
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic effect of hierarchical nanopores in Co-doped cobalt oxide 3D flowers for electrochemical energy storage†
Xia Deng‡
a,
Hong Zhang‡b,
Junwei Zhangb,
Dongsheng Leib and
Yong Peng *b
*b
aSchool of Life Sciences, Electron Microscopy Center of Lanzhou University, Lanzhou, 730000, P. R. China
bElectron Microscopy Centre of Lanzhou University, Key Laboratory of Magnetism and Magnetic Materials of the Ministry of Education and School of Physical Science and Technology, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: pengy@lzu.edu.cn
First published on 9th December 2020
Abstract
Hybridizing hierarchical porous transition oxides composed of nanoscale building blocks is highly desirable for improving the electrochemical performance of energy storage. Herein, we contribute a fabrication of novel hierarchically nanoporous flower-shaped metal/transition oxide (Co/Co3O4–CoO) with controllable three-dimensional structure. The designed Co/Co3O4–CoO 3D flowers (3DFs) are made of petal-shaped nanoporous Co3O4–CoO nanosheets with tunable pore sizes, in which metallic Co nanoparticles tend to attach to the edge of larger ones. The hierarchically nanoporous 3DFs with bimodal pore size distribution and higher fraction of small nanopores exhibit a higher specific capacitance (902.3 F g−1 at current density of 2 A g−1) and better cyclability than the uniformly nanoporous 3DFs with unimodal pore size distribution and larger BET surface area. The enhanced capacitance is mainly derived from the synergistic effect of hierarchical nanopores, in which large nanopores disproportionately facilitate osmotic solution flux and diffusive solute transport, whilst small nanopores supply faster channels for electron transportation and ion diffusion. Our work should provide a strategy to fabricate a smart functional hierarchical nanoporous architecture with 3DF structures for the development of electrochemical energy storage materials.
1. Introduction
The morphologies and structures of materials can effectively tune their physical and chemical properties, such as shape which determines surface atomic arrangement/coordination, and size which controls surface area.1–6 In order to improve the performances of materials in energy storage and delivery, considerable efforts7,8 have been devoted to fabricating various electrode materials with both a large specific surface area for increasing activity sites and a stable structure for facilitating the electron transportation and ion diffusion.9–14 Among the reported electrode materials, transition metal oxides are considered to be one group of the most promising and practical materials because of their high theoretical capacitance values and variable oxidation states of metal ions.12,15–18 Low-cost transition metal oxide materials, including WO3,19 MoO2,20 MnO2,21 Fe2O3,22 NiO,23 Co3O4,24 Co/Ni binary metal oxide25 and Co–Ni–Cu ternary metal oxide.26 with controlled microstructures, such as nanoparticles,27 nanotubes,28 nanowires,29 nanosheets,30 and 3D hierarchical microstructures,31 have been developed and utilized for energy storage.Cobalt oxides as one of transition metal oxides are considered to be high priority candidates of ideal electrode materials because of their intrinsic advantages including low-cost, relatively environmental friendliness and theoretical high specific capacitance (∼3560 F g−1 for Co3O4 and 2467 F g−1 for CoO). However, similar to other transition metal oxides, cobalt oxides have poor electrical conductivity due to the relative wide band gap or large volume change modulated by ions insertion/extraction during the redox reaction, which prevents them from practical applications.32,33 Numerous efforts have been devoted to overcome this disadvantage. One approach is to incorporate cobalt oxides with highly conductive materials such as hydroxides MoO2/Co(OH)2,32 carboneous materials Co3O4/graphane,34 conducting polymers CoO@Polypyrrole35 or other transition metal oxides Co3O4@MnO2.36 Another approach is to construct porous architectural nanostructure such as mesoporous Co3O4 nanosheet,37 multishelled Co3O4 hollow spheres38 and mesoporous Co3O4 architectures.39 The above approaches have somewhat improved the performances of cobalt oxide materials, but there is still a lot of room to further improve.
Construction of porous structure with defined pore size and shape (length–diameter ratio) is one important approach to improve the electrochemical properties40–43 of cobalt oxide materials for their applications in energy storage. In widely used energy storage systems, such as electric double-layer supercapacitors, Faraday pseudocapacitors and lithium batteries, the ions are selected to transport among the electrode materials and electrochemical reactions where localize at selective area.44 Due to the sieving of the electrolyte ions, adequate pore architecture and size distribution is more important to achieve higher electrochemical performance.45 As the pore size reduces to nanometer, the transport of ions through pores is modulated by various factors, such as the hydrophilic or hydrophobic inner surface, surface charge properties, size effect, and ion–ion interactions.46 Both experimental and theoretical studies have showed that micropores provide high accessible surface area and mesopores offer fast ion transport channels. Therefore, hierarchically porous materials with both micropores and mesopores could have relatively high power and energy density simultaneously, which has been validated by using carbon materials.47–49 The same strategy has been also employed in the development of hybrid transition metal oxide materials. However, since the ion insertion/extraction during redox reactions could result in large volume change,50 the relationship between the pore size/architechture and the electrochemical performance of transition metal oxide materials are still unclear.
In this work, we present a strategy to synthesize hybridizing hierarchically nanoporous cobalt oxide material (Co/Co3O4–CoO) with controllable pore size, and studied the relationship between pore size/architechture and the electrochemical performance of this material. The material has a three-dimensional flower-shaped (3DF) structure composed of nanoscale building blocks. Pores within the blocks could have precisely controlled size and architecture, in which Co nanoparticles can be embedded into the edge of large nanopores by straightforward annealing Co(OH)2 3DFs in Ar atmosphere (Co/Co3O4–CoO). In electrochemical tests, the bimodally nanoporous Co/Co3O4–CoO 3DFs with mainly smaller nanopores show the best performance. Consistently, EIS study verifies that, compared with 3DFs with unimodal nanopores, the bimodally nanoporous Co/Co3O4–CoO 3DFs exhibits faster redox reaction and ions/electrons transportation/diffusion, smaller charge resistance and larger diffusion coefficient and slope. The enhancement of electrochemical performance is likely dominated by the synergistic effect of hierarchical nanopores, in which large nanopores disproportionately facilitate osmotic solution flux and diffusive solute transport, while small nanopores supply fast channels for electron transportation and ion diffusion. This hierarchically nanoporous architecture opens up an opportunity for the development and design of energy storage device.
2. Results and discussion
2.1 Synergistic effect of hierarchical nanopores in Co-doped cobalt oxides 3D flowers for electrochemical energy storage
The pore size distribution and stable 3D nanostructure of the hierarchical nanoporous Co-doped cobalt oxides 3D flowers (Co/Co3O4–CoO 3DFs) can be controlled to synthesize by calcining Co(OH)2 3DFs at 300 °C in Ar atmosphere, which maintains the 3D morphologies of precursors Co(OH)2 3DFs (Fig. S1 and S2†). The mechanism of Co/Co3O4–CoO 3DFs, a battery-type electrode material for energy storage (Fig. 1a), is acknowledged to proceed via redox reaction. The corresponding reversible reactions of Co3+/Co4+ and Co2+/Co4+ associated with anions OH− (Fig. 1d) can be shown by the following chemical reaction eqn (1)–(4).12,17,35,51–54| Co3O4 + H2O + OH− ↔ 3CoOOH + e− | (1) |
| CoOOH + OH− ↔ CoO2 + H2O + e− | (2) |
| CoO + OH− ↔ CoOOH + e− | (3) |
| CoOOH + OH− ↔ CoO2 + H2O + e− | (4) |
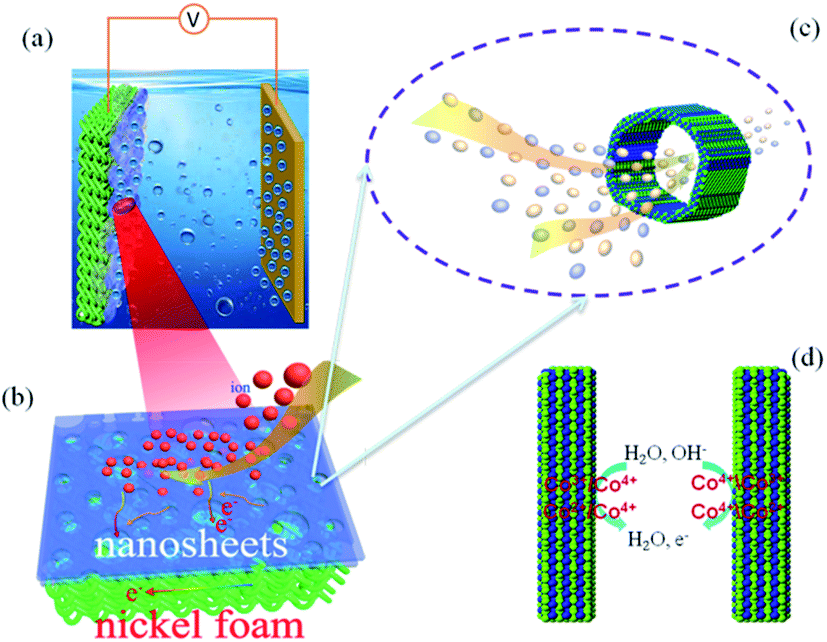 | ||
| Fig. 1 The schematic illustration of electrochemical energy storage: (a) the nanoporous 3DFs are loaded on nickel foam for electrochemical energy storage, (b) the zoom-in view of electron transport and ion diffusion in the nanosheet of 3DFs, (c) the ions transport diffusion among the nanopores of nanosheets, (d) redox reaction on the surface of materials. | ||
According to the mechanism of redox reaction, one dominant factor that affects electrochemical performance of the material is the number of activity cites and the transportation/diffusion coefficient of electrons/ions/molecules. Since the synthesized Co/Co3O4–CoO 3DFs is assembled from petaloid hierarchical nanoporous nanosheets consisting of small CoO and Co3O4 nanoparticles building blocks with metallic Co embedded in the edge of larger nanopores (Fig. S3† and 1a), the 3DFs can provide significantly enlarged specific surface area, thus shorten the ion-transportion/diffusion path and provide more activity cites for the reactions between electrode materials and electrolyte ions.45,55–58
The hierarchical nanoporous Co/Co3O4–CoO 3DFs with bimodal pore size (4.3 nm and 18.2 nm) distribution exhibit a high specific capacitance 902.3 F g−1 at current density of 2 A g−1 and good cyclability, suggesting a better electrochemical performance than the uniformly nanoporous Co/Co3O4–CoO 3DFs with unimodal pore size ditribution. It should be noted that the specific surface area of bimodally nanoporous 3DFs is lower than that of unimodally nanoporous 3DFs. This is consistent with previous reports in which a higher specific surface area is not always conducive to higher electrochemical performance due to the sieving of electrode ions.45,59,60 Therefore, the enhancement of electrochemical performance of hierarchical nanoporous Co/Co3O4–CoO 3DFs in this work is deduced from the pore architecture and size distribution.
The roughness and atomic occupancy of the pore surface, pore size and draw ratio directly affect the transportation of electrode ions, electrons and water molecules. The insertion/extraction of ions may occur during the redox reaction because of the peak of CV and platform of GCD curves,50 which would change the surface structure or volume of nanopores or cobalt oxides nanoparticles. However, the micro dynamic behaviour of ions coming from the change can not be tested or quantitatively simulated. The macro average dynamics parameters of ions or electrons can be calculated from the EIS measurement. The typical Nyquist plots consist of two parts, a high frequency area (semicircle) controlled by the kinetics reaction of electrode and a low frequency area (line) governed by diffusion of reactants or products of electrode reaction.61 For the kinetics reaction, the charge transfer resistance (Rct) of the hierarchically nanoporous Co/Co3O4–CoO 3DFs electrodes (0.13 Ω) is half of that of the uniformly nanoporous Co–Co3O4–CoO 3DFs electrodes (0.25 Ω), which confirms that the hierarchically nanoporous Co/Co3O4–CoO 3DFs electrodes have a faster electron transport. And the diffusion coefficient D of ions can be calculated from the low frequency line (the detail derivation has shown in electrochemical performance test part). The diffusion coefficient Dh of hierarchically nanoporous Co/Co3O4–CoO 3DFs is 6.86 × 10−8 cm2 s−1, which is twelve times faster than that of uniformly nanoporous Co–Co3O4–CoO 3DFs, revealing a faster ions diffusion during the electrochemical reactions. Therefore, the hierarchical nanoporous 3D nanostructure consisting of small Co3O4 and CoO nanoparticle building blocks with Co embedded in the edge of larger pores can facilitate the electrons transportation and ions diffusion (Fig. 1b and c), which is mainly dominated by the synergistic effect of hierarchical nanopores.
2.2 Morphologies and nanostructures of the hierarchically nanoporous Co/Co3O4–CoO 3DFs
To verify the correlation between nanostructure and electrochemical performance of Co–Co3O4–CoO 3DFs, the 3D morphologies, nanostructures and chemical properties of 3DFs were investigated. The 3DFs were first imaged by scanning electron microscope (SEM). Fig. 2 shows representative SEM images of nanoporous Co/Co3O4–CoO 3DFs prepared by calcining Co(OH)2 3DFs at 300 °C in Ar atmosphere, of which precursors Co(OH)2 3DFs were synthesized by using 10 ml (named as sample C1) or 20 ml (named as sample C2) ethanolamine. In these images, all particles show a 3D flower shape (Fig. 2a like Dahlia pinnata flower and Fig. 2c like Rosa chinensis flower), and are composed of petal-shaped nanoporous nanosheets as building blocks. These Co/Co3O4–CoO 3DFs particles (Fig. S1c and d†) have well preserved the morphologies of their precursor Co(OH)2 3DFs (see ESI Fig. S1a and b† for details). The average diameters (Fig. S2†) of these Co/Co3O4–CoO 3DFs prepared by above two conditions are ∼15.87 μm (C1 with precursors of CH1 (∼17.95 μm)) and ∼4.96 μm (C2 with precursors of CH2 (∼5.10 μm)), respectively. The thicknesses of the nanosheets within the 3DFs are typically less than 10 nm, which are smaller than that of the Co(OH)2 3DF precursors (Fig. S3†). The thickness shrinkage may originate from that the dynamic thermal motion makes the ions tending to aggregate at the surface of the nanosheets, which eventually cause each nanosheet to split into two pieces during the calcination. The average diameter and nanosheet numbers of individual Co/Co3O4–CoO 3DFs increased as the decrease of the ethanolamine stoichiometry. The intrinsic reason is deduced to come from the concentration of the OH− solution released from the ethanolamine. Lower concentration of OH− solution would provide more time and suitable conditions for the assembly of larger and denser precursor Co(OH)2 3DFs, in which the morphologies are eventually preserved in the product of Co/Co3O4–CoO 3DFs after calcination.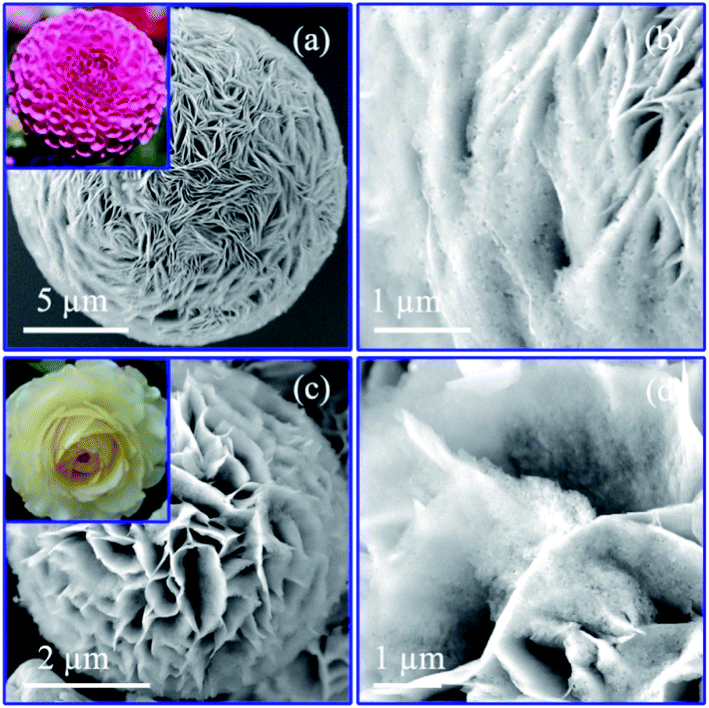 | ||
| Fig. 2 The morphologies of the nanoporous Co/Co3O4–CoO 3DFs at 300 °C calcined temperature: (a) SEM image of the specimen C1; (b) the magnified SEM image of C1; (c) SEM image of specimen C2; (d) the magnified SEM image of C2. | ||
The detailed nanostructures of individual nanosheets of the nanoporous Co/Co3O4–CoO 3DFs were further investigated by atomic resolution aberration-corrected electron microscopy (Cs-STEM). The representative TEM images show that individual nanosheets of Co/Co3O4–CoO 3DFs (C1, Fig. 3a) are hierarchically nanoporous thin films consisting of nanoscale building blocks with black nanoparticles embed in the edge of large pores. The nanopores size shows bimodal distribution with two peaks at ∼4.3 nm and ∼26 nm (Fig. S6b†). The nanoparticles are ∼31 nm in diameter, and prefer to embed into the edge of large-sized nanopores. The nanosheets of the Co–Co3O4–CoO 3DFs (sample C2, Fig. 3f) acquiring by calcining the precursors CH2 are made of nanoparticles and form a nearly uniform nanopores with ∼3.5 nm (Fig. S6d†).
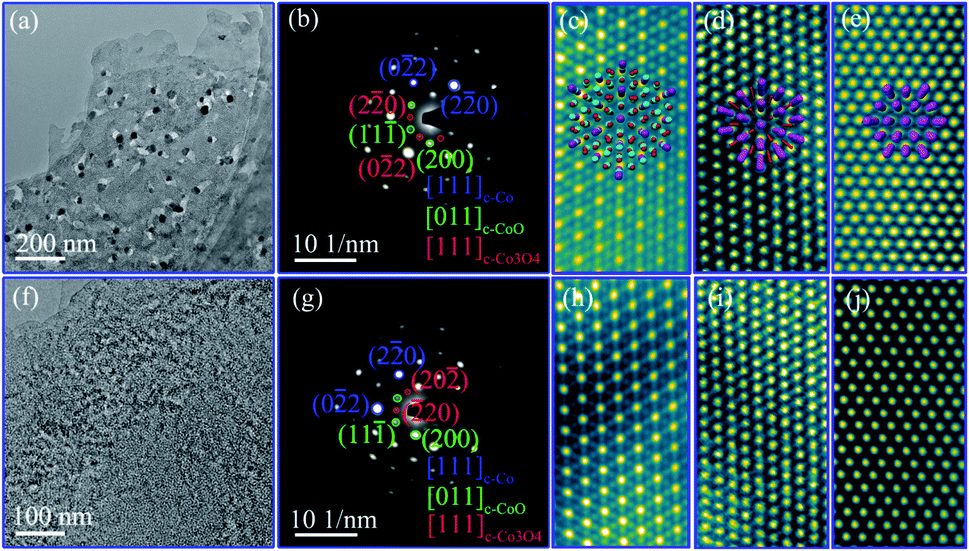 | ||
| Fig. 3 The nanostructure of the nanosheet of the nanoporous Co/Co3O4–CoO 3DFs: (a–e), bright field (BF) TEM, SAED pattern of the hierarchically nanoporous nanosheet (C1), HAADF-HRSTEM image of [111] Co3O4, [011] CoO and [111] Co, respectively. Inset of (c–e) the perspective view of the [111] oriented cell of Co3O4, [011] of CoO and [111] of Co showing the atomic columns distribution. (f–j), BF TEM, SAED pattern of the uniformly nanoporous nanosheet (C2), HAADF-HRSTEM image of [111] Co3O4, [011] CoO and [111] Co, respectively. | ||
The crystal structures of the hierarchically nanoporous nanosheets of Co/Co3O4–CoO 3DFs (C1) were further characterized by SAED and HAADF-HRSTEM. The corresponding SAED pattern of a single nanosheet is shown in Fig. 3b, which can be indexed into [011] orientation of fcc CoO, [111] of fcc Co3O4, and [111] of fcc Co. This observation reveals that each nanosheet has three crystal phases with preferential orientation distribution. The phase configuration of the three crystal phases, fcc CoO, fcc Co3O4 and fcc Co, is imaged with HAADF-HRSTEM. Fig. 3c–e show the atomic resolution HAADF-HRSTEM images of fcc Co3O4, fcc CoO and fcc Co nanoparticles projected from the [111], [011] and [111] orientation, respectively. The consistent of SAED and HAADF-HRSTEM studies demonstrates that the nanosheet was assembled from nanoscale fcc CoO and fcc Co3O4 building blocks with metallic Co nanoparticles embed in the edge of large nanopores. It is therefore observed that individual hierarchically nanoporous Co/Co3O4–CoO 3DFs (C1) are formed by Co3O4–CoO petal-like hierarchically nanoporous nanosheets skeleton with about 10 nm thickness and with two-level nanopore sizes to which metallic Co nanoparticles are directly attached at the edge of the large-level nanopores.
The crystal structure of the uniformly nanoporous Co–Co3O4–CoO 3DFs nanosheets were also characterized by SAED and HAADF-HRSTEM techniques. The SAED pattern (Fig. 3g) of the uniformly nanoporous Co–Co3O4–CoO 3DFs (C2) also can be indexed into [011] orientation of fcc CoO, [111] of fcc Co3O4, and [111] of fcc Co, demonstrating that each nanosheet has three crystal phases with preferential orientation distribution. Furthermore, the atomic resolution HAADF-HRSTEM images of [111] orientation of fcc Co3O4 (Fig. 3h), [011] of fcc CoO (Fig. 3i) and [111] of fcc Co (Fig. 3j) show that the nanosheet is made of small nanoscale fcc Co3O4, fcc CoO and fcc Co building blocks. Thus the individual uniformly nanoporous Co–Co3O4–CoO 3DFs (C2) are formed by Co–Co3O4–CoO petal-like nanoporous nanosheets skeleton and with one-level nanopore size.
In order to double confirm above observations, the crystal structures of Co/Co3O4–CoO 3DFs were further investigated by XRD technique. Fig. S4† shows the XRD spectra of two Co/Co3O4–CoO 3DFs. The corresponding XRD diffraction peaks can be indexed to (111), (220), (311), (400), (422), (511), (440) planes of fcc Co3O4, (111), (200), (220), (222) planes of fcc CoO, and (111), (200), (220) planes of fcc Co, respectively. This result is well consistent with the structure characterization by using SEAD and HAADF-HRSTEM techniques.
2.3 Chemical analysis of the hierarchically nanoporous Co/Co3O4–CoO 3DFs
The chemistry states of the hierarchically nanoporous Co/Co3O4–CoO 3DFs were characterized by using XPS, EDX and HAADF-STEM mapping on a 300 kV HRTEM. The chemical distributions of individual hierarchically nanoporous Co/Co3O4–CoO nanosheet were studied by EDX elemental mapping analysis technique. Fig. 4a shows the HAADF-STEM image of a single piece of nanosheet (C1), in which the region used for the elemental mapping analysis is marked by red rectangular. The EDX spectrum (Fig. 4b) collected from the area in Fig. 4a shows the peaks of Co and O, proving the existence of cobalt and oxygen elements in the nanosheet. Fig. 4c–e show the corresponding EDX elemental mappings of cobalt (Kα, 6.93 keV), oxygen (Kα, 0.52 keV) elements and their combination, respectively. The elements Co and O are nearly evenly distributed throughout the skeleton of the nanosheet except the place where the nanoparticles locate. The signal of element Co is much stronger than that of element O inside the nanoparticles, which indicates that the nanoparticles are metallic Co and their surfaces may be slightly oxidized. The EDX elemental mappings of the uniformly nanoporous Co/Co3O4–CoO shows that elements O and Co are evenly distributed throughout the whole nanosheets (Fig. S5c and d†), which do not show a clear existence of Co nanoparticles. The intrinsic reason is believed to originate from the uniformly nanoporous nanosheet is composed of Co, Co3O4 and CoO nanoparticles stacked randomly.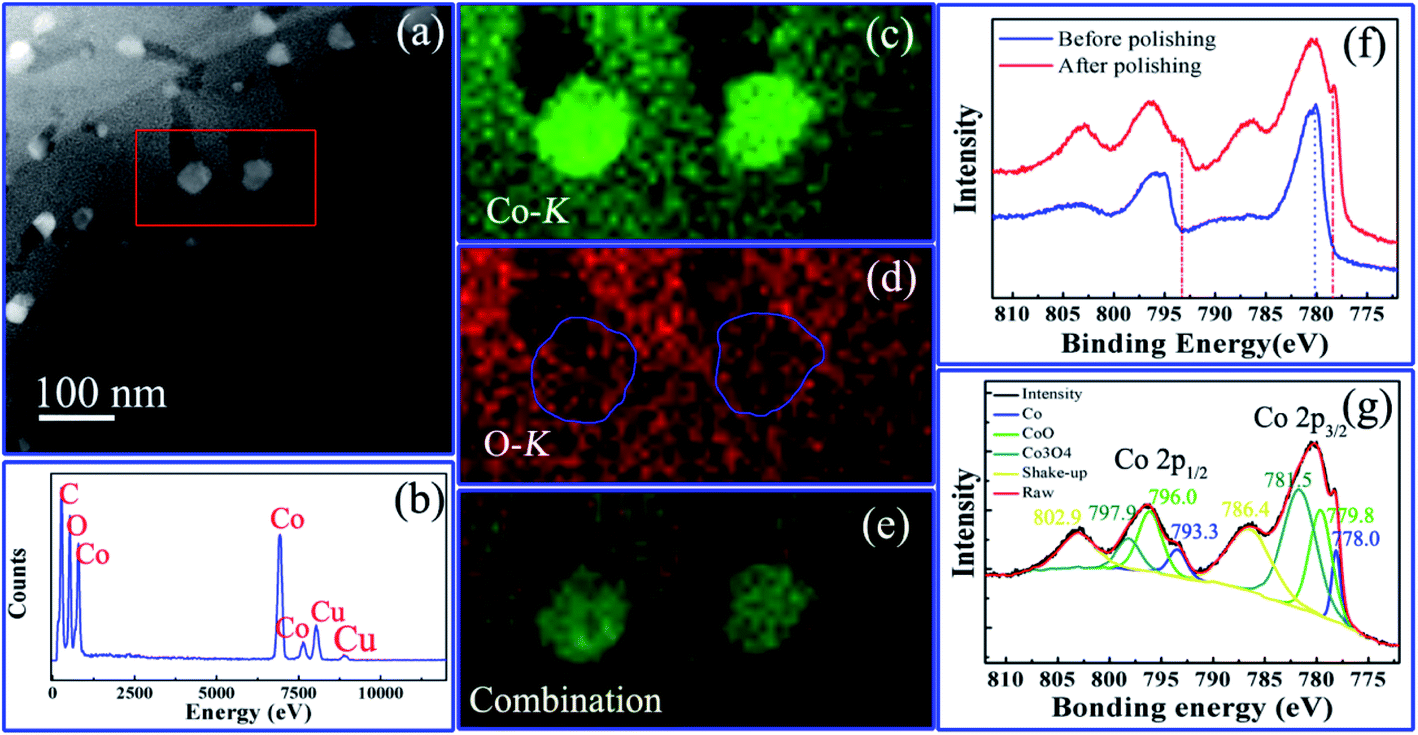 | ||
| Fig. 4 Chemical characterization of the hierarchically nanoporous Co/Co3O4–CoO 3DFs (C1): (a) HAADF-STEM image of the single nanosheet used for elemental mapping analysis; (b) EDX spectrum; (c) cobalt mapping; (d) oxygen mapping; (e) their combination; (f) the XPS spectra before and after polishing; (g) the fitted XPS spectra after polishing. | ||
To further verify the chemical states and composition of the hierarchically nanoporous Co/Co3O4–CoO 3DFs (C1), XPS spectra were obtained before polishing (or ion beam etching) and after polishing, respectively, of which binding energy was calibrated with the C 1s peak at 284.6 eV. The XPS spectrum of the hierarchically nanoporous Co/Co3O4–CoO 3DFs obtained before polishing (the blue curve in Fig. 4f) shows that the surface is composed of Co3O4 and CoO. After Ar+ ion polishing for 600s, a new peak (red curve in Fig. 4f) located at ∼778.0 eV appears in the Co 2p XPS spectrum, which confirms the existence of metallic Co. The XPS spectrum after polishing was well fitted with characteristic peaks of Co3O4, CoO and metallic Co as displayed in Fig. 4g. The Co 2p3/2 peak located at 778.0 eV and the Co 2p1/2 peak located at 793.3 eV verify the existence of the metallic Co.62,63 The 779.8 eV Co 2p3/2 peak combing with the shake-up satellite peak 786.4 eV and the 796.0 eV Co 2p1/2 peak accompanying the prominent shake-up satellite peak 802.9 eV indicate the existence of the CoO phase.64–66 The peaks located at 781.5 eV with shake-up satellite peak 786.4 eV and 797.9 eV with shake-up satellite peak 802.9 eV confirm the existence of Co3O4 phase.23,67 Quantitative analysis of the XPS spectrum indicates a 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40:48 mass ratio of Co, CoO and Co3O4. This analysis confirms that the hierarchically nanoporous Co/Co3O4–CoO 3DFs is consisted of hierarchically nanoporous Co3O4–CoO petal-shaped nanosheets (within thickness of 10 nm) with two-level size of nanopores and the metallic Co nanoparticles are directly attached at the edge of large level nanopores. The XPS spectrum of the uniformly nanoporous Co/Co3O4–CoO 3DFs (Fig. S5f and g†) also reveals the present of Co, CoO and Co3O4 with a 10:40:50 mass ratio, which is approximately equal to that of the hierarchically nanoporous Co/Co3O4–CoO 3DFs.
:40:48 mass ratio of Co, CoO and Co3O4. This analysis confirms that the hierarchically nanoporous Co/Co3O4–CoO 3DFs is consisted of hierarchically nanoporous Co3O4–CoO petal-shaped nanosheets (within thickness of 10 nm) with two-level size of nanopores and the metallic Co nanoparticles are directly attached at the edge of large level nanopores. The XPS spectrum of the uniformly nanoporous Co/Co3O4–CoO 3DFs (Fig. S5f and g†) also reveals the present of Co, CoO and Co3O4 with a 10:40:50 mass ratio, which is approximately equal to that of the hierarchically nanoporous Co/Co3O4–CoO 3DFs.
2.4 Electrochemical performances of hierarchically nanoporous Co/Co3O4–CoO 3DFs
The electrochemical performances of hierarchically nanoporous Co/Co3O4–CoO 3DFs were studied in detail by using cyclic voltammograms (CV) and galvanostatic charging/discharging (GCD) techniques. Fig. 5a–c show the CV curves of the two nanoporous Co/Co3O4–CoO 3DFs in 6 M NaOH electrolyte at three different scan rate (2 mV s−1, 10 mV s−1 and 50 mV s−1) within the potential ranging from −0.1 to 0.5 V. Their loading mass of the active materials were 1.39 mg cm−2 (C1) and 1.42 mg cm−2 (C2). Two redox peaks at about 0.13 V and 0.29 V (vs. SCE) are clearly observed for the two nanoporous Co/Co3O4–CoO 3DFs electrodes, which correspond to the reversible reactions of Co3+/Co4+ and Co2+/Co4+ associated with anions OH−. As the increase of scan rate, the electrochemical response current increases. It is also found (Fig. 4b–c) that the shapes of CV curves nearly keep same except that the peak positions are slightly shifted, indicating a good electrochemical reversibility of these nanoporous Co/Co3O4–CoO 3DFs electrodes.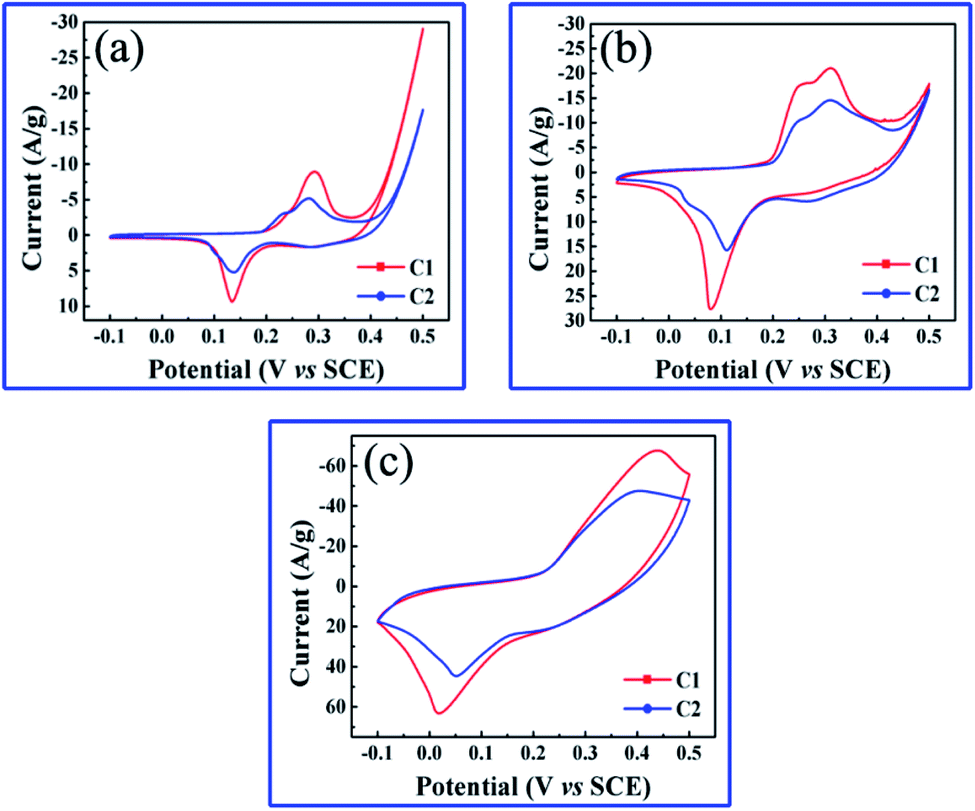 | ||
| Fig. 5 (a–c) CVs of nanoporous Co/Co3O4–CoO 3DFs electrodes at scan rate of 2 mV s−1, 10 mV s−1 and 50 mV s−1. | ||
Fig. 6a–c shows the galvanostatic charging/discharging (GCD) curves of the two nanoporous Co/Co3O4–CoO 3DFs electrodes measured at different discharge current densities of 0.2 A g−1, 1 A g−1, and 2 A g−1 within 0.35 V electrochemical voltage window. Fig. 6d displays the specific capacitances calculated from the GCD curves as a function of discharge current. The specific capacitances of the hierarchically nanoporous Co/Co3O4–CoO 3DFs electrodes (the red line in Fig. 6d) are 1083.6 F g−1, 965.1 F g−1, 902.3 F g−1 and 760 F g−1 at the discharge current densities of 0.2 A g−1, 1 A g−1, 2 A g−1 and 10 A g−1, respectively. The specific capacitance value of 965.1 F g−1 at 1 A g−1 discharge current density (the condition is widely used in many reports) is higher than the reported values, as shown in Table S1.†18,51,67–78 In addition, the specific capacitance value of 760 F g−1 at the discharge current density of 10 A g−1 shows a capacitance retention of 78.7% in comparison with that of 2 A g−1, demonstrating a stable current density. The specific capacitances of the uniformly nanoporous Co–Co3O4–CoO 3DFs electrodes (the blue line in Fig. 6d) are 799.1 F g−1, 753.7 F g−1, 721.7 F g−1 and 525.7 F g−1 at the discharge current densities of 0.2 A g−1, 1 A g−1, 2 A g−1 and 10 A g−1, respectively, which are all smaller than the corresponding capacitances of the hierarchically nanoporous Co/Co3O4–CoO 3DFs electrodes. The specific capacitance value of 525.7 F g−1 at the discharge current density of 10 A g−1 shows a capacitance retention of 69.7% in comparison with that of 2 A g−1. This capacitance retention value is smaller than that of hierarchically nanoporous (78.7%), indicating that the hierarchically nanoporous structure can improve the electrode current density stability in comparison with uniformly nanoporous structure, although they are made of the same Co/Co3O4–CoO 3DFs materials.
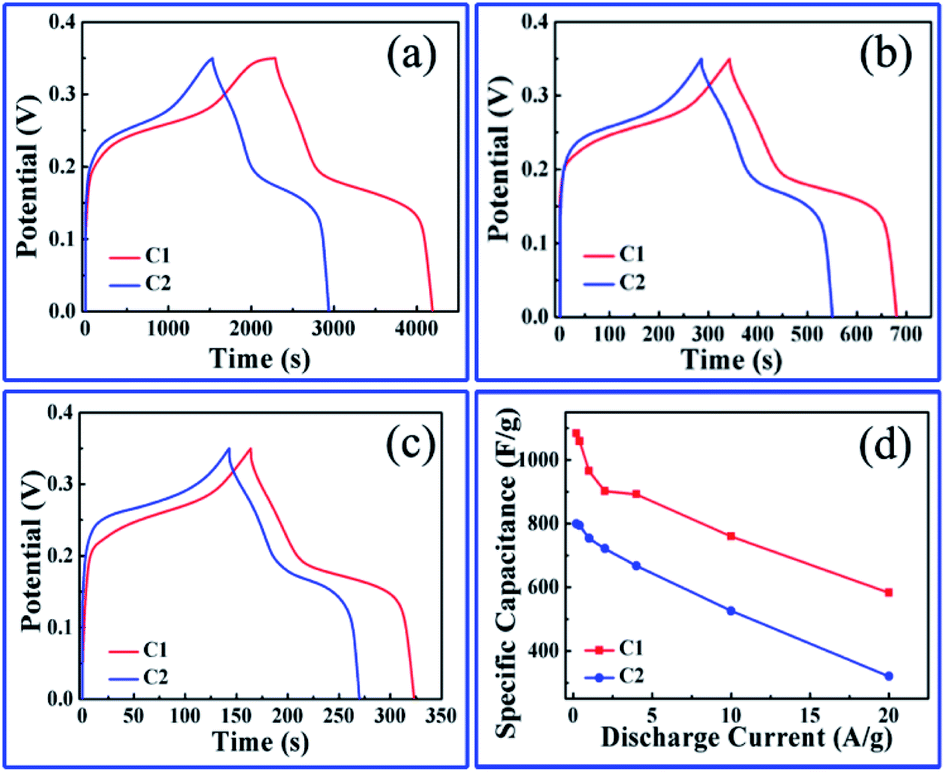 | ||
| Fig. 6 (a–c) GCD curves of nanoporous Co/Co3O4–CoO 3DFs electrodes at different current densities of 0.2 A g−1, 1 A g−1 and 2 A g−1. (d) Specific capacitance vs. discharge current for the above two samples calculated from their corresponding GCDs. | ||
The above electrochemical performance tests prove that the hierarchically nanoporous Co/Co3O4–CoO 3DFs could be a promising high-performance electrode material for electrochemical energy storage. The structure–function relationship between mcrostructure and electrochemical performance should be then analyzed comprehensively. Moreover, the hierarchically nanoporous Co/Co3O4–CoO 3DFs show similar 3D architecture and phase composition with these of uniformly nanoporous Co/Co3O4–CoO 3DFs, but differ in BET surface area and pore size distribution. It is interesting to note that, although the hierarchically nanoporous Co/Co3O4–CoO 3DFs have smaller BET surface area (83.63 m2 g−1, Fig. S6a†) thus provide less efficient contacts between electrode materials and electrolyte ions than that of uniformly nanoporous Co–Co3O4–CoO 3DFs (92.60 m2 g−1, Fig. S6c†), they show an improved electrochemical performance. This is consistent with previous reports that a higher BET surface area is not always benefit to electrochemical performance. Therefore, the improved electrochemical performance of hierarchically nanoporous Co/Co3O4–CoO 3DFs is deduced to result from the bimodal pore size distribution.
To explore the role of different sized pore in redox reactions, EIS measurement79,80 were performed on hierarchically and uniformly nanoporous Co/Co3O4–CoO 3DFs, which could provide us information about redox reaction (electron transportation and ion diffusion). Moreover, the hierarchically nanoporous Co/Co3O4–CoO 3DFs show similar pore size distribution around 4.3 nm to that of uniformly nanoporous Co/Co3O4–CoO 3DFs, but differ from 26 nm. The two-level sizes nanopores of the hierarchically nanoporous Co/Co3O4–CoO 3DFs electrodes should perform different roles during the redox reactions. The typical Nyquist plots (Fig. 7a) of the electrodes show that charge transfer resistance (Rct) of the hierarchically nanoporous Co/Co3O4–CoO 3DFs electrodes (0.13 Ω) is smaller than that of the uniformly nanoporous Co/Co3O4–CoO 3DFs electrodes (0.25 Ω), which confirms that the hierarchically nanoporous Co/Co3O4–CoO 3DFs electrodes have a faster electron transport. To calculate the ions diffusion coefficient D, the resistance R0, the point at which the straight extension line intersects the real axis is equal to R1 + Rct − σ2/Rct (where R1 is the solution resistance and σ is the Warburg coefficient), were tested as 1.78 Ω of hierarchically nanoporous 3DFs and 2.04 Ω of uniformly nanoporous 3DFs. Meanwhile, the σ can be described as: σ = RT/[Z2F2AC*(2D)1/2] (where R is the gas constant (8.31 J (K−1 mol−1)), T is the absolute temperature, Z is the number of transferred electron charges per diffusing species, F is Faradays constant (96500 C mol−1), A is the surface area of electrode, C* is the bulk concentration of the diffusing species).61 Therefore, the ions diffusion coefficient D can be derived as:
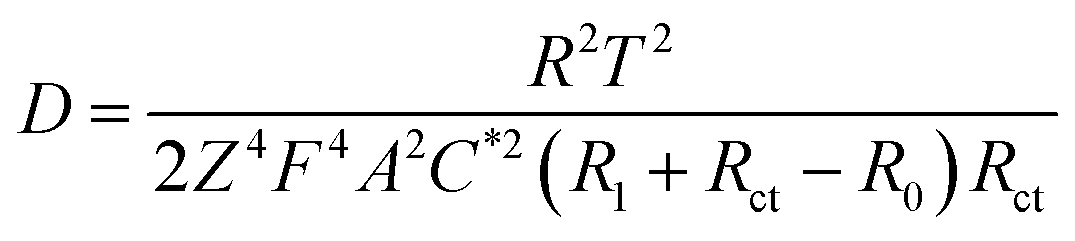
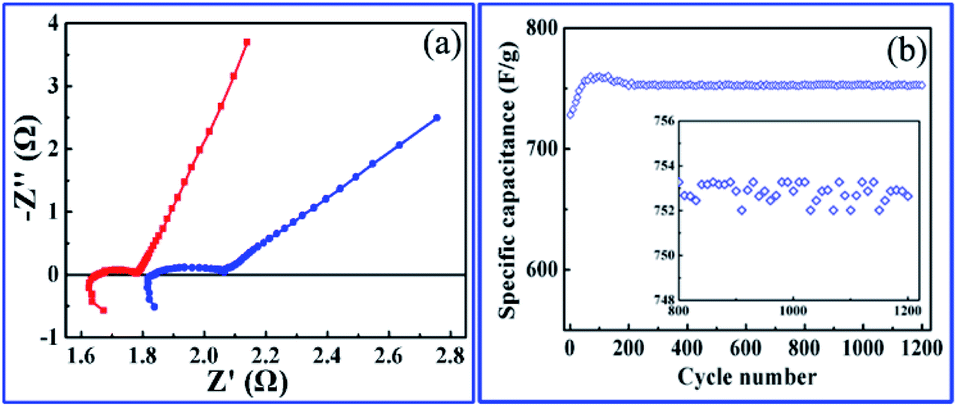 | ||
| Fig. 7 (a) Nyquist plots, red curve for the hierarchically nanoporous Co/Co3O4–CoO 3DFs and blue for the uniformly nanoporous 3DFs. (b) The cycling performance of the hierarchically nanoporous Co/Co3O4–CoO 3DFs (C1) electrode with scan rate of 20 mV s−1. | ||
Accordingly, the ion diffusion coefficient D of hierarchically nanoporous 3DFs is 6.86 × 10−8 cm2 s−1 and that of uniformly nanoporous 3DFs is 0.53 × 10−8 cm2 s−1, revealing a faster ion diffusion.
Long cycle life performance is another critical parameter for electrochemical energy storage, which determines their practical applications. To examine the long-term cyclability of the hierarchically nanoporous Co/Co3O4–CoO 3DFs, charge/discharge cycling test with a scan rate of 20 mV s−1 was performed (Fig. 7b). During the first 70 cycles, the specific capacitance of the hierarchically nanoporous Co/Co3O4–CoO 3DFs electrode gradually increased from 728.2 F g−1 to 760.4 F g−1, which may be attributed to the gradual activation process of the electroactive Co/Co3O4–CoO 3DFs surface. As cycle numbers increase even more than 1200, the specific capacitance only shows a ∼1% negligible decay, which demonstrates a remarkable electrochemical stability of Co/Co3O4–CoO 3DFs.
3. Conclusion
In conclusion, we present a controllable synthesis of a novel architecture, hierarchically nanoporous Co/Co3O4–CoO 3DFs by straightforward annealing Co(OH)2 3DFs in Ar atmosphere, and the synthesized 3DFs show remarkable electrochemical properties for energy storage. Individual hierarchically nanoporous Co/Co3O4–CoO 3DF is consisted of hierarchically nanoporous Co3O4–CoO petal-shaped nanosheets (thickness within 10 nm) skeleton with bimodal pore size distribution and the metallic Co nanoparticles are directly attach to the edge of large sized nanopores. The hierarchically nanoporous Co/Co3O4–CoO 3DFs shows a high specific capacitance (902.3 F g−1 at current density of 2 A g−1) with good cyclability. The high capacitor performance is resulted from the novel nanostructure and hierarchically nanopores, which offer more electroactive sites for efficient redox reactions and facilitate electron transport and ion diffusion. The improved capacitance mainly originates from the novel nanostructures and the hierarchical nanopores coordinating role. Our work should provide a smart way for synthesize functional hierarchically nanoporous 3D architecture, which can be used as electrochemical electrodes for improved energy storage capacity.4. Experimental section
4.1 Synthesis of Co(OH)2 3DFs
10 ml of ethanolamine was added to a 1 L of 0.1 M Co(CH3COO)2·4H2O solution in a glass vial. The uniformity blue-green mixture was kept at room temperature for 24 h. After complete reaction, the pink precipitates (sample CH1) were generated at the bottom of the glass vial and the color of the upper solution changed to pink. The precipitates were collected and washed several times with water and ethanol. Then the ethanolamine was changed to 20 ml (sample CH2), the other steps followed as upon, and another three samples were prepared.4.2 Synthesis of nanoporous Co/Co3O4–CoO 3DFs
Co/Co3O4–CoO 3DFs were synthesized by straightforward annealing the four Co(OH)2 3DFs at 300 °C for 4 h under a Ar flow, where the heating rate was 4 °C min−1 and the Ar flow rate was 20 ml min−1.4.3 Characterization
The morphological, crystal structural and chemical characterization of the Co/Co3O4–CoO 3DFs were analyzed at the nanoscale using a field emission scanning electron microscope (FESEM, Hitachi S-4800, Japan), spherical aberration correction electron microscope (Cs-STEM, Titan Cubed Themis G2 300, FEI), high angle annular dark field and high resolution scanning transmission electron microscope (HAADF-HRSTEM), selected area electron diffraction (SAED), EDX mapping and X-ray diffraction (XRD) instrument using Cu Kα radiation (λ = 1.5418 Å) (X'pert powder, Philips). The chemical composition and atomic bonding states were analyzed by X-ray photoelectron spectroscopy (XPS, Kratos Axis Ultra DLD, Japan). The Nitrogen adsorption–desorption isothermals were measured at 77 K using Brunauer–Emmett–Teller (BET, ASAP2020, Micromeritics, USA).4.4 Electrochemical tests
The electrochemical studies were tested in a three-electrode system with a 6 M NaOH electrolyte solution at room temperature, where a platinum electrode was used as counter electrode, a saturated calomel electrode as reference electrode and the nanoporous Co/Co3O4–CoO 3DFs (80 wt%) compounded with acetylene black (15 wt%) and polyvinylidene fluoride (5 wt%) then pasted on to a piece of nickel foam (1 × 1 cm2) as working electrode.33 The cyclic voltammogram (CV), galvanostatic chaging/discharging (GCD) and electrochemical impedance spectroscopy (EIS) were measured with a RST5200 electrochemical workstation (Zhengzhou Shiruisi Instrument Technology Co., Ltd. China), and the window voltage was set between −0.1 and 0.5 V at scan rates of 1, 2, 5, 10, 20, 50 and 100 mV s−1, respectively.The specific capacitance (Cs) of the nanoporous Co/Co3O4–CoO 3DFs electrode could be calculated from the GCD curves with the following equations:
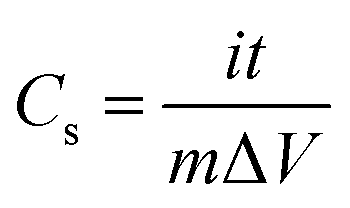
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (51771085, 51571104, 51801087, and 91962212).References
- X. Q. Huang, Z. P. Zhao, Y. Chen, C. Chiu, L. Y. Ruan, Y. Liu, M. F. Li, X. F. Duan and Y. Huang, Nano Lett., 2014, 14, 3887–3894 CrossRef CAS.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981–984 CrossRef CAS.
- J. Oliver-Meseguer, J. R. Cabrero-Antonino, I. Dominguez, A. Leyva-Perez and A. Corma, Science, 2012, 338, 1452–1455 CrossRef CAS.
- Z. L. Wang, J. Phys. Chem. B, 2000, 104, 1153–1175 CrossRef CAS.
- N. Tian, Z. Y. Zhou and S. G. Sun, J. Phys. Chem. C, 2008, 112, 19801–19817 CrossRef CAS.
- Y. H. Xiao, S. J. Liu, F. Li, A. Q. Zhang, J. H. Zhao, S. M. Fang and D. Z. Jia, Adv. Funct. Mater., 2012, 22, 4052–4059 CrossRef CAS.
- M. S. Qin, W. H. Ren, J. S. Meng, X. P. Wang, X. H. Yao, Y. J. Ke and L. Q. Mai, ACS Sustainable Chem. Eng., 2019, 7, 11564–11570 CrossRef CAS.
- W. H. Ren, M. S. Qin, Z. X. Zhu, M. Y. Yan, Q. Li, L. Zhang, D. N. Liu and L. Q. Mai, Nano Lett., 2017, 17, 4713–4718 CrossRef CAS.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS.
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS.
- A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef CAS.
- P. J. Hall, M. Mirzaeian, S. I. Fletcher, F. B. Sillars, A. J. R. Rennie, G. O. Shitta-Bey, G. Wilson, A. Cruden and R. Carter, Energy Environ. Sci., 2010, 3, 1238–1251 RSC.
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS.
- D. R. Rolison, J. W. Long, J. C. Lytle, A. E. Fischer, C. P. Rhodes, T. M. McEvoy, M. E. Bourg and A. M. Lubers, Chem. Soc. Rev., 2009, 38, 226–252 RSC.
- S. Kim, J. S. Lee, H. J. Ahn, H. K. Song and J. H. Jang, ACS Appl. Mater. Interfaces, 2013, 5, 1596–1603 CrossRef CAS.
- G. H. Yu, L. B. Hu, N. A. Liu, H. L. Wang, M. Vosgueritchian, Y. Yang, Y. Cui and Z. A. Bao, Nano Lett., 2011, 11, 4438–4442 CrossRef CAS.
- R. B. Rakhi, W. Chen, D. Cha and H. N. Alshareef, Nano Lett., 2012, 12, 2559–2567 CrossRef CAS.
- S. K. Meher and G. R. Rao, J. Phys. Chem. C, 2011, 115, 15646–15654 CrossRef CAS.
- L. N. Gao, X. F. Wang, Z. Xie, W. F. Song, L. J. Wang, X. Wu, F. Y. Qu, D. Chen and G. Z. Shen, J. Mater. Chem. A, 2013, 1, 7167–7173 RSC.
- J. Rajeswari, P. S. Kishore, B. Viswanathan and T. K. Varadarajan, Electrochemistry Communications, 2009, 11, 572–575 CrossRef CAS.
- T. Zhai, S. L. Xie, M. H. Yu, P. P. Fang, C. L. Liang, X. H. Lu and Y. X. Tong, Nano Energy, 2014, 8, 255–263 CrossRef CAS.
- D. W. Wang, Q. H. Wang and T. M. Wang, Nanotechnology, 2011, 22, 135604 CrossRef.
- Y. B. Zhang and Z. G. Guo, Chem. Commun., 2014, 50, 3443–3446 RSC.
- Y. Y. Wang, Y. Lei, J. Li, L. Gu, H. Y. Yuan and D. Xiao, ACS Appl. Mater. Interfaces, 2014, 6, 6739–6747 CrossRef CAS.
- A. Biswal, P. Panda, Z. T. Jiang, B. Tripathy and M. Minakshi, Nanoscale Adv., 2019, 1, 1880–1892 RSC.
- A. Biswal, P. K. Panda, A. N. Acharya, S. Mohapatra and N. Swain, ACS Omega, 2020, 5, 3405–3417 CrossRef CAS.
- C. Z. Yuan, L. Yang, L. Hou, L. Shen, F. Zhang, D. Li and X. Zhang, J. Mater. Chem., 2011, 21, 18183–18185 RSC.
- J. A. Xu, L. Gao, J. Y. Cao, W. C. Wang and Z. D. Chen, Electrochim. Acta, 2010, 56, 732–736 CrossRef CAS.
- X. H. Xia, J. Tu, Y. Mai, X. Wang, C. Gu and X. Zhao, J. Mater. Chem., 2011, 21, 9319–9325 RSC.
- Y. Q. Fan, H. Shao, J. Wang, L. Liu, J. Zhang and C. Cao, Chem. Commun., 2011, 47, 3469–3471 RSC.
- S. K. Meher and G. R. Rao, J. Phys. Chem. C, 2011, 115, 15646–15654 CrossRef CAS.
- K. M. Hercule, Q. L. Wei, A. M. Khan, Y. L. Zhao, X. C. Tian and L. Q. Mai, Nano Lett., 2013, 13, 5685–5691 CrossRef CAS.
- J. L. Zhang, J. C. Fu, J. W. Zhang, H. B. Ma, Y. M. He, F. S. Li, E. Q. Xie, D. S. Xue, H. L. Zhang and Y. Peng, Small, 2014, 10, 2618–2624 CrossRef CAS.
- C. H. Wu, Q. Shen, R. Mi, S. X. Deng, Y. Q. Shu, H. Wang, J. B. Liu and H. Yan, J. Meter. Chem. A, 2014, 2, 15987–15994 RSC.
- C. Zhou, Y. W. Zhang, Y. Y. Li and J. P. Liu, Nano Lett., 2013, 13, 2078–2085 CrossRef CAS.
- J. P. Liu, J. Jiang, C. W. Cheng, H. X. Li, J. X. Zhang, H. Gong and H. J. Fan, Adv. Mater., 2011, 23, 2076–2081 CrossRef CAS.
- W. J. Hao, S. M. Chen, Y. J. Cai, L. Zhang, Z. X. Li and S. J. Zhang, J. Mater. Chem. A, 2014, 2, 13801–13804 RSC.
- X. Wang, X. L. Wu, Y. G. Guo, Y. T. Zhong, X. Q. Cao, Y. Ma and J. N. Yao, Adv. Funct. Mater., 2010, 20, 1680–1686 CrossRef CAS.
- N. Venugopal, D. J. Lee, Y. J. Lee and Y. K. Sun, J. Mater. Chem. A, 2013, 1, 13164–13170 RSC.
- G. Huang, F. F. Zhang, X. C. Du, Y. L. Yin and L. M. Wang, ACS Nano, 2015, 9, 1592–1599 CrossRef CAS.
- J. W. Zhou, R. Li, X. X. Fan, Y. F. Chen, R. D. Han, W. Li, J. Zheng, B. Wang and X. G. Li, Energy Environ. Sci., 2014, 7, 2715–2724 RSC.
- S. Kondrat, C. R. Pérez, V. Presser, Y. Gogotsi and A. A. Kornyshev, Energy Environ. Sci., 2012, 5, 6474–6479 RSC.
- D. Jang, J. C. Idrobo, T. Laoui and R. Karnik, ACS Nano, 2017, 11, 10042–10052 CrossRef CAS.
- R. Tan, A. Q. Wang, R. Malpass-Evans, R. Williams, E. W. Zhao, T. Liu, C. C. Ye, X. Q. Zhou, B. P. Darwich, Z. Y. Fan, L. Turcani, E. Jackson, L. J. Chen, S. Y. Chong, T. Li, K. E. Jelfs, A. I. Cooper, N. P. Brandon, C. P. Grey, N. B. McKeown and Q. L. Song, Nat. Mater., 2020, 19, 195–202 CrossRef CAS.
- X. J. Wei, X. Q. Jiang, J. S. Wei and S. Y. Gao, Chem. Mater., 2016, 28, 445–458 CrossRef CAS.
- J. Ma, K. Li, Z. W. Li, Y. H. Qiu, W. Si, Y. Y. Ge, J. J. Sha, L. Liiu, X. Xie, H. Yi, Z. H. Ni, D. Y. Li and Y. F. Jie, J. Am. Chem. Soc., 2019, 141, 4264–4272 CrossRef CAS.
- K. Q. Qin, J. L. Kang, J. J. Li, E. J. Liu, C. S. Shi, Z. J. Zhang, X. X. Zhang and N. Q. Zhao, Nano Energy, 2016, 24, 158–164 CrossRef CAS.
- C. Prehal, C. Koczwara, H. Amenitsch, V. Presser and O. Paris, Nat. Commun., 2018, 9, 1–8 CrossRef CAS.
- C. Merlet, B. Rotenberg, P. A. Madden, P. L. Taberna, P. Simon, Y. Gogotsi and M. Salanne, Nat. Mater., 2012, 9, 306–310 CrossRef.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS.
- J. C. Deng, L. D. Kang, G. L. Bai, Y. Li, P. Y. Li, X. G. Liu, Y. Z. Yang, F. Gao and W. Liang, Electrochimica Acta, 2014, 132, 127–135 CrossRef CAS.
- Z. Song, Y. Zhang, W. Liu, S. Zhang, G. Liu, H. Chen and J. Qiu, Electrochimica Acta, 2013, 112, 120–126 CrossRef CAS.
- C. Guan, J. P. Liu, C. W. Cheng, H. X. Li, X. L. Li, W. W. Zhou, H. Zhang and H. J. Fan, Energy Environ. Sci., 2011, 4, 4496–4499 RSC.
- C. Guan, X. H. Xia, N. Meng, Z. Y. Zeng, X. H. Cao, C. Soci, H. Zhang and H. J. Fan, Energy Environ. Sci., 2012, 5, 9085–9090 RSC.
- H. Zhang, X. Yu and P. V. Braun, Nat. Nanotechnol., 2011, 6, 277–281 CrossRef CAS.
- C. H. Tang, X. S. Yin and H. Gong, ACS Appl. Mater. Interfaces, 2013, 5, 10574–10582 CrossRef CAS.
- L. L. Liu, H. P. Guo, Y. Y. Hou, J. Wang, L. J. Fu, J. Chen, H. K. Liu, J. Z. Wang and Y. P. Wu, J. Mater. Chem. A, 2017, 5, 14673–14681 RSC.
- Y. Li, C. X. Lu, S. C. Zhang, F. Y. Su, W. Z. Shen, P. C. Zhou and C. L. Ma, J. Mater. Chem. A, 2015, 3, 14817–14825 RSC.
- S. Shrestha and W. E. Mustain, J. Electrochem. Soc., 2010, 157, B1665–B1672 CrossRef CAS.
- W. Lu, R. Hartman, L. T. Qu and L. M. Dai, J. Phys. Chem. Lett., 2011, 2, 655–660 CrossRef CAS.
- V. F. Lvovich, Impedance Spectroscopy Applications to Electrochemical and Dielectric Phenomena, Published by John Wiley & Sons, Inc., Hoboken, New Jersey, 2012 Search PubMed.
- N. Q. Wu, L. Fu, M. Su, M. Aslam, K. C. Wong and V. P. Dravid, Nano Lett., 2004, 4, 383–386 CrossRef CAS.
- A. Y. Khodakkov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1774 CrossRef.
- S. L. Xiong, J. S. Chen, X. W. Lou and H. C. Zeng, Adv. Funct. Mater., 2012, 22, 861–871 CrossRef CAS.
- H. Tüysüz, Y. Liu, C. Weidenthaler and F. Schüth, J. Am. Chem. Soc., 2008, 130, 14108–14110 CrossRef.
- X. Deng, D. Z. Yang, G. G. Tan, X. H. Li, J. W. Zhang, Q. F. Liu, H. L. Zhang, N. J. Mellors, D. S. Xue and Y. Peng, Nanoscale, 2014, 6, 13710–13718 RSC.
- M. M. Liu, S. J. He and W. Chen, Nanoscale, 2014, 6, 11769–11776 RSC.
- X. H. Xia, J. P. Tu, Y. J. Mai, X. L. Wang, C. D. Gu and X. B. Zhao, J. Mater. Chem., 2011, 21, 9319–9325 RSC.
- X. H. Xia, J. P. Tu, X. L. Wang, C. D. Gu and X. B. Zhao, Chem. Commun., 2011, 47, 5786–5788 RSC.
- X. H. Xia, J. P. Du, Y. Q. Zhang, Y. J. Mai, X. L. Wang, C. D. Gu and X. B. Zhao, RSC Adv., 2012, 2, 1835–1841 RSC.
- C. Z. Yuan, L. Yang, L. R. Hou, J. Y. Li, Y. X. Sun, X. G. Zhang, L. F. Shen, X. J. Lu, S. L. Xiong and X. W. Lou, Adv. Funct. Mater., 2012, 22, 2560–2566 CrossRef CAS.
- F. Zhang, C. Z. Yuan, J. J. Zhu, J. Wang, X. G. Zhang and X. W. Lou, Adv. Funct. Mater., 2013, 23, 3909–3915 CrossRef CAS.
- F. L. Meng, Z. G. Fang, Z. X. Li, W. W. Xu, M. J. Wang, Y. P. Liu, J. Zhang, W. R. Wang, D. Y. Zhao and X. H. Guo, J. Mater. Chem. A, 2013, 1, 7235–7241 RSC.
- L. Cao, M. Lu and H. L. Li, J. Electrochem. Soc., 2005, 152, A871–A875 CrossRef CAS.
- C. Zheng, C. Cao, Z. Ali and J. Hou, J. Mater. Chem. A, 2014, 2, 16467–16473 RSC.
- S. Baskar, D. Meyrick, K. S. Ramakrishnan and M. Minakshi, Chemical Engineering Journal, 2014, 253, 502–507 CrossRef CAS.
- X. Liu, D. Ji, J. Li, L. Chen, D. Zhang, T. Liu, N. Zhang, R. Ma and G. Qiu, RSC Adv., 2015, 5, 41627–41630 RSC.
- M. J. Barmi and M. Minakshi, ChemPlusChem, 2016, 81, 964–977 CrossRef CAS.
- D. W. Wang, F. Liu, M. Liu, G. Lu and H. M. Cheng, Angew. Chem., Int. Ed. Engl., 2008, 47, 373–376 CrossRef CAS.
- G. J. Lee and S. I. Pyun, Langmuir, 2006, 22, 10659–10665 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08319d |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2020 |