
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing bifunctional catalytic activity of cobalt–nickel sulfide spinel nanocatalysts through transition metal doping and its application in secondary zinc–air batteries†
Yijie Xu ab,
Afriyanti Sumboja*c,
Alexandra Grovesa,
Thomas Ashtona,
Yun Zong*b and
Jawwad A. Darr*a
ab,
Afriyanti Sumboja*c,
Alexandra Grovesa,
Thomas Ashtona,
Yun Zong*b and
Jawwad A. Darr*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: j.a.darr@ucl.ac.uk
bInstitute of Materials Research and Engineering (IMRE), A*STAR (Agency for Science, Technology and Research), 2 Fusionopolis Way, Innovis #08-03, 138634, Singapore. E-mail: Zong_Yun@hq.a-star.edu.sg
cMaterial Science and Engineering Research Group, Faculty of Mechanical and Aerospace Engineering, Institut Teknologi Bandung, Jl. Ganesha 10, Bandung 40132, Indonesia. E-mail: afriyanti.sumboja@material.itb.ac.id
First published on 17th November 2020
Abstract
Developing large-scale and high-performance OER (oxygen evolution reaction) and ORR (oxygen reduction reaction) catalysts have been a challenge for commercializing secondary zinc–air batteries. In this work, transition metal-doped cobalt–nickel sulfide spinels are directly produced via a continuous hydrothermal flow synthesis (CHFS) approach. The nanosized cobalt–nickel sulfides are doped with Ag, Fe, Mn, Cr, V, and Ti and evaluated as bifunctional OER and ORR catalyst for Zn–air battery application. Among the doped spinel catalysts, Mn-doped cobalt–nickel sulfides (Ni1.29Co1.49Mn0.22S4) exhibit the most promising OER and ORR performance, showing an ORR onset potential of 0.9 V vs. RHE and an OER overpotential of 348 mV measured at 10 mA cm−2, which is attributed to their high surface area, electronic structure of the dopant species, and the synergistic coupling of the dopant species with the active host cations. The dopant ions primarily alter the host cation composition, with the Mn(III) cation linked to the introduction of active sites by its favourable electronic structure. A power density of 75 mW cm−2 is achieved at a current density of 140 mA cm−2 for the zinc–air battery using the manganese-doped catalyst, a 12% improvement over the undoped cobalt–nickel sulfide and superior to that of the battery with a commercial RuO2 catalyst.
Introduction
Increased global attention towards the adverse effects of climate change and an accompanying desire to reduce dependencies on fossil fuels has led the drive to expand the use of sustainable energy sources.1,2 While alternative energy sources have been successfully demonstrated as ecological alternatives, their intermittent nature demands inexpensive, safe, grid-scale energy storage solutions to be developed to handle varying loads. Thus, research into affordable grid-scale rechargeable energy storage solutions is growing in importance. Of these solutions, metal–air systems such as zinc–air batteries show particular promise due to their high energy densities, environmental benignity, and relatively low cost. They also possess excellent safety features by being non-flammable (i.e., using aqueous electrolytes) and having an open system to release the built-up internal pressure.3,4 Structurally, a zinc–air battery consists of an air-reactive cathode, a zinc anode, and typically a potassium hydroxide (KOH)-based electrolyte. At the cathode, molecular oxygen undergoes reduction, catalysed by the bifunctional catalyst to form hydroxide ions. Simultaneously, electrolyte hydroxides react with zinc anode to form zincate [Zn(OH)4]2−, releasing electrons to travel to the cathode through an external circuit. The zincate then decays into ZnO and releases water to the electrolyte.A crucial requirement to facilitate high-performance zinc–air cells is the presence of a highly active bifunctional catalyst capable of promoting both the Oxygen Evolution Reaction (OER) and Oxygen Reduction Reaction (ORR) that drive the charge and discharge processes, respectively.3 The sluggish kinetics of ORR and OER originate from the multi-electron transfer process, leading to high overpotentials and low round-trip energy efficiencies, thus hindering commercial development rechargeable zinc–air batteries at grid scale.5,6
The benchmarks for OER and ORR catalysts are the precious-metal-based catalysts, such as Pt/C (for ORR) and RuO2 or IrO2/C (for OER). However, their high cost and scarcity make them unsuitable for industrial or commercial applications.3,5 Researches have shown inexpensive earth-abundant transition metal compounds to be suitable substitute catalyst candidates.6–10 These materials generally possess comparably low intrinsic catalytic activities, which can be enhanced by several methods. These include incorporating conductive supports or additives to improve the electron transfer kinetics, as well as introducing hetero-atom dopant and chemical activation to increase the number of exposed active sites and the electrochemically active surface area.11 It has been established that certain transition metal hydroxides and oxides have displayed excellent OER property, while transition metal sulfides, selenides, nitrides, and phosphides have shown promising ORR activities.12–17 Ternary transition metal chalcogenides are known for their good electrocatalytic activities, benefitting from the synergistic coupling between their host cations.13,18 They can tolerate the changes in oxidation states, aiding in resisting the detrimental effects of repeated shifts between oxidative and reductive environments, which result in the improved catalyst stability during battery operation.5,9 For instance, as compared to their monometallic counterparts, the coupling of cobalt with nickel in their bimetallic chalcogenide spinels resulted in the improved catalytic activities during methane and water oxidation.19,20
Spinel bimetallic sulfides (e.g., NiCo2S4) are of interest in oxygen electrocatalysis due to their rich multivalent oxidation state chemistry, good thermal and mechanical stability, exceptional electronic conductivity, large population of exposed octahedral catalytically active sites, as well as a favourable electronic structure.7,19,21,22 NiCo2S4 has been synthesized and evaluated in various morphologies in Li-ion batteries, supercapacitors, dye-sensitized solar cells, and as OER, ORR, and Hydrogen Evolution Reaction (HER) catalysts.23–29 The reported morphologies include nanospheres, nanowires, and hydrangea-like shaped structures, all of which have exhibited bifunctional catalytic activity in ORR and OER.5,6,8,9,13,30–32 Their catalytic activity can be further enhanced by improving on the electron transfer kinetics through surface morphology engineering and/or incorporating structural supports, as well as by doping with tertiary atoms to affect the electronic structure.5,33–39
Previous studies on the bifunctional ORR/OER activity of cobalt–nickel oxides have shown a rich mixed-valence redox chemistry of bimetallic components.5,13,19 The intrinsic restrictions of the spinel structure help distinguish the individual contribution from each cationic species by tailoring occupancies in the bimetallic spinels and their correlation to catalytic activities.40–42 This was achieved by altering the ratio of metal cation precursors during synthesis and observing differences in composition and catalytic activity. Recent studies on the effect of compositional tuning of spinel cobalt–nickel oxides and sulfides on the electrocatalytic performance have resulted in new insights into each cationic species' roles.43,44 This information could be used for the rational design of optimum electrocatalysts with high domain-specific catalytic activity.40,45,46
In our previous work, the influence of the relative proportions of nickel and cobalt cations on oxygen electrocatalysis in undoped spinel cobalt–nickel sulfides was discussed. Among other factors, we concluded that the activities of Ni(III) in OER together with that of Co(II) and Ni(II) and cations in ORR, allowed for the optimization of the bimetallic sulfide to target excellent bifunctional catalytic performance. As such, the influence of nickel and cobalt on catalyst activity will not be discussed in this work, and the reader is kindly directed to the aforementioned work for associated literature on the catalytic performance of the two species.47
However, the introduction of foreign atomic elements into the spinel sulfide structure remains relatively unexplored, in sharp contrast to high-performance doped oxide catalysts that are extensively documented in the literature for oxygen electrocatalysis.10,43,48 Of particular concern to catalysis is the capability of atomic doping to tailor both the physical and electronic attributes of the catalyst. While the former is often observed as changes in surface area or pore distribution, the latter is exhibited through surface-level structural changes in the catalyst by altering the electronic structure of neighbouring atomic active sites, thereby changing the energetics of oxygen absorption of possible reactant and intermediate compounds.10,49 Changes in composition through replacement by dopant species of the host cations have also been observed,43 which can be realized as improved kinetics, increased electronic conductivity, and long-term stability over their corresponding component metallic oxide counterparts.50
In order to develop viable catalysts, it is essential to utilise reproducible, tailorable, and scalable routes to manufacture. Amongst the many processes for the manufacture of well-defined nanoparticle oxide catalysts, Continuous Hydrothermal Flow Synthesis (CHFS) methods offer a rapid and scalable synthesis technology which is particularly suited for systematic doping of materials.51 CHFS syntheses are essentially hydrothermal flow processes that feature a well-defined mixing point in which nanoparticle products such as metal oxides or sulfides are precipitated at elevated temperatures in a fraction of a second and then collected as a slurry in water after inline cooling to room temperature via a series of reactions. Due to the rapid, instantaneous precipitation inflow and well-defined conditions, the technology offers high consistency and control over particle properties. The technology can also rapidly deliver libraries of nanomaterials in hours via high throughput syntheses, as has been detailed in and in previous publications on CHFS.47,52
Herein, nanosized doped cobalt–nickel sulfide spinels were made using CHFS technology, and the electrochemical properties of the resulting nanopowders were evaluated as bifunctional electrocatalysts for OER, ORR, and as cathodes for zinc–air batteries. The effect of introducing titanium, vanadium, chromium, manganese, iron, and silver as dopants into spinel cobalt–nickel sulfide was evaluated. The choice of dopant species was based on those previously successfully demonstrated examples in literature, for either ORR or OER. The total metallic composition in the catalysts was quantified using X-ray fluorescence (XRF), while the surface composition ratios of Ni(II)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni(III) and Co(II):Co(III) were determined using X-ray Photoelectron Spectroscopy (XPS).
:Ni(III) and Co(II):Co(III) were determined using X-ray Photoelectron Spectroscopy (XPS).
Experimental
Synthesis of doped cobalt–nickel-sulfide nanoparticles
Nanosized metal-doped cobalt–nickel sulfides were synthesised using a laboratory-scale CHFS reactor, the design, and setup of which are shared with our previous cobalt–nickel sulfide study.47 The ambient aqueous sources of cobalt and nickel nitrates were kept at 0.09 M each, with the concentration of dopant precursors kept at 0.02 M. Titanium(IV) bis(ammonium lactato)dihydroxide (TiBald, 50% wt Sigma Aldrich, UK), ammonium vanadate (NH4VO3, Sigma Aldrich, UK), chromium nitrate (Cr(NO3)3·9H2O, Sigma Aldrich, UK), manganese nitrate (Mn(NO3)2·xH2O, Sigma Aldrich, UK), iron nitrate (Fe(NO3)3·9H2O) and silver nitrate (AgNO3, Sigma Aldrich, UK) were used as dopant precursors for this study. After formation in the CHFS process, the dark-coloured nanoparticle slurry was collected at ca. 40 °C at an outlet, allowed to sediment under gravity, and then cleaned by washing in DI water. The resulting wet solids were then freeze-dried using a Virtis Genesis 35XL freeze-drier by warming the samples for a 24 h period from −60 °C to 25 °C under a vacuum (<13.3 Pa), yielding dark brown colored powders. These were stored in a cool and dry place to minimize moisture absorption. The synthesized cobalt–nickel sulfides were named by combining the prefix NC11, representing the equimolar proportion of nickel and cobalt precursors, together with the dopant metallic salt's chemical symbols precursors during their synthesis, such as NC11 (V) for the vanadium doped sample.Materials characterization
Powder X-ray diffraction (XRD) data of the samples were collected on a Stöe diffractometer using Mo-Kα radiation (λ = 0.7093 Å) over a 2θ range of 2 to 40° with a step size of 0.5° and an interval of 10 s for data collection. To observe the particles' detailed features (i.e., morphology, interlayer spacing, and size), high-resolution transmission electron microscopy (HR-TEM) was performed on the samples using a Jeol JEM 2100 microscope fitted with a LaB6 filament. Elemental compositional quantification was performed using the attached Energy-Dispersive X-ray Spectroscopy (EDS). Digital images of the sulfide samples were captured using an attached digital camera (Gatan Orius, Gatan, United States). X-ray Photoelectron Spectroscopy (XPS, Thermo Scientific Theta Probe monochromatized spectrometer with Al-Kα (hν = 1484.6 eV) radiation) was used to collect surface-level elemental quantification, chemical bonding, and valence information. High-resolution regional scans for Co 2p, Ni 2p, C 1s, S 2p, and each metal dopant 2p orbitals were conducted at 50 eV, with the XPS data processed and fitted using CasaXPS™ software (version 2.3.16). The specific surface area was determined using nitrogen adsorption in a Hyden BET instrument and calculated according to the Brunauer–Emmett–Teller (BET) theory. The samples were degassed at 150 °C (for 12 h) under a flow of N2 before the measurement. Elemental quantification analysis of the metals in each compound was also obtained from X-ray fluorescence (XRF) spectrometry measurements of the sample powders, collected on a Brüker M4 Micro XRF Spectrometer, using a 30 W Rh source.Electrochemical characterization
The catalytic activities of the cobalt–nickel sulfides were examined using a potentiostat (Autolab model PGSTAT302N) fitted with a three-electrode Rotating-Disk Electrode (Model RDE-2, Metrohm AG, Herisau, Switzerland) setup. ORR and OER measurements were performed in 0.1 M or 1 M KOH electrolyte, respectively. Prior to ORR measurements, the setup was continuously purged by O2 gas, while for the OER measurements, the electrolyte was first saturated with O2 gas to establish an equilibrium. Pt foil (Model 3.109 Metrohm AG, Herisau, Switzerland) and Ag/AgCl (Model 6.0726, Metrohm AG, Herisau, Switzerland) served as counter and reference electrodes, respectively. To prepare the catalyst inks, active material and carbon black (Vulcan XC-72, Cabot, Alpharetta Georgia, USA) were combined at a ratio of 80:20 wt% together with a mixture of 10 mL DI H2O, isopropyl alcohol (IPA), and Nafion™ (10% solution, Sigma Aldrich, Dorset UK) solution at a mass ratio of 2.5:1:0.094 to reach an active material concentration of 3.75 mg mL−1. The final mixture was sonicated for 30 minutes, after which 10.68 μL of sample catalyst ink was drop-cast onto a polished glassy carbon electrode to give an active material loading of 0.1 mg cm−2. The electrodes were air-dried for 30 min at room temperature before use. For benchmarking, Pt/C (99.9% purity Sigma Aldrich, Dorset UK) and RuO2 (99.9% purity, Sigma Aldrich, Dorset UK) were also prepared in the same manner as above.
The performance of the electrocatalysts in Zn–air batteries was evaluated using an in-house designed and built Perspex-based cell. A Neware battery tester (model V5, Shenzen Neware Technology Company, China, Fig. S2†) was used to evaluate the cells' performance. Air cathodes were prepared for the cobalt–nickel sulfide samples by pipetting sample solutions onto a carbon paper (SIGRACELL, SGL Carbon, Wiesbaden, Germany) to achieve a dry solid loading of ca. 1.0 mg cm−2 (±0.1 mg cm−2), while a polished 70 × 60 × 5 mm Zn plate served as the anode (Alfa Aesar, United Kingdom), with a hydrophilic 50 μm thick PTFE membrane (pore size 0.2 μm, Merck Millipore, Burlington Massachusetts, USA) serving as a separator. Titanium mesh current collector (Ti, 80 mesh, Alfa Aesar, Haverhill Massachusetts, USA) was placed next to the air cathode, with a Teflon-coated hydrophobic carbon paper backing layer (SIGRACELL, SGL Carbon, Wiesbaden, Germany) preventing electrolyte from leaking out or flooding the cell under high humidity environments. 6.0 M KOH aqueous solution containing 0.1 M Zn(O2CCH3)2 was used as the electrolyte and injected into the cell after its assembly. The exposed area of the cathode was ca. 0.79 cm2.
Results and discussion
Physical characterization
The XRD patterns of the as-synthesized cobalt–nickel sulfide samples are displayed in Fig. 1, together with a reference pattern of NiCo2S4 (JCPDS 073-1704), which is indistinguishable with CoNi2S4 (JCPDS 073-1297) due to the similar size of the cobalt and nickel cations.53 The XRD patterns of the doped and undoped cobalt–nickel sulfides all displayed characteristic peaks at 2θ corresponding to indices (in parentheses) at 7.5° (111), 12.3° (220), 14.4° (311), 17.4° (400), 21.3° (422), 22.6° (511), and 24.7° (440), respectively. For the manganese- (NC11 Mn) and silver-doped (NC11 Ag) samples, small impurity peaks were detected, which were attributed to MnS and Ag2S. The average crystallite as-synthesized sulfide samples (Table S3†) was estimated via application of the Scherrer equation to the pronounced (001) and (220) XRD peaks revealed the undoped sulfide sample crystallite size at ca. 12 nm and the majority of transition doped sulfide sample exhibiting sizes in the range of 7–12 nm, with NC11 Mn alone exhibiting a larger size of ca. 15 nm.54 Generally, a spinel structure such as NiCo2S4 consists of cubic close-packed S2− anions with four octahedral and eight tetrahedral sites and occupied by Ni(II) and Co(III) and cations, respectively, in an Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group.13,55 CoNi2S4 adopts the same cobalt structure according to powder neutron diffraction studies, with Ni(III) and Co(II) occupying the tetrahedral and octahedral sites, respectively.55,56 Hence, it is impossible to use reference XRD patterns to distinguish between CoNi2S4, NiCo2S4, and other possible intermediate states, such as the cobalt–nickel sulfide samples herein.
m space group.13,55 CoNi2S4 adopts the same cobalt structure according to powder neutron diffraction studies, with Ni(III) and Co(II) occupying the tetrahedral and octahedral sites, respectively.55,56 Hence, it is impossible to use reference XRD patterns to distinguish between CoNi2S4, NiCo2S4, and other possible intermediate states, such as the cobalt–nickel sulfide samples herein.
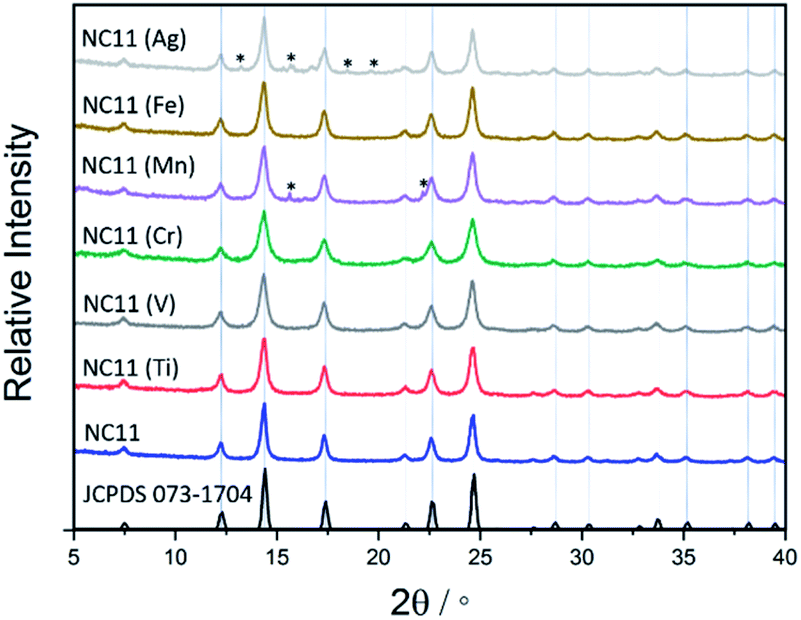 | ||
| Fig. 1 XRD patterns of the as-synthesized undoped and doped cobalt–nickle sulfides, displayed together with the JCPDS pattern 073-1704 (space group Fdm) for NiCo2S4 (black). Impurity peaks for Ag2S and MnS are indicated with (*), based on reference patterns JCPDS 014-0072 and 089-4952, respectively. | ||
Analysis of the materials via Transmission Electron Microscopy (TEM) showed that they formed agglomerated nanostructures, with individual particle sizes of around 10–20 nm (Fig. 2). Closer examination of some of the crystals of chromium- (NC11 Cr), manganese- (NC11 Mn), and iron-doped (NC11 Fe) samples, revealed d-spacings of the (440) planes of 0.30, 0.32, and 0.30 nm (±0.1 nm), respectively; these are similar to values that Kim and Liang reported in the literature for NiCo2S4.57,58 Energy-Dispersive (X-ray) Spectroscopy (EDS) mapping (Fig. S3†) suggested that the dopant cations were evenly distributed across the near-surface of the cobalt–nickel sulfide samples. The BET surface areas of the cobalt–nickel sulfide samples were all found to increase significantly following dopant introduction, with a minimum increase of 54%, and the top-three samples highest surface areas were for chromium- (NC11 Cr, 33 m2 g−1), manganese- (NC11 Mn, 22 m2 g−1), and titanium-doped (NC11 Ti, 20 m2 g−1) samples. The full BET characterization is listed in Table S1.†
 | ||
| Fig. 2 TEM images of (top) and particle spacings (bottom) of the (a) undoped (NC11) (b) chromium-doped (NC11 Cr) (c) manganese-doped (NC11 Mn) (d) iron-doped (NC11 Fe) cobalt–nickel sulfide samples. | ||
The elemental composition of the cobalt–nickel sulfides was quantified by X-ray fluorescence (XRF), with cobalt, nickel, and sulfur suggested to be present for all samples. The relative percentages of the metallic components are shown in Table 1, with the full characterization shown in Table S2.† Interestingly, the amount of sulfur detected was lower than expected for the spinel structure, which was attributed to the inherent reduced sensitivity of XRF to lighter elements. The amount of dopant relative to the total metal concentration in most cobalt–nickel sulfides was in the range of 7 to 8 at%, except for the vanadium-doped sample (NC11 V) being at 2.7 at%. To determine the bulk composition of our samples, the relative ratio of the metal components was used in combination with the structural match for NiCo2S4 derived from X-ray diffraction.
| Sample | [Ni]/% | [Co]/% | [Dopant]/% | Target composition |
|---|---|---|---|---|
| NC11 | 46.0 | 54.0 | N/A | Ni1.4Co1.6S4 |
| NC11 (Ti) | 44.6 | 47.7 | 7.7 | Ni1.35Co1.45Ti0.2S4 |
| NC11 (V) | 44.9 | 52.4 | 2.7 | Ni1.35Co1.57V0.08S4 |
| NC11 (Cr) | 42.8 | 49.1 | 8.1 | Ni1.29Co1.47Cr0.24S4 |
| NC11 (Mn) | 43.0 | 49.8 | 7.2 | Ni1.29Co1.49Mn0.22S4 |
| NC11 (Fe) | 42.4 | 49.0 | 8.6 | Ni1.27Co1.47Fe0.26S4 |
| NC11 (Ag) | 42.7 | 49.9 | 7.4 | Ni1.28Co1.5Ag0.22S4 |
The chemical valence and surface composition of the doped cobalt–nickel sulfides were characterized by X-ray Photoemission Spectroscopy (XPS), quantifying the presence of cobalt, nickel, and sulfur in all doped cobalt–nickel sulfide samples. Along with the respective dopant for each sample. Fig. 3 displays the fitted Co 2p, Ni 2p, and S 2p XPS spectra for the chromium- (NC11 Cr), manganese- (NC11 Mn), and iron-doped (NC11 Fe) cobalt–nickel sulfides. The remaining sample spectra are displayed in Fig. S4.† The deconvoluted two spin-orbital doublets can be assigned to M(II) and M(III) species of nickel and cobalt, together with their shake-up satellites. The peak positions of nickel and cobalt were similar across all considered samples, with a maximum variation of 0.2 eV. In undoped cobalt–nickel sulfide, the doublet pair for Co 2p3/2 and 2p1/2 states was located at 780.6 and 778.7 eV and 793.8 and 796.4 eV, respectively; while for Ni 2p, the 2p3/2 a doublet was located at 855.9 and 853.4 eV. The corresponding 2p1/2 doublet was at 873.8 and 870.7 eV. For sulfur, the single S 2p state was confirmed with peaks at 161.1 and 162.9 eV for 2p3/2 and 2p1/2, respectively, which is in good agreement with the reported values of transition-metal sulfides.22,59 The detected XPS binding energies of all dopant species orbitals in each of the doped cobalt–nickel sulfides are shown in Table 2, together with the predicted oxidation states and d-numbers of the dopant species within the host cobalt–nickel sulfide. For the samples herein, all dopant 2p3/2 signals were weak, with 2p1/2 signals undetected for the majority of dopants, attributed to their low doped surface concentrations. Furthermore, no responses were detected for the vanadium 2p orbitals, which was associated with their lower doped concentration, as evidenced by XRF.
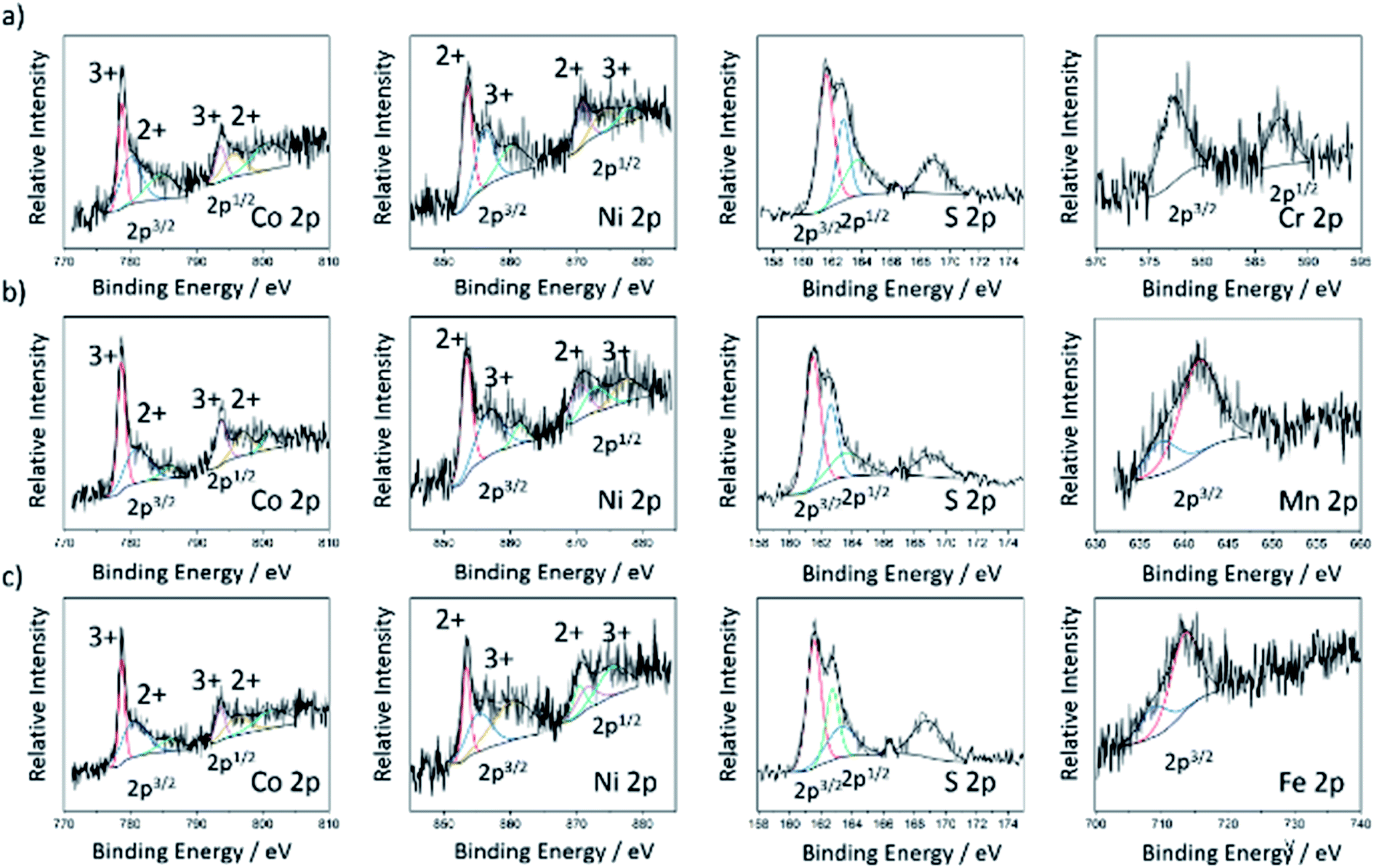 | ||
| Fig. 3 XPS spectra for chromium- (a), manganese- (b), and iron-doped (c) cobalt–nickel sulfides (NC11 Cr, NC11 Mn, and NC11 Fe, respectively) exhibiting 2p orbital belonging to the cobalt, nickel, sulfur, and their respective dopant species. | ||
| Dopant orbital | Binding energy/eV | Oxidation state | d-Number |
|---|---|---|---|
| Ti 2p [3/2, 1/2] | 459.0, 464.6 | Ti(I) | 3 |
| Cr 2p [3/2, 1/2] | 577.5, 587.3 | Cr(III) | 3 |
| Mn 2p [3/2] | 637.4, 641.6 | Mn(II), Mn(III) | 5, 4 |
| Fe 2p [3/2] | 708.8, 713.5 | Fe(III) | 5 |
| Ag 3d [5/2, 3/2] | 367.9, 373.9 | Ag(I) | 10 |
As the oxidation states of surface-level nickel and cobalt cations have been shown to affect the catalytic activities of the overall spinel cobalt–nickel catalysts by affecting the energy of oxygen species adsorption, a detailed understanding of the cationic surface composition of the cobalt–nickel sulfides is imperative.43 Table 3 displays the relative ratios of the intra-species oxidation states of nickel and cobalt in the doped cobalt–nickel sulfides obtained through XPS. As previously demonstrated in literature studies, nickel and cobalt were found to co-exist in both oxidation states in the cobalt–nickel sulfide samples.55 Across of our samples, the maximum intra-species oxidation state differences were small, with a maximum difference of roughly 10% being observed. It was observed that the majority of the samples exhibited a higher proportion of Co(II), with the manganese-doped sample (NC11 Mn) being the only exception. The majority of cobalt–nickel sulfide samples contained a higher proportion of Ni(III) species, with the vanadium- (NC11 V), chromium- (NC11 Cr), and manganese-doped (NC11 Mn) samples exhibiting a higher proportion of Ni(II). Differences in host framework cationic composition following doping have also been observed previously for cobalt–nickel oxides.43 Furthermore, XPS quantification revealed that none of the examples exhibited any surface-level sulfide deficiencies.
| Sample | Ni(II)/Ni(III)/% | Co(II)/Co(III)/% |
|---|---|---|
| NC11 | 46/54 | 57/43 |
| NC11 (Ti) | 48/52 | 51/49 |
| NC11 (V) | 53/47 | 56/44 |
| NC11 (Cr) | 55/45 | 57/43 |
| NC11 (Mn) | 52/48 | 45/55 |
| NC11 (Fe) | 43/57 | 59/41 |
| NC11 (Ag) | 48/52 | 57/43 |
Electrochemical characterization
The OER catalytic activities of the undoped and doped cobalt–nickel sulfides were evaluated in 1 M KOH using a Rotating Disc Electrode (RDE, model GC50, Metrohm, Switzerland) setup at a scan rate of 5 mV s−1. Fig. 4a displays the OER polarization data of the electrodes made using the doped cobalt–nickel sulfides. Linear Sweep Voltammetry (LSV) polarization curves suggested that cobalt–nickel sulfide samples doped with iron (NC11 Fe), manganese (NC11 Mn), and titanium (NC11 Ti) perform well in OER LSV tests, achieving a current density of 10 mA cm−2 at low overpotentials of 358, 348, and 353 mV, respectively (where overpotential is defined as the potential value over the theoretical OER potential of 1.23 V vs. Standard Hydrogen Electrode, SHE). However, only the manganese-doped sample (NC11 Mn) exhibited superior overpotential over the undoped NC11 sample (351 mV), suggesting more improved OER kinetics for the former. Furthermore, the manganese- (NC11 Mn) and iron-doped (NC11 Fe) samples exhibited the highest maximum current density, reaching 49 and 52 mA cm−2 at 1.636 V, respectively.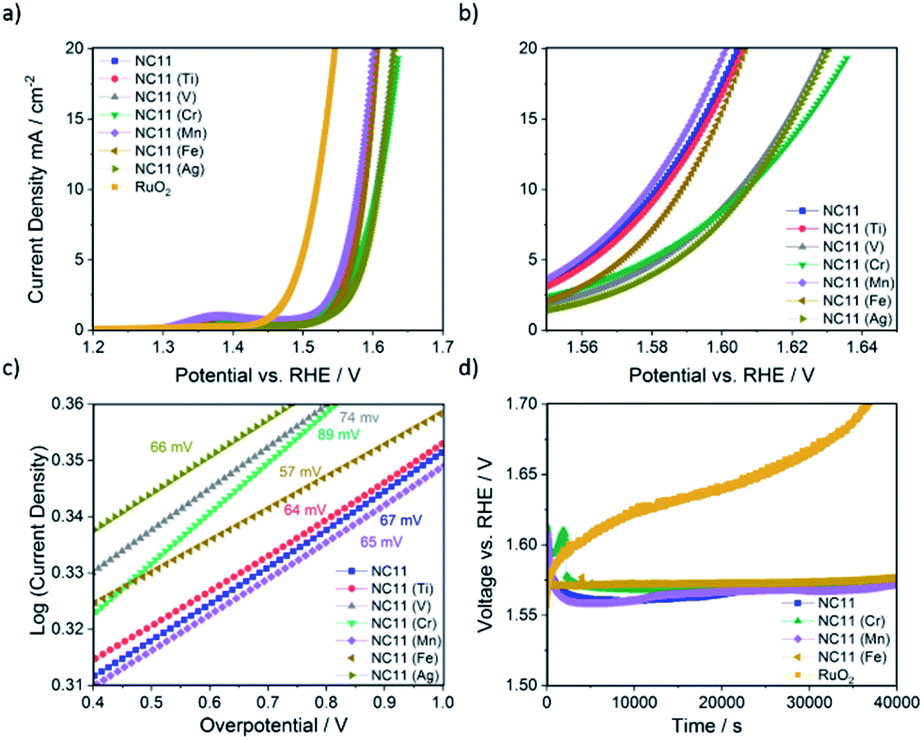 | ||
| Fig. 4 Catalytic activity of the samples during OER (a) linear sweep voltammograms (LSVs) of the samples, with a scan rate of 5.0 mV s−1 from 1.2 to 1.7 V (vs. RHE). (b) Close-up of the LSVs, from 1.55 to 1.65 V. (c) Tafel slopes of the samples derived from LSV curves. (d) Stability test of NC11, NC11 Cr, NC11 Mn, and NC11 Fe compared to the standard RuO2 catalyst, measured at a constant current density of 10 mA cm−2. All of the measurements were conducted in 1 M KOH electrolyte at an RDE rotating speed of 1600 rpm. | ||
Tafel slopes measure the amount of overpotential necessary to increase the OER currents by a decade. Across our doped materials, NC11 doped with iron (NC11 Fe), manganese (NC11 Mn), and titanium (NC11 Ti) exhibited slightly lower Tafel slope values, at 57, 65, and 64 mV, respectively, in contrast to the undoped NC11 sample (66 mV) and the commercial RuO2 catalyst (57 mV, Fig. S5c†). These values are similar or lower slopes than a number of promising OER catalysts reported in the literature, including Co3O4 (74 mV dec−1),60 CuCo2O4 (65 mV dec−1),61 graphene-supported Co3O4 (67 mV dec−1),62 graphene-supported copper-based MOFs (65 mV dec−1),63 and nickel foam-supported NiSe (64 mV dec−1).64 Furthermore, these values are also comparable or superior to those obtained for literature reports on manganese-, iron-, copper-, and zinc-doped cobalt–nickel oxides.43
The stability of the undoped (NC11), chromium (NC11 Cr)-, manganese- (NC11 Mn), and iron-doped (NC11 Fe) cobalt–nickel sulfides was examined by chronopotentiometry, where the voltage change was recorded as a factor of time under constant current of 10 mA cm−2. It was observed that all of the considered cobalt–nickel sulfide samples exhibited excellent stability during the OER following prolonged exposure to 1 M KOH (Fig. 4d), with ca. 0.03 V deterioration being observed for all considered sulfide samples after ∼40000 (Fig. 4c) seconds. In contrast, a 0.17 V deterioration is observed for the benchmark RuO2 catalyst.
The ORR catalytic activity of the cobalt–nickel sulfide samples was evaluated in 0.1 M KOH (Fig. 5). LSV polarisation curves demonstrated that the chromium- (NC11 Cr), manganese- (NC11 Mn), and iron-doped (NC11 Fe) cobalt–nickel sulfides had the highest saturating currents at 0.25 V vs. RHE. The onset and half-wave potentials of the undoped and doped cobalt–nickel sulfides were measured using the multiple tangent intersection method (as demonstrated in Fig. S5a and b in the ESI†) and displayed in Table 4.
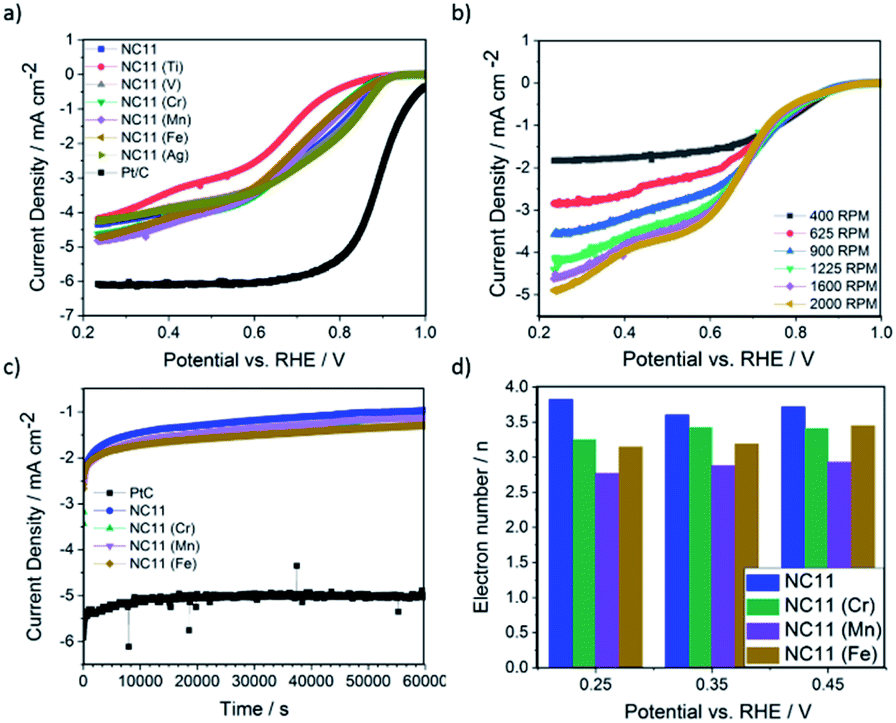 | ||
| Fig. 5 Catalytic activity of the samples during ORR (a) LSVs of the cobalt–nickel sulfide samples and Pt/C reference, measured at an RDE rotation speed of 1600 rpm with a scan rate 5.0 mV s−1 across a potential range of 0.2 to 1.0 V (vs. RHE). (b) LSVs of NC11 with a scan rate of 5.0 mV s−1 between RDE rotation speeds of 400–2000 rpm. (c) Long term stability test performance of NC11 and Pt/C, at a potential of 0.635 V (vs. RHE). (d) Electron transport numbers of undoped (NC11), chromium-doped (NC11 Cr), manganese-doped (NC11 Mn), and iron-doped (NC11 Fe) samples, calculated at 0.25, 0.35, and 0.45 V (vs. RHE). All of the measurements were conducted in 0.1 M KOH electrolyte. | ||
| Sample | Eonset/V (±0.02) | E1/2/V (±0.02) |
|---|---|---|
| NC11 | 0.90 | 0.75 |
| NC11 (Ti) | 0.82 | 0.70 |
| NC11 (V) | 0.91 | 0.76 |
| NC11 (Cr) | 0.89 | 0.75 |
| NC11 (Mn) | 0.90 | 0.75 |
| NC11 (Fe) | 0.90 | 0.74 |
| NC11 (Ag) | 0.92 | 0.78 |
| Pt/C | 0.99 | 0.88 |
The small changes in onset potential for most samples were within acceptable statistical variation and were not considered significant. Most of our cobalt–nickel sulfide samples shared similar onset and half-wave potentials centered around 0.91 V or 0.75 V, with only the titanium-doped sample exhibiting notably poorer performance. The rest of the samples' measured potentials were only slightly lower than to that of the commercial Pt/C benchmark, at 0.99 and 0.88 V, respectively. The long-term stability of the chromium (NC11 Cr), manganese (NC11 Mn), and iron-doped (NC11 Fe) cobalt–nickel sulfide samples were examined concurrently with the undoped NC11 sample via chronoamperometry in 1 M KOH over 19 hours; a decrease in current density of 59% was observed over 17 hours of continuous operation for the undoped nickel sulfide sample, in contrast to a 10% decrease for Pt/C. Transition metal doping was shown to improve stability in this test, with the chromium (NC11 Cr), manganese (NC11 Mn), and iron-doped (NC11 Fe) samples exhibiting a decrease in current density of 49%, 54%, and 43%, respectively. While graphitized carbons have been used in literature to improve the electrochemical stability of transition metal-based catalysts, this was not investigated as the primary focus was on evaluating the effect of dopant introduction.65
Analysis of the electron kinetics of the chromium- (NC11 Cr), manganese- (NC11 Mn), and iron-doped (NC11 Fe) cobalt–nickel sulfides revealed that tertiary metal doping altered the electron transfer pathway during ORR. While NC11 exhibited an electron transport number of around ca. 3.7, implying the co-occurrence of a two-electron transfer pathway in ORR for this sample. The introduction of dopants to NC11 resulted in a shift toward an electron transport number of ca. 3, which implied a greater proportion of two-electron pathways during the ORR. For energy storage applications, four-electron electron transfer pathways are preferred over two-electron pathways. The latter is known to suffer from a reduction in energy conversion efficiency and is also known to produce carbon-corroding hydroperoxide groups detrimental to the air-cathode structure.66,67 The poor electrochemical stability of our undoped- and doped cobalt–nickel sulfide samples compared to Pt/C (observed during long-term ORR stability tests), was ascribed to peroxide-based oxidative attacks arising from the partial reliance on the 2-electron transfer pathway (Fig. 5c).68
The improved activity of transition metal-based catalysts following transition metal-doping can be associated with their increased surface area and the inherent higher activity of the dopant cations (and the altered host nickel and cationic cobalt composition). An increased electrochemical surface area has been shown to accompany an increased overall catalytic activity previously, due to an increased number of available active sites.49 However, although the introduction of dopants into the host structure did increase the surface area of the host cobalt–nickel sulfides for all samples, it was not a deciding factor for activity values. The samples exhibiting the highest saturated current (NC11 Mn, NC11 Fe, and NC11 Cr) ranked first, second, and fourth in terms of surface area. However, NC11 Ti, which possessed the third-highest surface area (19.2 m2 g−1), exhibited similar or poorer performance to its undoped counterpart (NC11), despite possessing an 84% larger surface area, suggesting that surface area alone is not responsible for differences in activity.
The altered ratios of the host cations may be a significant factor in the improved performance observed for the doped cobalt–nickel sulfides herein. In the literature, it has been shown that while the intrinsic high conductivity of NiCo2S4 makes it an excellent bifunctional catalyst, compositional tuning could be utilized to improve OER and ORR activity further.21,69,70 Moreover, a previous study by the authors, determined that tuning the composition to achieve a high proportion of Ni(III) was key for improved OER performance and that high proportions of Ni(II) and Co(II) facilitated improved ORR performance.44 Similar patterns have been observed in literature for cobalt–nickel oxides by Lu et al., who showed that dopants altered the ratio of host cations, with a low Ni(II)/Ni(III) ratio and a high Co(II)/Co(III) ratio giving an optimal balance for bifunctional OER/ORR activity.43,71 Ni(III) and Co(II) have been shown to have a favourable electronic structure in spinels that facilitate the improved catalytic activity, while Ni(II) has been theorized to support neighbouring cation–adsorbate interactions via by improving conductivity.65,71–79
Suntivich et al. observed a correlation of catalytic activities of spinel metal oxides, with the electronic structure at their low-spin octahedral centres.74 An eg orbital occupancy level close to unity at the octahedral centre was found to promote electron transfer from the surface metal cations to the adsorbed intermediates, with occupancy values slightly lower or higher than unity promoting ORR and OER activity, respectively. This was attributed to the change in energy of the rate-determining step of the adsorption process.74,80 In NiCo2S4, the t62ge1g configuration is shared by low-spin Co(II) and Ni(III) cations, with Co(III) believed to be able to adopt the intermediate t52ge1g configuration through the adoption of a square-pyramidal crystal field at a particle surface due to nearby anionic vacancies.74 Among the cobalt–nickel sulfides, the undoped (NC11), titanium- (NC11 Ti), iron- (NC11 Fe), and silver-doped (NC11 Ag) samples all exhibited a low Ni(II)/Ni(III) ratio, explaining their activity in OER. In contrast, the high OER activity of the chromium- (NC11 Cr), and manganese-doped (NC11 Mn) sulfide samples, can be primarily attributed to their higher surface area, with a 203% and 107% increase over the undoped cobalt–nickel sulfide (NC11), respectively. The high bifunctional activity of the manganese-doped sulfide sample (NC11 Mn) is also supported by the presence of Mn(III), which possesses the favourable t32ge1g high-spin structure, suggesting its possible role as an active site. This trait was not exhibited by the other dopant cations. Conversely, with the possible exceptions of the titanium- (NC11 Ti) and manganese-doped (NC11 Mn) sulfide samples, all of the samples herein exhibited a Co(II)/Co(III) ratio value greater than 1, which would be expected to support good ORR performance. As previously mentioned, the Mn-doped sulfide's high activity may be attributed to its higher surface area and the favourable electronic structure of the dopant.
The role of the dopant species in affecting the activity of the cobalt and nickel species (through altering the kinetics and energetics of the transport processes) has been examined in several studies. For example, Xu et al. demonstrated how iron doping improved the OER performance of ABO3-type barium cobalt oxides, which was attributed to the fine-tuning of the OH− adsorption and O2 desorption processes through dopant introduction.81 Thin-film nickel oxide-based catalysts exhibited improved stability and ORR performance with <1% of Fe doping, which was linked to the suppression of the nickel state transition through stabilizing interactions with neighbouring iron centres.82,83 Similarly, the introduction of Ni2+ ions into alpha-MnO2 to decrease the pore size and volume of the catalyst with an accompanying increase in conductivity, resulted in the improved ORR performance.84 Computational ab initio simulations by Kondov et al. of Cr-modified Ni-based catalysts suggested that the improved ORR activity and long-term chemical stability was attributed to increased oxygen adsorption energies and the formation of a protective co-adsorbed hydroxide layer, respectively.48 Similar conclusions were obtained by Faubert et al., who demonstrated that Cr-doped nickel catalysts had a 50% increase in oxygen kinetic transfer coefficients, which was associated with more favourable thermodynamic conditions at the catalyst surface.85
Following RDE evaluation herein, the three doped cobalt–nickel sulfides and their undoped counterpart samples were deposited onto carbon papers and separately evaluated as bifunctional catalysts in secondary Zn–air cells. Pt/C and RuO2 were used as benchmark catalysts at a similar mass loading of 1.5 ± 0.1 mg cm−2 on the respective electrode. Fig. 6a displays the charge–discharge profiles Zn–air batteries using cobalt–nickel sulfides, RuO2, and Pt/C as bifunctional catalysts on their air-cathodes. The manganese-doped cobalt–nickel sulfide (NC11 Mn) outperformed the other sulfide samples by exhibiting a higher discharge voltage across the majority of current densities. Despite the slightly inferior performance at lower current densities (<30 mA cm−2) to RuO2 in ORR, the manganese-doped (NC11 Mn) sample exhibited superior activity once the current density was above 30 mA cm−2, which was accompanied by with superior OER performance to both benchmark reference catalysts (Fig. 6b).
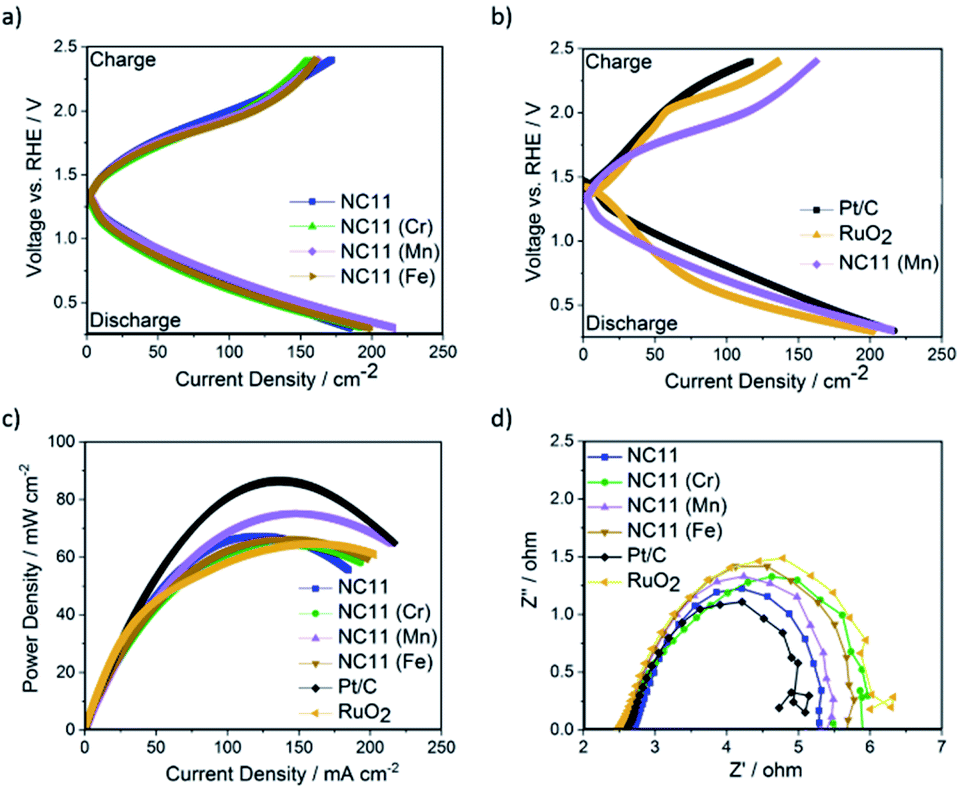 | ||
| Fig. 6 Performance of undoped and doped cobalt–nickel sulfides as air-cathode catalysts in Zn–air batteries. (a) Galvanodynamic charge–discharge curves of zinc–air cells with undoped NC11, and NC11 doped with chromium (NC11 Cr), manganese (NC11 Mn), and iron (NC11 Fe), respectively. (b) Galvanodynamic charge–discharge plots of NC11 Mn against reference samples of RuO2, Pt/C and 50–50% physical mixture of RuO2 and Pt/C catalysts. (c) Power density plots extracted from galvanodynamic discharge curves. (d) Nyquist plots obtained from electrochemical impedance spectroscopy results of cobalt–nickel sulfides along with that of RuO2 and Pt/C. | ||
In terms of the power density (Fig. 6c), Zn–air batteries with air electrodes made from undoped NC11 were able to achieve 66 mW cm−2 (±0.05 mW cm−2) at a current density of 140 mA cm−2; notably, this value is similar to the RuO2 (64 mW cm−2). While cobalt–nickel sulfide samples doped with iron (NC11 Fe) and chromium (NC11 Cr) yielded similar power density over NC11, the sample doped with manganese (NC11 Mn) exhibited a superior power density of 75 mW cm−2 (±0.05 mW cm−2) at a current density of 140 mA cm−2. This value was higher or similar to that of the zinc–air batteries using air cathodes made from several high-performance metal oxide-based zinc–air catalysts, including Co3O4/stainless steel,86 carbon black/MnO2,87 MnO2–LaNiO3/carbon nanotube (CNT).88 NiCo2S4/N-doped carbon nanotubes were reported to deliver a higher power density of 140 mW cm−2 at 150 mA cm−2 (at an active material loading of 1 g cm−2), benefitting from its high conductivity and high electrochemically active surface area that was provided by the N-doped CNT catalyst support network.89 In literature, iron-doped spinel cobalt–nickel oxide with the composition Fe0.1Ni0.9Co2O4 has been shown to exhibit a high power density of 120 mW cm−2 at 150 mA cm−2 (at an active material loading of 3 g cm−2), significantly outperforming its manganese-, copper-, and zinc-doped counterparts in zinc–air batteries.43
The Nyquist plots of EIS measurements displayed in Fig. 6d suggest that the impedance of zinc–air cells made with undoped NC11 cobalt–nickel sulfide exhibits similar an impedance compared to those made with Pt/C and RuO2, with all of the considered doped examples yielding similar the measured impedance to both benchmark catalysts (with all considered sulfides exhibiting lower impedance to RuO2). The impedance data were also fitted to equivalent circuits and were found to follow the structure R1 + Q2/R2 + Q3/R3, where R and Q stand for the resistor and Constant Phase Elements (CPE), respectively. The resistance for each circuit is shown in Table 5 below, with the equivalent circuit model shown in Fig. S5d.†
| Sample | R1/Ω (±0.02 Ω) | R2/Ω (±0.02 Ω) | R3/Ω (±0.02 Ω) | Rtotal (±0.04 Ω) |
|---|---|---|---|---|
| Pt/C | 2.60 | 0.62 | 1.97 | 5.19 |
| RuO2 | 2.48 | 0.87 | 2.88 | 6.23 |
| NC11 | 2.7 | 0.26 | 2.39 | 5.35 |
| NC11 (Cr) | 2.61 | 1.04 | 2.31 | 5.96 |
| NC11 (Mn) | 2.54 | 0.39 | 2.53 | 5.46 |
| NC11 (Fe) | 2.52 | 0.54 | 2.71 | 5.77 |
The results demonstrate that the cells featuring chromium- (NC11 Cr), manganese- (NC11 Mn), and iron-doped (NC11 Fe) cobalt–nickel sulfides exhibited lower impedance than those made with RuO2, with the manganese- and iron-doped samples exhibiting lower impedance than their chromium-doped counterpart. However, these values are still higher than that of undoped NC11, suggesting that these dopants do not improve conductivity, which has also been observed in cobalt–nickel spinel oxides.90 Moreover, the double-layer capacitance (Cdl, see ESI S5c†) of our sulfide samples was calculated to be 7.8 mF, 9.6 mF, 8.6 mF, and 7.8 mF for the undoped- (NC11), chromium-doped (NC11 Cr), manganese-doped (NC11 Mn), and iron-doped (NC11 Fe) samples, respectively. For materials of similar compositions, Cdl has been demonstrated to be linearly correlated with the Electrochemically Active Surface Area (ECSA) in literature.91 The results suggest that the introduction of selected transition metal dopants into metal sulfides is accompanied by changes in ECSA, a pattern also observed in transition metal oxides.92,93 However, detailed analysis suggests that the increase in Cdl (and by proxy, ECSA) is not solely responsible for the improved performance of the best cobalt–nickel sulfide samples. For example, the chromium-doped (NC11 Cr) sulfide sample exhibited the highest Cdl of the considered sulfides, with a 23% higher Cdl value than its undoped counterpart (NC11), but failed to exhibit the highest improved discharge current density, with only a modest 5% improvement being observed versus its undoped counterpart. Similarly, the manganese- (NC11 Mn) and iron-doped (NC11 Fe) sulfides exhibited a 10% and 0% higher Cdl values than their undoped counterpart (NC11) while displaying an improved discharge current density of 16% and 7%, respectively. These results support the theory that the dopants improved catalytic activity of the host cobalt–nickel sulfide by effects outside of conductivity and enhanced surface area, possibly through changes in the energetics of the oxygen adsorption process by the host cations or by serving as active sites themselves, as has been suggested in the literature.91,92
The stability of each catalyst in zinc–air batteries over the continuous charge and discharge cycling was evaluated by galvanostatic pulse cycling measurements at a discharge and charge currents of 4 mA cm−2 (Fig. 7) for 10 minutes per step per cycle. All of the cells made with cobalt–nickel sulfide catalysts displayed stability over 2000 minutes, or approximately 100 cycles, before signs of cell failure were observed. To account for possible side reactions with the electrolyte, the cells were allowed to cycle for 5 cycles (100 minutes) before initial voltage readings were taken. As voltage range directly affects the energy density of a cell, the differences in voltage ranges were examined (Fig. 7b). Cells featuring undoped NC11 catalysts as cathodes exhibited an initial discharge and charge voltages of 1.12 and 2.04 V, respectively, similar to RuO2 (1.10 and 1.94 V), with Pt/C exhibiting higher values of 1.10 and 2.61 V, respectively. Cobalt–nickel sulfide doped with manganese (NC11 Mn, 1.11 and 2.10 V), chromium (NC11 Cr, 1.09 and 2.08 V), and iron (NC11 Fe, 1.12 and 2.00 V), yielded similar discharge and charge voltages compared to undoped NC11. The batteries with nickel sulfide catalysts continued to exhibit an almost constant discharge and charge voltage for 100 cycles. By the end of the 101th cycle, NC11 exhibited a discharge voltage of 1.16 V and a charge voltage of 2.09 V, representing a voltage gap increase of 0.02 V. Similarly, cobalt–nickel sulfides doped with manganese (NC11 Mn, 1.17 and 2.11 V), chromium (NC11 Cr, 1.16 and 2.10 V), and iron (NC11 Fe, 1.17 and 2.03 V) yielded similar performance, with negligible change in voltage gap. In contrast, Pt/C exhibited a shift to 1.14 and 1.97 V, while RuO2 exhibited a smaller shift to 1.13 and 1.96 V, corresponding to an increase in voltage gap from 0.86 to 0.98 V. The steady voltages observed across our undoped and doped cobalt–nickel sulfides suggest high energy efficiency and overall bifunctional catalytic activity, which would, in turn, decrease the overpotentials observed for their respective processes. The reduction in overpotentials, in turn, has been shown to decrease the catalyst degradation rate, resulting in improved cell-life.8
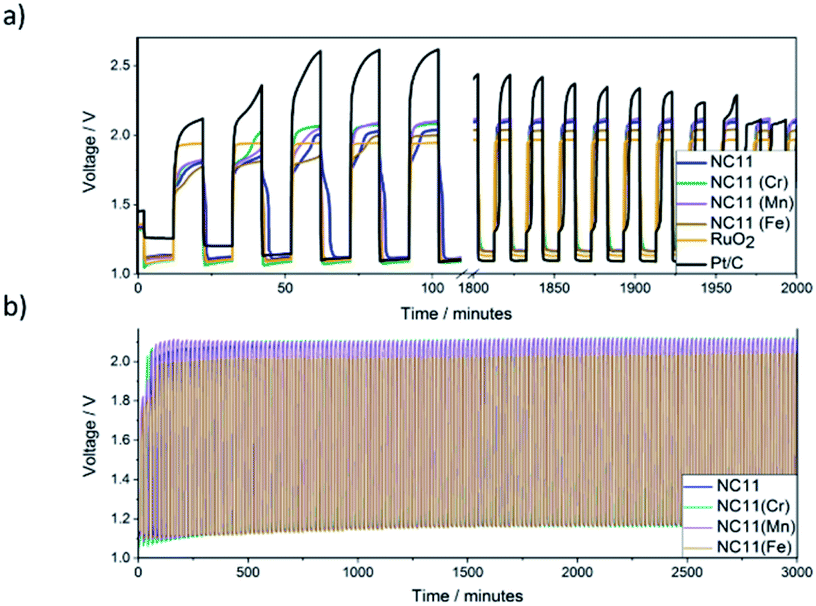 | ||
| Fig. 7 (a) Voltage range comparison of NC11 alongside the chromium (NC11 Cr)-, manganese- (NC11 Mn), iron-doped (NC11 Fe) cobalt–nickel sulfide samples, and Pt/C and RuO2 benchmarks during the first and last 100 minutes during galvanostatic pulse cycling tests at a current of 4 mA cm−2 with a discharge and charge time of 10 minutes each per cycle, across 100 cycles. (b) Cycling stability of reference catalysts vs. undoped (NC11), chromium- (NC11 Cr), manganese- (NC11 Mn), and iron-doped (NC11 Fe) cobalt–nickel sulfide samples during galvanostatic pulse cycling tests at a current of 4 mA cm−2 with a discharge and charge time of 10 minutes each per cycle. | ||
Conclusion
Transition metal-doped cobalt–nickel sulfide spinels were directly produced via a CHFS synthesis process incorporating a double confined jet mixer arrangement. The resulting materials from CHFS were evaluated electrochemically for their OER and ORR activities and subsequently tested in secondary zinc–air batteries as bifunctional air-electrode catalysts. It was found that the Zn–air battery utilizing an air-cathode containing cobalt–nickel sulfide sample doped with 7.2% manganese exhibited a power density of 75 mW cm−2 at a current density of 140 mA cm−2, which outperformed all of the other cobalt–nickel sulfide samples as well as the benchmark RuO2 catalyst, with a power density increase of 12% over its undoped counterpart. Interestingly, it was observed that the introduction of dopants increased overall cell impedance. The beneficial catalytic activities following dopant introduction could be attributed to three components: the high surface area following dopant introduction, the favourable tuning of existing nickel and cobalt cation concentrations, and the inherent activity of the dopant cations themselves.The catalysts were adequately stable in alkaline media of zinc–air batteries, with only 1 to 2% deterioration in electrochemical performance observed after 100 cycles. This study further reinforces the development of an understanding of the roles of specific cations in each catalytic process and demonstrates the synergies of transition metal-doping. Future studies should focus on further refining dopant concentrations in host cobalt–nickel sulfides, better understand the role of dopant species as active sites, and a detailed understanding of the morphological effects of dopant introduction. Finally, with the possibility of industrial scale-up by the CHFS process, this study contributes to the advancement of producing high-performance catalysts to enable the construction of high-energy, reliable Zn–air batteries at a commercial scale.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank the Engineering and Physical Sciences Research Council (EPRSC) for funding the Centre for Doctoral Training in Molecular Modelling & Materials Science (CDT, UCL, UK; EPSRC reference EP/L015862/1). The CDT and A*STAR (Singapore NUS) are thanked for studentship support for Y. X. The authors thank the EPSRC for funding The JUICED Hub (Joint University Industry Consortium for Energy (Materials) and Devices Hub, EP/R023662/1). The authors also acknowledge the financial support from Research, Community Service and Innovation Program (P3MI) Institut Teknologi Bandung, grant 2020.References
- M. Pablo-Romero, R. Pozo-Barajas and R. Yñiguez, Energy Policy, 2017, 101, 342–352 CrossRef CAS.
- D. Larcher and J.-M. Tarascon, Nat. Chem., 2015, 7, 19 CrossRef CAS.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- A. Sumboja, X. Ge, F. Goh, B. Li, D. Geng, T. Hor, Y. Zong and Z. Liu, ChemPlusChem, 2015, 80, 1341–1346 CrossRef CAS.
- A. Sivanantham, P. Ganesan and S. Shanmugam, Adv. Funct. Mater., 2016, 26, 4661–4672 CrossRef CAS.
- Q. Liu, J. Jin and J. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 5002–5008 CrossRef CAS.
- X. Wu, S. Li, B. Wang, J. Liu and M. Yu, New J. Chem., 2017, 41, 115–125 RSC.
- M. Wang, Y. Lai, J. Fang, F. Qin, Z. Zhang, J. Li and K. Zhang, Catal. Sci. Technol., 2016, 6, 434–437 RSC.
- D. Liu, Q. Lu, Y. Luo, X. Sun and A. M. Asiri, Nanoscale, 2015, 7, 15122–15126 RSC.
- H. Osgood, S. V. Devaguptapu, H. Xu, J. Cho and G. Wu, Nano Today, 2016, 11, 601–625 CrossRef CAS.
- W. Xu, Z. Lu, X. Lei, Y. Li and X. Sun, Phys. Chem. Chem. Phys., 2014, 16, 20402–20405 RSC.
- A. Sumboja, J. Chen, Y. Zong, P. S. Lee and Z. Liu, Nanoscale, 2017, 9, 774–780 RSC.
- Z. Zhang, X. Wang, G. Cui, A. Zhang, X. Zhou, H. Xu and L. Gu, Nanoscale, 2014, 6, 3540–3544 RSC.
- F. Lai, D. Yong, X. Ning, B. Pan, Y. E. Miao and T. Liu, Small, 2017, 13, 1602866 CrossRef.
- Y. Pan, Y. Lin, Y. Chen, Y. Liu and C. Liu, J. Mater. Chem. A, 2016, 4, 4745–4754 RSC.
- M. Lübke, A. Sumboja, L. McCafferty, C. F. Armer, A. D. Handoko, Y. Du, K. McColl, F. Cora, D. Brett and Z. Liu, ChemistrySelect, 2018, 3, 2613–2622 CrossRef.
- K. Ojha, S. Saha, B. Kumar, K. S. Hazra and A. K. Ganguli, ChemCatChem, 2016, 8, 1218–1225 CrossRef CAS.
- J. Wu, S. Dou, A. Shen, X. Wang, Z. Ma, C. Ouyang and S. Wang, J. Mater. Chem. A, 2014, 2, 20990–20995 RSC.
- M. Sun, J. Tie, Y. Li and L. Yu, Gen. Chem., 2017, 3, 202–206 Search PubMed.
- F. F. Tao, J.-j. Shan, L. Nguyen, Z. Wang, S. Zhang, L. Zhang, Z. Wu, W. Huang, S. Zeng and P. Hu, Nat. Commun., 2015, 6, 7798 CrossRef CAS.
- C. Xia, P. Li, A. Gandi, U. Schwingenschlögl and H. Alshareef, Chem. Mater., 2015, 27, 6482–6485 CrossRef CAS.
- L. Hou, H. Hua, R. Bao, Z. Chen, C. Yang, S. Zhu, G. Pang, L. Tong, C. Yuan and X. Zhang, ChemPlusChem, 2016, 81, 557–563 CrossRef CAS.
- D. P. Dubal, P. Gomez-Romero, B. R. Sankapal and R. Holze, Nano Energy, 2015, 11, 377–399 CrossRef CAS.
- Z. Wu, Y. Zhu and X. Ji, J. Mater. Chem. A, 2014, 2, 14759–14772 RSC.
- L. Croguennec and M. R. Palacin, J. Am. Chem. Soc., 2015, 137, 3140–3156 CrossRef CAS.
- X. Xu, W. Liu, Y. Kim and J. Cho, Nano Today, 2014, 9, 604–630 CrossRef CAS.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- J. Xiao, X. Zeng, W. Chen, F. Xiao and S. Wang, Chem. Commun., 2013, 49, 11734–11736 RSC.
- J.-Y. Lin and S.-W. Chou, Electrochem. Commun., 2013, 37, 11–14 CrossRef CAS.
- Y. Xue, Z. Zuo, Y. Li, H. Liu and Y. Li, Small, 2017, 13, 1700936 CrossRef.
- L. Ma, Y. Hu, R. Chen, G. Zhu, T. Chen, H. Lv, Y. Wang, J. Liang, H. Liu and C. Yan, Nano Energy, 2016, 24, 139–147 CrossRef CAS.
- J. Jiang, C. Yan, X. Zhao, H. Luo, Z. Xue and T. Mu, Green Chem., 2017, 19, 3023–3031 RSC.
- X. Y. Yu, L. Yu and X. W. D. Lou, Adv. Energy Mater., 2016, 6, 1501333 CrossRef.
- Z. Ai, Z. Hu, Y. Liu and M. Yao, ChemPlusChem, 2016, 81, 322–328 CrossRef CAS.
- F. Deng, J. Tie, B. Lan, M. Sun, S. Peng, S. Deng, B. Li, W. Sun and L. Yu, Electrochim. Acta, 2015, 176, 359–368 CrossRef CAS.
- A. Pramanik, S. Maiti, M. Sreemany and S. Mahanty, Electrochim. Acta, 2016, 213, 672–679 CrossRef CAS.
- Y. Zhu, X. Chen, W. Zhou, K. Xiang, W. Hu and H. Chen, Electrochim. Acta, 2017, 249, 64–71 CrossRef CAS.
- J. Shen, J. Ji, P. Dong, R. Baines, Z. Zhang, P. M. Ajayan and M. Ye, J. Mater. Chem. A, 2016, 4, 8844–8850 RSC.
- G. He, M. Qiao, W. Li, Y. Lu, T. Zhao, R. Zou, B. Li, J. A. Darr, J. Hu and M. M. Titirici, Adv. Sci., 2017, 4, 1600214 CrossRef.
- B. Lu, D. Cao, P. Wang, G. Wang and Y. Gao, Int. J. Hydrogen Energy, 2011, 36, 72–78 CrossRef CAS.
- J.-P. Jacobs, A. Maltha, J. G. Reintjes, J. Drimal, V. Ponec and H. H. Brongersma, J. Catal., 1994, 147, 294–300 CrossRef CAS.
- W. King and A. Tseung, Electrochim. Acta, 1974, 19, 493–498 CrossRef CAS.
- Y.-T. Lu, Y.-J. Chien, C.-F. Liu, T.-H. You and C.-C. Hu, J. Mater. Chem. A, 2017, 5, 21016–21026 RSC.
- Y. Xu, A. Sumboja, Y. Zong and J. A. Darr, Catal. Sci. Technol., 2020, 10, 2173–2182 RSC.
- I. Nikolov, R. Darkaoui, E. Zhecheva, R. Stoyanova, N. Dimitrov and T. Vitanov, J. Electroanal. Chem., 1997, 429, 157–168 CrossRef CAS.
- G. Li, M. A. Mezaal, K. Zhang and L. Lei, Int. J. Electrochem. Sci., 2015, 10, 5395–5404 CAS.
- Y. Xu, A. Sumboja, Y. Zong and J. A. Darr, Catal. Sci. Technol., 2020, 10, 2173–2182 RSC.
- I. Kondov, P. Faubert and C. Müller, Electrochim. Acta, 2017, 236, 260–272 CrossRef CAS.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef.
- W. Liu, E. Hu, H. Jiang, Y. Xiang, Z. Weng, M. Li, Q. Fan, X. Yu, E. I. Altman and H. Wang, Nat. Commun., 2016, 7, 10771 CrossRef CAS.
- J. A. Darr, J. Zhang, N. M. Makwana and X. Weng, Chem. Rev., 2017, 117, 11125–11238 CrossRef CAS.
- J. A. Darr, J. Zhang, N. M. Makwana and X. Weng, Chem. Rev., 2017, 117, 11125–11238 CrossRef CAS.
- Y. Tao, L. Ruiyi and L. Zaijun, Mater. Lett., 2016, 167, 234–237 CrossRef CAS.
- U. Holzwarth and N. Gibson, Nat. Nanotechnol., 2011, 6, 534 CrossRef CAS.
- O. Knop, K. Reid, Sutarno and Y. Nakagawa, Can. J. Chem., 1968, 46, 3463–3476 CrossRef CAS.
- X. Zhang and A. Zunger, Adv. Funct. Mater., 2010, 20, 1944–1952 CrossRef CAS.
- D. Kim, G. Ghodake, N. Maile, A. Kadam, D. Lee, V. Fulari and S. Shinde, Sci. Rep., 2017, 7, 9764 CrossRef.
- K. Liang, W. He, X. Deng, H. Ma and X. Xu, J. Alloys Compd., 2018, 735, 1395–1401 CrossRef CAS.
- X. Chen, D. Chen, X. Guo, R. Wang and H. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 18774–18781 CrossRef CAS.
- M. Li, Y. Xiong, X. Liu, X. Bo, Y. Zhang, C. Han and L. Guo, Nanoscale, 2015, 7, 8920–8930 RSC.
- R. Singh, J. Pandey, N. Singh, B. Lal, P. Chartier and J.-F. Koenig, Electrochim. Acta, 2000, 45, 1911–1919 CrossRef CAS.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780 CrossRef CAS.
- M. Jahan, Z. Liu and K. P. Loh, Adv. Funct. Mater., 2013, 23, 5363–5372 CrossRef CAS.
- C. Tang, N. Cheng, Z. Pu, W. Xing and X. Sun, Angew. Chem., 2015, 127, 9483–9487 CrossRef.
- X. Yu, T. Hua, X. Liu, Z. Yan, P. Xu and P. Du, ACS Appl. Mater. Interfaces, 2014, 6, 15395–15402 CrossRef CAS.
- E. Fabbri, R. Mohamed, P. Levecque, O. Conrad, R. Kötz and T. Schmidt, ChemElectroChem, 2014, 1, 338–342 CrossRef.
- I. Katsounaros, W. B. Schneider, J. C. Meier, U. Benedikt, P. U. Biedermann, A. Cuesta, A. A. Auer and K. J. Mayrhofer, Phys. Chem. Chem. Phys., 2013, 15, 8058–8068 RSC.
- D. Banham, S. Ye, K. Pei, J.-i. Ozaki, T. Kishimoto and Y. Imashiro, J. Power Sources, 2015, 285, 334–348 CrossRef CAS.
- W. Fu, C. Zhao, W. Han, Y. Liu, H. Zhao, Y. Ma and E. Xie, J. Mater. Chem. A, 2015, 3, 10492–10497 RSC.
- X. Wu, S. Li, B. Wang, J. Liu and M. Yu, Phys. Chem. Chem. Phys., 2017, 19, 11554–11562 RSC.
- X. Gao, X. Long, H. Yu, X. Pan and Z. Yi, J. Electrochem. Soc., 2017, 164, H307–H310 CrossRef CAS.
- Z. Liu, N. Li, H. Zhao, Y. Zhang, Y. Huang, Z. Yin and Y. Du, Chem. Sci., 2017, 8, 3211–3217 RSC.
- M. Hamdani, R. Singh and P. Chartier, Int. J. Electrochem. Sci., 2010, 5, 556–577 CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546 CrossRef CAS.
- D. Kan, Y. Orikasa, K. Nitta, H. Tanida, R. Kurosaki, T. Nishimura, T. Sasaki, H. Guo, Y. Ozaki and Y. Uchimoto, J. Phys. Chem. C, 2016, 120, 6006–6010 CrossRef CAS.
- A. Aijaz, J. Masa, C. Rösler, W. Xia, P. Weide, A. Botz, R. Fischer, W. Schuhmann and M. Muhler, Angew. Chem., 2016, 55, 4087–4091 CrossRef CAS.
- H. Wang, R. Liu, Y. Li, X. Lü, Q. Wang, S. Zhao, K. Yuan, Z. Cui, X. Li, S. Xin, R. Zhang, M. Lei and Z. Lin, Joule, 2018, 337–348 CrossRef.
- J. O. Bockris and T. Otagawa, J. Phys. Chem., 1983, 87, 2960–2971 CrossRef CAS.
- A. Trunov, Electrochim. Acta, 2013, 105, 506–513 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS.
- X. Xu, C. Su, W. Zhou, Y. Zhu, Y. Chen and Z. Shao, Adv. Sci., 2016, 3, 1500187 CrossRef.
- D. A. Corrigan, J. Electrochem. Soc., 1987, 134, 377–384 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS.
- T. N. Lambert, J. A. Vigil, S. E. White, C. J. Delker, D. J. Davis, M. Kelly, M. T. Brumbach, M. A. Rodriguez and B. S. Swartzentruber, J. Phys. Chem. C, 2017, 121, 2789–2797 CrossRef CAS.
- P. Faubert, I. Kondov, D. Qazzazie, O. Yurchenko and C. Müller, MRS Commun., 2018, 8, 160–167 CrossRef CAS.
- D. U. Lee, J. Y. Choi, K. Feng, H. W. Park and Z. Chen, Adv. Energy Mater., 2014, 4, 1301389 CrossRef.
- C. Z. Luo, Y. Zhu, Y. Xu, Y. Liu, T. Gao, J. Wang and C. Wang, J. Power Sources, 2014, 250, 372–378 CrossRef CAS.
- H. Ma and B. Wang, RSC Adv., 2014, 4, 46084–46092 RSC.
- X. Han, X. Wu, C. Zhong, Y. Deng, N. Zhao and W. Hu, Nano Energy, 2017, 31, 541–550 CrossRef CAS.
- V. Maruthapandian, M. Mathankumar, V. Saraswathy, B. Subramanian and S. Muralidharan, ACS Appl. Mater. Interfaces, 2017, 9, 13132–13141 CrossRef CAS.
- X.-Z. Song, F.-F. Sun, Y.-L. Meng, Z.-W. Wang, Q.-F. Su and Z. Tan, New J. Chem., 2019, 43, 3601–3608 RSC.
- G. Zhang, Y.-S. Feng, W.-T. Lu, D. He, C.-Y. Wang, Y.-K. Li, X.-Y. Wang and F.-F. Cao, ACS Catal., 2018, 8, 5431–5441 CrossRef CAS.
- D. Xu, M. B. Stevens, Y. Rui, G. DeLuca, S. W. Boettcher, E. Reichmanis, Y. Li, Q. Zhang and H. Wang, Electrochim. Acta, 2018, 265, 10–18 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08363a |
| This journal is © The Royal Society of Chemistry 2020 |