
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZero-field slow relaxation of magnetization in cobalt(II) single-ion magnets: suppression of quantum tunneling of magnetization by tailoring the intermolecular magnetic coupling†
Ryoji Mitsuhashi *a,
Satoshi Hosoyab,
Takayoshi Suzukic,
Yukinari Sunatsukid,
Hiroshi Sakiyamae and
Masahiro Mikuriyab
*a,
Satoshi Hosoyab,
Takayoshi Suzukic,
Yukinari Sunatsukid,
Hiroshi Sakiyamae and
Masahiro Mikuriyab
aInstitute of Liberal Arts and Science, Kanazawa University, Kakuma, Kanazawa, Ishikawa 920-1192, Japan. E-mail: mitsuhashi@staff.kanazawa-u.ac.jp
bSchool of Science and Technology, Kwansei Gakuin University, 2-1 Gakuen, Sanda, Hyogo 669-1337, Japan
cResearch Institute for Interdisciplinary Science, Okayama University, 3-1-1 Tsushima-naka, Kita-ku, Okayama 700-8530, Japan
dDepartment of Chemistry, Faculty of Science, Okayama University, 3-1-1 Tsushima-naka, Kita-ku, Okayama 700-8530, Japan
eDepartment of Science, Faculty of Science, Yamagata University, 1-4-12 Kojirakawa, Yamagata 990-8560, Japan
First published on 9th December 2020
Abstract
The correlation between magnetic relaxation dynamics and the alignment of single-ion magnets (SIMs) in a crystal was investigated using four analogous cobalt(II) complexes with unique hydrogen-bond networks. The hydrogen-bonding interactions in the crystals resulted in a relatively short intermolecular Co⋯Co distance, which led to non-zero intermolecular magnetic coupling. All the complexes with a Co⋯Co distance shorter than 6.5 Å exhibited zero-field slow magnetic relaxation as weak magnetic interactions split the ground ±Ms levels and suppressed quantum tunneling of magnetization (QTM). In particular, antiferromagnetically coupled one-dimensional chain SIM networks effectively suppressed QTM when the two intrachain Co⋯Co distances were non-equivalent. However, when the two distances in a chain were equivalent and each molecular symmetry axis aligned parallell within the chain, QTM suppression was insufficient because magnetic coupling from the adjacent molecules was virtually cancelled. Partial substitution of the CoII ion with the diamagnetic ZnII ion up to 33% for this complex resulted in complete QTM suppression in the absence of an external field. These results show that the manipulation of intermolecular distances and alignments is effective for suppressing undesired QTM events in SIMs.
Introduction
Single-molecule magnets (SMMs) are individual molecules that display a bistable spin ground state.1–3 Since their first discovery, substantial effort has been devoted to this research field owing to the potential application in areas such as high-density information storage, spintronics, and quantum computing.4–6 The magnetic bistability of SMMs arises from the negative axial zero-field splitting (ZFS) and the spin reversal-barrier (U). The U can be described as |D|S2 for an integer spin system and |D|(S2 − 1/4) for a half-integer spin system, where D denotes the axial ZFS parameter and S is the spin of the ground state of the molecule. In the early days of SMM studies, many polynuclear clusters were synthesized and characterized to achieve a large U.1–3,7–11 However, this task appears to be difficult because the D value is inversely proportional to S.12Single-ion magnets (SIMs) are a subclass of SMMs with a single magnetic center. Since the first discovery of a lanthanide-based SIM,13 SIMs have taken the place of polynuclear cluster-based SMMs because of their simplicity and facile synthesis in controlling and enhancing the axial magnetic anisotropy. Mononuclear complexes with first-row transition metal are also good candidates for SIMs.14–28 Within the 3d SIMs, particular attention has been paid to the pseudo-tetrahedral cobalt(II) complexes because of their strong magnetic anisotropy. The strong magnetic anisotropy is originated from a second order spin–orbit coupling interaction between ground and excited electronic states that yields a large ZFS.25
Although a number of 3d SIMs with metals, such as Mn, Fe, Co, and Ni, have been reported, most of these do not show slow magnetic relaxation in the absence of an external direct current (dc) field because of fast relaxation via quantum tunneling of magnetization (QTM).29–32 Therefore, for future SMM applications, QTM suppression is one of the most important topics that needs to be addressed.
QTM arises from the mixing of ±Ms levels through hyperfine interactions, transverse ZFS or dipolar interactions.31 The Kramers theorem predicts that the mixing of the ground ±Ms levels by transverse ZFS for half-integer spin system is forbidden.33 To avoid QTM induced by dipolar interactions, SMMs have to be discrete and kept separated by a long distance in the crystal. By contrast, Christou and co-workers reported that a weak intermolecular magnetic coupling can suppress QTM in a hydrogen-bonded dimer of Mn4 clusters in the absence of a dc field.34 Such a weak coupling between SMMs can work as a bias to split the ground ±Ms levels. Since the first report of such an exchange-biased SMM, several supramolecular dimers, chains, and three-dimensional networks of weakly coupled SMMs connected by hydrogen bonds have been reported.35–39 However, most such supramolecular aggregates of SMMs have been unintentional discoveries. To control and design such a SMM aggregate by hydrogen bonds, a mononuclear SIM is more advantageous than with polynuclear clusters because of its simplicity.
With the above in mind, we recently reported on hydrogen-bonded supramolecular aggregates of pseudo-tetrahedral cobalt(II) SIMs with three bidentate ligands, [Co(Himl)2]·CH3OH (1·CH3OH), [Co(Himn)2] (2), and [Co(Hthp)2] (3), where Himl−, Himn−, and Hthp− are 2-(2-imidazolyl)phenolate, 2-(2-imidazolinyl)phenolate, and 2-(1,4,5,6-tetrahydropyrimidin-2-yl)phenolate, respectively (Scheme 1).40 It was indicated that one-dimensional (1-D) chain structures with intrachain Co⋯Co distances of 6.0–6.3 Å suppress the QTM of the SIMs by intermolecular magnetic coupling. However, the correlation between the SIM alignments in the chain and the quenching of QTM remained unclear. In this study, we prepared a new complex with a dimeric hydrogen-bond structure to elucidate the magnetic relaxation dynamics of the SIMs in supramolecular hydrogen-bonded structures. The magnetic relaxation dynamics in CoII SIMs with hydrogen-bond networks were considerably varied depending on the intrachain Co⋯Co distances. Furthermore, we succeeded in tailoring the magnetic coupling by a partial substitution of CoII ions with ZnII, which resulted in complete suppression of QTM in 3.
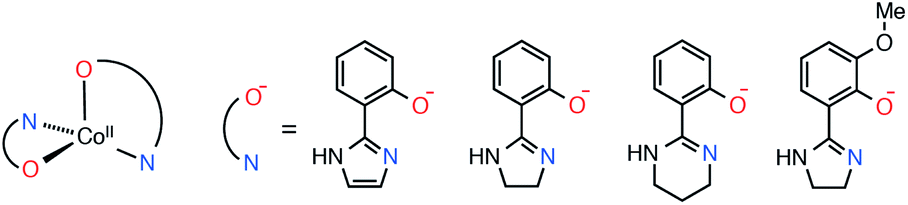 | ||
| Scheme 1 Chemical structures of the CoII complex and the ligands Himl−, Himn−, Hthp−, and Hmimn−. | ||
Results and discussion
Synthesis
The cobalt(II) complexes, 1·CH3OH, 2, and 3, were synthesized according to previously reported methods.40 An analogous complex, [Co(Hmimn)2]·CH3OH (4·CH3OH), was obtained as red crystals by a reaction of CoCl2·6H2O and the ligand in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio in methanol under an Ar atmosphere (Hmimn− = 2-(2-imidazolinyl)-6-methoxyphenolate).
:2 ratio in methanol under an Ar atmosphere (Hmimn− = 2-(2-imidazolinyl)-6-methoxyphenolate).
The isomorphous zinc(II) complexes for 1–3 were reported by He and by our group.41–43 The synthesis of the Hthp− analogue, [Zn(Hthp)2], using the BF4− salt resulted in precipitation because the formation of the product was too fast. Alternatively, the desired crystals were obtained from a reaction with ZnCl2, in which the coordination of Cl− ions may hinder the fast formation of [Zn(Hthp)2].
The ZnII-doped samples for 1–3 were prepared by mixing the chloride or tetrafluorobarate salt of CoII and ZnII ion in the starting material under an Ar atmosphere. The identity of the crystals was confirmed by powder X-ray diffraction (PXRD) measurements (Fig. S32–S34†). As the Co:Zn ratio (x = molar fraction of CoII ion) of mixed crystals may deviate from the expected value, the accurate molar fractions of the crystals were determined by inductively-coupled plasma atomic emission spectroscopy (ICP-AES). The composition of [CoxZn(1−x)(Himn)2] (2′x) was consistent with the expected value. The molar fractions of the CoII ion in the [CoxZn(1−x)(Himl)2]·CH3OH (1′x·CH3OH) compounds were smaller than expected, presumably because of the more facile formation of the Zn complex. By contrast, for [CoxZn(1−x)(Hthp)2] (3′x), the molar fraction of Co ion was higher than expected. Upon reaction of the metal salts mixture and the Hthp− ligand, a colorless precipitate of [Zn(Hthp)2] was immediately formed.43 Thirty minutes later, pale red crystals of 3′x started to form. As the colorless [Zn(Hthp)2] precipitate was removed upon purification, it resulted in a larger CoII ion composition than expected (xexpected = 0.49 and xactual = 0.67). An attempt to prepare 3′x with x < 0.5 was unsuccessful, and only a colorless precipitate was obtained as solid.
Crystal structures
Single-crystal X-ray analysis of the CoII complexes indicated the formation of bis-bidentate complexes with O–N chelate. The molecular structures and bonding parameters of the complexes are shown in Fig. 1 and Tables S1, S2.† In the CoII complexes with Himl− and Hmimn−, a methanol molecule of crystallization was contained in the crystal, whereas the others contained no solvent molecule. The bond distances around the CoII ion were comparable among the series. The bite angle in 3 was larger by ca. 3° than the others because of the steric restriction of the ligand.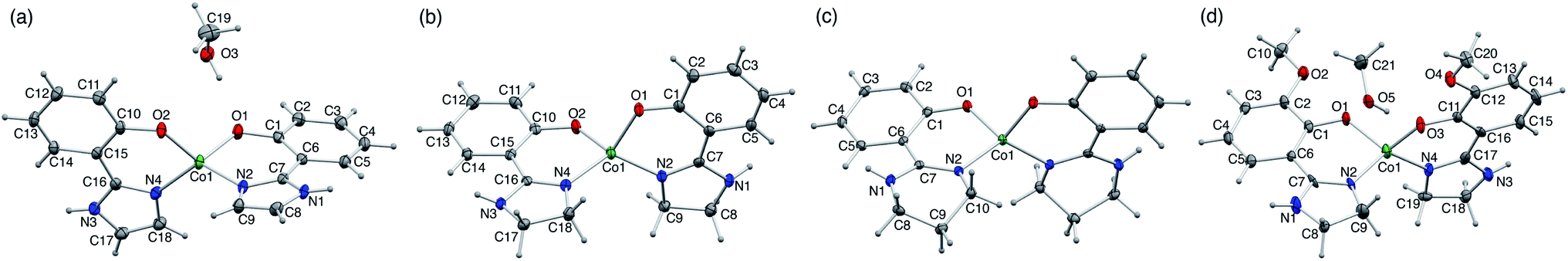 | ||
| Fig. 1 Molecular structures of (a) 1·CH3OH, (b) 2, (c) 3, (d) 4·CH3OH (50% probability levels). | ||
Owing to the non-coordinating N–H bond in the ligands, all the complexes formed supramolecular structures by intermolecular hydrogen bonds (Fig. 2). In the crystal, 1 formed hydrogen-bond networks via a methanol molecule of crystallization to construct a two-dimensional sheet structure. The shortest intrasheet Co⋯Co distance was 7.03 Å, and the intersheet distance was 7.51 Å.
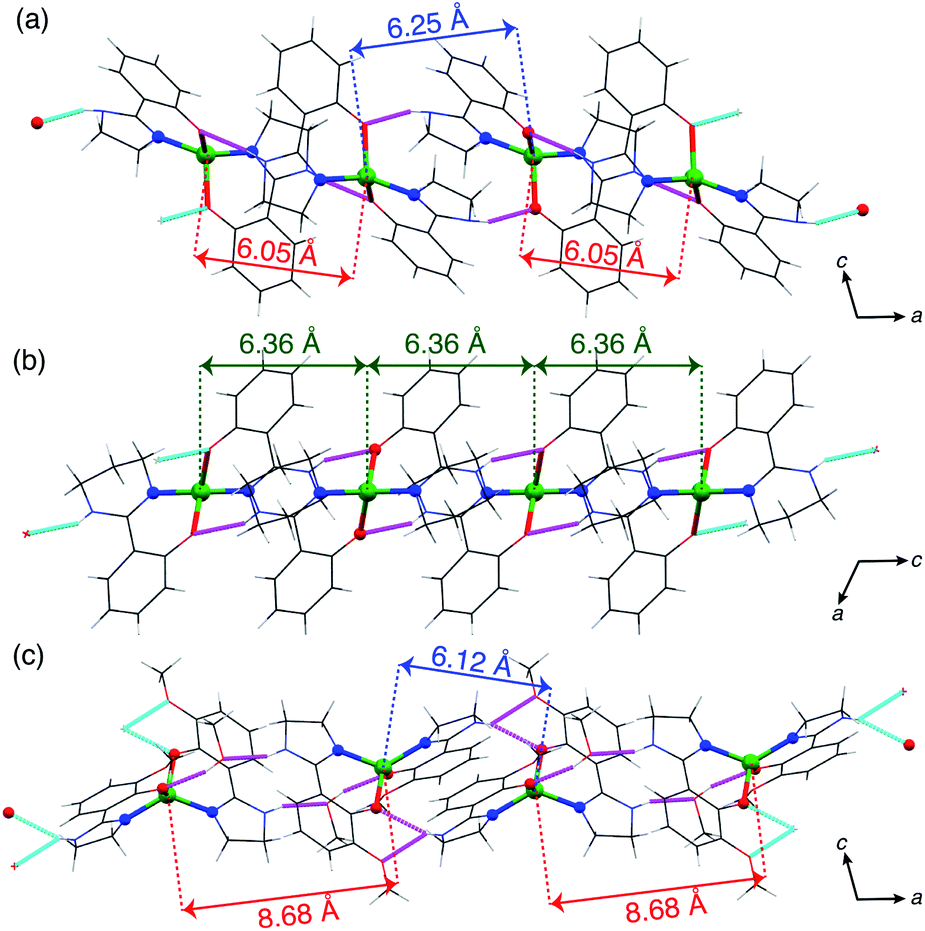 | ||
| Fig. 2 1-D hydrogen-bonded chain structures of (a) 2 along the a axis, (b) 3 along the c axis and (c) 4·CH3OH along the a axis. The magenta and cyan dashed lines indicate the hydrogen bonds. | ||
By contrast, the other complexes formed 1-D chain structures in the crystals. In the crystal of 2, the Co⋯Co distances between two adjacent molecules in the chain were 6.05 and 6.25 Å, which are significantly shorter than the Co⋯Co distances in 1·CH3OH. The shortest interchain distance of 7.67 Å was considerably longer than the intrachain distance, which suggests that the intermolecular magnetic coupling in 2 is dominated by the intrachain interactions.
In the case of 3, the intrachain distances were crystallographically equivalent, 6.36 Å, and the shortest interchain distance was 7.93 Å. As the interchain Co⋯Co distance is ca. 8 Å, the interchain magnetic interactions must be negligible, like in 2.
In 4·CH3OH, the two intrachain Co⋯Co distances were differed by over 2.5 Å (6.15 and 8.68 Å). In the chain, two discrete 4 molecules were directly connected by a double hydrogen bond to form a dimeric structure. The dimers were connected by hydrogen bonds via two methanol molecules of crystallization unlike in 2 and 3. The shortest interchain distance was 7.55 Å, also much longer than that in the dimer. Thus, the supramolecular structure in 4·CH3OH can be regarded as a dimer structure in terms of intermolecular magnetic interactions.
Static magnetic properties
The temperature and field dependent magnetic data of 4·CH3OH is depicted in Fig. 3. The features of χMT vs. T and M vs. H plots were similar to those of 1·CH3OH, 2 and 3 (Fig. S1†).40 The effective χMT product at room temperature were ca. 2.5 cm3 mol−1 K, which coincides to previously reported values for tetrahedral CoII complexes.44–47 The χMT product remained approximately constant upon lowering the temperature down to ca. 70 K, and it decreased abruptly below that, which is suggestive of non-zero ZFS. In the field-dependent measurements, the magnetization values did not saturate at 5 T and the isothermal magnetization curves at different temperatures did not coincide. To determine the g-factors and the axial ZFS parameter (D), the temperature dependence of the χMT data and the field dependence of the magnetization were simultaneously fitted to the following spin Hamiltonian:where D and zJ are axial ZFS parameter and the intermolecular magnetic interaction, respectively. The transversal ZFS parameter E was not considered in the spin Hamiltonian because it is difficult to determine only from magnetic data. The obtained parameters are summarized in Table 1. The data could not be fitted without considering the intermolecular interactions. For all the compounds, the determined zJ values for all compounds imply the existence of intermolecular antiferromagnetic interactions. Furthermore, negative D values were obtained although both negative and positive signs are possible for a pseudo-tetrahedral CoII complex.22 The large negative axial ZFS parameters are suggestive of SIM behavior with a spin-reversal barrier of ca. 60–80 cm−1 (U = 2|D|).
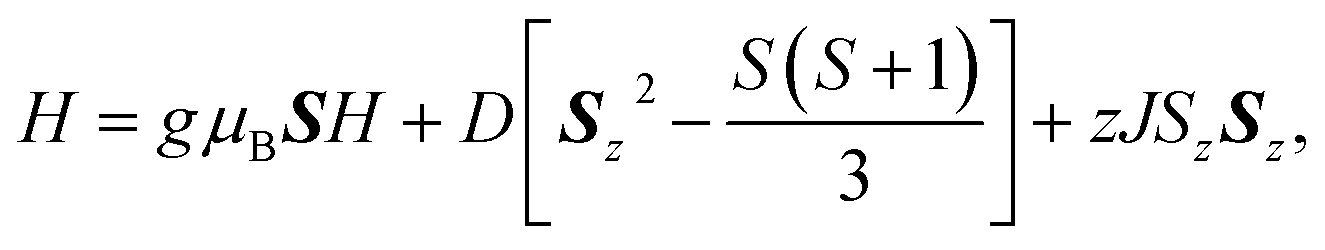
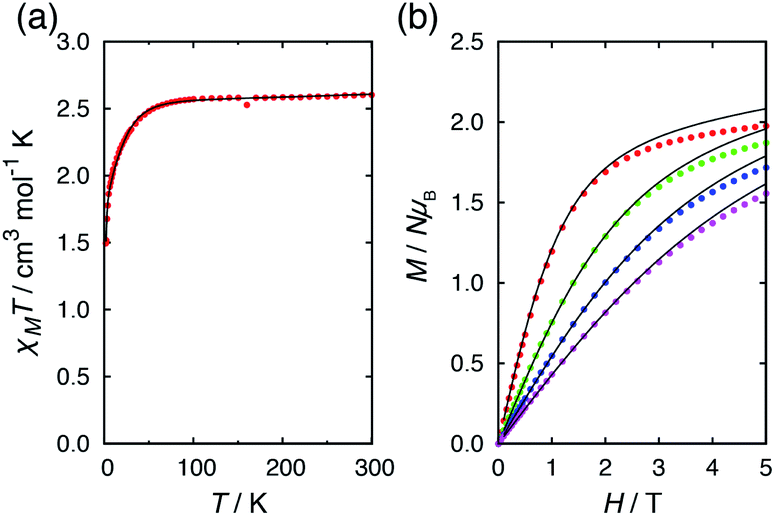 | ||
| Fig. 3 (a) Temperature dependence of the χMT product for 4·CH3OH in an applied field of 5.0 kOe, and (b) field dependence of the magnetization for 4·CH3OH at 2, 4, 6, and 8 K (red, green, blue, and magenta points, respectively). The solid lines correspond to the fit using the MagSakiTetra W0913 program.60 | ||
| 1·CH3OHb | 2b | 3b | 4·CH3OH | |
|---|---|---|---|---|
| a The temperature-independent paramagnetism for all fits was fixed at 0.0003 cm−1 mol−1.b The experimental parameters for 1·CH3OH, 2, and 3 were obtained by fitting the previously reported data with the latest fitting program MagSakiTetra W0913.40,60 | ||||
| gx, gy | 2.11(1) | 2.19(1) | 2.18(1) | 2.18(1) |
| gz | 2.48(1) | 2.52(1) | 2.48(1) | 2.55(1) |
| D/cm−1 | −41(1) | −32(1) | −30(4) | −30(1) |
| zJ/cm−1 | −0.15(3) | −0.19(1) | −1.5(3) | −0.25(8) |
Dynamic magnetic properties
Alternating current (ac) magnetic susceptibility measurements for 1·CH3OH, 2 and 3 have previously reported.40 1·CH3OH did not exhibit a slow magnetic relaxation behavior in the absence of an external field presumably due to the fast relaxation via QTM. In the presence of an external field, a slow relaxation behavior was observed in 1·CH3OH (Fig. S2†). The relaxation dynamics was reported to be predominated by the combination of a phonon-bottleneck-limited direct process (1.9–2.5 K) and a Raman process (4.0–6.5 K): τ−1 = ApbT2 + CTn (Apb = 1.96 s−1 K−2, C = 1.35 × 10−3 s−1 K−8.0 and n = 8.0).48,51 For a Kramers ion, the n value should be ≤9 when both an acoustic and an optical phonon are considered.52,53 The obtained n value is consistent with the expected value.In contrast to 1·CH3OH, compounds 2, 3, and 4·CH3OH, which form 1-D chain structures, exhibited slow relaxation of the magnetization in the absence of an external dc field (Fig. 4a, S4 and S7†). It should be noted that zero-field 3d SIMs are still rare. The τ vs. T plot of 2 in the absence of an external field was nicely overlapped with the one under 0.4 kOe.40 Both plots were well-fitted with the Raman process: τ−1 = CTn (C = 1.18 × 10−2 s−1 K−6.6, n = 6.6, Table S21†). Thus, in the relaxation dynamics of 2, the Raman relaxation process is predominant.
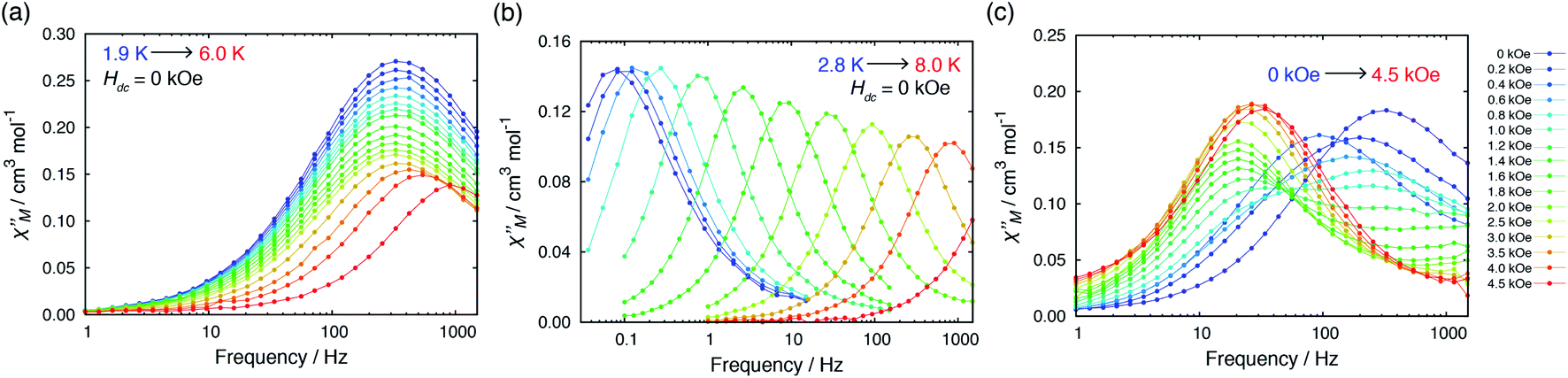 | ||
Fig. 4 Temperature dependence of out-of-phase 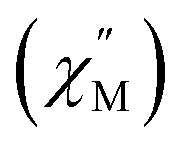 susceptibilities for (a) 4·CH3OH and (b) 3′0.67 in the absence of the dc field. (c) Dc field-dependence of susceptibilities for (a) 4·CH3OH and (b) 3′0.67 in the absence of the dc field. (c) Dc field-dependence of 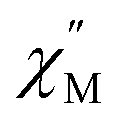 for 4·CH3OH with an dc field of 0–4.5 kOe. for 4·CH3OH with an dc field of 0–4.5 kOe. | ||
In 3, by contrast, the τ vs. T plot under 0 kOe was independent of temperature between 1.9 and 5 K, which is a characteristic feature of QTM (Fig. 5b).40 This temperature independent feature was completely removed upon application of an external field. The τ vs. T plot under 0.8 kOe fit well with the combination of two relaxation processes, Raman (≤4 K) and Orbach (>4 K) with τ−1 = CTn + τ0−1exp(−ΔOrbach/kBT) (C = 4.69 × 10−4 s−1 K−6.7, n = 6.7, τ0 = 4.77 × 10−10 s, ΔOrbach = 63.9 cm−1, Table S22†). The relaxation dynamics under zero field were reproduced by the combination of the QTM and with Orbach process with the same ΔOrbach and with a τ0 value (τ0 = 2.40 × 10−10 s) similar to that under 0.8 kOe. It should be noted that the obtained ΔOrbach value was consistent with the expected spin-reversal barrier for 3 (U = 2|D| = 60 cm−1). Thus, slow magnetic relaxation was observed for 3 under a zero dc field, although the relaxation via QTM remained at low temperature.
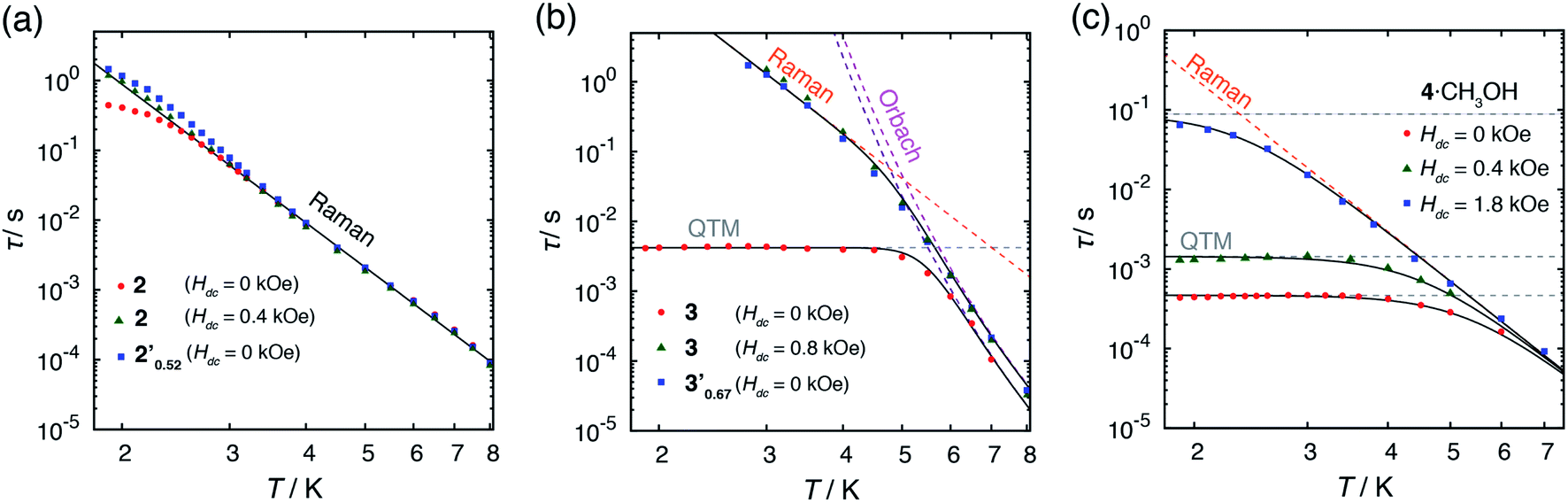 | ||
| Fig. 5 Temperature dependence of relaxation time τ. (a) 2 under fields of 0 and 0.4 kOe, and 2′0.52 under 0 kOe. (b) 3 under fields of 0 and 0.8 kOe, and 3′0.67 under 0 kOe. (c) 4·CH3OH under fields of 0, 0.4, and 1.8 kOe. The dashed lines indicate fitted lines for a single relaxation process of Raman τ−1 = CTn, Orbach τ−1 = τ0−1exp(−ΔOrbach/kBT) and QTM τ−1 = τQTM−1. The solid black lines indicate the sum of the relaxation processes. | ||
In the absence of a dc field, 4·CH3OH also exhibited slow magnetic relaxation (Fig. 4a and c). Upon application of a dc field, the relaxation pathway started to shift to a lower frequency, reaching minimum frequency at 0.4 kOe. With further increase of the applied field, another relaxation pathway appeared at a lower frequency, reaching minimum frequency at 1.8 kOe. The τ vs. T plots under 0, 0.4, and 1.8 kOe are shown in Fig. 5c. The measurements under these fields exhibited similar relaxation features: temperature independence, which is a characteristic of QTM at low temperature, and the power law of τ ∝ T−n at higher temperature. The relaxation dynamics under all the fields fit well with a combination of QTM and the Raman process with the same C and n values (τ−1 = τQTM−1 + CTn; C = 4.68 × 10−2 s−1 K−6.4, n = 6.4; τQTM = 4.67 × 10−4, 1.43 × 10−3, and 8.82 × 10−2 s for Hdc = 0, 0.4, and 1.8 kOe, respectively).
Magnetic relaxation dynamics and intermolecular interactions
As described above, in the absence of an external field, superparamagnet-like slow magnetic relaxation was observed in 2, 3, and 4·CH3OH. One of the possible reasons for the zero-field SIM behavior in these complexes is the short intermolecular Co⋯Co distance. In some hydrogen-bonded structures of SMM, QTM is suppressed because a static exchange bias field splits the ground ±Ms levels.34–39 The shortest Co⋯Co distance in 2, 3, and 4·CH3OH was 6.0–6.4 Å, whereas the distance in 1·CH3OH was ≥7 Å. In general, the strength of a magnetic coupling depends on the distance between the magnetic centers. To suppress QTM by a dipolar field, a weak but a certain degree of intermolecular magnetic interaction is necessary. Hence, the individual dipolar field from the neighboring SIMs might be too weak in 1·CH3OH.40QTM at ≥2.5 K was completely suppressed in 2, but a significant QTM contribution was observed in the relaxation dynamics of 3. Unlike the other complexes, 3 molecule is C2 symmetric in the crystal. In the hydrogen-bonded chain, each molecule is equally spaced along the c-axis and the molecular C2 axis oriented along the b-axis. As each molecular symmetry axis is parallelly aligned in a chain, the dipolar field and magnetic anisotropy axis can be mutually oriented and the bias field from adjacent molecule in the chain is canceled. Consequently, the weakly coupled 1-D networks in 3 suppress QTM only partially. By contrast, in the 1-D networks of 2, the dipolar field from the adjacent molecule is not canceled because the magnetic anisotropy axis is not mutually oriented and the two intrachain Co⋯Co distances differ by ca. 0.2 Å. Thus, QTM in 2 is completely suppressed even in the absence of an external field.
Considering the intrachain Co⋯Co distances, the intermolecular magnetic interaction in 4·CH3OH should be originated from the dimeric structure. In the magnetic relaxation dynamics, two relaxation pathways were observed. At Hdc = 0 kOe, each molecule of 4 was under the magnetic perturbation (±Hdip) of the other molecule in the dimer. This Hdip splits the ±Ms levels that results in partial QTM suppression. In the presence of a dc field, this Hdip is stochastically directed to be either parallel or antiparallel to the field (H = Hdc + Hdip or Hdc − Hdip). When Hdc = Hdip, Hdip in the parallel state suppresses QTM by magnetic perturbation, whereas Hdip in the antiparallel state is virtually canceled by Hdc, which results in the occurrence of QTM. Under Hdc ≫ Hdip, QTM is suppressed irrespective of the direction of Hdc and Hdip. Consequently, the field dependence of the 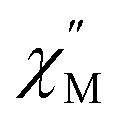 vs. frequency plot showed two peaks when Hdc was between 0.4 and 1.8 kOe because the relaxation process of 4·CH3OH should be influenced by both parallel and antiparallel states (Fig. 4c).
vs. frequency plot showed two peaks when Hdc was between 0.4 and 1.8 kOe because the relaxation process of 4·CH3OH should be influenced by both parallel and antiparallel states (Fig. 4c).
Magnetic relaxation dynamics in magnetically diluted crystals
As discussed above, it is suggested that a weak magnetic coupling between SIMs induces a zero-field slow relaxation of the magnetization. To elucidate the effect of the intermolecular coupling in more detail, we conducted partial magnetic dilution experiments. In a common magnetic dilution experiment, the concentration of a paramagnetic ion is diluted down to ca. 5% to remove the dipolar interaction.54–56 In our experiment, we first aimed to prepare the sample with a CoII ion concentration of ca. 50%. However, control of the molar fractions of the CoII ion in 1′x·CH3OH and 3′x was difficult, and 1′0.33·CH3OH and 3′0.67 were obtained from the 1:1 mixture of Co and Zn salts (see above).
Upon magnetic dilution of 1·CH3OH, frequency dependence of the 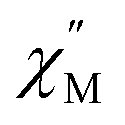 was observed under 0 kOe, and the peak of the
was observed under 0 kOe, and the peak of the 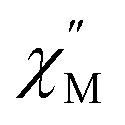 vs. frequency plots was shifted to a low frequency, although the temperature independence of the peak was still apparent (Fig. S19 and S22†). This suggests that the relaxation time was prolonged through reduced dipolar interactions by extension of the average Co⋯Co distances in 1′0.33·CH3OH and 1′0.12·CH3OH. However, temperature independence was observed for both 1′0.33·CH3OH and 1′0.12·CH3OH. As the relaxation time reached a plateau with the application of an external field, a significant contribution of QTM remained in 1′x·CH3OH (Fig. S21 and S24†). Thus, the partial substitution of the paramagnetic CoII center only reduced the influence of the weak dipolar interactions, and QTM was not completely suppressed. This suggests that a relatively short Co⋯Co distance is important for effective QTM suppression.
vs. frequency plots was shifted to a low frequency, although the temperature independence of the peak was still apparent (Fig. S19 and S22†). This suggests that the relaxation time was prolonged through reduced dipolar interactions by extension of the average Co⋯Co distances in 1′0.33·CH3OH and 1′0.12·CH3OH. However, temperature independence was observed for both 1′0.33·CH3OH and 1′0.12·CH3OH. As the relaxation time reached a plateau with the application of an external field, a significant contribution of QTM remained in 1′x·CH3OH (Fig. S21 and S24†). Thus, the partial substitution of the paramagnetic CoII center only reduced the influence of the weak dipolar interactions, and QTM was not completely suppressed. This suggests that a relatively short Co⋯Co distance is important for effective QTM suppression.
The τ vs. T plot of 2′0.52 measured under zero field overlapped well with that of 2 under both 0 and 0.4 kOe (Fig. 5a). Below 2.3 K, the value of τ in 2 under 0 kOe was slightly smaller than the expected value for the Raman process, but the τ vs. T plot of 2′0.52 under 0 kOe obeyed the model in the whole temperature range. Thus, the magnetic dilution down to ca. 50% in 2 afforded almost no change in the relaxation dynamics under zero field.
By contrast, the magnetic dilution of 3 drastically changed the magnetic relaxation dynamics (Fig. 4b). The temperature independence observed below 5 K in 3 disappeared completely in 3′0.67 under a zero dc field. The τ vs. T plot of 3′0.67 under 0 kOe perfectly overlaps with that of 3 under 0.8 kOe (Fig. 5b). Substitution of the CoII ion with a diamagnetic ZnII ion up to ca. 30% breaks the mutual orientation of the dipolar field and the magnetic anisotropy in the hydrogen-bonded chain. As a result, the dipolar field is not canceled within the chain, and QTM was effectively quenched. These results indicate that a relatively short distances between SIMs and a non-symmetric alignment of them are important for achieving a zero-field SIM behavior.
Conclusions
Static and dynamic magnetic properties of four structurally analogous bis-bidentate cobalt(II) complexes were analyzed. In all the complexes in which the intermolecular distances were less than 6.5 Å, QTM was either partially or completely suppressed because of intermolecular magnetic coupling in the absence of an external field. In 4·CH3OH, the hydrogen-bonded dimer structure of SIMs resulted in the partial suppression of QTM. Upon application of an external field, QTM was suppressed in two steps because the magnetic perturbation is canceled when the direction of the external field and the perturbation are antiparallel. QTM was suppressed more effectively in the 1-D chain network, as the lowest-energy state is the alternating alignment of ±Ms levels. The non-symmetric alignment of SIMs in the chain in 2 led to a complete suppression of QTM. However, the symmetric alignment of SIMs in 3 caused cancelation of the magnetic perturbation, which resulted in partial occurrence of the QTM event. A partial substitution of the CoII ion in 3 with the diamagnetic ZnII ion (3′0.67) resulted in complete suppression of QTM in the absence of an external field. This is a clear evidence that tailoring the intermolecular distances of SIMs and symmetry of the alignments can prevent the occurrence of QTM.Experimental
General considerations
All chemicals were used as purchased without further purification. The ligand precursors H2iml, H2imn and H2thp were prepared according to previously reported methods.57 1·CH3OH, 2 and 3 were prepared according to a previously reported method.40 The Co:Zn ratio of the magnetically diluted samples was determined by ICP-AES using a Shimadzu ICPE-9000. The 5 mg samples were digested in 60 μL of concentrated HNO3, and then diluted to 10 mL using ca. 1.2% HNO3 solution. The solution underwent a further 50 times dilution by ca. 1.2% HNO3 solution to obtain the desired sample solution. The standard solutions were prepared using Co(NO3)2·6H2O and Zn(NO3)2·6H2O.
Syntheses
X-ray crystallography
X-ray diffraction data were obtained at 90(2) K with a Bruker SMART APEX equipped with CCD detector with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). A single crystal was mounted with a glass capillary and flash-cooled with a cold N2 gas stream. Data were processed using the SMART software packages. Structures were solved using the intrinsic phasing methods employing the SHELXT software packages and refined on F2 (with all independent reflections).58,59 The non-hydrogen atoms were refined anisotropically. All H atoms at C–H bonds were located using a riding model, and H atoms at N–H bonds were located from the electron-density difference maps and refined isotropically.PXRD measurement
PXRD data were collected at room temperature on a Rigaku RINT 2100 powder diffractometer using Cu Kα radiation (λ = 1.5418 Å). The sample was ground in an agate mortar and placed on a silicon sample holder. The simulated diffraction pattern was calculated from the CIF using Mercury 3.8 software.Magnetic measurement
Magnetic susceptibility measurements were performed with a MPMS-XL7 or MPMS-7 SQUID magnetometer. Susceptibility data were obtained in the temperature range from 1.9 to 300 K with a static field of 0.5 T. The polycrystalline samples were ground into fine powders in an agate mortar. The samples were then loaded into a gelatin capsule and covered in liquid paraffin to prevent field-induced orientation of crystals. All data were corrected for the diamagnetism of the sample by means of Pascal's constants. The temperature and field-dependent magnetic data were fitted using the MagSakiTetra W0913 program.60 Dynamic susceptibility was measured with ac fields of 3 Oe magnitude and a constant dc field of 0–5000 Oe in the frequency range from 0.03 to 1500 Hz.Analysis of ac susceptibility measurements
The relaxation time τ was extracted from fitting to the following generalized Debye model:where χT and χS are the isothermal and adiabatic susceptibilities, ω is the angular frequency, and α indicates the deviation from a pure Debye model.49,50 The fit was performed using the CC-Fit program.61 The best fit parameters are summarized in Tables S7–S19.†
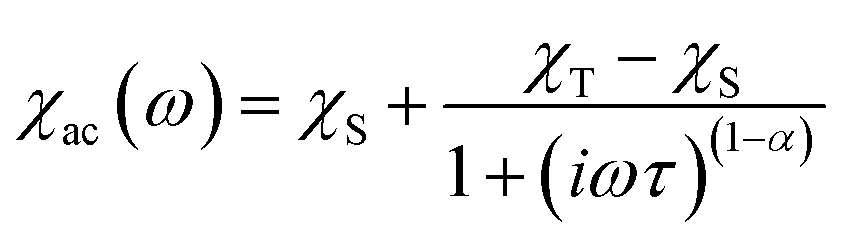
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Takahiro Iwai (Kwansei Gakuin University) for the help with the ICP-AES measurements. A part of the magnetic measurements was conducted at the Institute of Molecular Science, supported by the Nanotechnology Platform Program (Molecule and Material Synthesis). This work was supported by a Grant-in-Aid for Scientific Research No. 19K15525 from MEXT, Japan.Notes and references
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- H. J. Eppley, H.-L. Tsai, N. de Vries, K. Folting, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1995, 117, 301–317 CrossRef CAS.
- G. Arom, S. M. J. Aubin, M. A. Bolcar, G. Christou, H. J. Eppley, K. Folting, D. N. Hendrickson, J. C. Huffman, R. C. Squire, H.-L. Tsai, S. Wang and M. W. Wemple, Polyhedron, 1998, 17, 3005–3020 CrossRef.
- M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M.-A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Nat. Mater., 2009, 8, 194–197 CrossRef CAS.
- A. Ardavan, O. Rival, J. J. L. Morton, S. J. Blundell, A. M. Tyryshkin, G. A. Timco and R. E. P. Winpenny, Phys. Rev. Lett., 2007, 98, 057201 CrossRef.
- P. C. E. Stamp and A. Gaita-Ariño, J. Mater. Chem., 2009, 19, 1718–1730 RSC.
- A. L. Barra, A. Caneschi, A. Cornia, F. Fabrizi de Biani, D. Gatteschi, C. Sangregorio, R. Sessoli and L. Sorace, J. Am. Chem. Soc., 1999, 121, 5302–5310 CrossRef CAS.
- D. Gatteschi, R. Sessoli and A. Cornia, Chem. Commun., 2000, 725–732 RSC.
- H. Oshio, N. Hoshino and T. Ito, J. Am. Chem. Soc., 2000, 122, 12602–12603 CrossRef CAS.
- C. Boskovic, H. U. Güdel, G. Labat, A. Neels, W. Wernsdorfer, B. Moubaraki and K. S. Murray, Inorg. Chem., 2005, 44, 3181–3189 CrossRef CAS.
- T. Glaser, M. Heidemeier, T. Weyhermüller, R. Hoffmann, H. Rupp and P. Müller, Angew. Chem., Int. Ed., 2006, 45, 6033–6037 CrossRef CAS.
- O. Waldmann, Inorg. Chem., 2007, 46, 10035–10037 CrossRef CAS.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS.
- J. Vallejo, A. Pascual-Álvarez, J. Cano, I. Castro, M. Julve, F. Lloret, J. Krzystek, G. De Munno, D. Armentano, W. Wernsdorfer, R. Ruiz-García and E. Pardo, Angew. Chem., Int. Ed., 2013, 52, 14075–14079 CrossRef CAS.
- R. Ishikawa, R. Miyamoto, H. Nojiri, B. K. Breedlove and M. Yamashita, Inorg. Chem., 2013, 52, 8300–8302 CrossRef CAS.
- W. H. Harman, T. D. Harris, D. E. Freedman, H. Fong, A. Chang, J. D. Rinehart, A. Ozarowski, M. T. Sougrati, F. Grandjean, G. J. Long, J. R. Long and C. J. Chang, J. Am. Chem. Soc., 2010, 132, 18115–18126 CrossRef CAS.
- A. K. Bar, C. Pichon, N. Gogoi, C. Duhayon, S. Ramasesha and J.-P. Sutter, Chem. Commun., 2015, 51, 3616–3619 RSC.
- S. Mossin, B. L. Tran, D. Adhikari, M. Pink, F. W. Heinemann, J. Sutter, R. K. Szilagyi, K. Meyer and D. J. Mindiola, J. Am. Chem. Soc., 2012, 134, 13651–13661 CrossRef CAS.
- Y. Rechkemmer, F. D. Breitgoff, M. van der Meer, M. Atanasov, M. Hakl, M. Orlita, P. Neugebauer, F. Neese, B. Sarkar and J. van Slageren, Nat. Commun., 2016, 7, 10467 CrossRef CAS.
- V. V. Novikov, A. A. Pavlov, Y. V. Nelyubina, M.-E. Boulon, O. A. Varzatskii, Y. Z. Voloshin and R. E. P. Winpenny, J. Am. Chem. Soc., 2015, 137, 9792–9795 CrossRef CAS.
- J. M. Zadrozny and J. R. Long, J. Am. Chem. Soc., 2011, 133, 20732–20734 CrossRef CAS.
- S. Gomez-Coca, E. Cremades, N. Aliaga-Alcalde and E. Ruiz, J. Am. Chem. Soc., 2013, 135, 7010–7018 CrossRef CAS.
- Y.-Y. Zhu, C. Cui, Y.-Q. Zhang, J.-H. Jia, X. Guo, C. Gao, K. Qian, S.-D. Jiang, B.-W. Wang, Z.-M. Wang and S. Gao, Chem. Sci., 2013, 4, 1802–1806 RSC.
- M. S. Fataftah, J. M. Zadrozny, D. M. Rogers and D. E. Freedman, Inorg. Chem., 2014, 53, 10716–10721 CrossRef CAS.
- J. M. Frost, K. L. M. Harriman and M. Murugesu, Chem. Sci., 2016, 7, 2470–2491 RSC.
- T. J. Ozumerzifon, I. Bhowmick, W. C. Spaller, A. K. Rappé and M. P. Shores, Chem. Commun., 2017, 53, 4211–4214 RSC.
- Y. Peng, V. Mereacre, C. E. Anson, Y. Zhang, T. Bodenstein, K. Fink and A. K. Powell, Inorg. Chem., 2017, 56, 6056–6066 CrossRef CAS.
- R. Mitsuhashi, K. S. Pedersen, T. Ueda, T. Suzuki, J. Bendix and M. Mikuriya, Chem. Commun., 2018, 54, 8869–8872 RSC.
- P. Politi, A. Rettori, F. Hartmann-Boutron and J. Villain, Phys. Rev. Lett., 1995, 75, 537–540 CrossRef CAS.
- B. Barbara, W. Wernsdorfer, L. C. Sampaio, J. G. Park, C. Paulsen, M. A. Novak, R. Ferré, D. Mailly, R. Sessoli, A. Caneschi, K. Hasselbach, A. Benoit and L. Thomas, J. Magn. Magn. Mater., 1995, 140–144, 1825–1828 CrossRef CAS.
- D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268–297 CrossRef CAS.
- M. A. Sørensen, U. B. Hansen, M. Perfetti, K. S. Pedersen, E. Bartolomé, G. G. Simeoni, H. Mutka, S. Rols, M. Jeong, I. Zivkovic, M. Retuerto, A. Arauzo, J. Bartolomé, S. Piligkos, H. Weihe, L. H. Doerrer, J. van Slageren, H. M. Rønnow, K. Lefmann and J. Bendix, Nat. Commun., 2018, 9, 1292 CrossRef.
- H. A. Kramers, Proc. Amsterdam Akad., 1930, 33, 959–972 CAS.
- W. Wernsdorfer, N. Aliaga-Alcalde, D. N. Hendrickson and G. Christou, Nature, 2002, 416, 406–409 CrossRef.
- R. Tiron, W. Wernsdorfer, N. Aliaga-Alcalde and G. Christou, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 1–4 CrossRef.
- L. M. Wittick, K. S. Murray, B. Moubaraki, S. R. Batten, L. Spiccia and K. J. Berry, J. Chem. Soc., Dalton Trans., 2004, 4, 1003–1011 RSC.
- R. Bagai, W. Wernsdorfer, K. A. Abboud and G. Christou, J. Am. Chem. Soc., 2007, 129, 12918–12919 CrossRef CAS.
- R. Inglis, G. S. Papaefstathiou, W. Wernsdorfer and E. K. Brechin, Aust. J. Chem., 2009, 62, 1108–1118 CrossRef CAS.
- A. Das, K. Gieb, Y. Krupskaya, S. Demeshko, S. Dechert, R. Klingeler, V. Kataev, B. Büchner, P. Müller and F. Meyer, J. Am. Chem. Soc., 2011, 133, 3433–3443 CrossRef CAS.
- R. Mitsuhashi, S. Hosoya, T. Suzuki, Y. Sunatsuki, H. Sakiyama and M. Mikuriya, Dalton Trans., 2019, 48, 395–399 RSC.
- H.-S. He, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m3042–m3043 CrossRef CAS.
- R. Mitsuhashi and M. Mikuriya, X-Ray Struct. Anal. Online, 2019, 35, 15–16 CrossRef CAS.
- R. MitsuhashiI and M. Mikuriya, X-Ray Struct. Anal. Online, 2019, 35, 37–38 CrossRef.
- S. Sottini, G. Poneti, S. Ciattini, N. Levesanos, E. Ferentinos, J. Krzystek, L. Sorace and P. Kyritsis, Inorg. Chem., 2016, 55, 9537–9548 CrossRef CAS.
- S. Vaidya, S. K. Singh, P. Shukla, K. Ansari, G. Rajaraman and M. Shanmugam, Chem.–Eur. J., 2017, 23, 9546–9559 CrossRef CAS.
- R. C. Yang, D. R. Wang, J. L. Liu, Y. F. Wang, W. Q. Lin, J. D. Leng and A. J. Zhou, Chem.–Asian J., 2019, 14, 1467–1471 CrossRef CAS.
- T. Ishizaki, T. Fukuda, M. Akaki, A. Fuyuhiro, M. Hagiwara and N. Ishikawa, Inorg. Chem., 2019, 58, 5211–5220 CrossRef CAS.
- The relaxation time τ was extracted from the generalized Debye model fit of the ac susceptibility data at various temperatures.49,50.
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341–351 CrossRef CAS.
- Y.-N. Guo, G.-F. Xu, Y. Guo and J. Tang, Dalton Trans., 2011, 40, 9953–9963 RSC.
- R. Orbach, Proc. R. Soc. A, 1961, 264, 458–484 CAS.
- P. L. Scott and C. D. Jeffries, Phys. Rev., 1962, 127, 32–51 CrossRef CAS.
- K. N. Shrivastava, Phys. Status Solidi B, 1983, 117, 437–458 CrossRef CAS.
- J. M. Zadrozny, J. Telser and J. R. Long, Polyhedron, 2013, 64, 209–217 CrossRef CAS.
- S. Vaidya, S. Tewary, S. K. Singh, S. K. Langley, K. S. Murray, Y. Lan, W. Wernsdorfer, G. Rajaraman and M. Shanmugam, Inorg. Chem., 2016, 55, 9564–9578 CrossRef CAS.
- N. Ishikawa, M. Sugita and W. Wernsdorfer, Angew. Chem., Int. Ed., 2005, 44, 2931–2935 CrossRef CAS.
- R. Mitsuhashi, T. Suzuki and Y. Sunatsuki, Inorg. Chem., 2013, 52, 10183–10190 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- H. Sakiyama, J. Comput. Chem., 2016, 2, 1–4 Search PubMed.
- N. F. Chilton, CC-Fit, The University of Manchester, Manchester, U.K., 2014, http://www.nfchilton.com/cc-fit.html Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1986712. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra08286d |
| This journal is © The Royal Society of Chemistry 2020 |