
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCo-assembly of charge complementary peptides and their applications as organic dye/heavy metal ion (Pb2+, Hg2+) absorbents and arsenic(III/V) detectors†
Karabi Roy,
Monikha Chetia,
Ankan Kumar Sarkar and
Sunanda Chatterjee *
*
Department of Chemistry, Indian Institute of Technology Guwahati, Guwahati, Assam 781039, India. E-mail: sunanda.c@iitg.ac.in; Tel: +91-361-2583310
First published on 18th November 2020
Abstract
Learning from nature, molecular self-assembly has been used extensively to generate interesting materials using a bottom up approach. The enthusiasm in this field of research stems from the unique properties of these materials and their diverse applications. The field has not been limited to studying assembly of similar types of molecules but extended to multi component systems via the co-assembly phenomenon. We have designed two charge complementary peptides to study their co-assembly in mechanistic detail in the present work. The cooperative self-assembly is mainly driven by electrostatic interaction that is aided by aromatic interactions, hydrogen bonding interactions and hydrophobic interactions. The hydrogels obtained have been employed in waste water remediation. Both the self-assembled and co-assembled hydrogels are capable of removal of different kinds of organic dyes (cationic, anionic and neutral) and toxic metal ions (Ni2+, Co2+, Pb2+ and Hg2+) individually and as a mixture from water with high efficiency. Additionally, the peptides developed in this study can act as ion sensors and detect arsenic in its most toxic (III/V) oxidation states. Molecular understanding of the assembly process is of fundamental importance in the rational design of such simple, robust yet economically viable materials with versatile and novel applications.
1. Introduction
Molecular self-assembly has been a very active field of research in the last couple of decades owing to the several unique structures and properties of the products and the various applications they can be put into.1–4 It forms the basis of the bottom-up approach in nanotechnology. Self-assembly is ubiquitous in nature and scientists have employed this phenomenon to synthetic molecules to create materials of significance and value. The phenomenon which was initially discovered serendipitously, over the time can now be controlled, directed and manipulated to yield desirable products and applications. This ability stems from the fundamental understanding of the process at the molecular level. Simple peptide molecules with appropriate building blocks have been shown to store all the necessary information for the self-assembly process.5,6 To further expand the space of peptide assemblies in terms of structural complexities and functions, multicomponent supramolecular peptide assemblies have also been studied as a natural extension of the field.7–11 Co-assembly results in multifunctionality or a generation of some special properties by the combination of the two partners which cannot be achieved by any one of them.12,13 In addition to co-assembly between molecules of same kind, for example peptide molecules, several different kinds of molecules like deoxyribonucleic acid (DNA), porphyrin rings, proteins etc. also have been co-assembled with peptides.14–18 Co-assembly can take place in four possible ways, namely cooperative, orthogonal (self-sorting), random and destructive.12,19 In cooperative self-assembly, two peptide building blocks interact with each other to give rise to architectures in which both the components are arranged in alternate fashion. This kind of assembly usually occurs in cases where the individual components are very much similar to each other with minor differences. In contrast, orthogonal or self-sorted co-assembly occurs when the constituent partners assemble independently on their own in the presence of the other.20 Designing of orthogonally co-assembled systems is quite challenging as the two components involves different kinds of non-covalent interactions. Co-assembly in between different components can also occur in between the two extremes which involve various degrees of mixing and self-sorting. In random co-assembly, organization in between the partners does not occur in any precise manner while in destructive co-assembly, organization of one of the components is disrupted by the other. Controlling the mixing ratios of individual building blocks allows the adaptable co-assembly mechanism to tune the morphology and the resulting physical, chemical and mechanical properties leading to relevant applications of these multifunctional supramolecular architectures.12,21 Different kinds of non-covalent forces, most importantly aromatic interactions,19,22–24 electrostatic interactions25,26 and even anion–π interactions27 have been used in the design of co-assembled systems. Enantiomeric peptides,28,29 chemical30 and electrochemical stimuli31 and enzymatic reactions32 have also been exploited in various studies to generate co-assembled systems. Co-assembled systems have been vastly employed in controlling physical dimensions of nanostructures,33,34 generating non-canonical complex topologies,35 light harvesting systems,36 conducting architectures,37 biocatalytic assemblies38 and biomimetic scaffolds for cell cultures.39Peptide based hydrogels have been employed extensively is the area of waste water remediation.40–42 Industrial wastes, mining, oil spills and several other human activities contaminate drinking water with organic dyes, toxic ions, oils etc. which poses eminent threat to millions of people worldwide.43–45 Arsenic poisoning of the underground drinking water causes serious health issues which effects people from more than over 70 countries across the world.46,47 Thus, ways of detection and removal of the various contaminants from water is one of the greatest challenges faced by the humankind at the moment.
In the present study, we have rationally designed two charge complementary peptides A1 (NH2-WLVFFK-COOH) and A2 (NH2-WLVFFE-COOH) to form a cooperative hydrogel, employing electrostatic interaction as the main driving force for the co-assembly process. We have probed the mechanism of co-assembly to the molecular details using several experimental techniques.
In a previous study from our group,48 we have established the ability of A1 hydrogel to act as a delivery platform for a large range of therapeutic molecules of different sizes, chemistries, charges and bioactivities. All the cargoes were shown to retain their structure and bioactivity post release from the hydrogel. In the present study, we have extended the application of A1 hydrogel and the co-assembled gels from A1 and A2, to demonstrate that they can act as an excellent absorbent for several kinds of organic dyes (neutral, cationic or anionic) and metal ions (Ni2+, Co2+, Pb2+, Hg2+). Spectrum of contaminants that can be removed by our self-assembled and co-assembled hydrogelators recyclably, make these extremely promising for material engineering of the future. Additionally, A1 and A2 can act as ion sensors; they sense arsenic in their most toxic forms i.e. As(III) and (V) in their oxo anion states with a limit of detection (LOD) of 2.57 μM and 7.06 μM for A1 and 33.08 μM and 10.96 μM for A2 respectively. There are very few peptide based sensors46,47,49 for arsenic which makes our work quite unique.
2. Materials and methods
Materials procured
All Fmoc protected amino acids, hydroxybenzotriazole (HOBt) and benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) were purchased from GL Biochem; 4-dimethylaminopyridine (DMAP), N,N′-diisopropylcarbodiimide (DIC), dimethylformamide (DMF), dichloromethane (DCM), piperidine, N,N-diisopropylethylamine (DIPEA), acetic anhydride, pyridine, trifluoroacetic acid (TFA), tetrabutyl ammonium salts, metal ion salts, were purchased from Spectrochem, sodium meta arsenite was purchased from Novabiochem, arsenic solution was purchased from Sigma Aldrich.Synthesis and purification
The two peptides A1 (H2N-WLVFFK-COOH) and A2 (H2N-WLVFFE-COOH) were synthesized by standard solid phase peptide synthesis (SPPS) using Fmoc chemistry on Wang resin (0.1 millimolar scale). Deprotection of the Fmoc group was achieved by 20% piperidine in DMF. The amino acids were coupled using HOBt and PyBOP as coupling reagents in DMF. After attaching all the amino acids, the peptides were cleaved from the resin using 95% TFA and were precipitated in cold diethyl ether. The peptides were purified using reverse phase high performance liquid chromatography (HPLC) on a C18 column using acetonitrile/water gradient. After purification, the peptides were characterized using analytical HPLC on C18 column to check its purity using acetonitrile/water as solvent system (Fig. S1†). Further the peptides were characterized by electrospray ionization-mass spectrometry (ESI-MS) (Fig. S2 and S3†) and 1H nuclear magnetic resonance (NMR) spectroscopy (600 MHz) (Fig. S4 and S5†).A1: 1H NMR (600 MHz, DMSO-d6) δ 10.98 (s, 1H, –COO![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) at the C-terminus), 8.63 (s, 1H, –N of Trp indole ring), 8.15 (d, 1H, backbone N), 8.00 (d, 1H, backbone N), 7.93 (d, 1H, backbone N), 7.71 (d, 1H, backbone N), 7.36 (d, 1H, backbone N), 7.24–6.99 (m, 14 Hs, phenyl rings of Phe and Trp), 4.57–4.48 (m, 3Hs, α H of Phe and Lys), 4.43 (d, 1H, α H of Trp), 4.21–4.09 (m, 3Hs, α Hs of Leu, Val and Lys; merged), 3.91 (s, 1H, –C of indole ring) 3.05–3.00 (m, 2Hs, β Hs of Trp), 2.98–2.88 (3Hs, β Hs of Leu and Val), 2.83–2.68 (m, 4H, β Hs of Phe), 2.29–2.24 (m, 2Hs, ε Hs of Lys), 1.92–1.78 (m, 4H, –Ns at the N-terminus), 1.62–1.53 (m, 2H, β Hs of Lys), 1.48–1.41 (m, 2H, δ Hs of Lys), 1.41–1.35 (m, 2H, γ Hs of Lys), 0.90–0.70 (m, 12H, methyl Hs of Leu and Val), (δ 2.50 for solvent residual peak, δ 3.33 for H2O).
at the C-terminus), 8.63 (s, 1H, –N of Trp indole ring), 8.15 (d, 1H, backbone N), 8.00 (d, 1H, backbone N), 7.93 (d, 1H, backbone N), 7.71 (d, 1H, backbone N), 7.36 (d, 1H, backbone N), 7.24–6.99 (m, 14 Hs, phenyl rings of Phe and Trp), 4.57–4.48 (m, 3Hs, α H of Phe and Lys), 4.43 (d, 1H, α H of Trp), 4.21–4.09 (m, 3Hs, α Hs of Leu, Val and Lys; merged), 3.91 (s, 1H, –C of indole ring) 3.05–3.00 (m, 2Hs, β Hs of Trp), 2.98–2.88 (3Hs, β Hs of Leu and Val), 2.83–2.68 (m, 4H, β Hs of Phe), 2.29–2.24 (m, 2Hs, ε Hs of Lys), 1.92–1.78 (m, 4H, –Ns at the N-terminus), 1.62–1.53 (m, 2H, β Hs of Lys), 1.48–1.41 (m, 2H, δ Hs of Lys), 1.41–1.35 (m, 2H, γ Hs of Lys), 0.90–0.70 (m, 12H, methyl Hs of Leu and Val), (δ 2.50 for solvent residual peak, δ 3.33 for H2O).
A2: 1H NMR (600 MHz, DMSO-d6) δ 10.99 (s, 2Hs, –COO of Glu, –COO at the C-terminus), 8.63 (s, 1H, –N of Trp indole ring), 8.23 (s, 1H, backbone N), 8.11 (s, 1H, backbone N), 8.03 (d, 1H, backbone N), 7.94 (d, 1H, backbone N), 7.71 (d, 1H, backbone N), 7.43–6.94 (m, 14Hs, Phenyl rings of Phe and Trp), 4.59–4.50 (m, 2Hs, α H of Phe), 4.43 (s, 1H, α H of Trp), 4.16–4.10 (m, 3Hs, α H of Glu, Val, Leu), 3.91 (s, 1H, –C of indole ring), 3.03 (m, 1H, β H of Val), 2.94 (m, 2H, β Hs of Trp), 2.85–2.68 (m, 4H, β Hs of Phe), 1.89–1.69 (m, 2Hs, β Hs of Leu), 1.65–1.50 (m, 4H, β and γ Hs of Glu), 1.45 (m, 1H, γ H of Leu), 1.35 (q, 2H, –Ns at the N-terminus), 0.94–0.65 (m, 12Hs, methyl Hs of Leu and Val), (δ 2.50 for solvent residual peak, δ 3.33 for H2O).
Formation of hydrogels
First 1.0 mg of A1 peptide was taken in 500 μL of sodium phosphate buffer (0.2% wt/v; 2.38 mM) (pH = 7.0) and then heated on a hot plate till the peptide dissolved which was then kept undisturbed at room temperature (RT). It resulted in the formation of hydrogel after 2–3 hours of standing which was stable to inversion of the glass vial. We tried for the hydrogelation of A2 under same conditions but it did not form any gel. A1 also formed gel in 20% ethanol (EtOH)/phosphate buffer mixture at 0.2% wt/v. A1 and A2 when taken in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 and 1:1 (wt/wt) formed hydrogel in sodium phosphate buffer at pH = 7.0 whereas 1:2 and 1:3 resulted in the formation of viscous solution.
:0.5 and 1:1 (wt/wt) formed hydrogel in sodium phosphate buffer at pH = 7.0 whereas 1:2 and 1:3 resulted in the formation of viscous solution.
Determination of the gel to sol transition temperature for the hydrogels
For the hydrogels, the Tgel was obtained by placing the gel containing vial in an oil bath and then slowly raising the temperature of the bath at the rate of 1 °C per minute. The temperature was monitored using a thermometer and was recorded accordingly.Field emission scanning electron microscopy (FESEM)
FESEM images of the peptide samples were obtained on a FESEM Sigma 300 microscope. For the morphology of the hydrogels, preheated samples were casted on the silicon wafer and allowed to dry under vacuum before imaging. Peptide hydrogel A1 was taken at its minimum gelation concentration (MGC) in sodium phosphate buffer system and in EtOH/sodium phosphate buffer mixture. The viscous solution of A2 in sodium phosphate buffer at 5 mM concentration was used for imaging. The morphology of the co-assembled systems of A1 and A2 was taken in the ratio of 1:0.5, 1:1, 1:2 and 1:3 in sodium phosphate buffer.
Field-emission transmission electron microscopy (FETEM)
FETEM studies were performed on JEOL JEM (model 2100F) by casting 3 μL of the co-assembled hydrogels at 1:0.5 and 1:1 ratio in phosphate buffer on carbon-coated copper grids (300 mesh). The grid was allowed to dry under vacuum at room temperature before imaging.
Fourier transform infrared (FTIR) spectroscopy
IR spectra of the samples were recorded on a Spectrum Two PerkinElmer FTIR spectrometer using KBr pellet within the range of 1000–4000 cm−1. FTIR of peptides A1, A2 and xerogels obtained from freeze drying the hydrogels of A1, A1 + A2 (1:0.5 and 1:1) were recorded.
Rheology
The viscoelastic properties of hydrogels A1, A1 + A2 (1:0.5) and A1 + A2 (1:1) were determined by rheological studies using Anton Paar MCR102 Rheometer equipped with a 20 mm parallel-plate measuring system at 25 °C. For measuring rheology, A1 hydrogel was prepared at 4.5 mg mL−1 (0.45% wt/v) in sodium phosphate buffer (pH = 7.0) and in a mixture of EtOH/buffer system. The co-assembled hydrogels were prepared at 2:1 (wt/wt) and 2:2 (wt/wt) in 1 mL of sodium phosphate buffer (pH = 7.0). A strain sweep test was performed over a range from 0.1–100% strain at a fixed oscillatory frequency of 1 rad s−1. Furthermore, the mechanical strength of hydrogels was determined from the oscillatory test, i.e. frequency sweep, which was carried out under an appropriate strain 0.1% with the frequency ranging from 1–100 rad s−1.
Fluorescence spectroscopy
To look into the self-assembly pattern of the peptide A1 and A2, fluorescence experiment was performed on a Fluoromax-4 spectrophotometer by monitoring the fluorescence emission of the tryptophan (Trp) moiety. A concentration dependent fluorescence was performed for A1 in 20% EtOH/sodium phosphate buffer and A2 in 40% EtOH/sodium phosphate buffer system (0.00487–5 mM) keeping excitation wavelength at 280 nm.Powder X-ray diffraction (PXRD)
Wide angle X-ray diffraction analysis was done on a Bruker D2 Phaser X-ray diffractometer (Cu-Kα radiation, λ = 1.5406 Å) for both the powdered samples of A1, A2 and the xerogels obtained from lyophilising the hydrogels of A1 and the co-assembled hydrogels.Dye absorption studies
At first the hydrogel was prepared as mentioned in the gelation experiment (1 mg in 500 μL of sodium phosphate buffer, pH = 7.0). Then, aqueous solutions of Rhodamine B (RB) (20 μM), Crystal Violet (CV) (40 μM), Neutral Red (NR) (110 μM) and Methyl Orange (MO) (70 μM) were added into the preformed gel and allowed to absorb. Also, aqueous solutions of two metal ions, Ni2+ (100 mM) and Co2+ (100 mM) were added to absorb. The dye/metal ion absorption was measured using UV-Vis spectroscopy from time to time to know the trend in absorption by the hydrogel. Further, the amount of dye unabsorbed in the supernatant was checked by monitoring the UV of the supernatant aqueous solution after 48 hours and calculating the concentration from a standard curve of the dye/metal ion. The dye absorption by the co-assembled hydrogel A1 + A2 (1:1) was done in the similar fashion as described with two dyes RB and MO. The amount of dye/metal ion loaded in the hydrogel and the dye/metal ion loading efficiency were then calculated as follows:| Dye/metal ion absorbed = initial dye/metal ion − unabsorbed dye/metal ion |
| % Absorbed = dye/metal ion absorbed/initial dye/metal ion × 100 |
The dye and metal ion absorption from an all component mixture by the hydrogels was also checked using UV-Vis spectroscopy in which the aqueous solution of dyes (RB, MO, NR and CV) and the metal ion (Co2+ and Ni2+) was mixed together and was added to the preformed hydrogel to absorb.
Reusability of the hydrogel A1
The hydrogel from A1 was prepared as discussed earlier at its MGC in sodium phosphate buffer. Then 500 μL of 20 μM aqueous solution of RB was added into it and the dye was allowed to be absorbed for 24 hours. After 24 hours, the supernatant solution at the top was taken and its absorbance was measured from which the % of dye absorbed was calculated. Then to the dye absorbed gel ether was added as the release medium and it was kept undisturbed for 2 days after which the gel became clear. On the clear, dye free hydrogel, fresh solution of 20 μM RB was added again and left to be absorbed for 24 hours. This cycle continued for 2 times after which the hydrogel was no longer be used further for dye absorption.Anion sensing
A stock solution of 1 mM of the peptide A1 and A2 in 20% and 40% respectively in EtOH/water system was prepared. Stock solutions of 10 mM tetrabutylammonium salts of F−, Cl−, Br−, I−, H2PO4−, OH−, AcO− and HSO4−; sodium salt of arsenite (AsO2−) and arsenic acid were prepared in water. Then a solution of 0.2 mM of the A1 and A2 were prepared from the stock solution. Also, 0.2 mM of all the anion solutions was prepared from the 10 mM stock solution. For monitoring fluorescence spectra 500 μL of 0.2 mM of the peptide solution was taken and into it 500 μL of 0.2 mM anion solution was added so that the final concentration of both the solutions become 0.1 mM. The fluorescence was monitored at an excitation wavelength of 280 nm.Metal ion sensing
A stock solution of 1 mM of A1 and A2 in 20% and 40% respectively EtOH/water system was prepared was prepared. Stock solutions of 10 mM of metal ion salts (CoCl2, PbCl2, NiCl2, CuCl2 MnCl2, Zn(NO3)2, Hg(OAc)2, FeCl3) were prepared in water. Then a solution of 0.2 mM of A1 and A2 were prepared from the stock solution. Also, 0.2 mM of all the metal ion solutions was prepared from the 10 mM stock solution. For monitoring fluorescence spectra 500 μL of 0.2 mM of the peptide solution was taken and into it 500 μL of 0.2 mM metal ion solution was added so that the final concentration of the peptide and the metal ion solution became 0.1 mM. The fluorescence was then monitored at 280 nm.Titration of the gelator molecules with specific anions and metal ions
500 μL of 0.2 mM of peptide solution of A1 and A2 in 20% and 40% respectively in EtOH/water system was taken in different 1 mL volumetric flasks and into it 500 μL of different concentration of the anion and metal ion solution were added so that the final concentration of the peptide solution becomes 0.1 mM. The Trp fluorescence of these solutions was then monitored for the solutions at 280 nm.Detection limit
For detection limit (DL) calculation, fluorescence of 0.01 mM solution of A1 and A2 in EtOH/water solvent system was measured at 280 nm for ten times. Then standard deviation (σ) of the intensity at the emission wavelength was calculated. Then titration of A1 and A2 with the anions and metal ions at very low concentrations were monitored (0.002–0.025 mM) and a linear plot of concentration versus intensity was obtained. From the linear plot, the slope (k) was calculated and was put into the equation below:| DL = 3σ/k; n = 10 |
Pb2+ and Hg2+ removal by hydrogels A1 and A1 + A2
Hydrogel A1 and the co-assembled hydrogel A1 + A2 were prepared at 2 mg and 2:2 (wt/wt) in 2 mL respectively in phosphate buffer system. Into it 2 mL of 20 mg L−1 aqueous salt solution of Pb and 2 mL of 100 mg L−1 Hg was added and left to be absorbed for 24 hours. After that, the supernatant solution was analysed using atomic absorption spectroscopy (AAS) in AA240, Varian, Netherlands spectrophotometer. The experiment was done in duplicate and the values obtained were average of two readings. The % absorbed was then calculated using the following equation, | Concentration of metal ion absorbed = initial metal ion − unabsorbed metal ion |
| % Absorbed = metal ion absorbed/initial metal ion × 100 |
For Pb2+, 217 nm was used as the wavelength for analysis, with 1 nm slit width for an optimum working range of 0.1–30 μg mL−1 and for Hg2+, wavelength of 253 nm was used for analysis with 0.5 nm slit width with optimum working range of 2–400 μg mL−1.
3. Results and discussions
Peptide design
Charge complementary peptides A1 (NH2-WLVFFK-COOH) and A2 (NH2-WLVFFE-COOH) had identical sequences except for a positively charged Lys residue and a negatively charged Glu residues at the C terminal end of the peptides respectively (Fig. 1a and b). The LVFF moieties in the peptides were incorporated to act as the hydrophobic core while the Trp residue at the N terminus with its intrinsic fluorescence, acted as a reporter molecule. The unprotected termini of the peptides helped in solubilizing the peptide.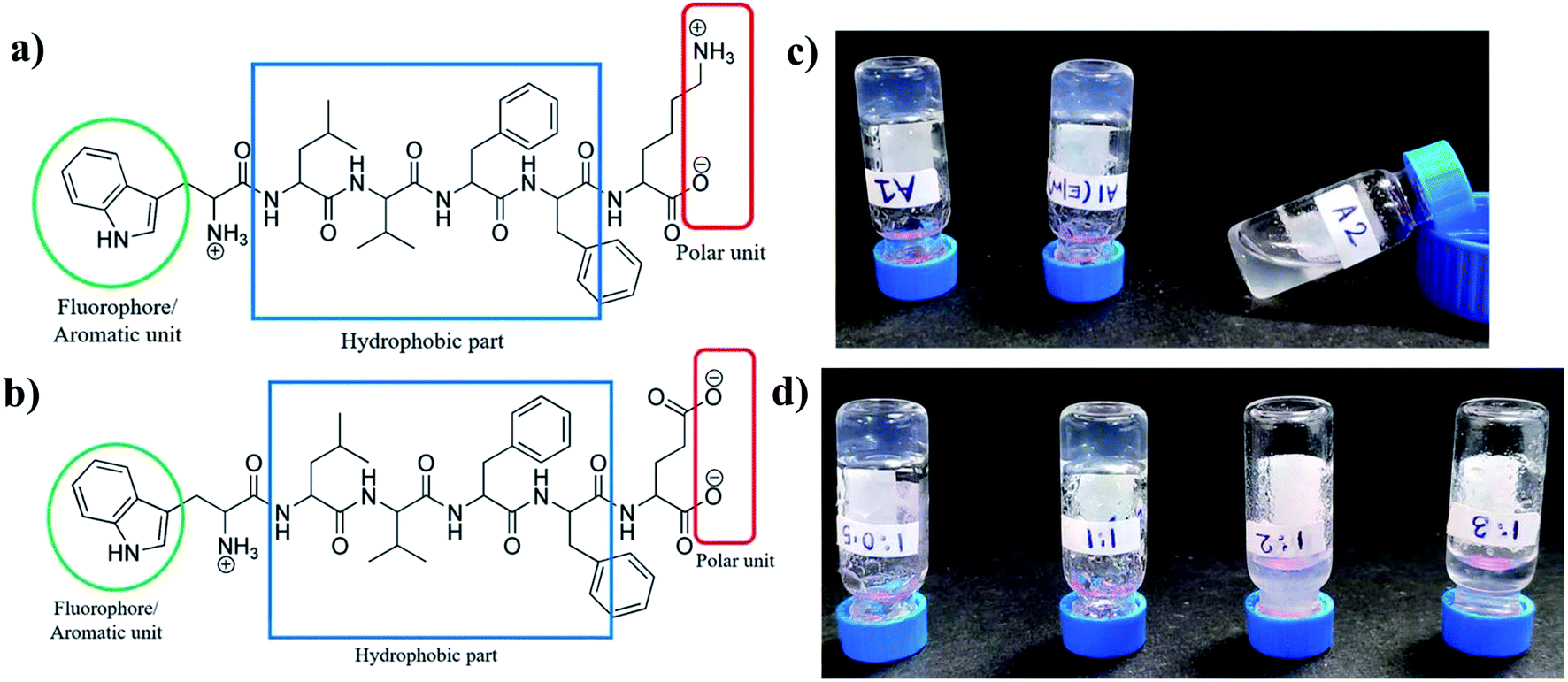 | ||
| Fig. 1 Chemical structures of (a) A1, (b) A2, (c) gel formation by A1 in sodium phosphate buffer (pH-7.0), EtOH/phosphate buffer system and viscous solution of A2 in sodium phosphate buffer (pH-7.0) and (d) gelation by different ratios of A1 and A2. A1, A2 mixtures of 1:1 and 1:0.5 ratios produced gels while those of 1:2 and 1:3 ratios did not. | ||
Hydrogelation and morphology
When subjected to gelation, A1 formed hydrogel in sodium phosphate buffer and also in 20% EtOH/sodium phosphate buffer upon heating and subsequent cooling (Fig. 1c), whereas A2 under similar conditions failed to form gel. Also, when A1 and A2 were taken together in 1:1 and 1:0.5 ratios and subjected to similar treatment as that of A1, co-assembled hydrogel was formed (Fig. 1d). It should be noted that the other ratios of A1 and A2, like 1:2 and 1:3 failed to form hydrogel under similar conditions. Thus it was obvious that either stoichiometric ratio or an excess of A1 favoured gelation. A mixture with excess of A2 failed to do so. Table S1† gives the characteristic features of the gels formed from A1 and A2.
At neutral pH, A1 is positively charged while A2 is negatively charged owing to the pKa's of the terminal and the side chain amino and carboxylic acid groups. Self-assembly of molecules requires their close approach. Approach of charged molecules induce electrostatic repulsion which disfavours the assembly process of A1 and A2 at neutral pH. This is overcame in sodium phosphate buffer where phosphate ions present might play a crucial role in neutralizing the electrostatic repulsive forces amongst the A1 molecules, leading to their close approach and formation of the hydrogel. The very small positive counterion Na+, present in sodium phosphate buffer, is most probably unable to diffuse the negative charge on A2 at neutral pH and induce assembly in them. The slight difference in the length of the hydrophobic side chains of the Lys and Glu residues might alter the hydrophobic–hydrophilic balance which brings about a difference in the gelation behaviour of A1 and A2. Additionally, the point charge in the side chain of Lys compared to the delocalized charge on the carboxylate group of Glu residue might also contribute to this difference. In the co-assembled systems, those that contain higher proportions of A2, resulting in an overall negative charge at the neutral pH, fail to form hydrogels proving the point further. Hence gelation is a very sensitive phenomenon and depends on a very fine balance of several parameters.
Morphological features of the gel formed by A1, A2 and (A1 + A2) have been studied by FESEM and FETEM. As mentioned in our earlier report,48 A1 hydrogel formed a dense and porous mesh like morphology in sodium phosphate buffer, pH 7.0 (Fig. S6a†), while it formed well entangled fibrous network in EtOH/sodium phosphate buffer mixture (Fig. S6b†). A2 formed irregular aggregate-like morphology in sodium phosphate buffer (pH = 7.0) (Fig. S6c†).
Fig. 2 shows the morphology of the co-assembled hydrogels formed from A1 and A2 taken in various ratios. It is found that, a well entangled uniform fibrous network was observed both in case of the co-assembled hydrogel formed by 1:0.5 and 1:1 ratio of A1 and A2. The viscous solutions obtained by other ratios gave an aggregate kind of morphology as visualised by FESEM.
 | ||
| Fig. 2 FESEM images of hydrogel of A1 + A2 in the ratio of (a) 1:0.5 wt/wt, (b) 1:1 wt/wt, (c) 1:2 wt/wt and (d) 1:3 wt/wt ratio in sodium phosphate buffer at pH = 7.0. | ||
To have a closer look at the morphology of the co-assembled hydrogels, FETEM was performed. Both 1:1 and 1:0.5 hydrogels contained fibre like morphology. The hierarchical self-assembly was clearly observable in the images where finer fibres were seen to form thicker fibre-bundles (Fig. 3). Formation of uniform fibre like morphology in both the co-assembled gels was a clear indication of cooperative self-assembly in the system.
 | ||
| Fig. 3 FETEM of A1 + A2 hydrogel in sodium phosphate buffer, pH = 7.0, (a) 1:0.5 and (b) 1:1 (wt/wt). | ||
Viscoelastic properties of the hydrogels
Viscoelastic property of the hydrogels was studied by rheology using 0.45 w/v% A1 hydrogel in (a) sodium phosphate buffer at pH 7.0 (Fig. S7a and b†) and (b) 20% EtOH/sodium phosphate buffer (Fig. S7c and d†). As mentioned previously,48 in the angular frequency sweep experiment performed at a constant strain of 0.1% at 25 °C, for both the hydrogels, G′ was found to be dominating over G′′ till about 100 rad s−1 (Fig. S7a and c†). Also, G′ and G′′ were found to be independent of angular frequency, in the region of 1–100 rad s−1 which indicated the formation of stable hydrogels. The value of storage moduli G′ was of the order of 102–103 Pa for A1 hydrogel in sodium phosphate buffer and in the EtOH/sodium phosphate buffer suggesting that the hydrogels formed in both the systems were mechanically robust. In strain sweep experiment, G′ was found to be higher than G′′ till a particular strain (Fig. S7b and d†), beyond which the two crossed each other, indicating loss of gel nature for both the systems.We have then checked the mechanical strength of the co-assembled hydrogels formed from A1 and A2 taken in ratios 1:0.5 (2 mg and 1 mg respectively for A1 and A2) and 1:1 (2 mg and 2 mg respectively for A1 and A2) in 1 mL of the solvent (Fig. 4). We observed that in the frequency sweep experiment, the G′ values for the co-assembled hydrogels 1:0.5 and 1:1 were between 100–101 and 102–103 Pa, which were lower and higher than the A1 hydrogel respectively, suggesting that the 1:0.5 and 1:1 co-assembled hydrogels were weaker and stronger than A1 respectively. The increased robustness of the 1:1 co-assembled hydrogel with respect to the A1 self-assembled hydrogel may be attributed to the additional electrostatic interactions in between A1 and A2 in the co-assembled system. In the A1 hydrogel, assembly was mediated through the phosphate ions while in the co-assembled hydrogel, this was not necessary. Complementary charges on A1 and A2 led to favourable interactions in the 1:1 co-assembled hydrogel. However, in the 1:0.5 co-assembled gel, there was an excess of positive charges and hence the assembly had to be mediated through the phosphate ions. Thus the 1:0.5 hydrogel was weaker than the 1:1 hydrogel. In strain sweep experiment, G′ was found to be higher than G′′ till a particular strain (Fig. 4b and d) for both the co-assembled gels, beyond which the two crossed each other, indicating loss of gel nature for both the systems.
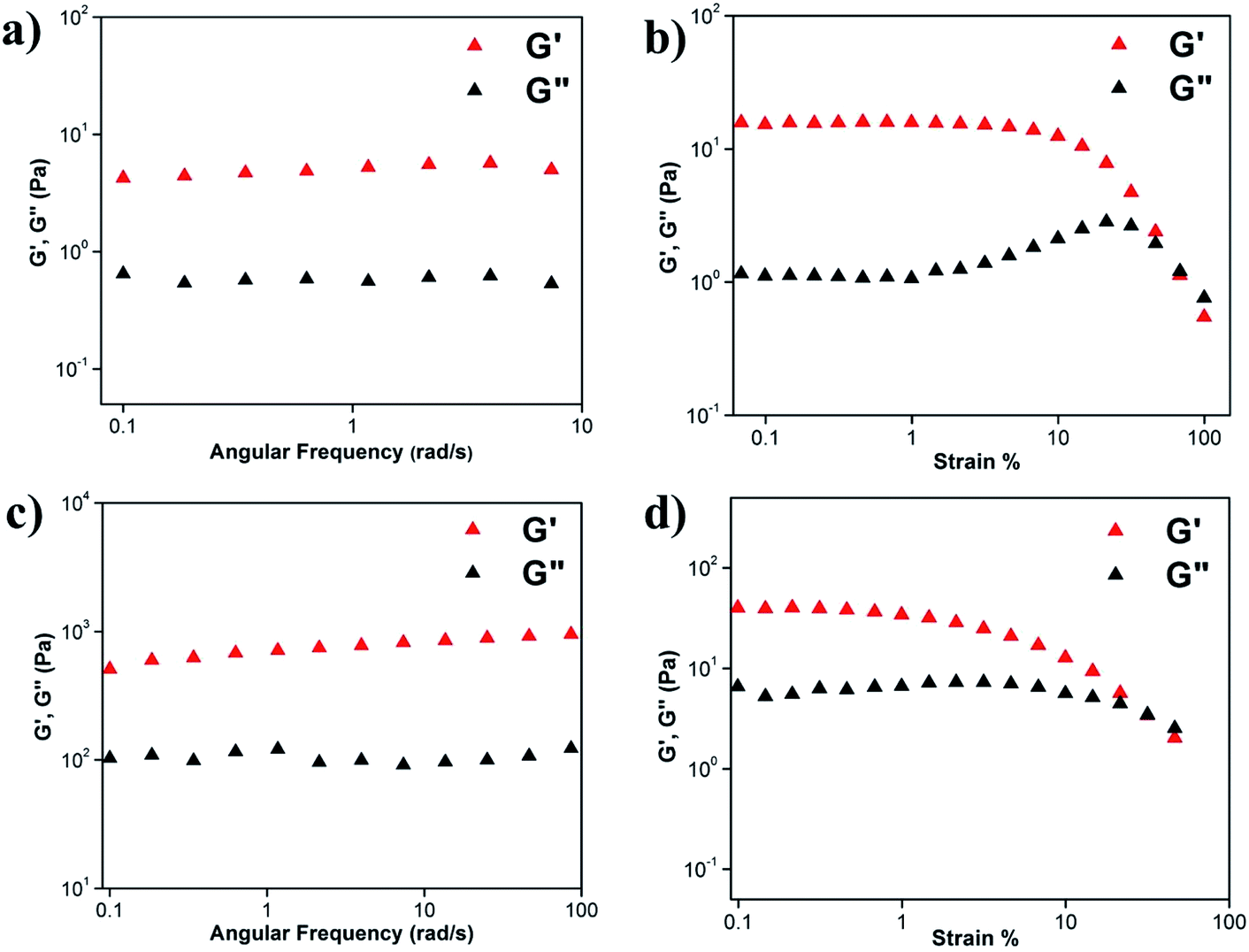 | ||
| Fig. 4 Angular frequency and strain dependence of the dynamic storage moduli (G′) and the loss moduli (G′′) of co-assembled hydrogels formed from (a and b) A1 + A2 = 1:0.5 and (c and d) A1 + A2 = 1:1 in sodium phosphate buffer (pH = 7.0). | ||
Factors driving self-assembly
Self-assembly of A1 was investigated earlier in details in a previous study from our group.48 Electrostatic interactions played the most crucial role in formation of the hydrogel from positively charged A1. Aromatic π–π stacking interactions, as observed from the fluorescence experiments and PXRD experiments, also contributed to the self-assembly of A1. In the A1 hydrogel, peptides adopted β-sheet backbone conformation which in turn involved hydrogen bonding between the amide groups present in the peptide backbone and hydrophobic interactions.In the present study, we wanted to understand the interactions involved in the formation of the co-assembled systems using various ratios of A1 and A2.
Backbone conformation and role of H-bonding
In order to understand the backbone conformation of A1 and A2 and the role of H-bonding in the co-assembled gel, solid state FTIR was performed on xerogels of (A1 + A2) hydrogels (1:1 and 1:0.5 ratios) (Fig. 5a) and compared it to powdered samples of A1 and A2 and A1 xerogel. There were no deviations in the peak positions in the self-assembled and the co-assembled xerogels indicating similar backbone conformation and hydrogen bonding status in both the systems. Presence of peaks at around 1631 and 1682 cm−1 in all the systems, clearly indicated antiparallel β-sheet conformation for the peptides in individual powdered form or in hydrogel state. Presence of NH stretching peak at 3279 cm−1 indicated presence of extensive H-bonding in all the systems.
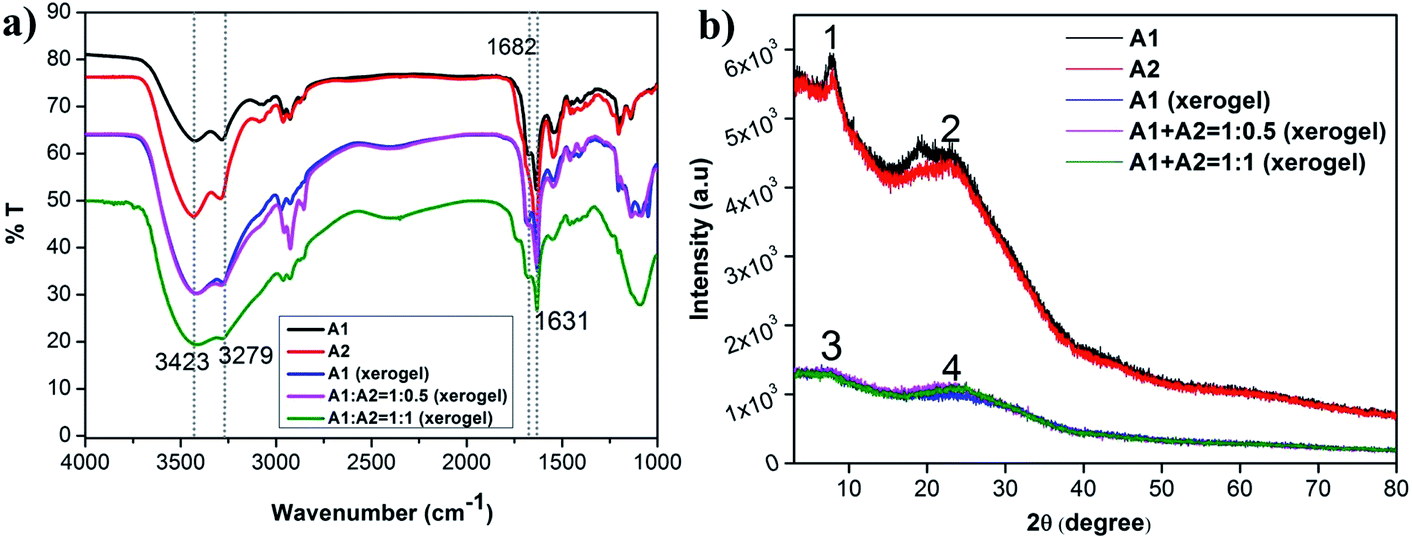 | ||
| Fig. 5 Overlay of (a) FTIR and (b) PXRD of A1, A2, A1 xerogel and 1:0.5 and 1:1 ratio of co-assembled xerogels of A1 + A2. | ||
Molecular packing
PXRD was performed to understand the ordered arrangement of the co-assembled systems (Fig. 5b and Table S2†). The results were compared with that of the powdered A1 and A2 samples and the A1 xerogel. A prominent peak at around 23° was obtained for all the systems, which corresponded to an interplaner distance of ∼3.8 Å, indicating the presence of a very strong aromatic π–π interaction present in all the systems. Additionally, in A1 and A2 another prominent interplaner distance of ∼11.5 Å was observed. This could be attributed to the distance in between the subsequent β-sheets. In all the xerogels studied, an interplaner distance of ∼12.4 Å was observed. This spacing was attributed to the distance in between the subsequent β-sheets in the xerogel structures. It must be remembered that the xerogels were derived from the hydrogels that were formed in sodium phosphate buffer. Presence of water, ions in between the subsequent β-sheet layers might be responsible for increasing inter-sheet spacings observed in the xerogels. Thus, in summary, A1 and A2 consistently formed β-sheet conformation in powdered, self-assembled or co-assembled xerogel state. As A1 and A2 have similar motifs in them (WLVFF), their co-assembly was cooperative in nature and occurred without perturbing the backbone conformation of the individual peptides. Secondly, strong aromatic π–π stacking interactions existed in all the systems and was another important interaction driving self-assembly.Mechanism of self-assembly and co-assembly
As seen from the various experiments, electrostatic interaction, aromatic π–π stacking interaction and H-bonding play important roles in the self-assembly and the co-assembly processes being studied here. At neutral pH, formation of hydrogel in the presence of sodium phosphate buffer, suggests that the phosphate ions play a crucial role in diffusing the positive charge on the A1 peptides, bringing them close together to form the hydrogel. In the case of A2, the positive counter ions of sodium phosphate buffer, being extremely small, may not have been able to diffuse the negative charge on A2 and bring about gelation. Difference in the nature of the charged groups and the slight difference in the hydrophobicity of the side chains of the charged amino acid residues might have contributed additionally to the difference in the gelation behaviour of the otherwise identical peptides A1 and A2. Hence electrostatic interactions play a very important role in these assembly processes. Fig. S8† depicts the molecular arrangement involved in the self-assembly of A1. A1 monomers form antiparallel β-sheets as evidenced from the FTIR spectroscopy. The sheets are amphipathic in nature, having two distinct faces. One of the faces is hydrophobic in nature which contains the Trp side chain while the other face contains the charged Lys side chains. The side chains of the intervening LVFF moiety alternately face opposite directions and point both towards the hydrophobic and charged faces of the β-sheets. Due to the formation of the antiparallel β-sheets, the positively charged amino terminus of one monomeric unit faces the negatively charged carboxylate end of the adjacent monomeric unit. This compensates the charge on the edges of the sheets. The sheets stack on each other in such a way that the hydrophobic face of one sheet confronts the hydrophobic face of the other. This stacking is favoured by the hydrophobic interaction in between the side chains of the LVFF moiety. Additionally, the Trp rings from one sheet form π–π stacking with that from the other sheet. The charged faces of the adjacent β-sheets face each other. The phosphate ions stack in between such positively charged faces and neutralize the repulsion, thus bringing stability to the system. As this charge stabilization is absent in water, hydrogelation of A1 does not occur there. The stacking of the sheets leads to formation of fibres. Such fibres come close together along the edges of the sheets. This is favoured by the electrostatic interactions in between the terminal charges, giving rise to thicker fibre bundles. The thick bundles entangle with each other and eventually give rise to the mesh like morphology that is observed by FESEM.Fig. 6 summarizes the mechanism of co-assembly of A1 and A2 in 1:1 ratio. Presence of an uniform fibre like morphology as seen from microscopic techniques like FESEM and FETEM, accompanied by the evidences that the backbone conformation of A1, A2 in the co-assembled gel was the same as in the self-assembled gel and interplaner spacings in the co- and self-assembled gels were identical, suggested that A1 and A2 underwent cooperative co-assembly. This was expected as peptides A1 and A2 were similar in their structure. As shown in the Fig. 6, A1 and A2 alternate to form β-sheets in which one face of the sheet is hydrophobic and contains the Trp side chain. The other surface of the sheet contains both the positive and the negative side chains of Lys and Glu residues from A1 and A2 respectively. Stacking of the β-sheets involve hydrophobic interactions and aromatic π–π stacking interactions in between the indole rings of the Trp chains in the hydrophobic faces. The charged face of the β-sheet contains positive and negative charges along the two edges. While stacking of the charged faces, the consecutive sheets approach an alternate fashion so that the oppositely charged edges face each other giving rise to favourable electrostatic interactions. It is due to this interaction, that the 1:1 co-assembled gel was stronger than the A1 hydrogel as observed from rheology. While in the later, phosphate ions mediated the interaction in between the positive faces of the β-sheets, in the former co-assembled system, complementary charges on the constituent peptides gave rise to the favourable electrostatic interactions that stabilized the system. The 1:0.5 hydrogel system underwent self-assembly pretty much the same way as the 1:1 system, with some minor differences (Fig. S8†). In the 1:0.5 co-assembled hydrogel, the peptides would repeat as A1:A2:A1 units within a sheet. This would give rise to β-sheets with edges containing mixed charges and the charged face containing overall positive charge. In this case, phosphate ions from the phosphate buffer helped in neutralizing the repulsive forces and bringing about stacking of the β-sheets as in the case of A1 hydrogel. This explained the relative weakness of the 1:0.5 co-assembled gel with respect to the 1:1 co-assembled gel. In the mixture ratios of A1 and A2, with higher amounts of A2, the system acquired excess negative charge which could be stabilized by the small positively charged counter ions of the sodium phosphate buffer and hence, there was no resultant hydrogelation just as in the case of A2.
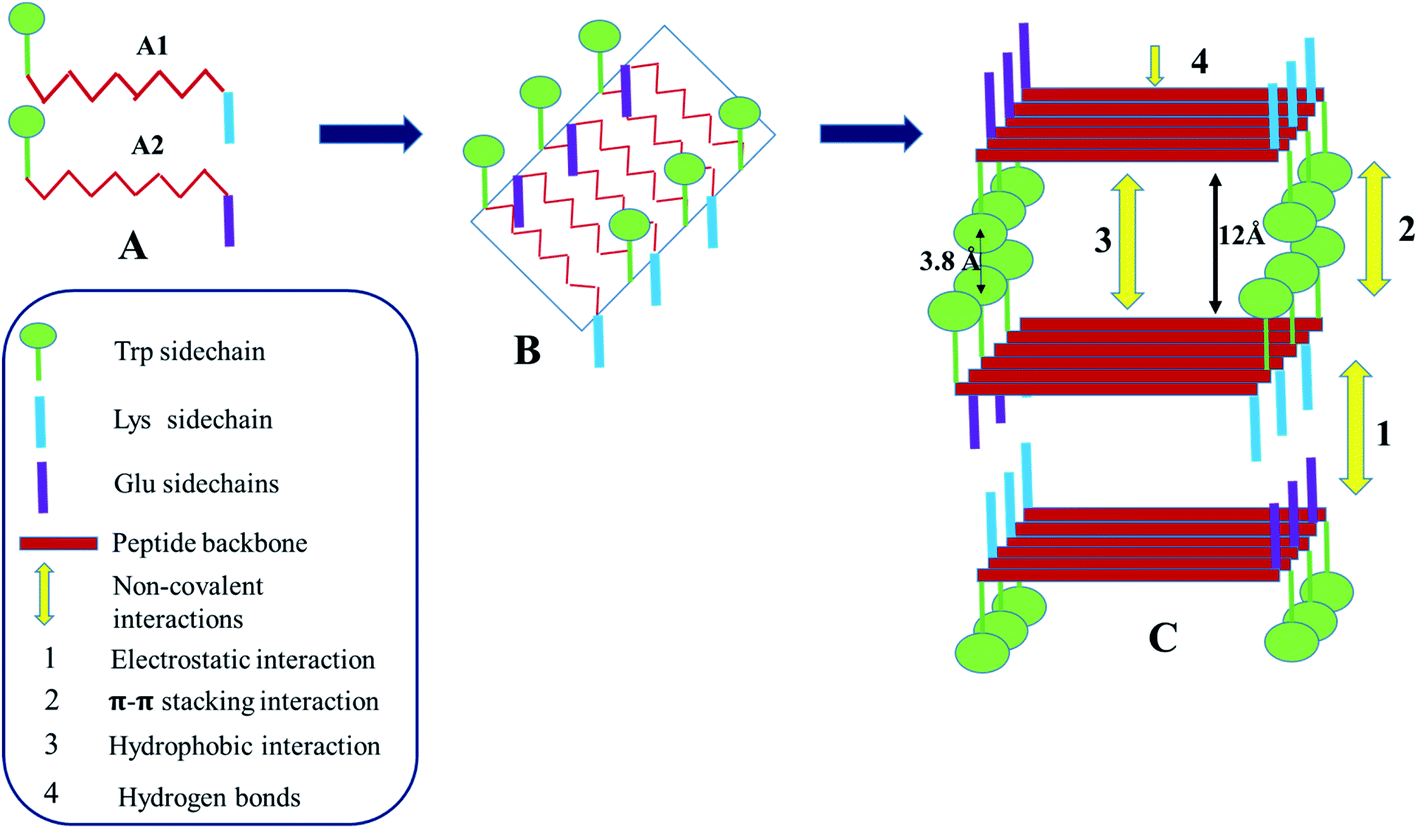 | ||
| Fig. 6 Schematic representation depicting the mechanism of co-assembly of A1 and A2 in 1:1 ratio. | ||
Application of the hydrogels in dye and metal ion absorption
We have used the A1 hydrogel and the co-assembled gels of A1 and A2 for dye and metal ion absorption studies. The aqueous solutions of the dyes or the metal ions were allowed to stand with the gel for 48 hours. The UV absorbance of the solution was monitored over the time to obtain the amount of unabsorbed dye (Fig. S9a–f, S10a and b†). Amount of absorbed dye was back calculated from the initial concentration of the solution as discussed in the experimental section. All our hydrogels (self and co-assembled) acted as very efficient absorbents for the organic dyes like RB, CV, NR, MO and divalent metal ions Ni2+ and Co2+ (Fig. 7a–f, 8a and b). It is mention worthy that the gels could absorb cationic, anionic and neutral organic dyes. Presence of diverse functionalities like cationic and anionic side chains, unprotected termini and the presence of aromatic group containing amino acid residues in the peptides promotes a wide spectrum of absorbed dyes through electrostatic and aromatic π–π stacking interactions. The absorption efficiency of different contaminants for the different hydrogels is tabulated in Table 1. | ||
| Fig. 7 Dye and metal ion absorption by hydrogel A1 (a) RB, (b) CV, (c) MO, (d) NR, (e) nickel chloride (Ni2+) and (f) cobalt chloride (Co2+). | ||
 | ||
| Fig. 8 Photograph of dye absorption by hydrogel A1 + A2 = 1:1. (a) RB and (b) MO. Photograph of the mixture of dye/metal ion absorbed by hydrogel formed from (c) A1 and (d) A1 + A2. | ||
| Dye/metal salt | Nature | Loaded | % absorbed | Hydrogel system |
|---|---|---|---|---|
| RB | Cationic | 20 μM | 96.92 | A1 |
| CV | Cationic | 40 μM | 79.93 | |
| MO | Anionic | 70 μM | 88.92 | |
| NR | Neutral | 110 μM | 81.52 | |
| Nickel chloride (Ni2+) | Metal ion | 100 mM | 83.32 | |
| Cobalt chloride (Co2+) | Metal ion | 100 mM | 93.65 | |
| RB | Cationic | 20 μM | 84.28 | A1 + A2 |
| MO | Anionic | 70 μM | 83.47 |
The high efficiency for absorption of individual dyes, prompted us to investigate the absorption efficiency of the hydrogels from a mixtures of dyes. As seen from the Fig. 8c and d, S10c and d,† both the self-assembled and the co-assembled hydrogels were capable of absorbing dyes (CV, NR, MO and RB) and metal ions (Co2+ and Ni2+) from an all component mixture. Thus such hydrogels can be used in the real life applications, where the water is contaminated by several contaminants simultaneously.
Reusability of the hydrogel A1
In order to be efficiently used as a material for water purification, the material should be economic and hence, reusable. A1 hydrogel could be reused over three cycles for subsequent loading and release of the dye, after which the quality of the hydrogel degraded. Fig. S11a† is the pictorial representation of the hydrogel loaded with dye RB, its subsequent release in ether and reloading of the dye for the next cycle. Fig. S11b† shows the UV-Vis studies performed to quantify the amount of dye that could be loaded to the hydrogel in each cycle.It was observed that in the first cycle, the hydrogel A1 was capable of absorbing about 89% of RB. Upon addition of ether, the release medium to the dye loaded gel, and leaving it for 48 hours, the hydrogel became colourless with the ether having turned pink. The integrity of the gel was intact as can be proved by the vial inversion test and FESEM studies (Fig. S12†). In the second and third cycles about 74% and 53% of dye was loaded onto the gel (Table S3†). After the third cycle, the gel degraded and could not be used further.
Removal of toxic metal ions: absorption of Pb2+ and Hg2+
After being successful in removing divalent metal ions from water, A1 and A1 + A2 hydrogels were utilized for the absorption of toxic heavy metal ions Pb2+ and Hg2+ from the aqueous solutions. For both the self-assembled and the co-assembled hydrogels, Pb2+ was absorbed better (∼80%) than the Hg2+ ions (∼60%). Both the metal ions were dipositive, and though Hg2+ was smaller in size than Pb2+ (Table 2), it was absorbed less. Pb2+ was efficiently absorbed irrespective of its counter ions, while absorption of Hg2+ depended on its counter anions (Table S4†). Smaller counter ion led to better absorption in case of Hg2+. This feature can be attributed to the greater charge density on the Hg2+ (owing to its smaller size) and its stronger association with its counter ion in comparison to Pb2+.| Hydrogels | Metal ion | Conc. loaded (mg L−1) | Conc. in the supernatant (mg L−1) | Conc. absorbed (mg L−1) | % absorbed |
|---|---|---|---|---|---|
| A1 | Pb2+ | 20 | 3.34 | 16.66 | 83.3 |
| Hg2+ | 100 | 41.20 | 58.80 | 58.8 | |
| A1 + A2 (1:1) |
Pb2+ | 20 | 4.31 | 15.69 | 78.45 |
| Hg2+ | 100 | 50.45 | 49.55 | 49.55 |
In summary, we have been able to prove that our self- and co-assembled hydrogels are highly efficient in absorbing different organic dyes and metal ions from water individually or from a mixture. It is worth mentioning that these hydrogels can also absorb toxic metal ions like Pb2+ and Hg2+ from water efficiently. Additionally, these materials can be used recyclably increasing their cost effectiveness. Thus these cheap and robust materials are extremely prospective in water remediation.
Ion sensing studies by peptides A1 and A2
We have employed our peptides A1 and A2 in sensing different kinds of ions. The fluorescence emission response of A1 and A2 was monitored in the absence and the presence of various anions like fluoride (F−), chloride (Cl−), bromide (Br−), iodide (I−), bisulphate (HSO4−), dihydrogenphosphate (H2PO4−), acetate (CH3COO−), hydroxide (OH−) (as their Bu4N+ salts), arsenite (AsO2−) and arsenate (AsO3−) (Fig. S13a and b†) and also various metal ions like Hg2+, Co2+, Pb2+, Ni2+, Cu2+, Mn2+, Zn2+ and Fe3+ (Fig. S14a and b†). The spectroscopic studies were performed in EtOH/water as the peptides are not completely soluble in water. The anion and the metal salts were dissolved in water.Peptides A1 and A2 responded for arsenite (AsO2−), arsenate (AsO3−) and hydroxide (OH−) anions. In the presence of other anions (F−, Cl−, Br−, I−, HSO4−, H2PO4−, AcO−), there was no change in the λmax of the fluorescence emission and only minor changes in peak intensities (Fig. S13a and b†). In the absence of anions, upon being excited at 280 nm, the fluorescence emission maxima for A1 and A2 were at 349 nm and 323 nm respectively. For, A1, upon addition of 0.1 mM AsO2− and OH−, the peak at 349 nm red shifted to 357 nm and 358 nm respectively, with an enhancement in fluorescence signal intensity (about 2 folds). In case of addition of 0.1 mM of AsO3− solution to A1, there was a slight blue shift of the emission peak to 343 nm, although the fluorescence intensity was not changed remarkably (Fig. S13a†). There was negligible effect on the fluorescence spectra of A1 in the presence of other ions. For A2, upon addition of 0.1 mM AsO2− and OH−, the peak at 323 nm was red shifted to 330 nm and 349 nm respectively. In case of addition of 0.1 mM of AsO3− solution to A2, there was a decrease in the fluorescent intensity without any change in the peak position (Fig. S13b†). There was negligible effect on the fluorescence spectra of A2 in the presence of other anions. Thus it can be concluded that molecules A1 and A2 selectively sensed AsO2−, AsO3− and OH− in solution.
To determine the sensitivity of the peptides towards the sensed anions, the fluorescence response of A1 and A2 was monitored as a function of anion concentration (Fig. 9a–c and S15a–c†). The concentration of the anions was varied from 0.02 mM to 2 mM. In case of A1, it was observed that for AsO2− and OH−, upon increasing the concentration of the anions, there was a steady enhancement in the intensity of fluorescence signal upto 0.07 mM, after which the signal abruptly intensified accompanied by a red shift (Fig. 9a and b). In case AsO3− solution, there was an irregular shuffle in fluorescent intensity although, it eventually decreased upon increasing the concentration of AsO3− solution. Also, there was a considerably little blue shift of the emission peak to 346 nm that was observed during the titration (Fig. 9c). In case of A2, similar trend in fluorescence signal was observed for AsO2− and OH− ions as observed for A1. When the concentration of AsO2− and OH− was increased from 0.02 mM, the fluorescence signal did not change much except a red shift to 330 nm till 0.1 mM, after which there was a sudden increase in the signal along with a major red shift to 356 nm beyond which the fluorescence signal got saturated till 2 mM (Fig. S15a and b†). In case of increasing concentration of AsO3−, at first the fluorescence intensity slightly increased, after which there was an irregular intensity shift of the signal followed by an eventual decrease in the signal intensity (at ∼0.4 mM) accompanied by a red shift to 347 nm (Fig. S15c†). The mode of interaction of the anions OH− and AsO2− seem to be similar from their fluorescence response. This might be due to the similarity in the size of these two anions in comparison to AsO3−. The detection limit of A1 and A2 for the anions is tabulated in Table 2. A1 had a lower LOD for all the anions in comparison to A2. This most probably was owing to the negative charge on the anions electrostatically favouring the positively charged A1 molecule over the negatively charged A2 molecule. LODs for the detection of AsO2− and AsO3− by A1 are 2.57 μM and 7.06 μM and by A2 are 33.08 μM and 10.96 μM respectively. AsO2− and AsO3− contain arsenic in the most toxic oxidation states which have very grave health implications causing serious diseases and death. In several parts of the world, contamination of the underground drinking water with arsenic causes severe problems. Though the limits of detection of As(III) and As(V) by the molecules A1 and A2 is above the recommended safety levels of arsenic in drinking water (10 μg L−1, 0.13 μM), it is still highly relevant. There are very few examples of peptide based arsenic detectors which make these sensors even more prospective candidates for selective arsenic detection.
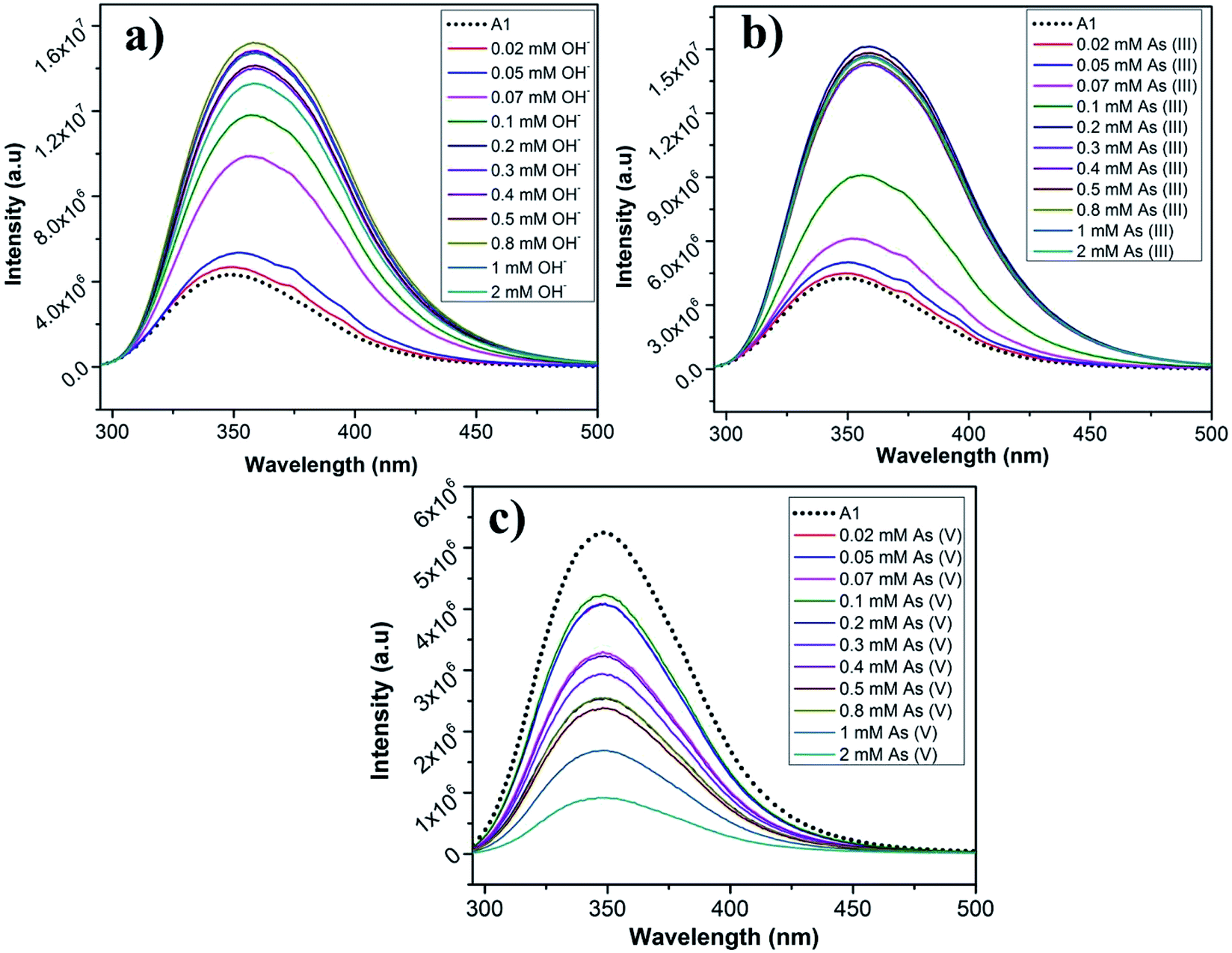 | ||
| Fig. 9 Anion sensing by A1: titration of A1 with (a) OH−, (b) As(III), and (c) As(V). | ||
Response towards metal ions
We have studied the response of our peptides A1 and A2 towards different metal ions (Cu2+, Hg2+, Fe3+, Pb2+, Zn2+, Ni2+, Co2+ and Mn2+). A decrease in fluorescence intensity for A1 and A2 was observed upon addition of 0.1 mM of Hg2+ and Fe3+ without any change in the peak position (λem = 349 nm and 323 nm for A1 and A2 respectively). In the presence of other metal ions Co2+, Pb2+, Ni2+, Cu2+, Mn2+, Zn2+ there was no change in peak positions or intensity of the signal (Fig. S14a and b†). To find out the sensitivity of Fe3+ ion detection, the fluorescence response of A1 and A2 was monitored as a function of metal ion concentration (0.02 mM to 2 mM) (Fig. S16a and b†). It was observed that for both A1 and A2, there was a quenching in fluorescence signal upon increasing the concentration of Fe3+ without any change in the peak position. A1 and A2 had a detection limit of 2.19 μM and 18.82 μM respectively towards Fe3+ (Table 3).| Ions | A1 | A2 |
|---|---|---|
| As(III) | 2.57 μM | 33.08 μM |
| OH− | 2.86 μM | 39.68 μM |
| As(V) | 7.06 μM | 10.96 μM |
| Fe3+ | 2.19 μM | 18.82 μM |
4. Conclusions
In this study, we have designed two charge complementary peptides, A1 and A2, containing Lys and Glu residues for forming cooperatively co-assembled materials. We have studied the self-assembly and the co-assembly processes of these materials to molecular details. A1 formed hydrogel in sodium phosphate buffer (pH 7.0) while A2 did not. Co-assembled gels were formed only with equal or greater amounts of A1 in the mixture. Electrostatic interaction was the most important driver though H-bonding, π–π stacking interactions and hydrophobic interactions were also instrumental in the assembly process. Peptides adopted β-sheet conformation and the stacking of the sheets gave rise to the formation of the fibre like structure. The thinner fibres assembled together laterally giving rise to the thicker ones, which entangled to form the mesh like architecture that constituted the hydrogel. Similarity in the structure of the constituent peptides led to cooperative co-assembly in the system, wherein electrostatic interactions between the charge complementary peptides gave rise to the tougher co-assembled gels. Application of the self- and co-assembled hydrogels in waste water remediation was thoroughly probed. All the reported hydrogels were efficient absorbents of different types of organic dyes (RB, CV, NR, MO), small divalent metal ions (Ni2+ and Co2+) as well as heavy toxic metal ions like Pb2+ and Hg2+. These materials could be recyclably deployed making their use economically viable. Ability to remove contaminants individually and from a mixture of several components makes these hydrogels highly prospective for practical uses of water remediation. Peptides A1 and A2 can selectively detect arsenite and arsenate, the most toxic forms of arsenic responsible for contamination of drinking water, in addition to detection of OH− and Fe3+. The simple peptide based materials developed in the present study can thus be employed for versatile applications. Understanding of the molecular details of the assembly processes would empower scientists to develop such materials with versatile applications through rational design in the future.Abbreviations
| DNA | Deoxyribonucleic acid |
| RB | Rhodamine B |
| CV | Crystal violet |
| NR | Neutral red |
| MO | Methyl orange |
| PBS | Phosphate buffer saline |
| HPLC | High performance liquid chromatography |
| ESI-MS | Electrospray ionization mass spectrometry |
| NMR | Nuclear magnetic resonance |
| CD | Circular dichroism |
| FTIR | Fourier transform infrared |
| FESEM | Field emission scanning electron microscopy |
| PXRD | Powder X-ray diffraction |
| MALDI | Matrix-assisted laser desorption/ionization |
| EtOH | Ethanol |
| MGC | Minimum gelation concentration |
| Lys | Lysine |
| Phe | Phenylalanine |
| Leu | Leucine |
| Val | Valine |
| Trp | Tryptophan |
| Glu | Glutamic acid |
| Fmoc | Fluorenylmethyloxycarbonyl |
| CT | Charge transfer |
| RT | Room temperature |
| HOBt | Hydroxybenzotriazole |
| PyBOP | Benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate |
| DMAP | 4-Dimethylaminopyridine |
| DIC | N,N′-Diisopropylcarbodiimide |
| DIPEA | N,N-Diisopropylethylamine |
| DCM | Dichloromethane |
| DMF | Dimethylformamide |
| TFA | Trifluoroacetic acid |
| SPPS | Solid phase peptide synthesis |
| AAS | Atomic absorption spectrometry |
Author contributions
SC designed the project, KR performed most of the experiments, AKS helped in the synthesis of the materials. SC and KR analysed the data and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SC would like to acknowledge CSIR, India (01 (2984)/19/EMR-II) for the financial support required for this research. KR acknowledges IIT Guwahati for scholarship, and Central Instruments Facility, IITG for the FESEM, 600 MHz NMR and FETEM instruments facility. SC acknowledges Sumit Chowdhuri for helping with the rheology experiment. SC and KR acknowledges Centre for Environment, IIT Guwahati and Dept. of Civil Engineering, IIT Guwahati for AAS instrument facility.References
- G. M. Whitesides and M. Boncheva, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4769–4774 CrossRef CAS.
- K. Arigaa, M. Nishikawaa, T. Mori, J. Takeya, L. K. Shrestha and J. P. Hill, Sci. Technol. Adv. Mater., 2019, 20, 51–95 CrossRef.
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CrossRef CAS.
- S. Zhang, Interface Focus, 2017, 7, 20170028 CrossRef.
- F. Clerici, E. Erba, M. L. Gelmi and S. Pellegrino, Tetrahedron Lett., 2016, 57, 5540–5550 CrossRef CAS.
- X. Hu, M. Liao, H. Gong, L. Zhang, H. Cox, T. A. Waigh and J. R. Lu, Curr. Opin. Colloid Interface Sci., 2020, 45, 1–13 CrossRef CAS.
- D. Mandal, A. N. Shirazib and K. Parang, Org. Biomol. Chem., 2014, 12, 3544–3561 RSC.
- B. B. Gerbelli, S. V. Vassiliades, J. E. U. Rojas, J. N. B. D. Pelin, R. S. N. Mancini, W. S. G. Pereira, A. M. Aguilar, M. Venanzi, F. Cavalieri, F. Giuntini and W. A. Alves, Macromol. Chem. Phys., 2019, 1900085 CrossRef.
- C. J. C. Edwards-Gayle and I. W. Hamley, Org. Biomol. Chem., 2017, 15, 5867–5876 RSC.
- L. Adler-Abramovicha and E. Gazit, Chem. Soc. Rev., 2014, 43, 6881–6893 RSC.
- P. Makam and E. Gazit, Chem. Soc. Rev., 2018, 47, 3406–3420 RSC.
- D. Shen, J. A. Cooper, P. Li, Q.-H. Guo, K. Cai, X. Wang, H. Wu, H. Chen, L. Zhang, Y. Jiao, Y. Qiu, C. L. Stern, Z. Liu, A. C.-H. Sue, Y.-W. Yang, F. M. Alsubaie, O. K. Farha and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 2042–2050 CrossRef CAS.
- R. Ni and Y. Chau, J. Am. Chem. Soc., 2014, 136, 17902–17905 CrossRef CAS.
- Q. Zou, L. Zhang, X. Yan, A. Wang, G. Ma, J. Li, H. Mçhwald and S. Mann, Angew. Chem., Int. Ed., 2014, 53, 2366–2370 CrossRef CAS.
- K. E. Inostroza-Brito and E. Collin, et al., Nat. Chem., 2015, 7, 897–904 CrossRef CAS.
- X.-D. Xu, C.-S. Chen, B. Lu, S.-X. Cheng, X.-Z. Zhang and R.-X. Zhuo, J. Phys. Chem. B, 2010, 114, 2365–2372 CrossRef CAS.
- L. Mei, S. He, Z. Liu, K. Xu and W. Zhong, Chem. Commun., 2019, 55, 4411–4414 RSC.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, N. T. Hunt and R. V. Ulijn, Biomacromolecules, 2014, 15, 1171–1184 CrossRef CAS.
- J. K. Sahoo, M. A. VandenBerg, E. E. R. Bello, C. D. Nazareth and M. J. Webber, Nanoscale, 2019, 11, 16534–16543 RSC.
- N. L. Truex and J. S. Nowick, J. Am. Chem. Soc., 2016, 138, 13891–13900 CrossRef CAS.
- D. M. Ryan, T. M. Doran and B. L. Nilsson, Langmuir, 2011, 27, 11145–11156 CrossRef CAS.
- S.-M. Hsu, F.-Y. Wu, T.-S. Lai, Y.-C. Lin and H.-C. Lin, RSC Adv., 2015, 5, 22943–22946 RSC.
- M. Halperin-Sternfeld, M. Ghosh, R. Sevostianov, I. Grigoriants and L. Adler-Abramovich, Chem. Commun., 2017, 53, 9586–9589 RSC.
- S. Li, A. K. Mehta, A. N. Sidorov, T. M. Orlando, Z. Jiang, N. R. Anthony and D. G. Lynn, J. Am. Chem. Soc., 2016, 138, 3579–3586 CrossRef CAS.
- H. A. Behanna, J. J. J. M. Donners, A. C. Gordon and S. I. Stupp, J. Am. Chem. Soc., 2005, 127, 1193–1200 CrossRef CAS.
- Z. Yu, A. Erbas, F. Tantakitti, L. C. Palmer, J. A. Jackman, M. O. de la Cruz, N.-J. Cho and S. I. Stupp, J. Am. Chem. Soc., 2017, 139, 7823–7830 CrossRef CAS.
- R. J. Swanekamp, J. T. M. DiMaio, C. J. Bowerman and B. L. Nilsson, J. Am. Chem. Soc., 2012, 134, 5556–5559 CrossRef CAS.
- K. J. Nagya, M. C. Giano, A. Jin, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2011, 133, 14975–14977 CrossRef.
- K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. O'Reilly, L. C. Serpell and D. J. Adams, Nat. Commun., 2013, 4, 1480 CrossRef.
- J. Raeburn, B. Alston, J. Kroeger, T. O. McDonald, J. R. Howse, P. J. Cameron and D. J. Adams, Mater. Horiz., 2014, 1, 241–246 RSC.
- S. Zhang, W. Cortes and Y. Zhang, Langmuir, 2020, 36, 6261–6267 CrossRef CAS.
- L. Adler-Abramovich, P. Marco, Z. A. Arnon, R. C. G. Creasey, T. C. T. Michaels, A. Levin, D. J. Scurr, C. J. Roberts, T. P. J. Knowles, S. J. B. Tendler and E. Gazit, ACS Nano, 2016, 10, 7436–7442 CrossRef CAS.
- R. C. G. Creasey, I. Louzao, Z. A. Arnon, P. Marco, L. Adler-Abramovich, C. J. Roberts, E. Gazit and S. J. B. Tendler, Soft Matter, 2016, 12, 9451–9457 RSC.
- S. Maity, S. Nirab and M. Reches, J. Mater. Chem. B, 2014, 2, 2583–2591 RSC.
- K. J. Channon, G. L. Devlin and C. E. MacPhee, J. Am. Chem. Soc., 2009, 131, 12520–12521 CrossRef CAS.
- M. A. Khalily, G. Bakan, B. Kucukoz, A. E. Topal, A. Karatay, H. G. Yaglioglu, A. Dana and M. O. Guler, ACS Nano, 2017, 11, 6881–6892 CrossRef CAS.
- Z. Huang, S. Guan, Y. Wang, G. Shi, L. Cao, Y. Gao, Z. Dong, J. Xu, Q. Luo and J. Liu, J. Mater. Chem. B, 2013, 1, 2297–2304 RSC.
- V. Jayawarnal, A. Smith, J. E. Gough and R. V. Ulijn, Biochem. Soc. Trans., 2007, 35, 535–537 CrossRef.
- Y. Li and L. Wang, Chem. Lett., 2016, 45, 1253–1255 CrossRef CAS.
- S. Basak, N. Nandi, S. Paul, I. W. Hamley and A. Banerjee, Chem. Commun., 2017, 53, 5910–5913 RSC.
- S. Ray, A. K. Das and A. Banerjee, Chem. Mater., 2007, 19, 1633–1639 CrossRef CAS.
- F. I. Hai, K. Yamamoto and K. Fukushi, Crit. Rev. Environ. Sci. Technol., 2007, 37, 315–377 CrossRef CAS.
- D. Solpan and O. Guven, Radiat. Phys. Chem., 2002, 65, 549–558 CrossRef CAS.
- M. Schrope, Nature, 2011, 472, 152–154 CrossRef CAS.
- K. J. Parker, S. Kumar, D. A. Pearce and A. J. Sutherland, Tetrahedron Lett., 2005, 46, 7043–7045 CrossRef CAS.
- T. Yang, X.-X. Zhang, J.-Y. Yang, Y.-T. Wang and M.-L. Chen, Talanta, 2018, 177, 212–216 CrossRef CAS.
- K. Roy, G. Pandit, M. Chetia, A. K. Sarkar, S. Chowdhuri, A. P. Bidkar and S. Chatterjee, ACS Appl. Bio Mater., 2020, 3, 6251–6262 CrossRef CAS.
- N. Xia, Y. Shi, R. Zhang, F. Zhao, F. Liu and L. Liu, Anal. Methods, 2012, 4, 3937–3941 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Figures of analytical HPLC, ESI-MS, 1H NMR, FESEM of A1 and A2 in sodium phosphate buffer and EtOH/sodium phosphate buffer, rheology of A1 hydrogel, mechanism of assembly, absorption studies of organic dyes/metal ions using UV spectroscopy by A1 hydrogel and (A1 + A2) co-assembled gels as individual and a mixture of contaminants, reusability of A1 hydrogel, cation and anion sensing by A1 and A2, concentration dependent titration of anions (AsO2−, AsO3−, OH−) by A2 and Fe3+ by A1 and A2. Tables of gelation properties of A1 and various (A1 + A2) hydrogels, interplaner distances in A1, A2, A1 xerogel and (A1 + A2) xerogels, percentage of RB absorbed by the hydrogels, dependence of absorption of Pb2+ and Hg2+ by the A1 hydrogel on counter ions. See DOI: 10.1039/d0ra08407g |
| This journal is © The Royal Society of Chemistry 2020 |