2.343 | 0.04 |
| Ni/Zn0.3Zr0.7 | 14.136 | 9.478 | 0.06 |
| Ni/ZnO | 7.588 | 3.073 | 0.01 |
3.2 XPS characterization
The electronic structure of NiO/ZnxZr1−x catalyst was analyzed by XPS. The spectrum in Fig. 4a shows that the Ni catalyst is mainly composed of Ni in the form of metal, oxide and hydroxide.28,29 The three peaks of NiO/ZrO2 at 851.48, 854.68 and 860.58 eV can be attributed to Ni0/NiO, Ni(OH)2 and shake-up satellite peak, respectively.30,31 The slight shift of the binding energy of NiO/ZrO2 and NiO/ZnO to lower values indicates that the size of Ni particles is larger.32 This indicates that the ZnO–ZrO2 composite carrier can better disperse NiO, which is consistent with the results of TEM. The XPS spectrum of the O 1s region of the catalyst is shown in Fig. 4b. The photoelectron peak of BE near 530 eV is attributed to the surface lattice oxygen (OL) of ZnO or ZrO2, while the second photoelectron peak at 531 eV is attributed to surface adsorption oxygen (OA).33 It can be seen from Fig. 4b that the OL and OA peaks of NiO/ZnO are displayed at 530.02 and 531.48 eV, while the NiO/ZrO2 peaks are displayed at 529.31 and 530.98 eV. For the Ni catalyst modified with ZnO–ZrO2 composite support, the OL and OA peaks are 529.60 and 531.58 eV, respectively, and gradually move to lower binding energy with the increase of Zr concentration. In the NiO/ZnxZr1−x catalytic system, the number of oxygen vacancies will increase with the increase of the Zr loading.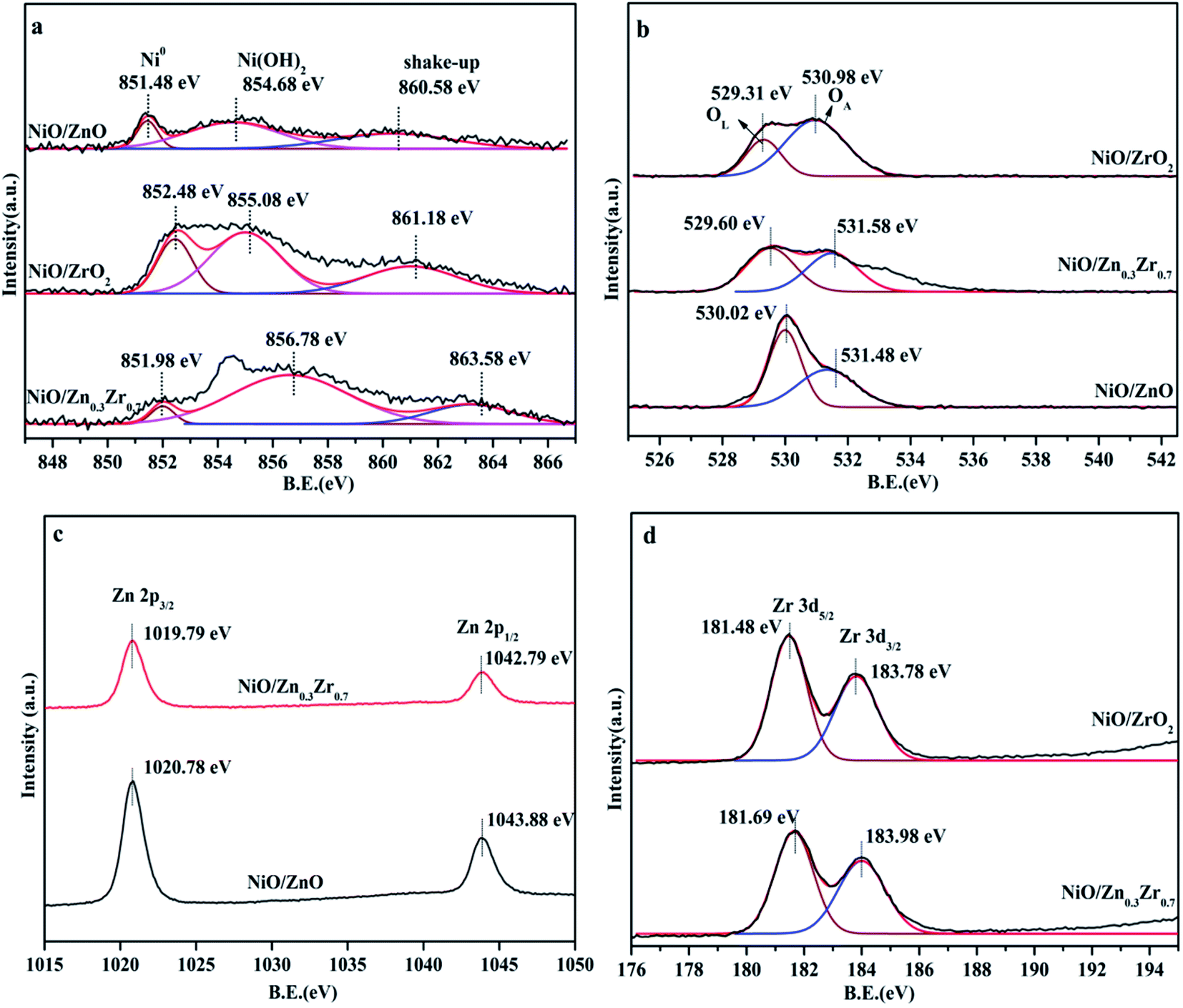 | ||
| Fig. 4 XPS spectrum of NiO/ZnxZr1−x catalyst, (a) Ni 2p; (b) XPS spectrum of O 1s; (c) Zn 2p; (d) Zr 3d. | ||
In Fig. 4c, all reduced Zn-containing catalysts show two strong peaks centered at 1019.79 eV and 1042.79 eV, which are attributed to Zn 2p3/2 and Zn 2p1/2, respectively. This indicates that the zinc species in the reduced Zn-containing catalyst mainly exist in the state of Zn2+.34 In Fig. 4d, the peaks at about 181.69 eV and 183.98 eV of the two centers of the reduced Zr-containing catalyst are assigned to Zr 3d5/2 and Zr 3d3/2 of Zr4+, respectively.35 The Zr 3d peak of the reduced Zr-containing catalyst also shows a slight shift to higher binding energy, which may be due to the fact that the incorporation of Zn into the NiO/ZrO2 catalyst reduces the number of oxygen vacancies in the ZrO2 lattice.36,37
3.3 Effect of Zn/Zr ratio on the reducibility of catalysts
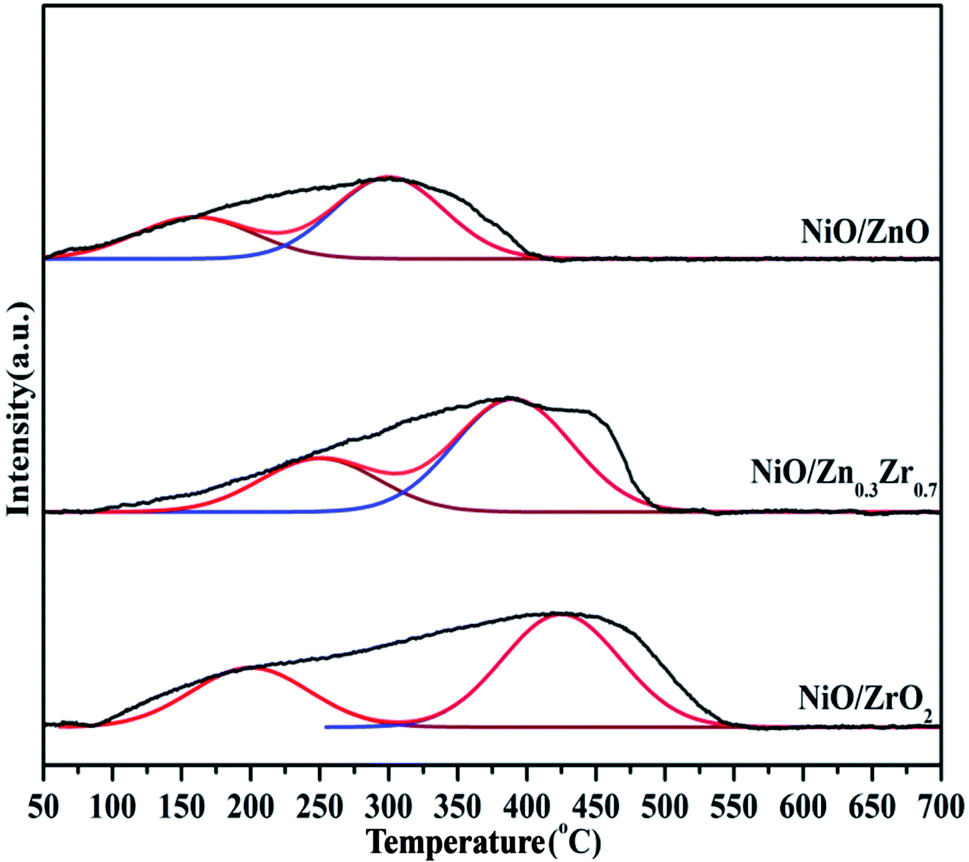
Temperature programmed reduction (TPR) analysis determined the reduction temperature and the type and strength of the metal–support interaction of the catalyst in different proportions to Zn/Zr. Fig. 5 shows the H2-TPR of the NiO/ZnxZr1−x catalyst. Since there was no hydrogen consumption on the ZnO–ZrO2 sample, this indicates that ZnO and ZrO2 are not reducible under the conditions applied for the analysis, and thus the collected H2-TPR curve represents only the NiO surface. The NiO/ZnO catalyst showed a major reduction peak at 551 °C, which was due to the reduction of NiO species that had a strong interaction with the ZnO support. The NiO/ZrO2 catalyst showed two reduction peaks at 581 and 668 °C, respectively, corresponding to the weak and strong interactions between NiO species and ZrO2.20 When Zn is added to the NiO/ZrO2 catalyst, the reduction peak of Ni slowly shifts to the low temperature direction. The reduction temperature of NiO gradually decreases from 581 °C of NiO/ZrO2 to 453 °C of NiO/Zn0.3Zr0.7, which indicates that the addition of Zn species to the NiO/ZrO2 catalyst forms a ZnO–ZrO2 solid solution and weakens the NiO–ZrO2. The interaction promotes the reduction of NiO.38 It shows that the combination of ZnO and ZrO2 promotes the reduction of NiO species by weakening the interaction of NiO–ZrO2. Compared with the ZnO–ZrO2 composite carrier supported NiO catalyst, the reduction of NiO on ZrO2 is poor, which easily leads to insufficient active NiO sites. When the Zn/Zr molar ratio is 3:7, the catalyst has the best reduction effect on NiO.
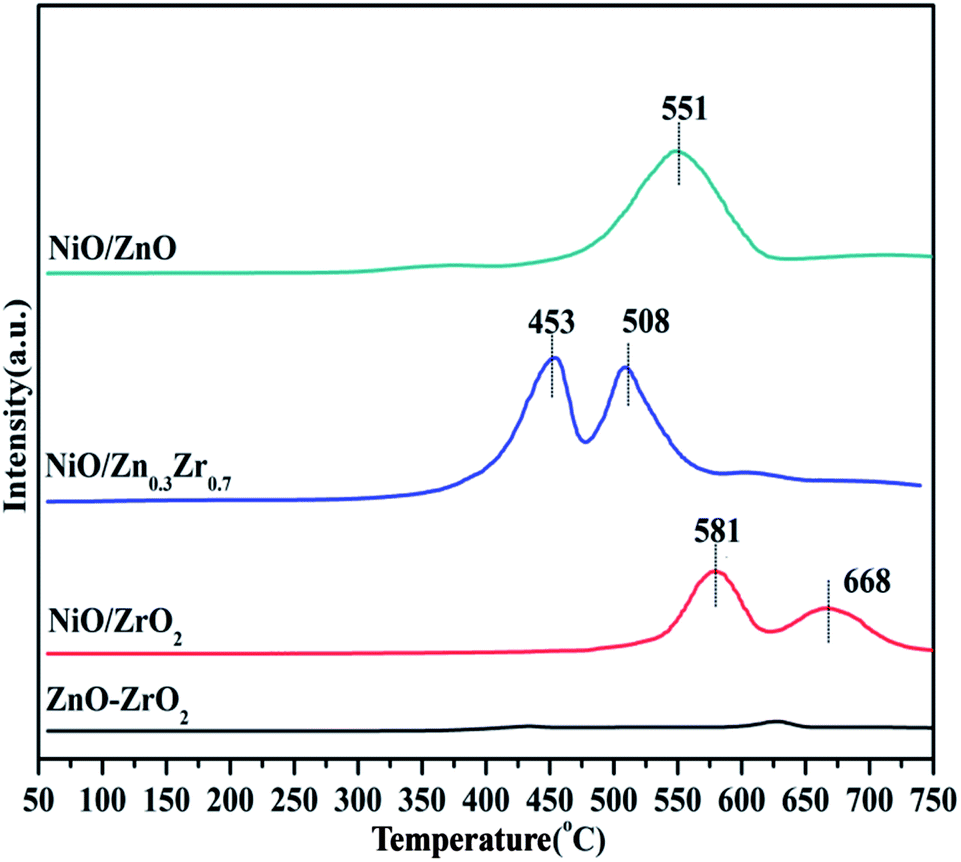 | ||
| Fig. 5 H2-TPR profiles of NiO/ZnxZr1−x catalysts. | ||
3.4 Effect of Zn/Zr ratio on the adsorption behaviors of catalysts
The H2 and CO2 adsorbed on the surface play a key role in the hydrogenation of CO2 to CH4. The basic sites of the NiO/ZnxZr1−x catalyst were measured by CO2-TPD. It can be seen from Fig. 6 that it is divided into three parts, including weak sites (<300 °C), moderate sites (300–500 °C) and strong sites (>500 °C). Weak, moderate and strong basic sites are respectively related to surface OH−, metal–oxygen pairs and low coordination surface O2−.39,40 The area and total area of the three corresponding basic sites are listed in Table 2. It is recognized that the CO2 methanation reaction is related to the moderately basic site.41 The weakly adsorbed CO2 cannot be fully activated, and the bond between C–O cannot be broken, and the too strong CO2 is difficult to desorb from the surface.42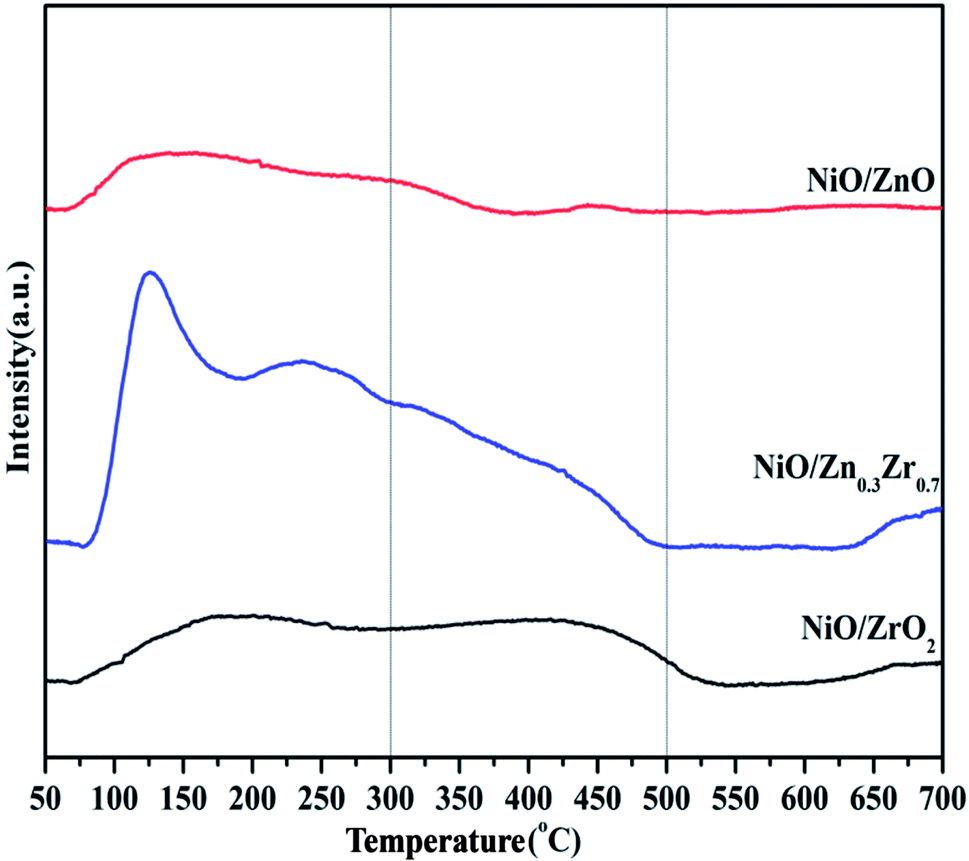 | ||
| Fig. 6 CO2-TPD profiles of NiO/ZnxZr1−x catalysts. | ||
| Sample | Weak sites (50–300 °C) | Moderate sites (300–500 °C) | Strong sites (500–700 °C) | Total sites |
|---|---|---|---|---|
| NiO/ZrO2 | 903.5 | 876.2 | 153.4 | 1933.1 |
| NiO/Zn0.3Zr0.7 | 2962.8 | 1303.5 | 167.9 | 4434.2 |
| NiO/ZnO | 745.7 | 82.5 | 21.6 | 846.8 |
There are abundant basic sites distributed on NiO/Zn0.3Zr0.7. Pan et al.39 proved that under the conditions of 300–700 °C, the adsorption of O2− sites and CO2 desorption significantly promoted the formation of carbonate, and promoted the subsequent hydrogenation reaction to form formate, and finally gaseous CH4. Therefore, the adsorption strength of CO2 on NiO/Zn0.3Zr0.7 is also one of the reasons for the significantly enhanced activity.
The H2-TPD of the NiO/ZnxZr1−x catalyst is shown in Fig. 7. The H2-TPD curves of all the catalysts show a low temperature desorption peak and a high temperature desorption peak. The low temperature peak is attributed to the desorption of hydrogen adsorbed on the metal surface, while the high temperature peak is attributed to the desorption of hydrogen overflowing from the hydrogen adsorbed on the oxide surface.7 The H2-TPD curve of NiO/ZnxZr1−x catalyst has desorption peaks at low temperature and high temperature. The low temperature can be attributed to the weak and strong adsorption of hydrogen on the nickel surface. The desorption peak at high temperature is attributed to the desorption peak of overflowing hydrogen.43 Under high temperature conditions, both NiO/ZrO2 and NiO/Zn0.3Zr0.7 have large desorption peaks, which indicates that there is a large amount of hydrogen overflow.7 It shows that both NiO/ZrO2 and NiO/Zn0.3Zr0.7 are beneficial to the overflow of atomic hydrogen on metal oxides.
 | ||
| Fig. 7 H2-TPD profiles of NiO/ZnxZr1−x catalysts. | ||
3.5 Catalytic activity study
The CO2 methanation performance of NiO/ZnxZr1−x catalyst is shown in Fig. 8. As shown in Fig. 8a, as the reaction temperature increases, the CO2 conversion rate of the catalyst gradually increases. In particular, at the same reaction temperature, the NiO/Zn0.3Zr0.7 catalyst showed the highest catalytic activity. The single NiO/ZrO2 catalyst has relatively low catalytic activity and CH4 selectivity, which is consistent with the results of Perkas et al.44 The traditional NiO/ZrO2 catalyst has a small surface area and pore size, resulting in a decrease in CO2 methanation performance. Fig. 8b shows the CH4 and CO selectivity of the NiO/ZnxZr1−x catalyst. As the temperature increases, only CH4 and CO are not detected as other by-products. It can be seen from the figure that NiO/Zn0.3Zr0.7 has the highest CH4 selectivity at the same temperature. The CO produced in the NiO/ZnO catalyst is more selective than CH4, which is consistent with the results of in situ FTIR, indicating that the NiO/ZnO catalyst is more conducive to the hydrogenation of CO2 to CO. Fig. 8c is the stability test of the NiO/ZnxZr1−x catalyst. It can be seen from the figure that the CO2 conversion rate and CH4 selectivity remain basically unchanged as time increases. The NiO/Zn0.3Zr0.7 catalyst showed the highest catalytic activity and CH4 selectivity at the same reaction temperature. For NiO/Zn0.3Zr0.7 catalyst, at 500 °C, the best CO2 conversion rate is 79%, and the CH4 selectivity is 94%. Therefore, in the NiO/ZnxZr1−x catalytic system, NiO/Zn0.3Zr0.7 is more helpful for CO2 hydrogenation to generate CH4.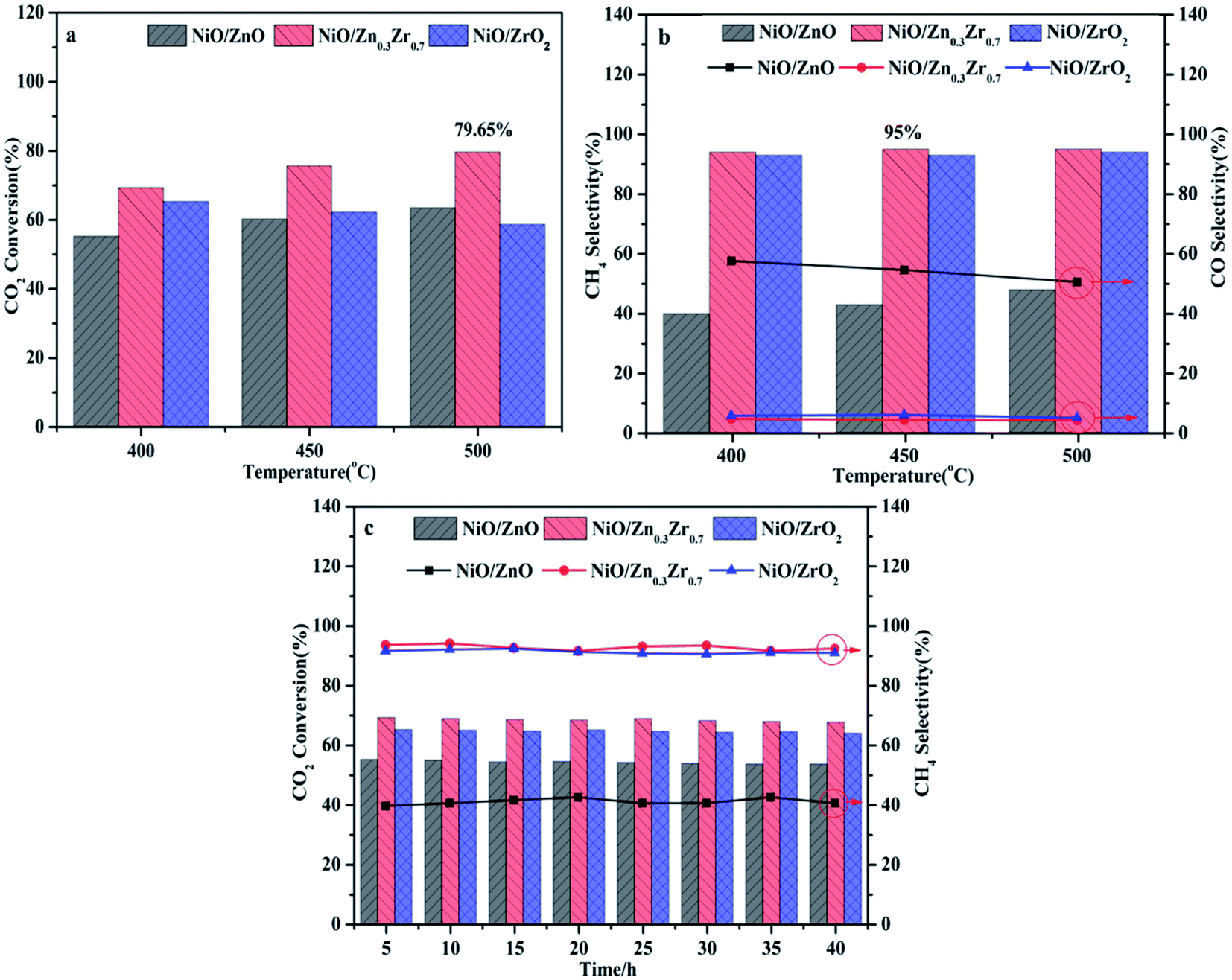 | ||
| Fig. 8 NiO/ZnxZr1−x catalyst activity test, (a) CO2 conversion rate; (b) CH4 selectivity; (c) catalyst stability. | ||
3.6 In situ FTIR analysis
According to previous reports, the bands observed in the range of 1700–1200 cm−1 can be attributed to carbonate-like substances adsorbed on the support.45–48 Since the thermal stability of carbonates depends on the type of carbonate, comparing their thermal stability is one way to distinguish one from the other. Fig. 9 shows the CO2 adsorption spectra of NiO/Zn0.3Zr0.7 and NiO/ZrO2 in the temperature programmed range of 200–500 °C. It appears at 1697, 1375 and 1205 cm−1 in Fig. 9a, which is attributed to bicarbonate. As the temperature increases, the intensity of the bicarbonate band decreases monotonically. However, formate 1595 and 1419 cm−1, bidentate carbonate 1674 cm−1, and monodentate carbonate 1641, 1519 and 1286 cm−1 appeared. At the same time, no disappearance above 400 °C indicates that its thermal stability is very good. It is worth noting that the CO adsorbed on the Ni surface appears at 2173 cm−1. The adsorbed CO should be formed by the direct decomposition of CO2 on the NiO surface (CO2 → CO + O).49 Fig. 9b is the CO2 adsorption spectrum of NiO/ZrO2. This is the peak of monodentate carbonate at 1323 cm−1, the peak of formate at 1423 cm−1, and the peak of bidentate carbonate at 1625 cm−1. As the temperature increased, the peaks of monodentate and bidentate carbonates remained almost unchanged, but the peaks of formate decreased significantly. The amount of formate directly affects the catalytic performance of CO2 methanation, which is the reason why the catalytic effect of NiO/ZrO2 catalytic system deteriorates at high temperature, which is consistent with the catalyst activity test results.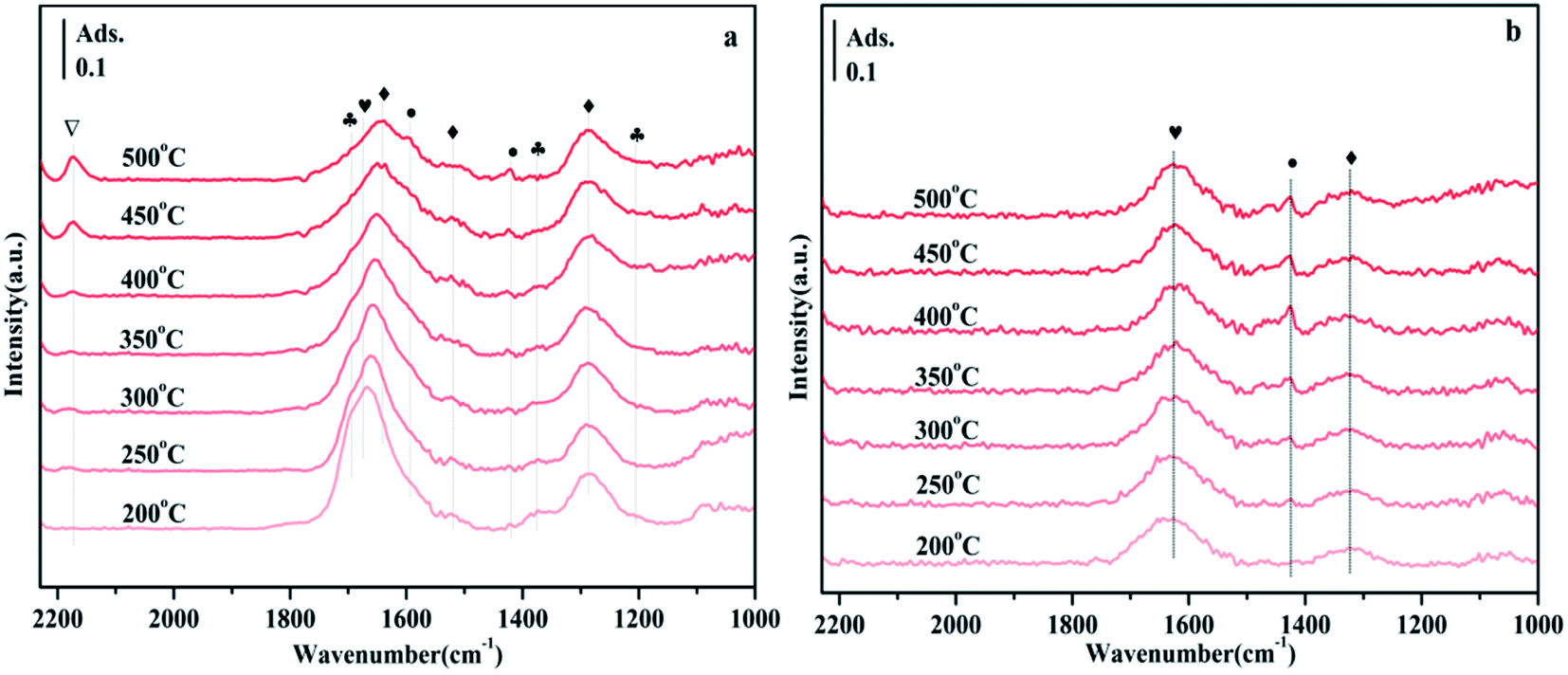 | ||
| Fig. 9 FTIR spectra of NiO/Zn0.3Zr0.7 and NiO/ZrO2 in 10% CO2/Ar gas with a flow rate of 30 ml min−1 and a heating rate of 10 °C min−1 during the temperature-programmed CO2 adsorption process. When CO2 adsorption reaches a steady state, each spectrum at the desired temperature (200–500 °C) will be collected. Symbol definition: bicarbonate (♣), monodentate carbonate (♦), bidentate carbonate (♥), formate (●), CO (▽), (a) NiO/Zn0.3Zr0.7; (b) NiO/ZrO2. | ||
Fig. 10 shows the FTIR spectra of CO2 methanation on NiO/Zn0.3Zr0.7 and NiO/ZnO catalysts. Fig. 10a is the FTIR spectrum of NiO/Zn0.3Zr0.7 catalyst in CO2/H2 (1:4) atmosphere with temperature change. At 300 °C, typical CO2 adsorbents, 1317, 1359, 1569, and 1614 cm−1 can be observed in Fig. 9a. As the temperature increased, the formate gradually decreased, while the peaks at 1305 and 3016 cm−1 gradually increased. These changes indicate that CH4 is produced by hydrogenation of formic acid.46 These are the peaks of CO at 2113 and 2190 cm−1, indicating that reverse water-gas shift (RWGS) have occurred on the catalyst. Fig. 10b is the FTIR spectrum of NiO/ZnO catalyst in CO2/H2 (1:4) atmosphere with temperature changes. There are CO peaks at 2113 and 2185 cm−1, and CH4 peaks at 3016 cm−1. It can be seen from Fig. 10b that the CO peak is obviously stronger than the CH4 peak, indicating that the NiO/ZnO catalytic system is more conducive to the reverse water-gas shift.
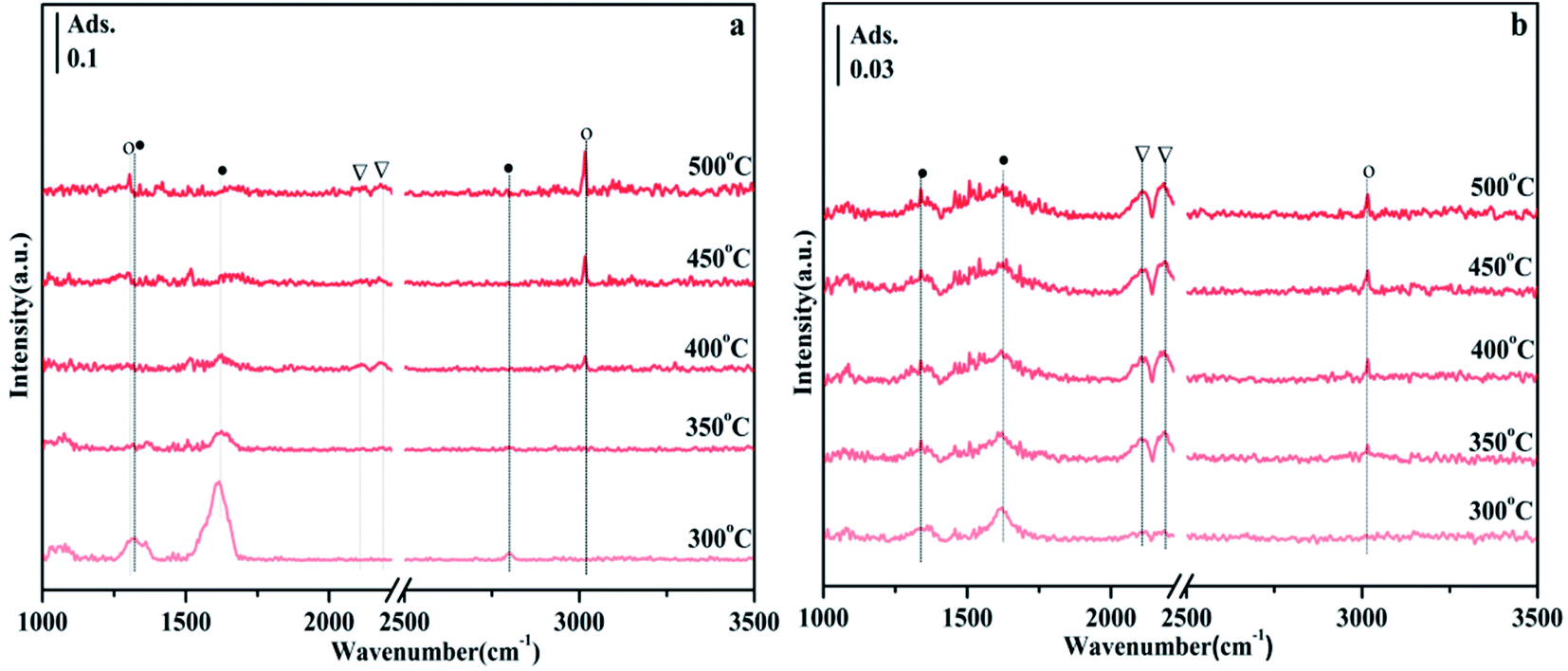 | ||
| Fig. 10 FTIR spectrum of the temperature-programmed CO2 methanation process on NiO/Zn0.3Zr0.7 with CO2/H2 (1:4) gas at a flow rate of 30 ml min−1 and a heating rate of 10 °C min−1. Symbol definition: formate (●), CO (▽), CH4 (○). (a) NiO/Zn0.3Zr0.7; (b) NiO/ZnO. | ||
Fig. 11 is the FTIR spectrum of NiO/Zn0.3Zr0.7 catalyst in CO2/H2 (1:4) atmosphere, held at 350 °C for 10 minutes, and then switched to H2. As shown in Fig. 11, in CO2/H2, the surface of the NiO/Zn0.3Zr0.7 catalyst has formate materials 1313 and 1614 cm−1, and then the CO2/H2 is converted to H2. As time changes, it can be found the formate substance in the image gradually weakened, while the 1303 and 3014 cm−1 substances of CH4 gradually increased. Therefore, it was confirmed that the formate was hydrogenated to methane. To sum up, the methanation of NiO/Zn0.3Zr0.7 follows the formate path.
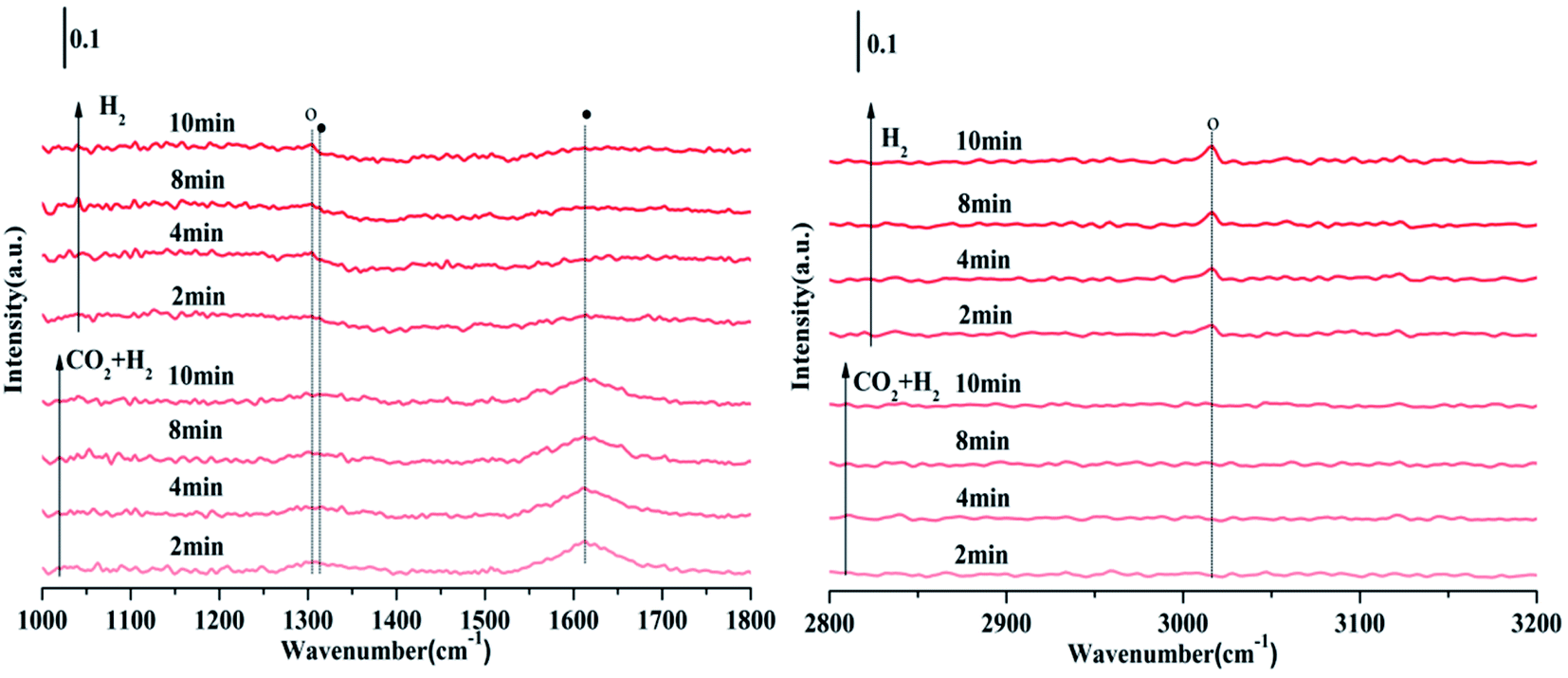 | ||
| Fig. 11 FTIR spectra of CO2/H2 (1:4) gas on NiO/Zn0.3Zr0.7 at a flow rate of 30 ml min−1, a temperature of 350 °C, and a duration of 10 min, switching CO2/H2 (1:4) to 10% H2/Ar for 10 min. Symbol definition: formate (●), CH4 (○). | ||
4. Conclusions
The performance of NiO/ZrO2, NiO/ZnO and NiO/Zn0.3Zr0.7 catalysts for CO2 hydrogenation at 400–500 °C was studied. Among the three groups of catalysts, NiO/Zn0.3Zr0.7 has higher CO2 conversion rate and CH4 selectivity. The results of XRD and H2-TPR show that adding ZnO to the NiO/ZrO2 system can form a ZnO–ZrO2 solid solution. The combination of solid solution will weaken the interaction between NiO and the oxide support, thereby promoting the reduction of NiO. Adding ZnO to the NiO/ZrO2 system will also promote the formation of surface oxygen vacancies, which also plays an important role in improving the catalytic activity and CH4 selectivity. FTIR results show that the methanation of CO2 in the NiO/Zn0.3Zr0.7 catalytic system is carried out through the hydrogenation route of formate.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation project “Functional Control of Methanol Catalyst Based on CO/CO2 Hydrogenation of Metallurgical Furnace Gas” No. 51304099 and the National Science and Technology Support Project “Key Technology for Utilization of Blast Furnace Gas Resources” 2011BAC01B03.References
- R. W. Dorner, D. R. Hardy and F. W. Williams, et al., Energy Environ. Sci., 2010, 3(7), 884–890, 10.1039/c001514h.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9(1), 62–73, 10.1039/c5ee02657a.
- W. Wei and G. V. Jinlong, Front. Chem. Sci. Eng., 2011, 5(1), 2–10, DOI:10.1007/s11705-010-0528-3.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148(3–4), 191–205, DOI:10.1016/j.cattod.2009.07.075.
- L. Jian, W. Aiqin and Q. Botao, et al., J. Am. Chem. Soc., 2013, 135(41), 15314–15317, DOI:10.1021/ja408574m.
- D. Li, K. Li and R. Xu, et al., ACS Appl. Mater. Interfaces, 2019, 11(21), 19227–19241, DOI:10.1021/acsami.9b05409.
- H. Zhang, Y. Dong and W. Fang, et al., Chin. J. Catal., 2013, 34(2), 330–335, DOI:10.1016/S1872-2067(11)60485-3.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007,(28), 2975–2992, 10.1039/b700658f.
- A. Beuls, C. Swalus and M. Jacquemin, et al., Appl. Catal., B, 2013, 113–114, 2–10, DOI:10.1016/j.apcatb.2011.02.033.
- G. Garbarino and D. Bellotti, et al., Catal. Today, 2016, 277(1), 21–28, DOI:10.1016/j.cattod.2015.12.010.
- M. Jacquemin, A. Beuls and P. Ruiz, Catal. Today, 2010, 157(1–4), 462–466, DOI:10.1016/j.cattod.2010.06.016.
- D. Li, K. Li and R. Xu, et al., Catal. Today, 2018, 318, 73–85, DOI:10.1016/j.cattod.2017.12.015.
- S. Abate and K. Barbera, et al., Ind. Eng. Chem. Res., 2016, 55(30), 8299–8308, DOI:10.1021/acs.iecr.6b01581.
- M. A. A. Aziz, A. A. Jalil and S. Triwahyono, et al., Appl. Catal., A, 2014, 486, 115–122, DOI:10.1016/j.apcata.2014.08.022.
- F. Song, Q. Zhong and Y. Yu, et al., Int. J. Hydrogen Energy, 2017, 42(7), 4174–4183, DOI:10.1016/j.ijhydene.2016.10.141.
- D. Li, R. Xu, Z. Gu, X. Zhu, S. Qing and K. Li, Energy Technol., 2019, 1900925, DOI:10.1002/ente.201900925.
- R. Razzaq, H. Zhu and L. Jiang, et al., Ind. Eng. Chem. Res., 2013, 52(6), 2247–2256, DOI:10.1021/ie301399z.
- G. Garbarino, P. Riani and L. Magistri, et al., Int. J. Hydrogen Energy, 2019, 2, 11557–11565, DOI:10.1016/j.ijhydene.2014.05.111.
- S. Sharma, Z. Hu and P. Zhang, et al., J. Catal., 2011, 278(2), 297–309, DOI:10.1016/j.jcat.2010.12.015.
- K. Zhao, W. Wang and Z. Li, J. CO2 Util., 2016, 16, 236–244, DOI:10.1016/j.jcou.2016.07.010.
- A. Cross, S. Roslyakov and K. V. Manukyan, et al., J. Phys. Chem. C, 2014, 118(45), 26191–26198, DOI:10.1021/jp508546n.
- A. Solis-Garcia, J. F. Louvier-Hernandez and A. Almendarez-Camarillo, et al., Appl. Catal., B, 2017, 218, 611–620, DOI:10.1016/j.apcatb.2017.06.063.
- L. Wen and N. Xiao, et al., Appl. Catal., B, 2018, 220, 397–408, DOI:10.1016/j.apcatb.2017.08.048.
- J. C. Lavalley, Catal. Today, 1996, 27(3–4), 377–401, DOI:10.1016/0920-5861(95)00161-1.
- J. Wang, G. Li and Z. Li, et al., Sci. Adv., 2017, 3(10), e1701290, DOI:10.1126/sciadv.1701290.
- A. Cimino and F. S. Stone, J. Cheminf., 2002, 141, 141–306, DOI:10.1002/chin.200317291.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32(5), 751–767, DOI:10.1515/ehs-2015-0001.
- L. Yang, L. Pastor-Pérez and S. Gu, et al., Appl. Catal., B, 2018, 232, 464–471, DOI:10.1016/j.apcatb.2018.03.091.
- C. Heine, B. A. J. Lechner and H. Bluhm, et al., J. Am. Chem. Soc., 2016, 138(40), 13246–13252, DOI:10.1021/jacs.6b06939.
- I. Czekaj, F. Loviat and F. Raimondi, et al., Appl. Catal., A, 2007, 329, 68–78, DOI:10.1016/j.apcata.2007.06.027.
- M. Németh, et al., Appl. Catal., A, 2015, 504, 608–620, DOI:10.1016/j.apcata.2015.04.006.
- J. G. Tao, J. S. Pan and C. H. A. Huan, et al., Surf. Sci., 2008, 602(16), 2769–2773, DOI:10.1016/j.susc.2008.06.034.
- D. Chen, D. He and J. Lu, et al., Appl. Catal., B, 2017, 218, 249–259, DOI:10.1016/j.apcatb.2017.06.053.
- K. M. Eblagon, et al., Appl. Catal., B, 2014, 154, 316–328, DOI:10.1016/j.apcatb.2014.02.032.
- G. Wu, L. Wang and Y. Liu, et al., Appl. Surf. Sci., 2006, 253(2), 974–982, DOI:10.1016/j.apsusc.2006.01.056.
- Y.-H. Wang and W.-G. Gao, et al., Rare Met., 2016, 35(10), 790–796, DOI:10.1007/s12598-015-0520-7.
- F. Cai, P. Lu, J. Ibrahim and J. Zhang, et al., Int. J. Hydrogen Energy, 2019, 44(23), 11717–11733, DOI:10.1016/j.ijhydene.2019.03.125.
- S. Han, Y. Bang and J. G. Seo, et al., Int. J. Hydrogen Energy, 2013, 38(3), 1376–1383, DOI:10.1016/j.ijhydene.2012.11.057.
- Q. Pan, J. Peng and T. Sun, et al., Catal. Commun., 2014, 45, 74–78, DOI:10.1016/j.catcom.2013.10.034.
- W. Deng and F. Zhang, et al., Catal. Sci. Technol., 2016, 6(5), 1530–1545, 10.1039/c5cy01712b.
- Y. Yan, Y. Dai and H. He, et al., Appl. Catal., B, 2016, 196, 108–116, DOI:10.1016/j.apcatb.2016.05.016.
- J. Tan, J. Wang and Z. Zhang, et al., Appl. Surf. Sci., 2019, 481, 1538–1548, DOI:10.1016/j.apsusc.2019.03.217.
- R. Prins, Chem. Rev., 2012, 112(5), 2714–2738, DOI:10.1021/cr200346z.
- N. Perkas, G. Amirian and Z. Zhong, et al., Catal. Today, 2009, 130(3–4), 455–462, DOI:10.1007/s10562-009-9952-8.
- Q. Pan, J. Peng and S. Wang, et al., Catal. Sci. Technol., 2014, 4, 502–509, 10.1039/c3cy00868a.
- X. Jia, X. Zhang and N. Rui, et al., Appl. Catal., B, 2019, 244, 159–169, DOI:10.1016/j.apcatb.2018.11.024.
- Y. Wang, S. Kattel, W. Gao, K. Li, P. Liu, J. G. Chen and H. Wang, Nat. Commun., 2019, 10(1), 1–10, DOI:10.1038/s41467-019-09072-6.
- M. Zhang, J. Zhang and Y. Wu, et al., Appl. Catal., B, 2019, 244, 427–437, DOI:10.1016/j.apcatb.2018.11.068.
- J. M. Lopes, et al., Appl. Catal., B, 2015, 174, 120–125, DOI:10.1016/j.apcatb.2015.02.026.
Footnote |
| † These authors (Shuai Chang and Wenshuo Mao) contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |