
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDinuclear thiolato-bridged arene ruthenium complexes: from reaction conditions and mechanism to synthesis of new complexes†
Hedvika Primasová‡
 *,
Silviya Ninova‡,
Mario de Capitani,
Jana Daepp,
Ulrich Aschauer* and
Julien Furrer*
*,
Silviya Ninova‡,
Mario de Capitani,
Jana Daepp,
Ulrich Aschauer* and
Julien Furrer*
Department of Chemistry and Biochemistry, Universität Bern, Freiestrasse 3, CH-3012, Switzerland. E-mail: hedvika.primasova@dcb.unibe.ch; ulrich.aschauer@dcb.unibe.ch; julien.furrer@dcb.unibe.ch
First published on 4th November 2020
Abstract
Several dinuclear thiophenolato-bridged arene ruthenium complexes [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4-R)3]+ (R = H, NO2, F) could so far only be obtained in fair yields using the synthetic route established in the early 2000s. With much less reactive aliphatic thiols or with bulky thiols, the reactions become even less efficient and the desired complexes are obtained with low yields or not at all. We employed density functional theory (DFT) calculations to gain a fundamental understanding of the reaction mechanisms leading to the formation of dithiolato and trithiolato complexes starting from the dichloro(p-cymene)ruthenium(II) dimer [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2. The results of the DFT study enabled us to rationalise the experimental results and allowed us, via a modified synthetic route, to synthesise previously unreported and hitherto considered as unrealistic complexes. Our study opens up possibilities for the synthesis of so far inaccessible thiolato-bridged dinuclear arene ruthenium(II) complexes but more generally, also the synthesis of other thiolato-bridged dinuclear group 8 and 9 metal complexes could be reexamined.
1. Introduction
Dinuclear tris(thiolato)-bridged arene complexes are typically obtained from the reaction of the precursor [(arene)MCl(μ2-Cl)2M(arene)Cl] (M = Fe, Ru, Rh, Os, Ir) with thiolate compounds and represent an interesting class of organometallic compounds. Iron complexes serve as carbon–halogen bond activation reagents, and carbon–halogen bond-cleavage agents,1–4 while osmium,5 iridium, rhodium6–12 and especially ruthenium complexes13–25 have in vitro antiproliferative activity against cancer cell lines and several protozoan parasites. Tris(thiolato)-bridged dimolybdenum complexes are also readily available but are synthesised using other strategies such as the direct oxidation of low-valence carbonyl precursors or reductive processes from higher-valence derivatives,26–28 which will not be discussed here.It is interesting to note that arene ruthenium(II) complexes were first obtained fortuitously about fifty years ago by Winkhaus and Singer and subsequently Zelonka and Baird.29–32 Only years later, these dimeric arene–ruthenium dichloride complexes [(η6-arene)Ru(μ2-Cl)Cl]2 were found to react with thiols to give cationic trithiolato complexes of the type [(η6-arene)2Ru2(μ2-SR)3]+, the first examples being the hexamethylbenzene derivative [(η6-C6Me6)2Ru2(μ2-SPh)3]+ reported by Rakowski DuBois and coworkers33 and the p-cymene derivative [(η6-p-MeC6H4Pri)2Ru2(μ2-SPh)3]+ reported by Nakamura and coworkers, both of which contain three thiophenolato bridges.34 Over the last fifteen years, the series of dinuclear trithiolato bridged arene ruthenium complexes was extended, including complexes of the general formula [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4–R)3]+ and so-called mixed complexes of the general formula [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4–R1)2(μ2-SC6H4–R2)]+, bearing two types of thiol ligands.13–15,18–20,22–24,35–39 Like many organometallic compounds, these dinuclear thiolato-bridged arene ruthenium complexes have originally been designed as catalysts, for instance for the carbonylation of methanol.40 While they did not attract much attention for this application, a revival started in 2008, when water-soluble arene ruthenium complexes were discovered to be cytotoxic.14,40–44 Remarkably, almost all tested trithiolato compounds are highly cytotoxic with IC50 values being in the submicromolar range, the most potent ones with IC50 values of 30 nM against A2780 cells and cisplatin-resistant A2780cisR cells.13,19,21,22 Recent in vivo studies have demonstrated that these complexes indeed have potential as anticancer drugs, since for instance the compound [(η6-p-MeC6H4Pri)2Ru2(μ2-S-p-C6H4But)3]Cl (termed diruthenium-1) significantly prolongs the survival of tumour-bearing mice25 and substantially influences metabolism of A2780cisR cells involving changes related to redox homeostasis, Warburg effect and lipid metabolism.45 Other derivatives appear less promising.44,46 Dinuclear thiolato-bridged arene ruthenium complexes are also promising as antiprotozoal agents, with IC50 values of up to 1.2 nM against T. gondii, N. caninum and T. brucei and IC50 values against human foreskin fibroblasts >800 μM, leading to selectivity indexes >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.16,17,47
000.16,17,47
The current synthesis route for obtaining dinuclear cationic trithiolato bridged arene ruthenium complexes of the general formula [(η6-p-MeC6H4Pri)2Ru2(μ2-S–R)3]+ with good yields dates back to the early 2000s and involves the reaction of the dimer [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2 with an excess of the corresponding thiol (see Scheme 1) usually in refluxing ethanol (EtOH).37 For thiophenolato complexes, depending on the thiophenol used, it is possible to adjust the conditions to direct the synthesis exclusively to the cationic trithiophenolato complex [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4–R)3]+,15 the neutral dithiophenolato complex [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4–R)2Cl2],48 or even the neutral monothiophenolato complex [(η6-p-MeC6H4Pri)2Ru2Cl2(μ-Cl)(μ2-SC6H4–R)].49 The reactivity of the thiol undoubtedly plays an important role and decides to a great extent the outcome of the reaction. For instance, the trithiophenolato complexes [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4–R)3]+ with the electron attracting substituents R = NO2 or R = F could so far be only obtained with moderate yields (48 and 45%, respectively) using the standard strategy described in Scheme 1.19 Similarly, when much less reactive aliphatic thiols are used, the reaction becomes more demanding and the desired trithiolato complexes are either obtained with modest yields or the reactions only give the neutral dithiolato complex. For instance, the trithiolato complex [(η6-p-MeC6H4Pri)2Ru2(μ2-SC8H17)3]+ was only obtained with a yield of 28%, despite a long 7 day reaction in EtOH under inert atmosphere and reflux conditions,50 and the trithiolato complex [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H11)3]+ could not be obtained from the neutral dithiolato complex [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H11)2Cl2], presumably due to steric reasons.48
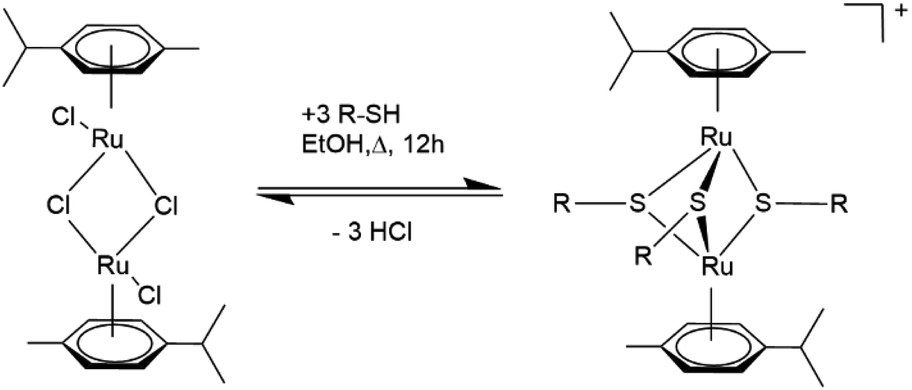 | ||
| Scheme 1 Synthesis of dinuclear cationic trithiolato-bridged arene ruthenium complexes.37 | ||
These experimental facts raise the question as to whether the formation of dinuclear trithiolato-bridged arene ruthenium complexes with different thiols is thermodynamically or kinetically hindered, which would give indications as to how conditions should be altered to enable reactions or to improve yields.
In the present work we aim at a fundamental understanding of the reaction mechanisms leading to the formation of trithiolato complexes starting from the dichloro(p-cymene)ruthenium(II) dimer [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2 and to modify, where necessary, the existing synthetic route to (i) increase the yields, (ii) reduce the overall reaction time (currently reflux in EtOH, 18 h), and to (iii) synthesise novel thiolato bridged complexes.
To this end, we employ density functional theory (DFT) calculations of the possible synthetic routes for trithiolato complexes [(η6-p-MeC6H4Pri)2Ru2(μ2-SR)3]+. The DFT results agree with new experimental results obtained for the trithiophenolato complexes [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4–R)3]+ with R = H (1), R = p-OMe (2), and R = p-NO2 (3), and the previously unreported trithiolato complexes [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H11)3]+ (4) and (5). Applying this approach further, we improved yield for (6) and were also able to synthesise two new dithiophenolato complexes (7) and (8) (Fig. 1), previously considered inaccessible, since only dithiobenzylato complexes could be obtained so far.18,46,48 These new dithiophenolato complexes intermediates could further be used for the synthesis of a novel mixed trithiolato complex (9) (see Fig. 1).
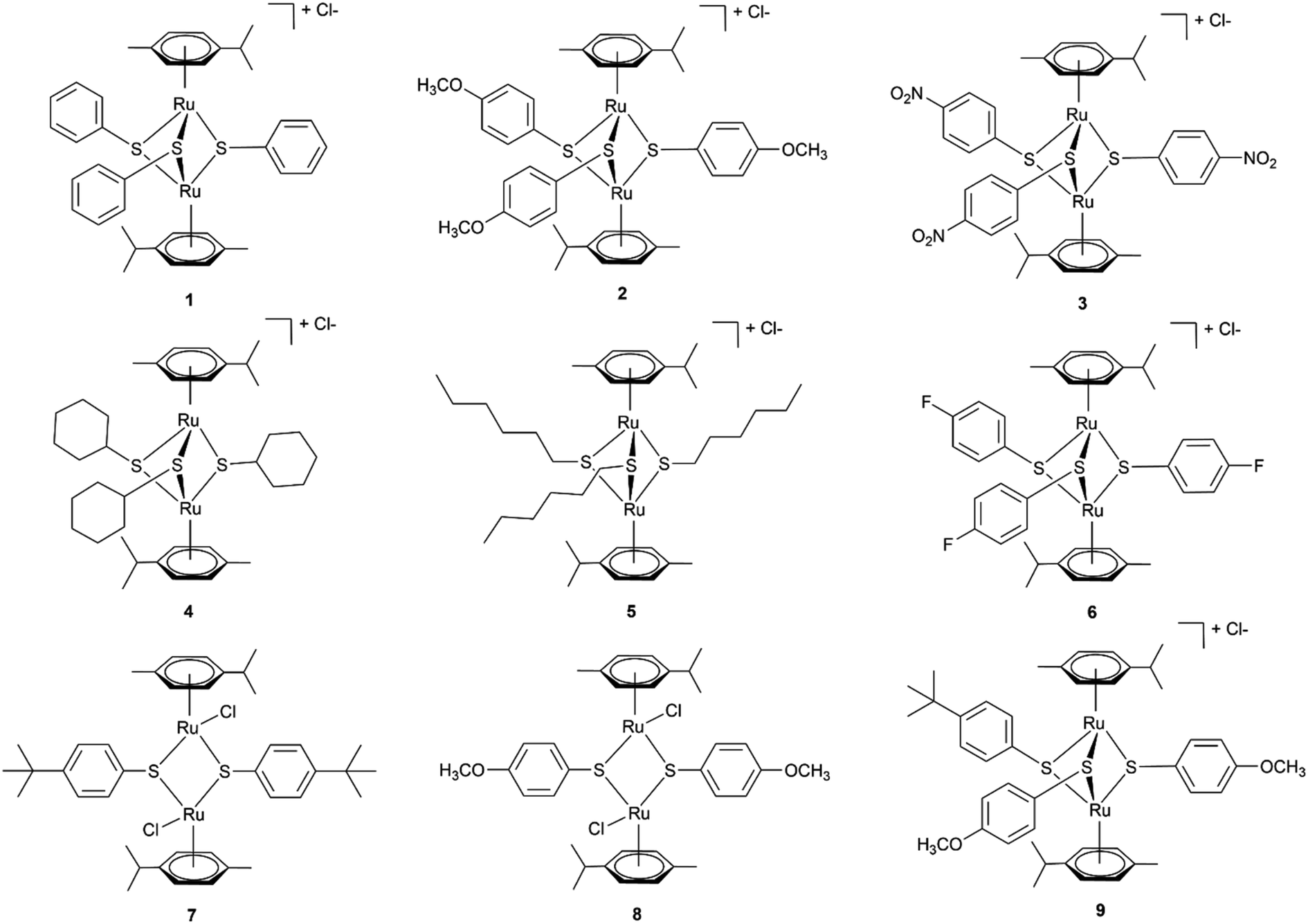 | ||
| Fig. 1 Structures of the six dinuclear trithiolato-bridged (1–6), the two dinuclear dithiolato-bridged arene ruthenium complexes (7–8) and the mixed complex (9) investigated and synthesised. | ||
2. Computational methods
All calculations were carried out with the CP2K package51 within the mixed Gaussian and Plane-Wave DFT formalism.52,53 The core-region of wave functions was smoothed out with norm-conserving dual-space Goedecker–Teter–Hutter pseudopotentials,54,55 whereas the valence pseudo-wave functions were expanded in molecularly optimised double-zeta valence polarised (DZVP) Gaussian basis sets for all elements.56 The auxiliary plane-wave basis set, used to calculate the Hartree potential, had a cut-off of 750 Ry. The BLYP functional was used57,58 with Grimme's D3 dispersion correction59 and Becke–Johnson (BJ) for the DFT-D3 damping function, which was reported to reduce the error for reaction barriers in BLYP calculations.60 Hybrid functionals, such as B3LYP, B3PW91 and PBE0, have been previously used to computationally investigate Ru-complexes.61–64 Due to the small geometry, energy and occupied electronic-structure differences between PBE0, B3LYP and BLYP, reported in the ESI (see Table S7†) and combined with the higher computational cost of hybrid-functional calculations in particular for barriers, we opt to carry out all calculations with BLYP. The wave function optimization was carried out with the orbital transformation method,65 while the geometry of complexes was relaxed until forces converged below 0.02 eV Å−1 and the energy difference between subsequent self-consistent steps was less than 10−6 Ha. Calculations were carried out in periodic simulation boxes of dimensions 30 × 30 × 30 Å that limit spurious interactions due to a distance of about 15 Å between periodic images of the complexes. The Ru(II) ion is considered to be non-magnetic as a geometry optimised ferromagnetic complex with spin multiplicity 9 lies 1.6 eV higher in energy. With this computational protocol, the average geometry deviation was determined to be about 0.81% with respect to experiment (see Table S7 in ESI†).All reaction mechanisms and their corresponding barriers were determined with the climbing image variant of the Nudged Elastic Band method.66 Each path was minimised until the energy difference converged below 0.002 eV.
The self-consistent continuum solvation model, SCCS,67 was used to implicitly account for the presence of a solvent. The cavity was defined as regions in the simulation cells with an electron density higher than 10−4 e bohr−3, while the continuum region is defined for densities smaller than 10−5 e bohr−3. The dielectric constants for dichloromethane and ethanol were set to 8.93 and 24.55, respectively. No further geometry relaxation was performed for structures in implicit solvent.68
3. Experimental section
The dichloro(p-cymene)ruthenium(II) dimer [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2, p-nitrothiophenol (technical grade), cyclohexylthiol, thiophenol and N,N-diisopropylethylamine (DIPEA) were purchased from Sigma Aldrich. p-methoxythiophenol was bought from Alfa Aesar. CDCl3 and MeOD were obtained from Cambridge Isotope Laboratories, CD2Cl2 was obtained from Eurisotop. All reactions were performed under inert atmosphere using standard Schlenk technique. NMR spectra were recorded on Bruker spectrometers: AVANCE III HD 300 MHz equipped with a 5 mm ATM BBFO probehead, AVANCE III HD 400 MHz equipped with a 5 mm ATM BBFO SmartProbe probehead, AVANCE II 400 MHz equipped with a 5 mm ATM Dual probehead and AVANCE II 500 MHz equipped with a 1.7 mm TXI 1H probehead, respectively. Unless specified otherwise NMR spectra were recorded at room temperature (25 °C) and processed using Topspin software (Bruker Biospin). MS and elemental analysis were performed, a LTQ Orbitrap XL with nano ESI (Thermo). All syntheses and purifications employing column chromatography are described in more detail in the ESI.† The temperatures were measured in the oil bath.3.1 Synthesis of 1
[(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H5)3]Cl (1) was obtained from the reaction of the dichloro(p-cymene)ruthenium(II) dimer [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2 with thiophenol.34 Four different reaction conditions were evaluated: (i) in DCM, 1 was obtained with 62% yield in 7 h, (ii) in DCM with addition of DIPEA, with 79% yield in 3 h, (iii) in EtOH with 69% yield in 23 h and (iv) in EtOH with addition of DIPEA with 80% yield in 3 h. Reactions in EtOH were performed under reflux at 78–83 °C while reactions in DCM were performed at 40–45 °C. The analytical data are provided in the ESI (Fig. S9–S11†) and are in agreement with the literature.343.2 Synthesis of 2
[(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4–OMe)3]Cl (2) was obtained in 73% yield from the reaction of [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2 with p-methoxythiophenol in DCM under reflux at 38–45 °C under N2 atmosphere in 9 h. The analytical data are provided in the ESI (Fig. S12–S14†) and are in agreement with the literature.193.3 Synthesis of 3
[(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4–NO2)3]Cl (3) was obtained in 73% yield from the reaction of [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2 with p-nitrothiophenol in DCM under reflux at 40–45 °C under N2 atmosphere in 2 h. The analytical data are provided in ESI (Fig. S15–S17†) and are in agreement with the literature.193.4 Synthesis of 4
[(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H11)3]Cl (4) was obtained from a 7-day reaction of [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2 with cyclohexanethiol in DCM with addition of DIPEA under reflux at 40–45 °C under Ar atmosphere. The desired complex 4 could only be obtained as an inseparable mixture with the dithiolato complex [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H11)2Cl2] as extensive purification procedures tend to contribute to degradation of the desired product. The purity of 4 was estimated with 1H NMR to be 65%. Analytical data (Fig. S18–S21†): 1H NMR (400.1 MHz, CDCl3): δH = 5.48 (m, 4H; H–Ar p-cymene), 5.35 (m, 2H; H–Ar p-cymene), 5.29 (d, JH,H = 5.0 Hz, 2H; H–Ar p-cymene), 2.57 (sept, JH,H = 6.7 Hz, 2H; C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) (CH3)2), 2.37 (m, 6H; H1 thiol), 2.21 (s, 6H; CH3), 1.73 (m, 6H; thiol), 1.51 (m, 6H; thiol), 1.39 (m, 6H; thiol), 1.31 (d, JH,H = 6.7 Hz, 6H; CH(CH3)2), 1.26 (d, JH,H = 6.7 Hz, 6H; CH(CH3)2), 0.96 ppm (m, 9H; thiol); 13C NMR (100 MHz; CDCl3): δC = 106.5 (C1 p-cymene), 101.4 (C4 p-cymene), 83.7 (Ar CH p-cymene), 83.2 (Ar CH p-cymene), 83.1 (Ar CH p-cymene), 82.9 (Ar CH p-cymene), 39.0 (thiol), 32.8 (thiol), 31.6 (
(CH3)2), 2.37 (m, 6H; H1 thiol), 2.21 (s, 6H; CH3), 1.73 (m, 6H; thiol), 1.51 (m, 6H; thiol), 1.39 (m, 6H; thiol), 1.31 (d, JH,H = 6.7 Hz, 6H; CH(CH3)2), 1.26 (d, JH,H = 6.7 Hz, 6H; CH(CH3)2), 0.96 ppm (m, 9H; thiol); 13C NMR (100 MHz; CDCl3): δC = 106.5 (C1 p-cymene), 101.4 (C4 p-cymene), 83.7 (Ar CH p-cymene), 83.2 (Ar CH p-cymene), 83.1 (Ar CH p-cymene), 82.9 (Ar CH p-cymene), 39.0 (thiol), 32.8 (thiol), 31.6 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H(CH3)2 + thiol), 28.9 (thiol), 24.0 (CH(H3)2), 22.6 (thiol), 22.4 (CH(H3)2), 18.3 (CH3), 14.1 ppm (thiol); ESI-MS (positive mode, MeOH): m/z = 817.2; Mw = 851.67 g mol−1.
H(CH3)2 + thiol), 28.9 (thiol), 24.0 (CH(H3)2), 22.6 (thiol), 22.4 (CH(H3)2), 18.3 (CH3), 14.1 ppm (thiol); ESI-MS (positive mode, MeOH): m/z = 817.2; Mw = 851.67 g mol−1.
3.5 Synthesis of 5
[(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H13)3]Cl (5) was obtained from the reaction [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2 and 1-hexanethiol in DCM under reflux at 40–45 °C with addition of DIPEA and Ar atmosphere in 68 h. An analogous reaction was performed in ethanol for comparison. The reaction in DCM gave the product in 62% yield, in EtOH in 42% yield. Analytical data (Fig. S22–S24†): 1H NMR (300.1 MHz, CDCl3): δH = 5.47 (m, 4H; H–Ar p-cymene), 5.34 (d, JH,H = 5.7 Hz, 2H; H–Ar p-cymene), 5.28 (d, JH,H = 5.7 Hz, 2H; H–Ar p-cymene), 2.56 (sept, JH,H = 7.0 Hz, 2H; C(CH3)2), 2.36 (m, 6H; SCH2), 2.19 (s, 6H; CH3p-cymene), 1.73 (m, 6H; CH2), 1.50 (p, JH,H = 7.5 Hz, 6H; CH2), 1.37 (m, 12H, CH2), 1.29 (d, JH,H = 7.0 Hz, 6H; CH(C3)2), 1.17 (d, JH,H = 7.0 Hz, 6H; CH(CH3)2), 0.95 ppm (t, JH,H = 6.8 Hz, 9H; CH3 thiol); 13C NMR (100 MHz, CDCl3): δC = 106.5 (Cq p-cymene), 101.4 (Cq p-cymene), 83.7 (Ar CH p-cymene), 83.2 (Ar CH p-cymene), 83.1 (Ar CH p-cymene), 82.9 (Ar CH p-cymene), 39.1 (SCH2), 32.9 (CH2), 31.7 and 31.6 (H(CH3)2 and CH2), 29.7, 28.9 (CH2), 24.0 and 22.6 (CH(H3)2), 22.5 (CH2), 18.3 (CH3p-cymene), 14.1 (CH3 thiol). ESI-MS (positive mode, EtOH): m/z = 823.2; elemental analysis calcd (%) for C38H67Ru2S3Cl·½H2O: C 52.66, H 7.91; found: C 52.57, H 7.95; Mw = 866.73 g mol−1.
3.6 Synthesis of 6
[(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4–F)3]Cl (6) was obtained in 80% yield from the reaction of [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2 and p-fluorothiophenol in DCM under Ar atmosphere and reflux at 40–45 °C with addition of DIPEA in 3.5 h. The analytical data are described in ESI (Fig. S25–S27†) and are in agreement with the literature.193.7 Synthesis of 7
[(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4-p-But)2Cl2] (7) was obtained in 98% yield from the reaction of [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2 and p-t-butylthiophenol in DCM cooled to 0 °C in 3.5 h. Analytical data (Fig. S28–S30†): 1H NMR (300.1 MHz, CDCl3): δH = 7.89 and 7.54 (br, 4H; H–Ar thiol), 7.34 (d, JH,H = 7.7 Hz, 4H; H–Ar thiol), 5.28, 5.18 and 5.04 (br, 8H; H–Ar p-cymene), 2.21 (br, 2H; C(CH3)2), 1.90 and 1.74 (br, 6H, CH3p-cymene), 1.30 (br, 18H; C(CH3)3), 0.99 and 0.91 ppm (br, 12H; CH(C3)2); 13C NMR (100 MHz, CDCl3): δC = 152.3 (C(CH3)3), 136.7 (Ar Cq thiol), 131.8 (CH thiol), 126.4 (CH thiol), 83.6–83.3 (Ar CH p-cymene), 34.9, 31.2 (C(H3)3), 30.6 (H(CH3)2), 22.1 (CH(H3)2), 17.9 (p-cymene CH3); ESI-MS (positive mode, EtOH): m/z 837.1; elemental analysis calcd (%) for C40H54S2Ru2Cl2·½CH2Cl2: C 53.19, H 6.06; found: C 53.24, H 6.05; Mw = 914.50 g mol−1.
3.8 Synthesis of 8
[(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4-p-OMe)2Cl2] (8) was obtained from the reaction of [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2 and p-methoxythiophenol in DCM stirred at room temperature for 4 h. Note that the extensive purification procedures lead to a slight degradation of the (8), leading to a final purity of about 85%. The final yield accounting for the degree of purity was 72%. Analytical data (Fig. S31–S34†): 1H NMR (400.1 MHz, CDCl3): δH = 7.93 and 7.56 (br, 4H; H–Ar thiol), 6.87 (d, JH,H = 8.4 Hz, 4H; H–Ar thiol), 5.18 and 5.00 (br, 8H; H–Ar p-cymene), 3.82 (s, 6H, OCH3), 2.31 (m, 2H; C(CH3)2), 1.89 and 1.73 (br, 6H, CH3p-cymene), 1.02 ppm (br, 12H; CH(C3)2); 13C NMR (100 MHz, CDCl3): δC = 160.4 (OCH3), 133.8 and 133.3 (CH thiol), 129.0 (CH thiol), 114.9 (CH thiol), 84.6–82.2 (Ar CH p-cymene), 55.7 (OCH3), 30.8 (H(CH3)2), 22.3 (CH(H3)2), 17.9 ppm (p-cymene CH3); ESI-MS (positive mode, EtOH): m/z 785.04; Mw = 819.86 g mol−1.
3.9 Synthesis of 9
[(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4-p-OMe)2(μ2-SC6H4-p-But)]Cl (9) was obtained in 29% yield from reaction of 8 with p-t-butylthiophenol in DCM at room temperature in 48 h. Analytical data (Fig. S35–S37†): 1H NMR (400.1 MHz, CDCl3): δH = 7.77 (m, 2H; H–Ar tBu-thiophenol), 7.71 (m, 4H; H–Ar MeOthiophenol), 7.31 (m, 2H; H–Ar tBu-thiophenol), 6.86 (m, 4H; H–Ar MeOthiophenol), 5.30, 5.13, 5.05 and 5.01 (d, JH,H = 5.3 Hz, 4 × 2H; H–Ar p-cymene), 3.81 (s, 6H; OC3), 1.85 (sept, JH,H = 6.8 Hz, 2H; C(CH3)2), 1.56 (s, 6H; CH3p-cymene), 1.28 (s, 9H; C(CH3)3), 0.83 (d, JH,H = 7.0 Hz, 6H; CH(C3)2), 0.71 (d, JH,H = 7.0 Hz, 6H; CH(C3)2); 13C NMR (100 MHz, CDCl3): δC = 159.0 (OCH3), 150.7 ((CH3)3), 133.4 (Cq tBu-thiophenol), 132.9 and 132.7 (CH MeOthiophenol), 131.1 (CH tBu-thiophenol), 127.6 and 127.3 (Cq MeOthiophenol), 125.0 (Ar CH tBu-thiophenol),113.7 (Ar CH MeOthiophenol), Ar CH 105.9 (Cq p-cymene), 98.9 (Cq p-cymene), 84.1, 84.0, 83.5 and 82.7 (all Ar CH p-cymene), 54.6 (OCH3), 33.7 ((CH3)3), 30.2(C(H3)3), 29.6 and 29.5 (H(CH3)2), 21.5 and 21.0 (CH(H3)2), 16.8 ppm (p-cymene CH3); ESI-MS (positive mode, MeOH): 915.15 m/z; elemental analysis calcd (%) for C44H55O2S3Ru2Cl·3/4CH2Cl2: C 53.04, H 5.62; found: C 53.04, H 5.84; Mw = 1013.39 g mol−1.
3.10 Reaction kinetics of 1–4 followed by 1H NMR
The kinetic of formation of complexes 1–4 was followed by NMR at 0 °C or 25 °C. For each reaction, [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2 was first dissolved in 500 μL CD2Cl2, and the NMR tube was sealed with a septum and flushed with stream of N2. Next, 4 eq. of the corresponding thiophenol dissolved in 300 μL CD2Cl2 was injected into the tube (Table S9†). We note that none of the kinetic experiments contain any DIPEA base. Once prepared, each tube was immediately inserted into the spectrometer and NMR measurements were started at a spinning rate of 10 Hz. For measurements at 0 °C, the spectrometer was pre-cooled and ice-cold CD2Cl2 was used for sample preparation. Processed 1H NMR spectra were transferred to dynamic center where normalised integrals of signals were plotted and fitted with built-in functions to find an optimal fit in order to obtain k.69–71 The reaction in DCM at 45 °C could not be performed directly in the tube. Instead, aliquots of each reaction mixture were collected at different time points, the original solvent immediately removed in vacuo, the residue dissolved in a suitable deuterated solvent and transferred into a new tube for NMR measurements. As such, only a low number of time points could be recorded, and k values could not be calculated, but an estimation of the kinetic of the reaction was still possible at this temperature (Fig. S38–S43†).4. Results and discussion
4.1 DFT calculations
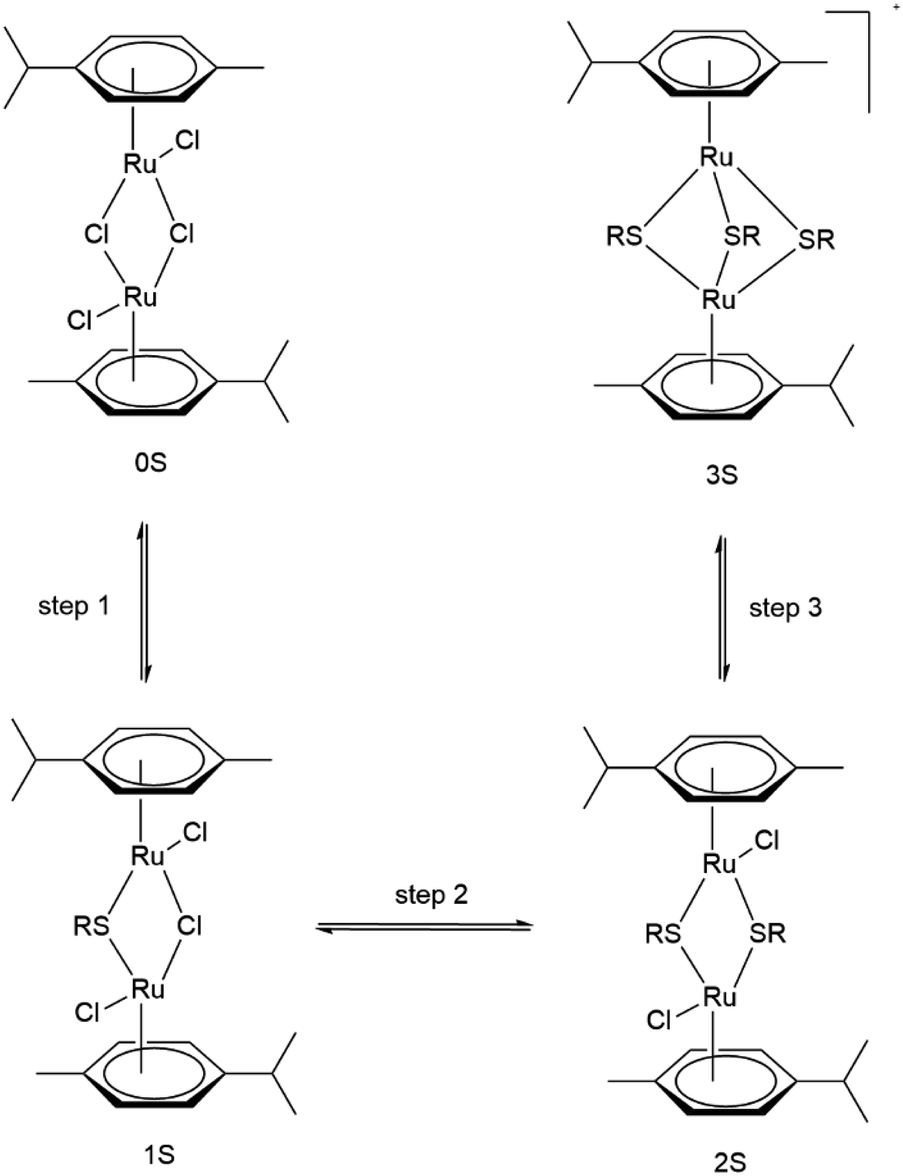 | ||
| Fig. 2 Stepwise formation of dinuclear trithiolato-bridged arene ruthenium complexes. We adopted the following abbreviations for the complexes – starting dimer [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2: 0S, neutral monothiolato complex [(η6-p-MeC6H4Pri)2Ru2Cl2(μ-Cl)(μ2-SR)]: 1S, neutral dithiolato complex [(η6-p-MeC6H4Pri)2Ru2(μ2-SR)2Cl2]: 2S and cationic trithiolato complex [(η6-p-MeC6H4Pri)2Ru2(μ2-SR)3]+: 3S. | ||
We tested two pathways for the insertion of the first thiol. The first pathway proceeds by initially forming a Ru–S bond with the subsequent release of HCl and the second one by reversing these steps with initial HCl formation followed by insertion of the thiolate. We find both pathway initializations to converge to the same final mechanism with two transition states (see Fig. S1a†). The first lower-energy transition state corresponds to the Ru–S bond formation (12.8 kcal mol−1), whereas the second higher-energy transition state is the thiol deprotonation (19.9 kcal mol−1). In protic solvents or in presence of a base thiols could exist as deprotonated anions and we would expect a reduced barrier for the second step. This is indeed confirmed by our calculations, where the barrier of insertion of the thiolate is reduced by 8.8 kcal mol−1 (see Fig. S2†). As these deprotonated systems are not charge neutral and computationally more challenging in our periodic setup, we will in the following consider only reaction mechanism for the neutral molecule but assume that protic solvents or addition of a base should systematically reduce reaction barriers.
The insertion of the second thiophenol follows the same mechanism as for the first one. However, the accommodation of the thiol in the Ru-complex core is hindered, which is reflected in the first transition state having a much higher activation barrier (24.0 kcal mol−1, see Fig. S1b†). The deprotonation step on the other hand requires less energy (10.9 kcal mol−1) than for the first insertion. The final step in the formation of the trithiolato Ru-complex is kinetically most demanding. While the barrier for the Ru–S bond formation is similar to the two preceding steps (17.5 kcal mol−1), the deprotonation is accompanied by rearrangement of the two other thiol ligands, so as to accommodate the third one, which results in a barrier of 31.4 kcal mol−1 (see Fig. S1c†). The overall pathway for thiol ligands is shown as the black curve in Fig. 3. It can be seen that the formation of all thiolato complexes is thermodynamically favourable. Nevertheless, the kinetic barriers continuously increase from formation of the monothiolato to the formation of the trithiolato complex, in correlation with lower experimental yield for the trithiolato complexes compared to mono- and dithiolato ones.
 | ||
| Fig. 3 Energy evolution and transition states along the formation pathway form the starting dimer dichloride (0S) to the trithiolato (3S) Ru complex via the mono- (1S) and dithiolato complexes. The black line represents thiophenol (1), while the red and blue lines are for thiophenols with –OCH3 (2) and –NO2 (3) substituents in para position and cyclohexanethiol (4) respectively. The values for the kinetic barriers are summarised in Table S1 (ESI†). | ||
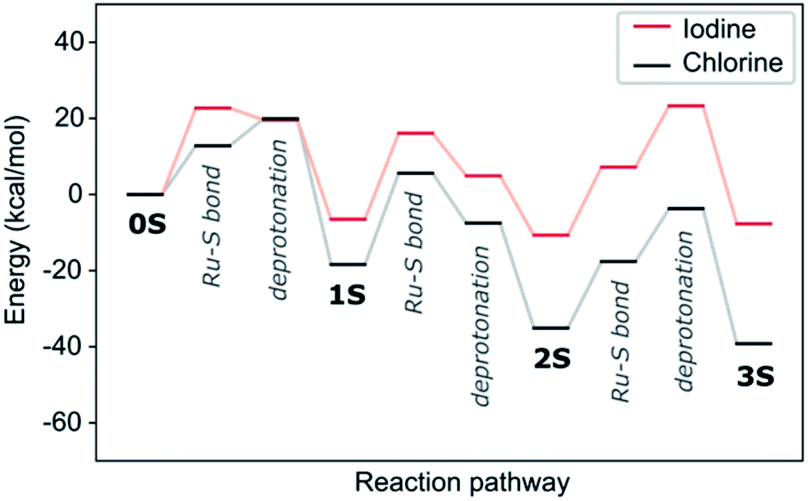
where kB is Boltzmann's constant, T the temperature, h Plank's constant, and E# is the computed barrier, listed in Table S1.† From the data shown in Fig. 5 and Table S4,† we can see that while steps 1 and 2 have reasonable reaction rates at room temperature, step 3 has very slow kinetics, which is further hindered by the substituents on the thiophenol. Only elevated temperatures lead to reaction rates that enable the complex formation on experimental timescales. These are in agreement with the experimental findings, that the 3rd step needs significant thermal energy to proceed.
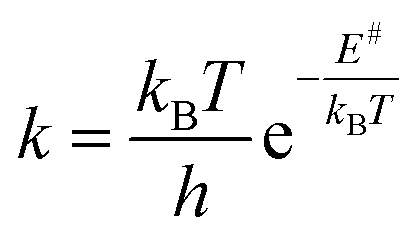
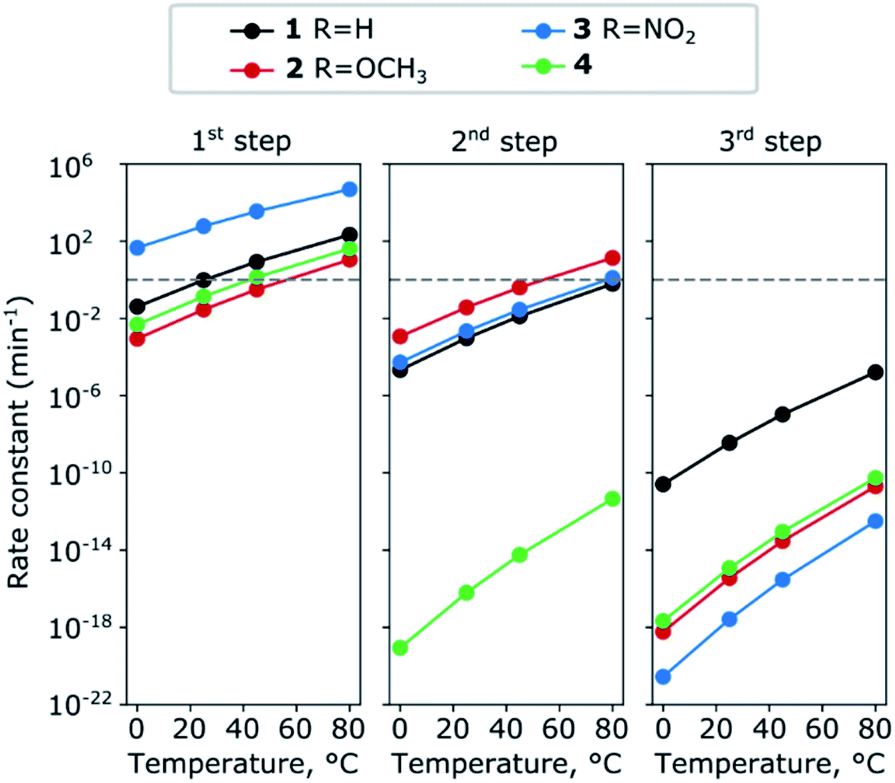 | ||
| Fig. 5 Change in the rate constants with temperature for the complexes 1–4 for the three steps. The numerical values are summarised in Table S4 (ESI†). | ||
Experimentally, the three-step reaction takes place in a polar solvent, ethanol or DCM (in the present work). We simulate the polarizability of the solvent with an implicit solvation model, which correctly describes the electrostatic effect of the medium. Accounting for any reaction with the solvent, however, such as H-bonding or proton transfer would require computationally expensive molecular dynamics calculations, where the solute is simulated with explicit solvent molecules. A compromise could be the addition of a few solvent molecules in combination with implicit solvent. Considering though the great number of possible solvent orientations and interactions with structurally flexible solutes as shown by test calculations, in particular for the transition states, there is a considerable risk of missing important information and thus of compromising the validity of the results. We therefore consider that the implicit model, despite its limitations, yields more reliable trends for solvated complexes than a mixed explicit/implicit scheme. We perform calculations with dielectric constants 8.93 and 24.55, corresponding to DCM and ethanol respectively and show the energy evolution in Fig. 6 and compare it with the one in vacuum. The higher dielectric constant makes the thiolato complexes thermodynamically more favourable but has a distinct destabilizing effect on the transition states corresponding to the insertion of the thiol group into the complex' core. This increase of the barriers is less marked for the lower dielectric constant that however also does not stabilise the complexes as much. This suggests that strongly polar solvents are kinetically unfavourable for the formation of the di- and especially the trithiophenolato complexes.
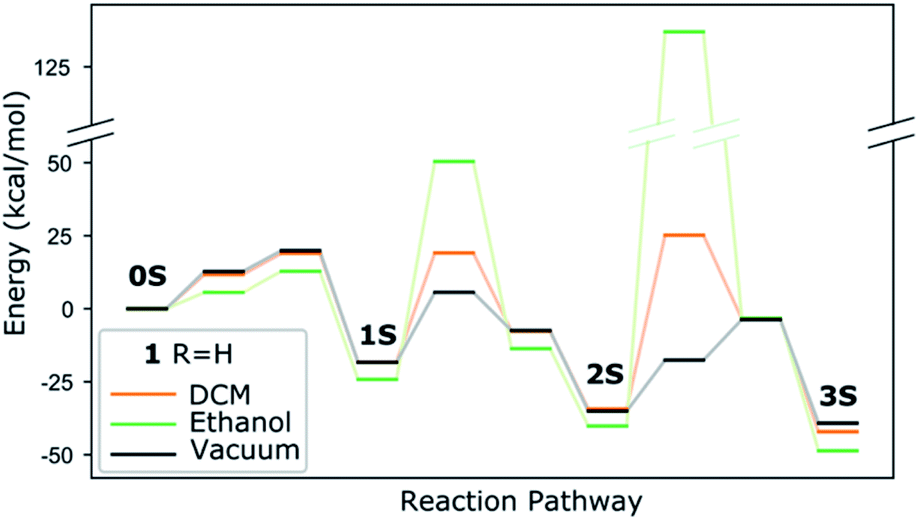 | ||
| Fig. 6 Effect of implicit DCM and ethanol solvents on the energy evolution of complex 1. The numerical values are in Table S3 (ESI†). | ||
The effect of the temperature on the reaction kinetics and the individual insertion of the thiol has already been experimentally demonstrated. For a given thiol, it was shown that the synthesis exclusively leads to the cationic trithiophenolato complex [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4–R)3]+. At 0 °C and for thiols with decreased reactivity, typically benzylthiols, but also for some thiophenols, the neutral dithiophenolato complex [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4–R)2Cl2], or even the neutral monothiophenolato complex [(η6-p-MeC6H4Pri)2Ru2Cl2(μ-Cl)(μ2-SC6H4–R)] can be obtained in pure forms.49
4.2 Experimental results
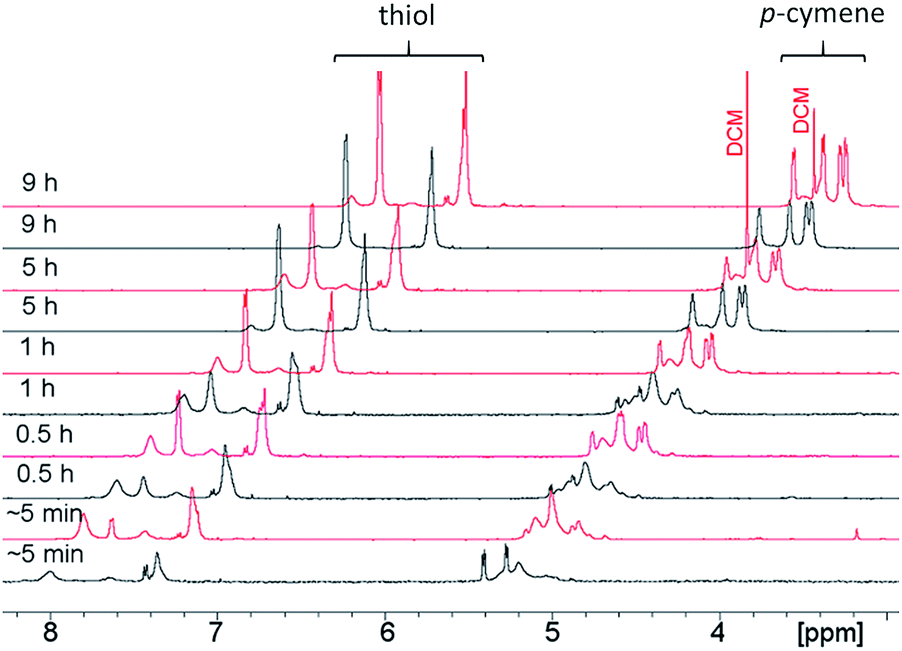 | ||
| Fig. 7 Synthesis of 1 followed by 1H NMR. The spectra show the reaction monitoring (thiol- (5–5.5 ppm) and p-cymene aromatic resonances (7–8 ppm) are shown) after ∼5 min, 0.5 h, 1 h, 5 h, and 9 h in EtOH (black line) at 80 °C and in DCM (red line) at 45 °C. Aliquots of reaction mixture samples at individual time points were dried and transferred to CDCl3 prior to measurement. | ||
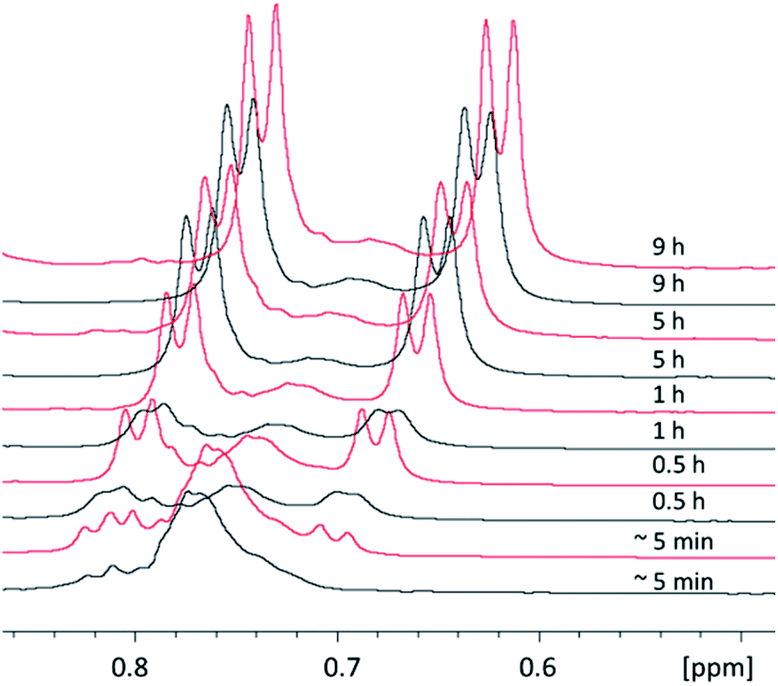 | ||
| Fig. 8 Synthesis of 1 followed by 1H NMR. The spectra show the reaction monitoring (the p-cymene methyl resonances are shown) after ∼5 min, 0.5 h, 1 h, 5 h, and 9 h in EtOH (black line) at 80 °C and in DCM (red line) at 45 °C. Aliquots of reaction mixture samples at individual time points were dried and transferred to CDCl3 prior to measurement. | ||
From the reactions performed at 0 °C and 25 °C, we were only able to estimate the rate constant for the dithiolato complex formation applying the Bodenstein approximation.75 The formation of the monothiolato complex is very fast and no reliable NMR data could be extracted. It turns out that the rate constant obtained from the kinetic data (see Table 1, Fig. S44 and S45†) was of the same order of magnitude as the one calculated for the reaction performed at 25 °C. Given that DFT yields the intrinsic rate, whereas experiment measures a concentration-dependent rate, and given the high variability of the rate constant for small changes in the barrier, an order of magnitude agreement is satisfactory.
| R | k steps 1 + 2 (min−1) | |||
|---|---|---|---|---|
| Calculated | Experimental | |||
| 0 °C | 25 °C | 0 °C | 25 °C | |
| H (1) | 8.6 × 10−7 | 9.2 × 10−2 | 7.5 × 10−4 | 2.9 × 10−3 |
| OCH3 (2) | 1.1 × 10−6 | 1.1 × 10−3 | 6.5 × 10−3 | 2.2 × 10−3 |
| NO2 (3) | 2.4 × 10−3 | 1.32 | 9.2 × 10−3 | 1.2 × 10−3 |
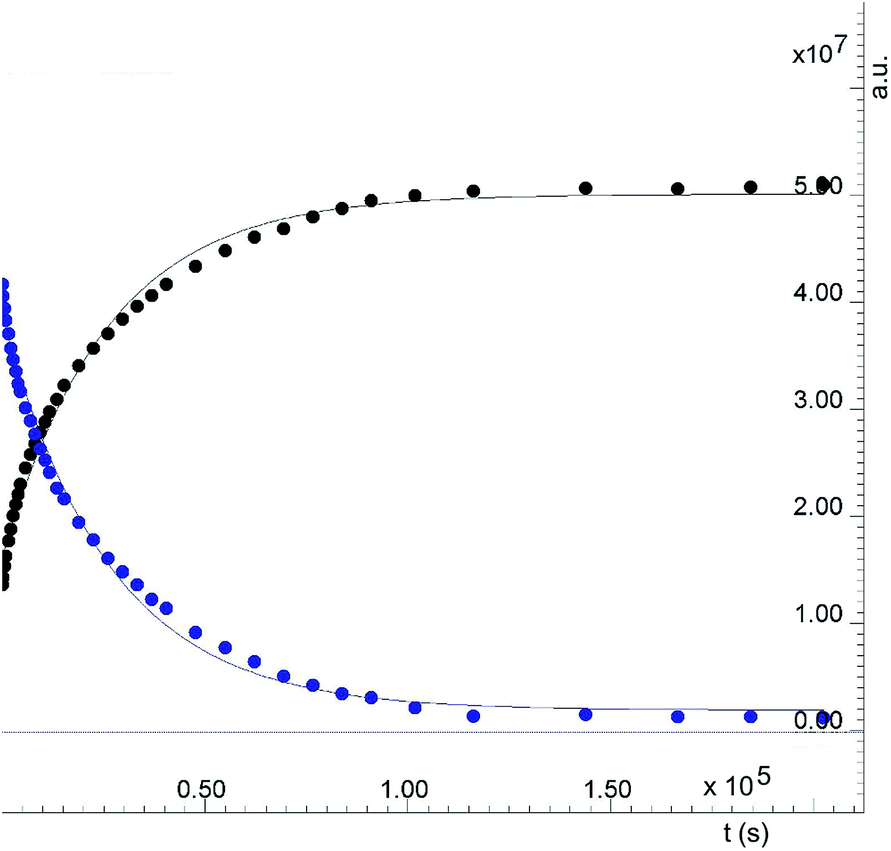 | ||
| Fig. 9 Formation of 2. Reaction between the dimer [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2 and p-methoxythiophenol in CD2Cl2 at 25 °C followed by 1H NMR. The data points represent normalised integrals. Black: resonance at 6.86 ppm (p-methoxythiophenol, 2) blue: resonance at 5.34 ppm (p-cymene ring, starting dimer). | ||
The new complex 5 with aliphatic thiol ligands has been obtained in 62% yield from the reaction in DCM with addition of DIPEA. An analogous reaction in ethanol with addition of DIPEA lead to 42% yield. Attempts to obtain 5 by performing the reaction in DCM without DIPEA were unsuccessful, only the neutral dithiolato complex [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H13)2Cl2] was observed, which can be ascribed to the low reactivity of 1-hexanethiol. Of note, a similar trithiolato complex [(η6-p-MeC6H4Pri)2Ru2(μ2-SC8H17)3]+ was obtained by Stibal et al. in 2016, who reported an analogous reaction with octanethiol under reflux in EtOH for 168 h and obtained 28% yield.50
5. Conclusions
In summary, we have conducted a DFT study aiming at a fundamental understanding of the reaction mechanisms leading to the formation of dinuclear thiolato-bridged arene ruthenium complexes [(η6-p-MeC6H4Pri)2Ru2(μ2-SC6H4–R)3]+ starting from the dichloro(p-cymene)ruthenium(II) dimer [(η6-p-MeC6H4Pri)Ru(μ2-Cl)Cl]2. Further, we studied variations in reaction conditions experimentally and followed the kinetics with NMR.The presence of electron-withdrawing or donating substituents on the thiol significantly influences the formation of the trithiolato complex, which is thermodynamically no longer favourable in presence of the former. In addition, the calculated reaction pathways suggest using a solvent with a lower dielectric constant could decrease the kinetic barriers for the formation of the di- and trithiolato complexes. Experimentally, changing the reaction solvent from EtOH to DCM indeed leads mostly to similar or better yields, but at lower temperature as compared to EtOH. Use of a base such as DIPEA allows to further increase the yield in a shorter reaction time. By this tuning of the reaction conditions, we were able to synthesise two new trithiolato complexes with aliphatic thiol ligands, improve the yields for two trithiolato complexes with aromatic thiol ligands and further synthesise two new dithiophenolato complexes, impossible to obtain so far. As such, our results and suggested adapted synthetic route open new possibilities for the synthesis of so far inaccessible dinuclear dithiophenolato- and especially trithiolato-bridged arene ruthenium(II) complexes that are known to possess very interesting anticancer and antiparasitic properties. More generally, the synthesis of other challenging thiolato-bridged dinuclear group 8 and 9 metal complexes could be reexamined.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the SNF Professorship Grant PP00P2_157615. Calculations were performed on UBELIX (http://www.id.unibe.ch/hpc), the HPC cluster at the University of Bern.Notes and references
- Y. Chen, Y. Peng, P. Chen, J. Zhao, L. Liu, Y. Li, S. Chen and J. Qu, Dalton Trans., 2010, 39, 3020–3025 RSC.
- Y. Chen, Y. Zhou, P. Chen, Y. Tao, Y. Li and J. Qu, J. Am. Chem. Soc., 2008, 46, 15250–15251 CrossRef PubMed.
- Y. Chen, Y. Zhou and J. Qu, Organometallics, 2008, 27, 666–671 CrossRef.
- J. Zhao, L. Wang, Y. Zhou, Y. Zhang, N. Zhang, C. Jia, F. Hu, Y. Chen, B. Wang and J. Qu, Inorg. Chem. Commun., 2013, 29, 179–182 CrossRef CAS.
- G. Gupta, N. Nagesh, B. S. Murray, P. J. Dyson and B. Therrien, Inorg. Chim. Acta, 2014, 423, 31–35 CrossRef CAS.
- J. Boudreau, J. Grenier-Desbiens and F.-G. Fontaine, Eur. J. Inorg. Chem., 2010, 2010, 2158–2164 CrossRef.
- F. Takagi, H. Seino, M. Hidai and Y. Mizobe, Organometallics, 2003, 22, 1065–1071 CrossRef CAS.
- F. Takagi, H. Seino, Y. Mizobe and M. Hidai, Can. J. Chem., 2001, 79, 632–634 CrossRef CAS.
- F. Takagi, H. Seino, Y. Mizobe and M. Hidai, Organometallics, 2002, 21, 694–699 CrossRef CAS.
- G. Gupta, A. Garci, B. S. Murray, P. J. Dyson, G. Fabre, P. Trouillas, F. Giannini, J. Furrer, G. Süss-Fink and B. Therrien, Dalton Trans., 2013, 42, 15457–15463 RSC.
- J. P. Johnpeter, G. Gupta, J. M. Kumar, G. Srinivas, N. Nagesh and B. Therrien, Inorg. Chem., 2013, 52, 13663–13673 CrossRef CAS PubMed.
- G. Gupta, B. S. Murray, P. J. Dyson and B. Therrien, J. Organomet. Chem., 2014, 767, 78–82 CrossRef CAS.
- M. Gras, B. Therrien, G. Süss-Fink, O. Zava and P. J. Dyson, Dalton Trans., 2010, 39, 10305–10313 RSC.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- J. Furrer and G. Süss-Fink, Coord. Chem. Rev., 2016, 309, 36–50 CrossRef CAS.
- A. P. Basto, J. Müller, R. Rubbiani, D. Stibal, F. Giannini, G. Süss-Fink, V. Balmer, A. Hemphill, G. Gasser and J. Furrer, Antimicrob. Agents Chemother., 2017, 61(9), e01031-17 CrossRef PubMed.
- A. P. Basto, N. Anghel, R. Rubbiani, J. Müller, D. Stibal, F. Giannini, G. Süss-Fink, V. Balmer, G. Gasser, J. Furrer and A. Hemphill, Metallomics, 2019, 11, 462–474 CrossRef CAS PubMed.
- F. Giannini, G. Süss-Fink and J. Furrer, Inorg. Chem., 2011, 50, 10552–10554 CrossRef CAS PubMed.
- F. Giannini, J. Furrer, A. F. Ibao, G. Süss-Fink, B. Therrien, O. Zava, M. Baquie, P. J. Dyson and P. Štěpnička, J. Biol. Inorg Chem., 2012, 17, 951–960 CrossRef CAS PubMed.
- F. Giannini, L. E. H. Paul and J. Furrer, Chimia, 2012, 66, 775–780 CrossRef CAS PubMed.
- F. Giannini, J. Furrer, G. Süss-Fink, C. M. Clavel and P. J. Dyson, J. Organomet. Chem., 2013, 744, 41–48 CrossRef CAS.
- F. Giannini, L. E. H. Paul, J. Furrer, B. Therrien and G. Süss-Fink, New J. Chem., 2013, 37, 3503 RSC.
- F. Giannini, M. Bartoloni, L. E. H. Paul, G. Süss-Fink, J. L. Reymond and J. Furrer, Medchemcomm, 2015, 6, 347–350 RSC.
- F. Giannini, L. Geiser, L. E. H. Paul, T. Roder, B. Therrien, G. Süss-Fink and J. Furrer, J. Organomet. Chem., 2015, 783, 40–45 CrossRef CAS.
- P. Tomšík, D. Muthná, M. Řezáčová, S. Mičuda, J. Ćmielová, M. Hroch, R. Endlicher, Z. Červinková, E. Rudolf, S. Hann, D. Stíbal, B. Therrien and G. Süss-Fink, J. Organomet. Chem., 2015, 782, 42–51 CrossRef.
- M. B. Gomes de Lima, J. E. Guerchais, R. Mercier and F. Y. Pétillon, Organometallics, 1986, 5, 1952–1964 CrossRef CAS.
- F. Y. Pétillon, P. Schollhammer, J. Talarmin and K. W. Muir, Coord. Chem. Rev., 1998, 178–180, 203–247 CrossRef.
- F. Y. Pétillon, P. Schollhammer and J. Talarmin, Coord. Chem. Rev., 2017, 331, 73–92 CrossRef.
- G. Winkhaus and H. Singer, J. Organomet. Chem., 1967, 7, 487–491 CrossRef CAS.
- R. A. Zelonka and M. C. Baird, J. Organomet. Chem., 1972, 44, 383–389 CrossRef CAS.
- R. A. Zelonka and M. C. Baird, J. Organomet. Chem., 1972, 35, C43–C46 CrossRef CAS.
- R. A. Zelonka and M. C. Baird, Can. J. Chem., 1972, 50, 3063–3072 CrossRef CAS.
- H. T. Schacht, R. C. Haltiwanger and M. R. DuBois, Inorg. Chem., 1992, 31, 1728–1730 CrossRef CAS.
- K. Mashima, A. Mikami and A. Nakamura, Chem. Lett., 1992, 21, 1795–1798 CrossRef.
- P. J. Dyson, G. Süss-Fink, F. Giannini, J. Furrer and C. M. Clavel, J. Organomet. Chem., 2013, 744, 41–48 CrossRef.
- D. Stíbal, L. Geiser, G. Süss-Fink and J. Furrer, RSC Adv., 2016, 6, 38332–38341 RSC.
- F. Chérioux, C. M. Thomas, T. Monnier and G. Süss-Fink, Polyhedron, 2003, 22, 543–548 CrossRef.
- F. Chérioux, B. Therrien and G. Süss-Fink, Inorg. Chim. Acta, 2004, 357, 834–838 CrossRef.
- G. Süss-Fink and B. Therrien, Organometallics, 2007, 26, 766–774 CrossRef.
- F. Chérioux, C. M. Thomas, B. Therrien and G. Süss-Fink, Chem.–Eur. J., 2002, 8, 4377–4382 CrossRef.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CrossRef CAS PubMed.
- E. S. Antonarakis and A. Emadi, Cancer Chemother. Pharmacol., 2010, 66, 1–9 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- D. Muthná, P. Tomšík, R. Havelek, R. Köhlerová, V. Kasilingam, E. Čermáková, D. Stíbal, M. Řezáčová and G. Süss-Fink, Anticancer Drugs, 2016, 27, 643–650 CrossRef PubMed.
- E. Primasová, H. Primasová, L. E. H. Paul, G. Diserens, P. Vermathen, M. Vermathen and J. Furrer, Metabolites, 2019, 9, 146 CrossRef PubMed.
- D. Stíbal, B. Therrien, G. Süss-Fink, P. Nowak-Sliwinska, P. J. Dyson, E. Čermáková, M. Řezáčová and P. Tomšík, J. Biol. Inorg Chem., 2016, 21, 443–452 CrossRef PubMed.
- J. Jelk, V. Balmer, D. Stibal, F. Giannini, G. Süss-Fink, P. Bütikofer, J. Furrer and A. Hemphill, Exp. Parasitol., 2019, 205, 107753 CrossRef CAS PubMed.
- A.-F. Ibao, M. Gras, B. Therrien, G. Süss-Fink, O. Zava and P. J. Dyson, Eur. J. Inorg. Chem., 2012, 2, 1531–1535 CrossRef.
- D. Stíbal, B. Therrien, F. Giannini, L. E. H. Paul, J. Furrer and G. Süss-Fink, Eur. J. Inorg. Chem., 2014, 2014, 5925–5931 CrossRef.
- D. Stíbal, T. Riedel, P. J. Dyson, G. Süss-Fink and B. Therrien, Inorg. Chim. Acta, 2016, 444, 51–55 CrossRef.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CAS.
- G. Lippert, J. Hutter and M. Parrinello, Mol. Phys., 1997, 92, 477–488 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703 CrossRef CAS PubMed.
- M. Krack, Theor. Chem. Acc., 2005, 114, 145–152 Search PubMed.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 4, 276–282 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- T. Zábojníková, R. Cajzl, J. Kljun, Z. Chval, I. Turel and J. V. Burda, J. Comput. Chem., 2016, 37, 1766–1780 CrossRef PubMed.
- I. Czerwinska, J. Far, C. Kune, C. Larriba-Andaluz, L. Delaude and E. De Pauw, Dalton Trans., 2016, 45, 6361–6370 RSC.
- G. Gupta, A. Garci, B. S. Murray, P. J. Dyson, G. Fabre, P. Trouillas, F. Giannini, J. Furrer, G. Süss-Fink and B. Therrien, Dalton Trans., 2013, 42, 15457 RSC.
- I. del Rosal, T. Gutmann, L. Maron, F. Jolibois, B. Chaudret, B. Walaszek, H.-H. Limbach, R. Poteau and G. Buntkowsky, Phys. Chem. Chem. Phys., 2009, 11, 5657 RSC.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2003, 118, 4365–4369 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga, H. Jónsson and G. Henkelman, J. Chem. Phys., 2011, 9901, 1–5 Search PubMed.
- O. Andreussi, I. Dabo and N. Marzari, J. Chem. Phys., 2012, 136, 064102 CrossRef PubMed.
- J. Riddick and W. Bunger, Organic Solvents: Physical Properties and Methods of Purification, Band 2, Wiley-Interscience, 3rd edn, 1970 Search PubMed.
- Dynamics Center 2.5.6, https://www.bruker.com/products/mr/nmr/software/dynamics-center.html, accessed 29 November 2019.
- H. Akaike, IEICE Trans. Fundam. Electron. Commun. Comput. Sci., 1974, 19, 716–723 Search PubMed.
- F. Susanne, D. S. Smith and A. Codina, Org. Process Res. Dev., 2012, 16, 61–64 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- L. Geiser, Synthesis, reactivity, and cytotoxicity of thiolato-bridged dinuclear arene ruthenium complexes, Master thesis, University of Bern, 2015 Search PubMed.
- H. Eyring, The Activated Complex in Chemical Reactions, 1935, vol. 3 Search PubMed.
- F. G. Helfferich, Kinetics of Multistep Reactions, Elsevier, 2004 Search PubMed.
- F. Giannini, J. Furrer, A. F. Ibao, G. Süss-Fink, B. Therrien, O. Zava, M. Baquie, P. J. Dyson and P. Štěpnička, J. Biol. Inorg Chem., 2012, 17, 951–960 CrossRef CAS PubMed.
- L. E. Revell and B. E. Williamson, J. Chem. Educ., 2013, 90, 1024–1027 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08146a |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |