
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bifunctional amino acid to study protein–protein interactions†
Tangpo Yang‡
,
Xin Li and
Xiang David Li*
and
Xiang David Li*
Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, China. E-mail: xiangli@hku.hk
First published on 18th November 2020
Abstract
Protein–protein interactions (PPIs) play crucial roles in regulating essentially all cellular processes. Photo-cross-linking represents a powerful method to study PPIs. To fulfil the requirements for the exploration of different PPIs, there is a continuous demand on the development of novel photo-reactive amino acids with diverse structural properties and functionalities. Reported herein is the development of a bifunctional amino acid termed dzANA, which contains a diazirine, for photo-cross-linking, and a terminal alkyne group, for bioorthogonal tagging. Using known PPIs between histone posttranslational modifications (PTMs) and their binding partners as models, we demonstrate that the dzANA-harbouring peptide-based photoaffinity probes could efficiently and selectively capture the weak and transient PPIs mediated by histone modifications. Our study indicates the potential of dzANA to identify and characterize unknown PPIs.
Introduction
Almost all biological processes, including DNA replication and gene expression, cellular transportation and secretion, signal transduction, and metabolism, rely on the exquisite regulation of interactions among their key protein components.1,2 Given the important roles played by protein–protein interactions (PPIs) in normal physiology, aberrant regulation of PPIs may lead to human diseases, such as Alzheimer's disease and cancer. Identification and characterization of PPIs are therefore critical for not only fundamental understanding of complex cellular processes but also the development new therapeutic strategies to treat human diseases. A variety of methods, including affinity purification, yeast two-hybrid screening, among others, have been developed to study PPIs.3 However, results obtained from these methods may suffer from a high false-positive rate. In addition, it remains a challenge to identify and characterize weak or transient PPIs, including those mediated by posttranslational modifications (PTMs).Photo-cross-linking represents a powerful method to study PPIs, which converts the noncovalent interactions between a protein of interest and its binding partners into covalent linkages, facilitating the following identification and characterization processes.4–6 Among all the frequently used photo-reactive groups, diazirine is known for its small size and high reactivity.7–10 These two features of diazirine ensure high cross-linking specificity and efficiency, as it can be installed close enough to the binding sites without inducing undesired steric hindrance, and thereby selectively and rapidly label the PPIs of interest upon photo-activation.
We and others have reported the development of various diazirine-based photo-reactive amino acids (Fig. 1).11–24 These unnatural amino acids are readily incorporated into peptides and proteins to offer versatile photoaffinity probes for PPIs mapping. To facilitate the detection and identification of the targeted PPIs, the peptide- or protein-based probes are usually decorated with epitope sequences or bioorthogonal chemical handles. Recognition of an epitope by antibodies may vary in affinity and selectivity case-by-case, leading to loss of target proteins or false-positive results. In contrast, the bioorthogonal handles, for example, alkyne and azide, are inert to different biomolecules and are covalently conjugated to fluorescent dyes (e.g., rhodamine) or affinity tags (e.g., biotin), enabling the subsequent visualization or isolation of the proteins labelled by photoaffinity probes with high reliability. The introduction of both photo-reactive and bioorthogonal functionalities into one probe at the same time, however, can sometimes be troublesome or even technically challenging.25,26
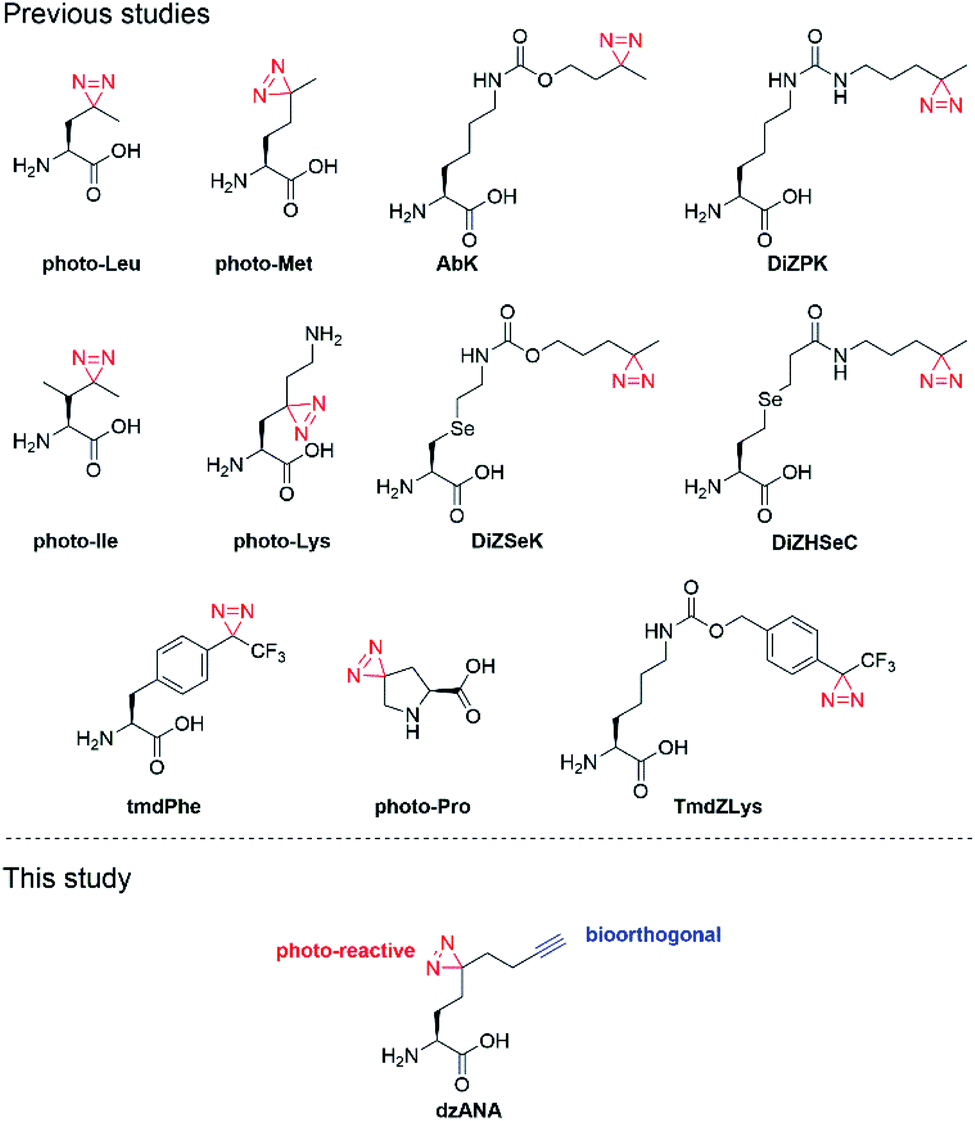 | ||
| Fig. 1 Structures of reported photo-reactive unnatural amino acids and dzANA developed in this study. | ||
Here, we report the development of dzANA (Fig. 1), a bifunctional amino acid containing both a diazirine moiety and a terminal alkyne group. Using a collection of PPIs mediated by histone posttranslational modifications (PTMs) as models, we showed that the peptide-based chemical probes armed with dzANA could robustly capture the known binding partners, suggesting the potential of this novel bifunctional amino acid to identify and characterize unknown PPIs.
Experimental
Reagents
Fmoc-protected amino acids were purchased from GL Biochem. Fmoc-N-ε-(trimethyl)-L-lysine chloride (Fmoc-Lys(Me3)-OH) were purchased from Chem-Impex International (Wood Dale, IL). All the other chemical reagents and solvents were purchased from Sigma-Aldrich and used without further purification. In-solution reactions were monitored by thin-layer chromatography (TLC) silica gel 60 F254 from Merck. Plates were visualized by UV light or 1% KMnO4. Flash column chromatography was carried out using silica gel purchased from Grace.Instrumentation
1H NMR (300 MHz or 400 MHz), 13C NMR (75 MHz, 100 MHz) were conducted on a Bruker spectrometer at 25 °C and were calibrated using residual undeuterated solvent as an internal reference. Chemical shifts were reported in ppm and coupling constants (J) were quoted to the nearest 0.1 Hz. High resolution mass spectrometry (HRMS) was recorded using a Bruker maXis II High Resolution QTOF. Peptides were analyzed by LC-MS with an Agilent 1260 Infinity HPLC system connected to a Thermo Finnigan LCQ DecaXP MS detector. Peptides were further purified by a preparative HPLC system with Waters 2535 Quaternary Gradient Module, Waters 515 HPLC pump, Waters SFO System Fluidics Organizer and Waters 2767 Sample Manager.Photo-cross-linking were performed with ENF-260C/FE hand-hang UV lamp (Spectroline). In-gel fluorescence scanning was performed using a Typhoon 9410 variable mode imager from GE Healthcare Life Sciences (excitation 532 nm, emission 580 nm). All images were processed by ImageJ software (National Institutes of Health), and contrast was adjusted appropriately.
Molecular cloning, protein expression and purification
Plasmids construction, protein expression and purification were performed as previously described:12,24 GST-SPIN1 (full-length), GST-ING2 (208–270), BHC80 (486–543), Sirt3 (102–399), BRD4 (44–168), Sirt5 (34–302), GST-HP1 (23–74).Photo-cross-linking
Indicated probes in the presence or absence of different concentrations of corresponding competitors were incubated with recombinant proteins (60 ng μL−1) in binding buffer (50 mM HEPES, 150 mM NaCl, 2 mM MgCl2, 0.1% Tween-20, 20% glycerol, pH 7.5) at 4 °C for 10 min. Then the samples were exposed to 365 nm UV irradiation for 20 min in 96-well (recombinant proteins, 75 μL per well) on ice.Cu(I)-catalyzed azide–alkyne cycloaddition/click chemistry
After photo-cross-linking, 100 μM rhodamine-N3 (10 mM stock in DMSO) was added, followed by 1 mM TCEP (freshly prepared 50 mM stock in H2O) and 100 μM TBTA (10 mM stock in DMSO). Finally, the reaction was initiated by the addition of 1 mM CuSO4 (freshly prepared 50 mM stock in H2O). The reactions were incubated at room temperature for 1 h with regular vortexing. After quenching by adding 5 volumes of ice-cold acetone, the samples were placed at −20 °C overnight to precipitate proteins.In-gel fluorescence scanning
After precipitation, proteins were spin down at 6000g for 5 min at 4 °C. The supernatant was discarded, and the pellet was washed with ice-cold methanol twice and air-dried for 10 min. The proteins were resuspended in 1× LDS loading buffer (Invitrogen) with 50 mM DTT and heated at 80 °C for 8 min before resolving by SDS-PAGE. The labelled proteins were visualized by scanning on a Typhoon 9410 model.Peptide synthesis
All peptides used in this research were synthesized on Rink-Amide MBHA resin followed standard Fmoc-based solid-phase peptide synthesis protocol. After the coupling of all amino acids, the removal of protecting groups and cleavage of peptides from the resin were done by incubating the resin with cleavage cocktail containing 95% trifluoroacetic acid (TFA), 2.5% triisopropylsilane, 1.5% water and 1% thioanisole for 2 h. Peptides were purified by preparative HPLC with an XBridge Prep OBDTM C18 column (30 mm × 250 mm, 10 μm, Waters). Mobile phase used were water with 0.1% TFA (buffer A) and 90% acetonitrile (ACN) in water with 0.1% TFA (buffer B).Cross-linking yield determination
The chemical conjugation of BSA to rhodamine and the determination of photo-cross-linking yield of selected proteins using the rhodamine-labelled BSA were performed as previously describe.Synthesis
The synthesis of dzANA and its derivatives was followed the synthetic route listed in Scheme 1.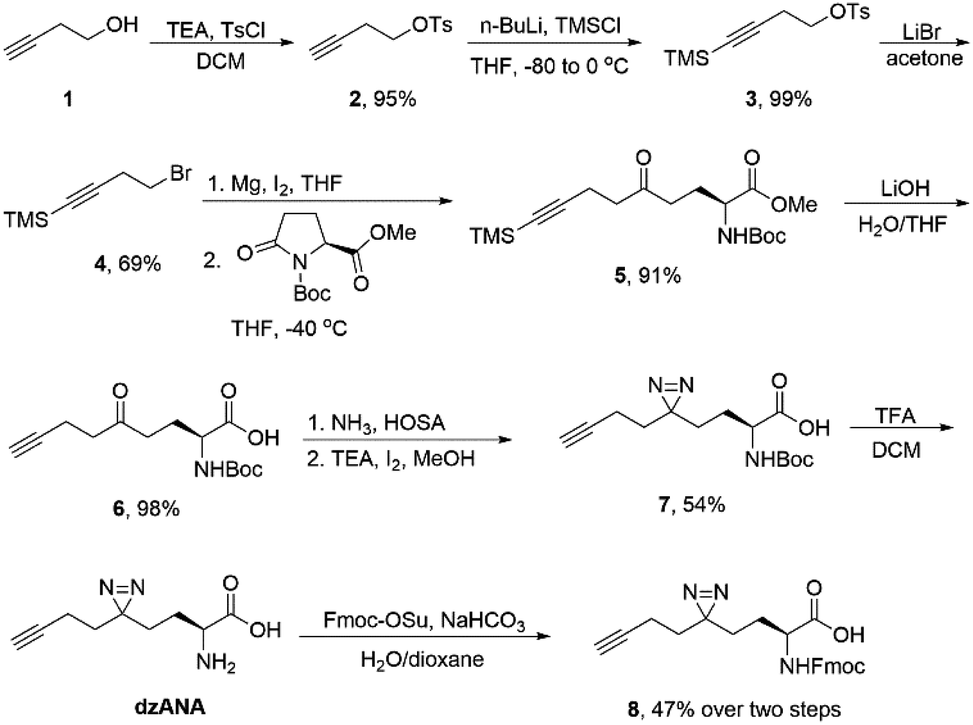 | ||
| Scheme 1 Synthetic route for free and Fmoc-protected dzANA. TEA, triethylamine; Ts, p-tosyl; DCM, dichloromethane; Bu, butyl; TMS, trimethylsilyl; THF, tetrahydrofuran; Me, methyl; Boc, tert-butyloxycarbonyl; HOSA: hydroxylamine-O-sulfonic acid; TFA, trifluoroacetic acid; Fmoc-OSu, 9-fluorenylmethyl N-succinimidyl carbonate. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2 mL in total). The mixture was stirred at room temperature for 3 h. Then solvent was removed under reduced pressure, the resulting white solid was used for next step without any further purification. The solid was dissolved in water (5 mL) and dioxane (10 mL) followed by adding NaHCO3 (213 mg, 2.5 eq.). To the mixture was added a solution of Fmoc-OSu (0.42 g, 1.2 eq.) in dioxane (2 mL) over 15 min. The solution was stirred for 24 h at room temperature. All the organic solvents were removed, and the residue was dissolved in 5 mL of water. The solution was acidified to pH 3–4 using 1 M HCl. The aqueous solution was extracted with ethyl acetate. The combined organic layers were dried over Na2SO4. The solution was concentrated and compound 8 was obtained by flash column chromatography as a white solid (200 mg, 47% yield). 1H NMR (400 MHz, d6-DMSO) δ 7.89 (d, J = 7.5 Hz, 2H), 7.71 (d, J = 7.4 Hz, 2H), 7.59 (d, J = 8.2 Hz, 1H), 7.42 (t, J = 7.5 Hz, 2H), 7.33 (t, J = 7.4 Hz, 2H), 4.33–4.20 (m, 3H), 3.88 (s, 1H), 2.81 (s, 1H), 2.05–1.94 (m, 2H), 1.71–1.28 (m, 6H); 13C NMR (100 MHz, d6-DMSO) δ 173.41, 156.11, 143.84, 143.77, 140.74, 127.64, 127.06, 125.23, 120.12, 71.74, 65.57, 53.17, 46.66, 31.41, 28.74, 28.06, 25.11, 12.71; LRMS (EI, 20 eV) m/z 389.2 (M+ − N2, 0.14), 178.1 (100); HRMS (EI, 20 eV) for C24H23NO4 (M+ − N2): calcd 389.1622, found 389.1630.
:1, 2 mL in total). The mixture was stirred at room temperature for 3 h. Then solvent was removed under reduced pressure, the resulting white solid was used for next step without any further purification. The solid was dissolved in water (5 mL) and dioxane (10 mL) followed by adding NaHCO3 (213 mg, 2.5 eq.). To the mixture was added a solution of Fmoc-OSu (0.42 g, 1.2 eq.) in dioxane (2 mL) over 15 min. The solution was stirred for 24 h at room temperature. All the organic solvents were removed, and the residue was dissolved in 5 mL of water. The solution was acidified to pH 3–4 using 1 M HCl. The aqueous solution was extracted with ethyl acetate. The combined organic layers were dried over Na2SO4. The solution was concentrated and compound 8 was obtained by flash column chromatography as a white solid (200 mg, 47% yield). 1H NMR (400 MHz, d6-DMSO) δ 7.89 (d, J = 7.5 Hz, 2H), 7.71 (d, J = 7.4 Hz, 2H), 7.59 (d, J = 8.2 Hz, 1H), 7.42 (t, J = 7.5 Hz, 2H), 7.33 (t, J = 7.4 Hz, 2H), 4.33–4.20 (m, 3H), 3.88 (s, 1H), 2.81 (s, 1H), 2.05–1.94 (m, 2H), 1.71–1.28 (m, 6H); 13C NMR (100 MHz, d6-DMSO) δ 173.41, 156.11, 143.84, 143.77, 140.74, 127.64, 127.06, 125.23, 120.12, 71.74, 65.57, 53.17, 46.66, 31.41, 28.74, 28.06, 25.11, 12.71; LRMS (EI, 20 eV) m/z 389.2 (M+ − N2, 0.14), 178.1 (100); HRMS (EI, 20 eV) for C24H23NO4 (M+ − N2): calcd 389.1622, found 389.1630.Results
To synthesize dzANA, a seven-step synthetic route was designed (Scheme 1). Briefly, 3-butyn-1-ol 1 was treated with p-tosyl chloride in the presence of triethylamine to give compound 2, whose terminal alkyne was further protected by trimethylsilyl group to yield 3. Nucleophilic substitution with LiBr converted the tosylate into organobromide 4, which was used for the preparation of corresponding Grignard reagent. The following ring-opening reaction of the Boc protected L-pyroglutamate methyl ester by the in situ generated organomagnesium species afforded the intermediate 5 with the aminononynoic acid skeleton. Simultaneous removal of the trimethylsilyl group and the methyl ester under an alkaline environment provided the ketone 6. Subsequent diazirine synthesis using the ketone as a precursor led to the formation of 7, which was further deprotected by trifluoroacetic acid to give the desired dzANA. To facilitate the solid-phase peptide synthesis, the Fmoc-protected dzANA (8) was also prepared. The overall yield of the whole synthetic route was 15%.We next sought out to test the ability of dzANA in capturing PPIs. To this end, the histone PTM-mediated PPIs were chosen as the models. Histones, the scaffold proteins for the package of DNA in eukaryotic cell nuclei, are extensively decorated by diverse PTMs.27 By recognizing the histone PTMs, many effector proteins bind to histones to regulate DNA-templated biological processes, such as gene expression.28–30 Because of the dynamic nature of histone PTMs, the PPIs mediated by these modifications are normally weak or even transient, rendering them stringent models to evaluate the performances of chemical probes to capture PPIs.
To develop dzANA-based photoaffinity probes for the study of histone PTMs, we first focused on a well-studied trimethylation mark at histone H3 Lys 4 (H3K4me3). Our design of probe 1 (Fig. 2a) was based on the H3K4me3 peptide (residues 1–15), in which the dzANA was incorporated at the original Ala 7 site, a position that is near the K4me3 binding pocket to ensure the cross-linking efficiency and specificity, but is not too close to interfere with the PPI of interest. Two known binding partners of H3K4me3, SPIN1 (ref. 31) and ING2,32,33 were used as positive controls to validate the capability of probe 1 in the identification of H3K4me3-mediated PPIs. The two proteins were first incubated with probe 1. After UV-induced photo-cross-linking, the probe 1-labelled proteins were conjugated to rhodamine-azide (Rho-N3) via ‘click chemistry’ and resolved by SDS-PAGE. Subsequent in-gel fluorescence scanning showed that both SPIN1 and ING2 could be labelled by probe 1, but not by a control probe 2 lacking the trimethylation mark (Fig. 2b). Interestingly, experiments using BHC80,34 an unmodified histone H3 tail binder, led to a completely reversed cross-linking pattern that the trimethylated probe 1 failed to capture the protein while the unmodified probe 2 induced robust labelling (Fig. 2b). This observation matched with the previous discovery that the K4me3 mark could disrupt the BHC80–H3 tail interaction. It was worth noting that the probe 1-induced labelling toward SPIN1 and ING2, and the probe 2-induced labelling toward BHC80, could be competed off by the addition of their corresponding native peptide ligands (Fig. 2b), further demonstrating that the cross-linking was modification-dependent instead of non-specific labelling.
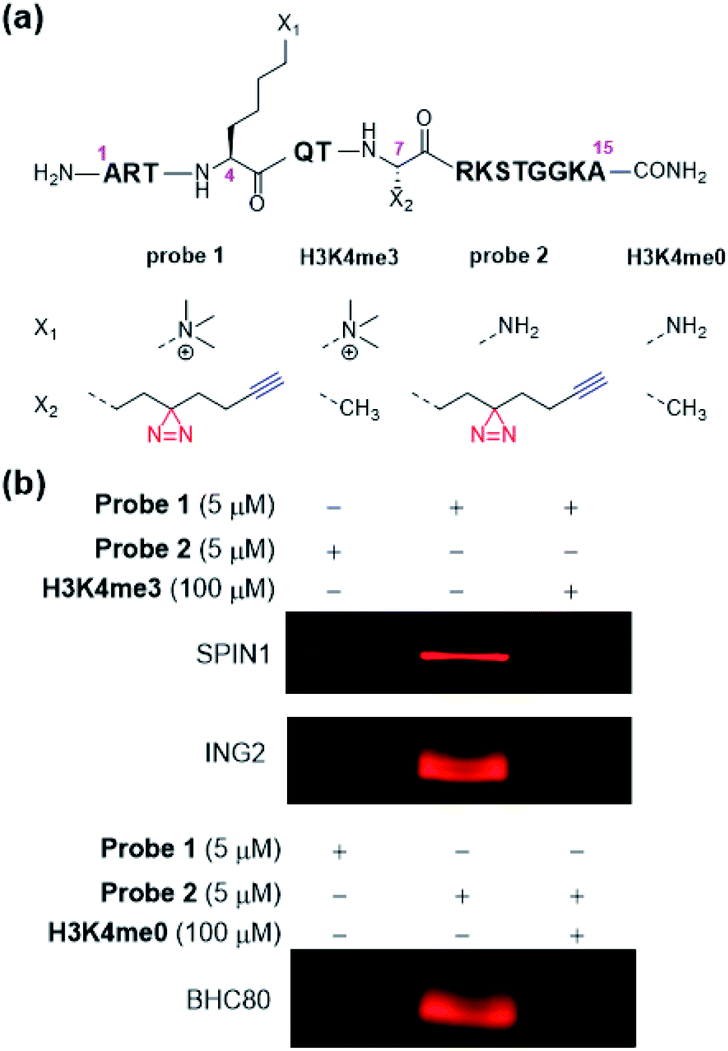 | ||
| Fig. 2 (a) Structures of probe 1, probe 2, H3K4me3 and H3K4me0 peptides. (b) Selective labelling of SPIN1 and ING2 by probe 1, and BHC80 by probe 2. For all the photo-cross-linking experiments, protein concentration used was 60 μg mL−1. After UV irradiation (365 nm) for 20 min on ice, the probe-labelled proteins were conjugated to Rho-N3 and visualized by in-gel fluorescence scanning. | ||
Encouraged by above results, we further designed and synthesized a series of dzANA-based probes (probe 3 to probe 6, Fig. 3a) to expand our tests to PPIs mediated by different histone PTMs at varied positions. As shown in Fig. 3b, all of these probes could robustly and selectively label their known binding proteins, which were the crotonylation mark at H3K4 (probe 3) toward Sirt3,35 the trimethylation mark at H3K9 (probe 4) toward HP1,36,37 the acetylation mark at H3K9 (probe 5) toward BRD4,38 and the malonylation mark at H3K9 (probe 6) toward Sirt5,39,40 while the corresponding control probe 2 and probe 7 without the modifications showed no cross-linking signals.
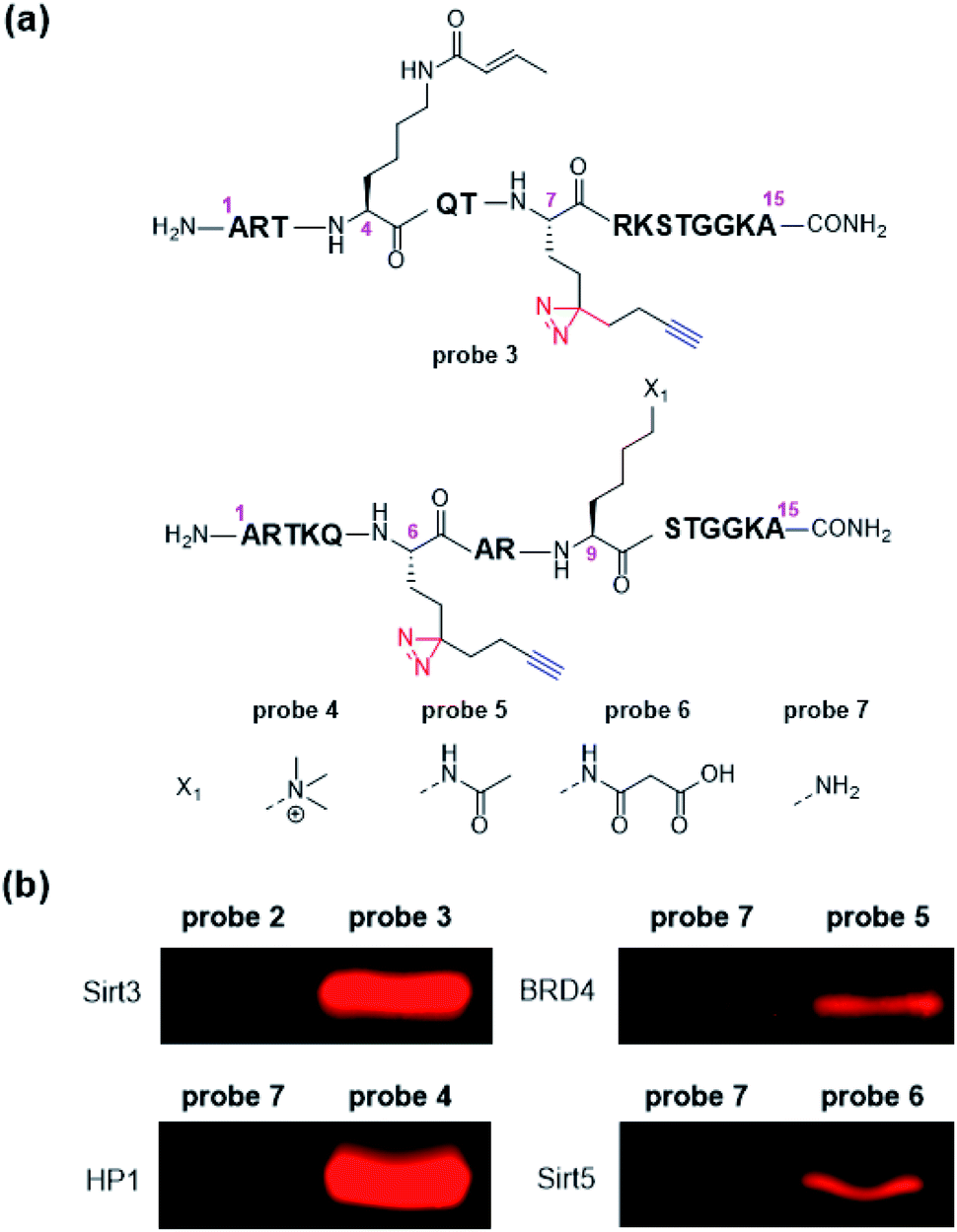 | ||
| Fig. 3 (a) Structures of probe 3 to probe 7, (b) selective labelling of Sirt3, HP1, BRD4, and Sirt5 by corresponding probes. Photo-cross-linking experiments followed the same conditions as listed in Fig. 2. | ||
To obtain a more quantitative assessment on how efficient the dzANA-harbouring probes captured their target proteins, we repeated the photo-cross-linking experiments of ING2, SPIN1, BHC80, and Sirt3 using the corresponding probes. To each of the resulting samples, we parallelly loaded a series of different amount of bovine serum albumin (BSA) pre-labelled by rhodamine in SDS-PAGE as standards.24 Based on the in-gel fluorescence scanning results (Fig. 4), all the four tested proteins showed satisfactory cross-linking yields, ranging from 13% to 50%.
 | ||
| Fig. 4 Quantification of photo-cross-linking yield of selected photoaffinity probes toward their corresponding binding partners. Photo-cross-linking experiments followed the same conditions as listed in Fig. 2. | ||
Discussion
To fulfil the requirements for the explorations of PPIs in various contexts and for obtaining different information about the PPIs, there is a continuous demand on the development of novel photo-reactive amino acids with diverse structural properties and functionalities. Compared with unnatural amino acids with only photo-reactivity, dzANA is expected to pose advantages in two different applications, including the mapping of protein–protein binding site and identification and characterization of PPIs in living cells.A comprehensive understanding of PPIs requires not only the identification of the binding partners but also the identification of the areas where two proteins interact with each other, i.e., mapping protein–protein binding regions. To build a photoaffinity probe for the identification of PPIs, a common practice is to incorporate two unnatural amino acids, one with photo-reactivity and another with bioorthogonality. Under most circumstances, such photoaffinity probes are incompetent to mapping protein–protein binding regions. This is because that the proteins labelled by the photoaffinity probes should be digested into short peptides for mass spectrometry-based protein identification. During this process, the peptide-based photoaffinity probes will also be digested. As a result, the bioorthogonal handles that are subsequently conjugated to affinity tags for protein purification will be separated from the photo-reactive groups that cross-linked to the regions mediated the PPIs, which will eventually lead to the loss of information on the binding regions. Incorporation of dzANA into peptide probes at the positions close to key residues for binding would allow efficient identification of both the PPIs involved and mapping of the binding sites (Fig. 5a).
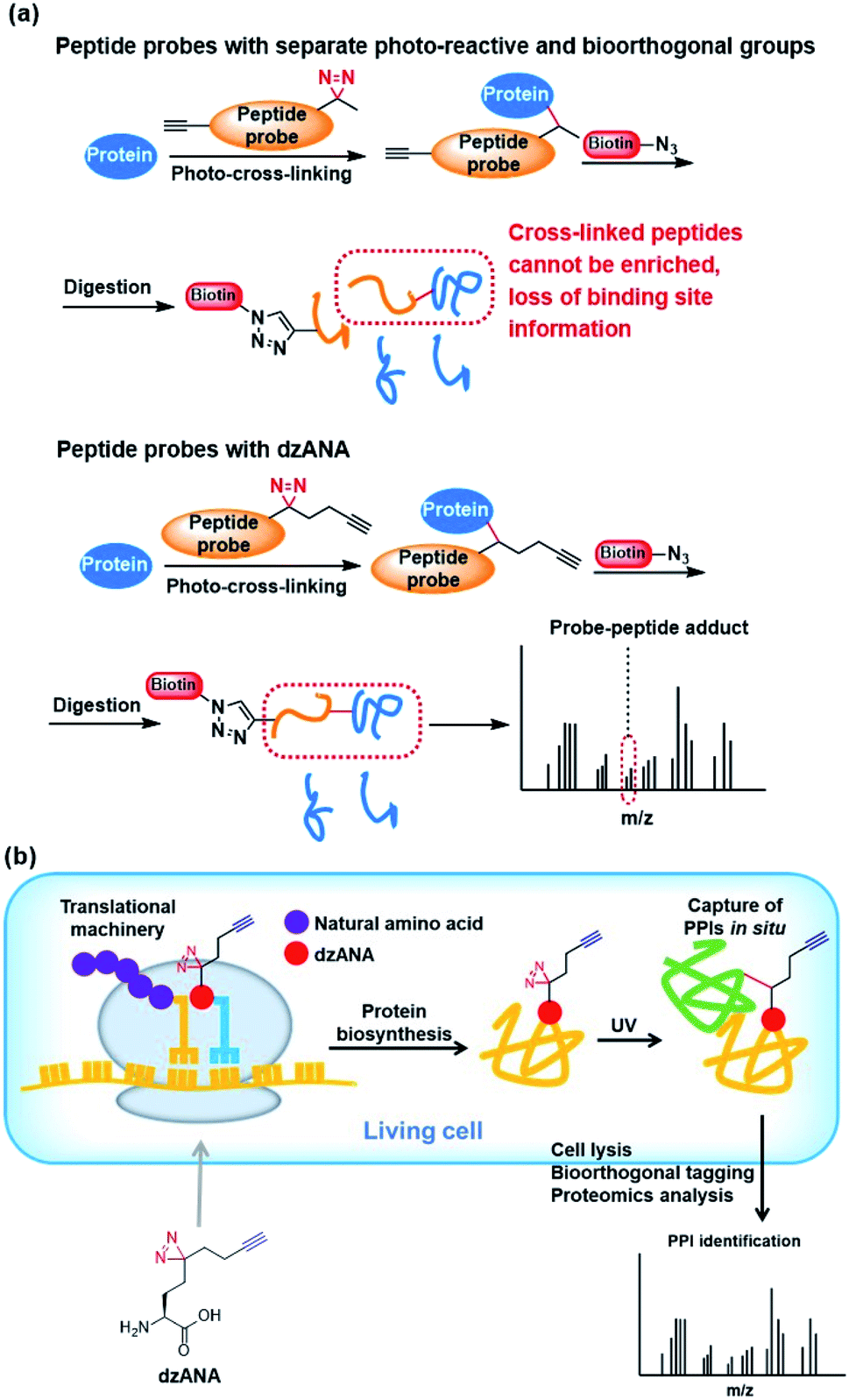 | ||
| Fig. 5 Potential applications of dzANA in (a) mapping PPIs binding sites, and (b) study PPIs in living cells. | ||
Most of the studies on PPIs have been carried out by using purified recombinant proteins or cell extracts in vitro. The PPIs are known to be dynamic and regulated by a series of cellular mechanisms in living cells, which are very different from the situation in vitro. The identification of PPIs in living cells should provide more direct information for unravelling their interaction networks. However, simultaneous incorporation of both a photo-reactive and a bioorthogonal unnatural amino acids into proteins co-translationally has been technically challenging. The newly developed bifunctional dzANA holds the potential to be incorporated into proteins by engineering the cellular translational machinery. It may facilitate the in situ fixing of PPIs in living cells and the isolation of the captured proteins for further investigations (Fig. 5b).
Conclusions
In summary, we have developed a novel bifunctional amino acid, dzANA, carrying both a diazirine moiety and a terminal alkyne handle, which is readily incorporated into peptides through standard Fmoc-based solid-phase peptide synthesis strategy. We showed that photoaffinity probes harbouring dzANA could effectively and specifically capture the weak and transient PPIs mediated by histone PTMs, suggesting the potential of this amino acid as a useful tool to study unknown PPIs. By integrating both photo-reactive and bioorthogonal functionalities into one amino acid, dzANA is expected to facilitate the mapping of PPI binding sites and the study of PPIs in living cells. Such explorations are our important next steps and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is supported by National Natural Science Foundation of China (91753130 to X. D. L.) and Excellent Young Scientists Fund of China (Hong Kong and Macau) (21922708 to X. D. L.). The authors acknowledge the support from the Hong Kong Research Grants Council (RGC) Collaborative Research Fund (CRF C7029-15G and C7017-18G to X. D. L.), the Areas of Excellence Scheme (AoE/P-705/16 to X. D. L.), the General Research Fund (GRF 17121120, 17126618, and 17125917 to X. D. L.), and the RGC Postdoctoral Fellowship (Scheme 2020/21 to X. L.).References
- L. Bonetta, Protein-protein interactions: interactome under construction, Nature, 2010, 468(7325), 851–854 CrossRef CAS.
- K. Venkatesan, J. F. Rual, A. Vazquez, U. Stelzl, I. Lemmens, T. Hirozane-Kishikawa, T. Hao, M. Zenkner, X. Xin, K. I. Goh, M. A. Yildirim, N. Simonis, K. Heinzmann, F. Gebreab, J. M. Sahalie, S. Cevik, C. Simon, A. S. de Smet, E. Dann, A. Smolyar, A. Vinayagam, H. Yu, D. Szeto, H. Borick, A. Dricot, N. Klitgord, R. R. Murray, C. Lin, M. Lalowski, J. Timm, K. Rau, C. Boone, P. Braun, M. E. Cusick, F. P. Roth, D. E. Hill, J. Tavernier, E. E. Wanker, A. L. Barabasi and M. Vidal, An empirical framework for binary interactome mapping, Nat. Methods, 2009, 6(1), 83–90 CrossRef CAS.
- T. Berggard, S. Linse and P. James, Methods for the detection and analysis of protein-protein interactions, Proteomics, 2007, 7(16), 2833–2842 CrossRef.
- N. D. Pham, R. B. Parker and J. J. Kohler, Photocrosslinking approaches to interactome mapping, Curr. Opin. Chem. Biol., 2013, 17(1), 90–101 CrossRef.
- D. P. Murale, S. C. Hong, M. M. Haque and J. S. Lee, Photo-affinity labeling (PAL) in chemical proteomics: a handy tool to investigate protein-protein interactions (PPIs), Proteome Sci., 2016, 15, 14 CrossRef.
- P. K. Mishra, C. M. Yoo, E. Hong and H. W. Rhee, Photo-crosslinking: An Emerging Chemical Tool for Investigating Molecular Networks in Live Cells, ChemBioChem, 2020, 21(7), 924–932 CrossRef CAS.
- L. Dubinsky, B. P. Krom and M. M. Meijler, Diazirine based photoaffinity labeling, Bioorg. Med. Chem., 2012, 20(2), 554–570 CrossRef CAS.
- Y. Tanaka, M. R. Bond and J. J. Kohler, Photocrosslinkers illuminate interactions in living cells, Mol. BioSyst., 2008, 4(6), 473–480 RSC.
- J. R. Hill and A. A. B. Robertson, Fishing for Drug Targets: A Focus on Diazirine Photoaffinity Probe Synthesis, J. Med. Chem., 2018, 61(16), 6945–6963 CrossRef CAS.
- M. W. Halloran and J. P. Lumb, Recent Applications of Diazirines in Chemical Proteomics, Chemistry, 2019, 25(19), 4885–4898 CrossRef CAS.
- M. Suchanek, A. Radzikowska and C. Thiele, Photo-leucine and photo-methionine allow identification of protein-protein interactions in living cells, Nat. Methods, 2005, 2(4), 261–267 CrossRef CAS.
- T. Yang, X. M. Li, X. Bao, Y. M. Fung and X. D. Li, Photo-lysine captures proteins that bind lysine post-translational modifications, Nat. Chem. Biol., 2016, 12(2), 70–72 CrossRef CAS.
- X. Xie, X. M. Li, F. Qin, J. Lin, G. Zhang, J. Zhao, X. Bao, R. Zhu, H. Song, X. D. Li and P. R. Chen, Genetically Encoded Photoaffinity Histone Marks, J. Am. Chem. Soc., 2017, 139(19), 6522–6525 CrossRef CAS.
- E. M. Tippmann, W. Liu, D. Summerer, A. V. Mack and P. G. Schultz, A genetically encoded diazirine photocrosslinker in Escherichia coli, ChemBioChem, 2007, 8(18), 2210–2214 CrossRef CAS.
- B. Van der Meijden and J. A. Robinson, Synthesis and application of photoproline - a photoactivatable derivative of proline, Arkivoc, 2011, 130–136 Search PubMed.
- C. J. Chou, R. Uprety, L. Davis, J. W. Chin and A. Deiters, Genetically encoding an aliphatic diazirine for protein photocrosslinking, Chem. Sci., 2011, 2(3), 480–483 RSC.
- R. E. Kleiner, L. E. Hang, K. R. Molloy, B. T. Chait and T. M. Kapoor, A Chemical Proteomics Approach to Reveal Direct Protein-Protein Interactions in Living Cells, Cell Chem. Biol., 2018, 25(1), 110–120.e3 CrossRef CAS.
- H. W. Ai, W. J. Shen, A. Sagi, P. R. Chen and P. G. Schultz, Probing Protein-Protein Interactions with a Genetically Encoded Photo-crosslinking Amino Acid, ChemBioChem, 2011, 12(12), 1854–1857 CrossRef CAS.
- M. Zhang, S. X. Lin, X. W. Song, J. Liu, Y. Fu, X. Ge, X. M. Fu, Z. Y. Chang and P. R. Chen, A genetically incorporated crosslinker reveals chaperone cooperation in acid resistance, Nat. Chem. Biol., 2011, 7(10), 671–677 CrossRef CAS.
- S. X. Lin, Z. R. Zhang, H. Xu, L. Li, S. Chen, J. Li, Z. Y. Hao and P. R. Chen, Site-Specific Incorporation of Photo-Cross-Linker and Bioorthogonal Amino Acids into Enteric Bacterial Pathogens, J. Am. Chem. Soc., 2011, 133(50), 20581–20587 CrossRef CAS.
- T. Yanagisawa, N. Hino, F. Iraha, T. Mukai, K. Sakamoto and S. Yokoyama, Wide-range protein photo-crosslinking achieved by a genetically encoded N-epsilon-(benzyloxycarbonyl)lysine derivative with a diazirinyl moiety, Mol. BioSyst., 2012, 8(4), 1131–1135 RSC.
- Y. Yang, H. P. Song, D. He, S. Zhang, S. Z. Dai, S. X. Lin, R. Meng, C. Wang and P. R. Chen, Genetically encoded protein photocrosslinker with a transferable mass spectrometry-identifiable label, Nat. Commun., 2016, 7, 12299 CrossRef CAS.
- S. X. Lin, D. He, T. Long, S. Zhang, R. Meng and P. R. Chen, Genetically Encoded Cleavable Protein Photo-Cross-Linker, J. Am. Chem. Soc., 2014, 136(34), 11860–11863 CrossRef CAS.
- T. P. Yang, Z. Liu and X. D. Li, Developing diazirine-based chemical probes to identify histone modification ‘readers’ and ‘erasers’, Chem. Sci., 2015, 6(2), 1011–1017 RSC.
- D. He, X. Xie, F. Yang, H. Zhang, H. M. Su, Y. Ge, H. P. Song and P. R. Chen, Quantitative and Comparative Profiling of Protease Substrates through a Genetically Encoded Multifunctional Photocrosslinker, Angew. Chem., Int. Ed., 2017, 56(46), 14521–14525 CrossRef CAS.
- J. E. Hoffmann, D. Dziuba, F. Stein and C. Schultz, A Bifunctional Noncanonical Amino Acid: Synthesis, Expression, and Residue-Specific Proteome-wide Incorporation, Biochemistry, 2018, 57(31), 4747–4752 CrossRef CAS.
- T. Kouzarides, Chromatin modifications and their function, Cell, 2007, 128(4), 693–705 CrossRef CAS.
- D. J. Patel and Z. X. Wang, Readout of Epigenetic Modifications, Annu. Rev. Biochem., 2013, 82, 81–118 CrossRef CAS.
- C. A. Musselman, M. E. Lalonde, J. Cote and T. G. Kutateladze, Perceiving the epigenetic landscape through histone readers, Nat. Struct. Mol. Biol., 2012, 19(12), 1218–1227 CrossRef CAS.
- S. D. Taverna, H. Li, A. J. Ruthenburg, C. D. Allis and D. J. Patel, How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers, Nat. Struct. Mol. Biol., 2007, 14(11), 1025–1040 CrossRef CAS.
- W. X. Wang, Z. Chen, Z. Mao, H. H. Zhang, X. J. Ding, S. Chen, X. D. Zhang, R. M. Xu and B. Zhu, Nucleolar protein Spindlin1 recognizes H3K4 methylation and stimulates the expression of rRNA genes, EMBO Rep., 2011, 12(11), 1160–1166 CrossRef CAS.
- X. B. Shi, T. Hong, K. L. Walter, M. Ewalt, E. Michishita, T. Hung, D. Carney, P. Pena, F. Lan, M. R. Kaadige, N. Lacoste, C. Cayrou, F. Davrazou, A. Saha, B. R. Cairns, D. E. Ayer, T. G. Kutateladze, Y. Shi, J. Cote, K. F. Chua and O. Gozani, ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression, Nature, 2006, 442(7098), 96–99 CrossRef CAS.
- P. V. Pena, F. Davrazou, X. B. Shi, K. L. Walter, V. V. Verkhusha, O. Gozani, R. Zhao and T. G. Kutateladze, Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2, Nature, 2006, 442(7098), 100–103 CrossRef CAS.
- F. Lan, R. E. Collins, R. De Cegli, R. Alpatov, J. R. Horton, X. B. Shi, O. Gozani, X. D. Cheng and Y. Shi, Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression, Nature, 2007, 448(7154), 718–722 CrossRef CAS.
- X. C. Bao, Y. Wang, X. Li, X. M. Li, Z. Liu, T. P. Yang, C. F. Wong, J. W. Zhang, Q. Hao and X. D. Li, Identification of ‘erasers’ for lysine crotonylated histone marks using a chemical proteomics approach, eLife, 2014, 3, e02999 CrossRef.
- S. A. Jacobs and S. Khorasanizadeh, Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail, Science, 2002, 295(5562), 2080–2083 CrossRef CAS.
- P. R. Nielsen, D. Nietlispach, H. R. Mott, J. Callaghan, A. Bannister, T. Kouzarides, A. G. Murzin, N. V. Murzina and E. D. Laue, Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9, Nature, 2002, 416(6876), 103–107 CrossRef CAS.
- R. Marmorstein and M. M. Zhou, Writers and Readers of Histone Acetylation: Structure, Mechanism, and Inhibition, Cold Spring Harbor Perspect. Biol., 2014, 6(7), a018762 CrossRef.
- C. Peng, Z. K. Lu, Z. Y. Xie, Z. Y. Cheng, Y. Chen, M. J. Tan, H. Luo, Y. Zhang, W. He, K. Yang, B. M. M. Zwaans, D. Tishkoff, L. Ho, D. Lombard, T. C. He, J. B. Dai, E. Verdin, Y. Ye and Y. M. Zhao, The First Identification of Lysine Malonylation Substrates and Its Regulatory Enzyme, Mol. Cell. Proteomics, 2011, 10(12), M111 012658 CrossRef.
- J. T. Du, Y. Y. Zhou, X. Y. Su, J. J. Yu, S. Khan, H. Jiang, J. Kim, J. Woo, J. H. Kim, B. H. Choi, B. He, W. Chen, S. Zhang, R. A. Cerione, J. Auwerx, Q. Hao and H. N. Lin, Sirt5 is a NAD-Dependent Protein Lysine Demalonylase and Desuccinylase, Science, 2011, 334(6057), 806–809 CrossRef CAS.
- H. M. Wisniewska, E. C. Swift and E. R. Jarvo, Functional-group-tolerant, nickel-catalyzed cross-coupling reaction for enantioselective construction of tertiary methyl-bearing stereocenters, J. Am. Chem. Soc., 2013, 135(24), 9083–9090 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09110c |
| ‡ Current address: Department of Cellular and Molecular Pharmacology, University of California, San Francisco, California 94158, United States. |
| This journal is © The Royal Society of Chemistry 2020 |