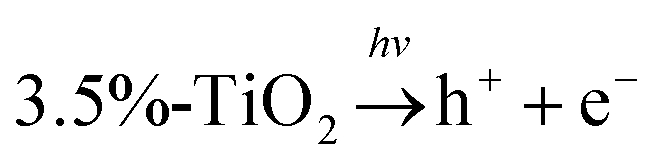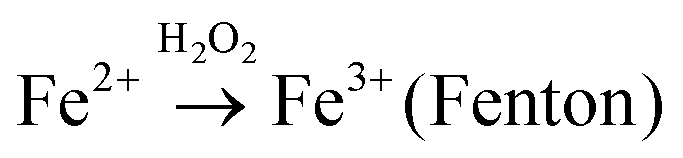DOI:
10.1039/D0RA08116G
(Paper)
RSC Adv., 2020,
10, 40619-40624
Enhanced active oxidative species generation over Fe-doped defective TiO2 nanosheets for boosted photodegradation†
Received
23rd September 2020
, Accepted 3rd November 2020
First published on 9th November 2020
Abstract
Semiconductor photocatalysis is widely proposed for decomposing multiple pollutants via photo-generated oxidative species. However, the photocatalytic degradation performance in practical settings still remains unsatisfactory due to the limited production of active oxidative species (AOS). In this work, a defect engineering strategy was developed to explore the superiority of oxygen vacancies (Vo) and their structural regulation to enhance AOS production for boosting photodegradation. Taking anatase TiO2 as a model photocatalyst, ultrathin TiO2 nanosheets containing abundant Vo and appropriate Fe doping exhibited an unprecedented 134 times higher activity in the degradation of Rhodamine B (RhB) (rate as high as 0.3073 min−1) than bulk anatase and were superior to most reported photocatalysts. The defect-rich ultrathin TiO2 nanosheets could be further applied in high-efficiency degradation of tetracycline hydrochloride (TC-HCl) with the degradation rate of 0.0423 min−1. The in situ electron paramagnetic resonance, advanced spectroscopic characterization and electrochemical measurement revealed the key role of Vo and Fe doping in facilitating the production of photo-generated holes and superoxide radicals (˙O2−) that were identified to be effective to decompose both RhB and TC-HCl. This research provides insight into defect engineering promoting AOS generation and gives inspiration for the design of efficient photocatalysts for photooxidation applications.
1. Introduction
With the rapid development of manufacturing and pharmaceutical industries, dyes and antibiotics from polluted water have attracted widespread attention in the field of public health.1–4 Commercial water disinfection strategies, such as physical adsorption and biodegradation,5,6 have unavoidable side effects, which would increase treatment cost and environmental problems. Solar photocatalysis technology uses highly active oxidative species (AOS) produced by semiconductors to drive the degradation of antibiotics and dye molecules, showing unique advantages and broad application prospects in water purification.7 Although AOS-driven degradation have been developed, its scalable application is still hampered by the intrinsic limitations of semiconductor photocatalysts, including the limited sunlight absorption, undesired photo-generated electron–hole recombination and inefficient reactant molecules (e.g. O2, H2O2) activation, severely restricting the generation of AOS and the efficiency of pollutants decomposition.8
Recent theoretical calculations and experimental results showed that surface anion defects can significantly affect the optical/electronic properties of semiconductor photocatalysts.9–11 Taking oxygen vacancies (Vo) on the surface of a semiconductor photocatalyst as a proof-of-concept, a certain concentration of surface Vo could induce defect energy levels between the valence band (VB) and conduction band (CB), that are deemed to be effective to narrow band gap of the photocatalytic material and promote separation and transfer of photo-generated electrons and holes.12 In addition, Vo and its derivative coordinatively unsaturated metal sites featuring with electron-rich property have been proven being efficient in activating chemically inert reactant molecules (e.g. O2, H2O2) and thereby facilitating the AOS production for pollutants degradation.13,14 However, when two or more reactant molecules co-exist on the surface of photocatalysts, it is still challenging to activate target reactants only by Vo for AOS generation with high selectivity and efficiency, in particular to advanced oxidation processes that involve in H2O2 or peroxosulfate.15 The competitive adsorption of O2 and other oxidants on the Vo would lead to low-efficiency and non-selective production of target AOS, thus lowering degradation efficiency.16 To overcome the aforementioned bottleneck, it is crucial to further regulate the defect structure for optimized catalytic activity.
Taken account of chemical reaction on the surface of photocatalysts, it generally requires the adsorption of reactants on the Vo and strong interaction between active metal sites and reactants (electrons transfer), realizing reactants activation for subsequent reactions. Hence, optimizing Vo-surrounding metal sites may represent an effective strategy to refine defect structure for enhanced catalytic activity and AOS production,17,18 especially for the metal species that show specific reactivity to reactant molecules. Among a variety of metal species for modifying photocatalysts, Fe-doping arose broad interest since its positive impact on the photocatalytic activity of materials.19,20 Zhang et al. reported that Fe-doping and highly exposed (001) facet furnished the anatase TiO2 hierarchical spheres with excellent photodegradation activity, in which subtle Fe-doping has been shown to enhance visible light absorption of anatase TiO2 and boost photo-generated charge separation/charge.19 More importantly, it is widely accepted that Fe2+/Fe3+ pair is the active specie for accelerating the selective conversion of H2O2 into hydroxyl radicals, whilst Vo and neighboring unsaturated metal sites (e.g. Tiδ+, δ < 4) could expedite O2 activation for the efficient generation of ˙O2−,21–23 probably establishing an access to selectivity-activity balance, in expectation to the high-efficiency AOS production and pollutants degradation. Thence, modulating the defect structure in Vo-containing anatase by introducing the Fe2+/Fe3+ pair is likely to bridge the selectivity-activity gap and represent an ideal model for exploring the role of Vo in AOS-driven degradation of organic pollutants.
In recent years, two-dimensional (2D) materials have demonstrated unique physicochemical properties with great potential for photocatalysis, lithium-ion storage and relevant application.24–26 To be specific, 2D materials offer the ultrahigh surface area for sufficient contact with reactants and shorter bulk-to-surface distance for efficient charge transportation. Cui et al. exploited a universal alkaline-free strategy to synthesize 2D TiO2 nanobelts, which exhibited greatly increasing electrolyte/electrode contact area and shorten diffusion distance of Li ions, in turn delivering excellent electrochemical performance.24 In addition, surface defects (e.g., Vo) can be easily created on 2D materials with resulting low-coordination metal sites, which were proven to be efficient in facilitating inert small molecules (e.g., O2, N2) conversion.8,18 Based on the above discussion, 2D materials serve as an ideal platform to explore the effect of surface defects and metal doping on the photocatalytic activity. Herein, we selected ultrathin TiO2 nanosheets as a model material for tunable Fe doping, in which both of Vo and Fe doping contribute to efficient AOS generation and boosted degradation of dye and antibiotics.
2. Results and discussion
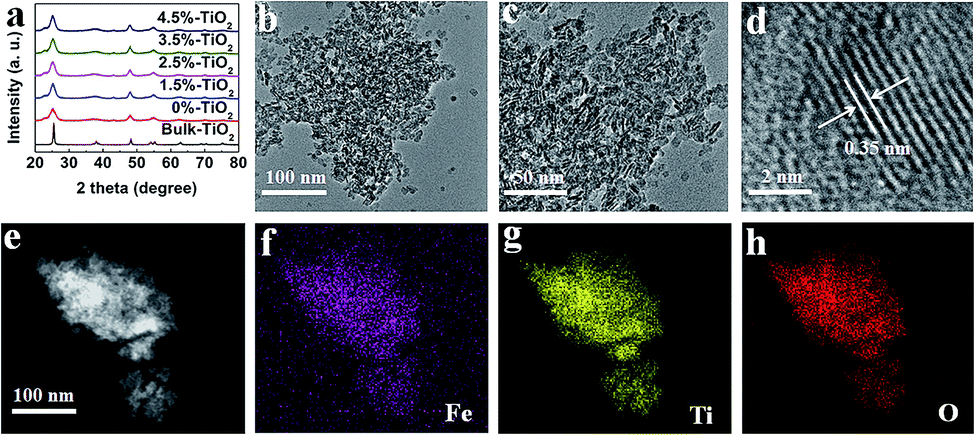
A facile hydrothermal method was used (details were offered in the ESI) to synthesize the ultrathin TiO2 nanosheets with different amounts of Fe ions (here expressed as X%-TiO2, where X is the Fe/Ti molar ratio in percentage). As shown in Fig. 1a, X-ray diffraction (XRD) patterns for X%-TiO2 nanosheets (X = 0, 1.5, 2.5, 3.5, 4.5) and a bulk anatase TiO2 (denoted as Bulk-TiO2) were consistent with anatase TiO2 (JCPDS-21-1272) and metallic Fe or Fe oxides were absent in all the X%-TiO2 nanosheets. High-resolution transmission electron microscope (HRTEM) images for 3.5%-TiO2 (Fig. 1b and c) and 0%-TiO2 (Fig. S1a, ESI†) exhibited the thinner and smaller nanosheets morphology relative to the bulk reference (Fig. S1b†). The average thickness of 3.5%-TiO2 nanosheets was determined to be ∼4.5 nm by atomic force microscopy (Fig. S2†). Further, 3.5%-TiO2 nanosheets showed a lattice fringe with a pitch of 0.35 nm (Fig. 1d), which can be attributed to the typical (101) plane of anatase TiO2. Scanning transmission electron microscope (STEM) equipped with energy dispersive X-ray (EDX) elemental mappings (Fig. 1e–h) confirmed the successful incorporation of Fe and the uniform distribution of Fe, Ti, and O elements in 3.5%-TiO2 nanosheets. The Fe content in all the X%-TiO2 was quantified by inductively coupled plasma atomic emission spectrometry (ICP-AES) (Table S1†), with the result that the actual Fe contents in the X%-TiO2 nanosheets were almost identical to the nominal contents of Fe doping, indicating the good control of the Fe contents in TiO2 nanosheets by a facile hydrothermal synthesis.
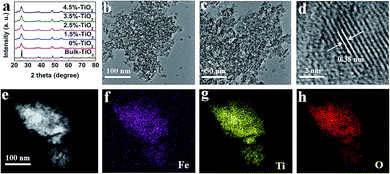 |
| | Fig. 1 (a) XRD patterns for different X%-TiO2 nanosheets (X = 0, 1.5, 2.5, 3.5, 4.5) and bulk-TiO2. (b–d) HRTEM images of 3.5%-TiO2 nanosheets at different magnifications. (e) STEM image and the corresponding EDX elemental mappings of (f) Fe, (g) Ti and (h) O in 3.5%-TiO2 nanosheets. | |
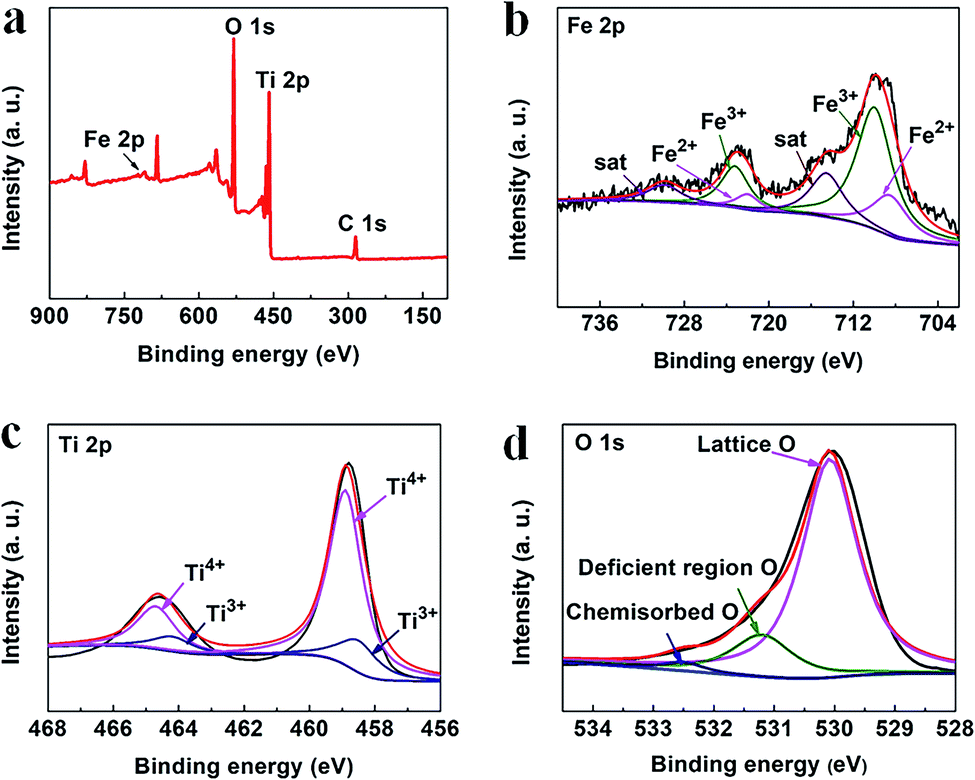
The chemical states of the elements present in the 3.5%-TiO2 were analyzed by X-ray photoelectron spectroscopy (XPS) (Fig. 2a). As shown in Fig. 2b, the main peaks located at around 709.58 and 722.89 eV were linked to the Fe 2p3/2 and Fe 2p1/2, respectively.27,28 They can be further fitted with six peaks: two peaks locating at 710 and 723.20 eV assigned to Fe3+ with the satellite peaks at 714.53 and 729.89 eV as the fingerprint of Fe3+,28,29 and the additional peaks at 708.50 and 722.00 eV associated with Fe2+.30 The XPS data for Ti 2p (Fig. 2c) showed the characteristic Ti 2p3/2 peak at 458.91 eV and 458.63 eV, corresponded to Ti4+ and Ti3+, and the other two Ti 2p1/2 peaks at 464.73 eV and 464.27 eV belonged to Ti4+ and Ti3+, respectively.31 The presence of Ti3+ probably resulted from Ti4+ gaining electrons from nearby Vo.31 For the O 1s spectrum (Fig. 2d), three peaks at 530.07 eV, 531.21 eV, 532.42 eV could be identified by spectral deconvolution, which were assigned to lattice oxygen, oxygen-deficient region, and chemisorbed oxygen species, respectively.32,33 According to XPS results, it could be inferred that Vo might be present in the ultrathin nanosheets and mixed-valent iron ions were potential to be converted into each other during the photochemical reaction.27,34
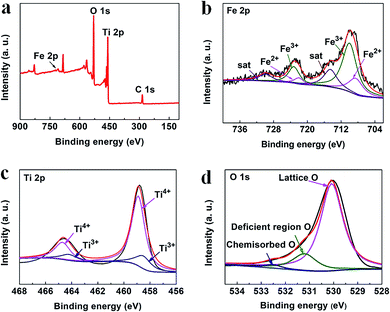 |
| | Fig. 2 XPS spectra of (a) survey, (b) Fe 2p, (c) Ti 2p, and (d) O 1s in the 3.5%-TiO2 nanosheets. | |
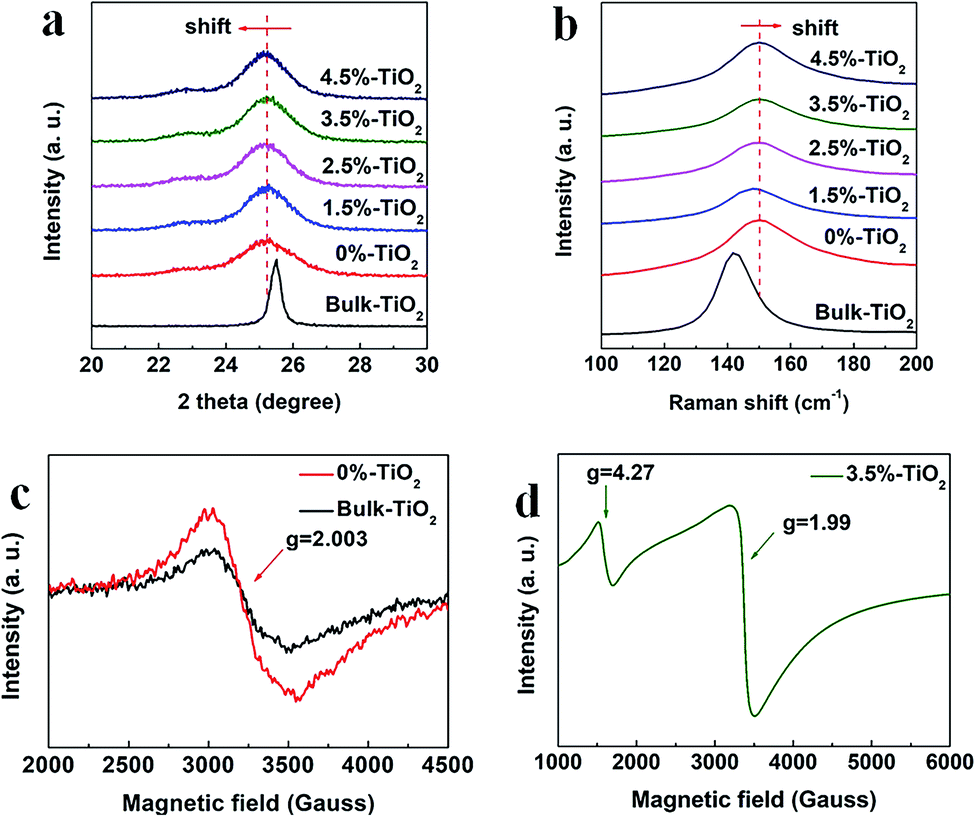
Compared to the bulk TiO2, the ultrathin TiO2 nanosheets intrinsically possess more defect structures (e.g. Vo) for decreasing surface energy of nanomaterials in theory, which was further verified by a series of characterizations. Based on the detail analysis of XRD patterns (Fig. 3a), it was found that the anatase (101) diffraction peak broadened and weakened relative to the bulk counterpart, reflecting the ultrathin TiO2 nanosheets (0%-TiO2) with relatively smaller and thinner nanostructures. Furthermore, the anatase (101) reflection for 0%-TiO2 shifted to lower 2θ angle than that for the bulk sample, such detectable variation could be explained as the presence of local structural distortion and Vo in the 0%-TiO2 nanosheets as reported previously.35 However, with the introduction of iron ions into TiO2 nanosheets, merely a slight shift of the (101) reflection peak was detected (Fig. 1a and 3a), it can be inferred that Fe doping would not induce the formation of Vo in the ultrathin TiO2 nanosheets. The anatase structures were further investigated by the Raman spectroscopy (Fig. 3b and S3†). The typical Raman peaks and consistent active modes for all the samples were observed at 143 cm−1, 515 cm−1 and 638 cm−1, respectively.18,35 Noticeably, the variation in the position and width of Raman peak for the anatase was related to quantum size effect of nanomaterials.35 In the case of 0%-TiO2 nanosheets, the Raman peak at 143 cm−1 was found to broaden and shift toward high frequency shift relative to that of Bulk-TiO2 (Fig. 3b), which could be interpreted to the effect of surface strain and Vo in the ultrathin nanosheets.18,35 As for the Fe-doped TiO2 nanosheets (X%-TiO2, X = 1.5, 2.5, 3.5, 4.5), Raman modes for iron bonds and obvious change in Raman peaks were not observed, which validated that Fe doping could not induce phase separation and additional Vo formation in ultrathin TiO2 nanosheets. The existence of Vo in X%-TiO2 was subsequently probed by electron paramagnetic resonance (EPR) spectroscopy. As displayed in the Fig. 3c, 0%-TiO2 exhibited distinguishing EPR signal at approximately g = 2.003 associated with Vo,18 further consolidating the existence of Vo in 0%-TiO2 nanosheets (in agreement with the XRD and Raman findings above). Moreover, compared with Bulk-TiO2, 0%-TiO2 exhibited higher EPR signal at g = 2.003 (Fig. 3c) and higher oxygen-deficient region at 531.21 eV in the O 1s XPS spectra (Fig. S4a and b†),32,33 which further proved that the concentration of Vo in 0%-TiO2 nanosheets is higher than that of bulk TiO2. The EPR spectrum of 3.5%-TiO2 nanosheets showed two distinct Fe3+ signals at g = 4.27 and 1.99 (Fig. 3d).36,37 These two signals could be ascribed to Fe3+ substituted for Ti4+ in the TiO2 lattice (g = 1.99) and to Fe3+ substituted in the lattice nearby a charge-compensating anion vacancy (g = 4.27).36–38 The abundance of Vo and its structural modulation by Fe doping were anticipated to facilitate photo-generated charge separation/transfer and reactants activation, thus providing the favorable requirements for enhanced photocatalytic activity.
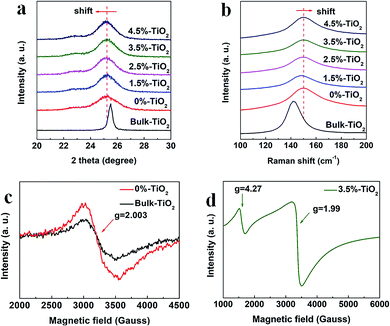 |
| | Fig. 3 Enlarged view of (a) XRD patterns and (b) Raman spectra for X%-TiO2 nanosheets and Bulk-TiO2. EPR spectra for (c) 0%-TiO2, Bulk-TiO2 and (d) 3.5%-TiO2 nanosheets. | |
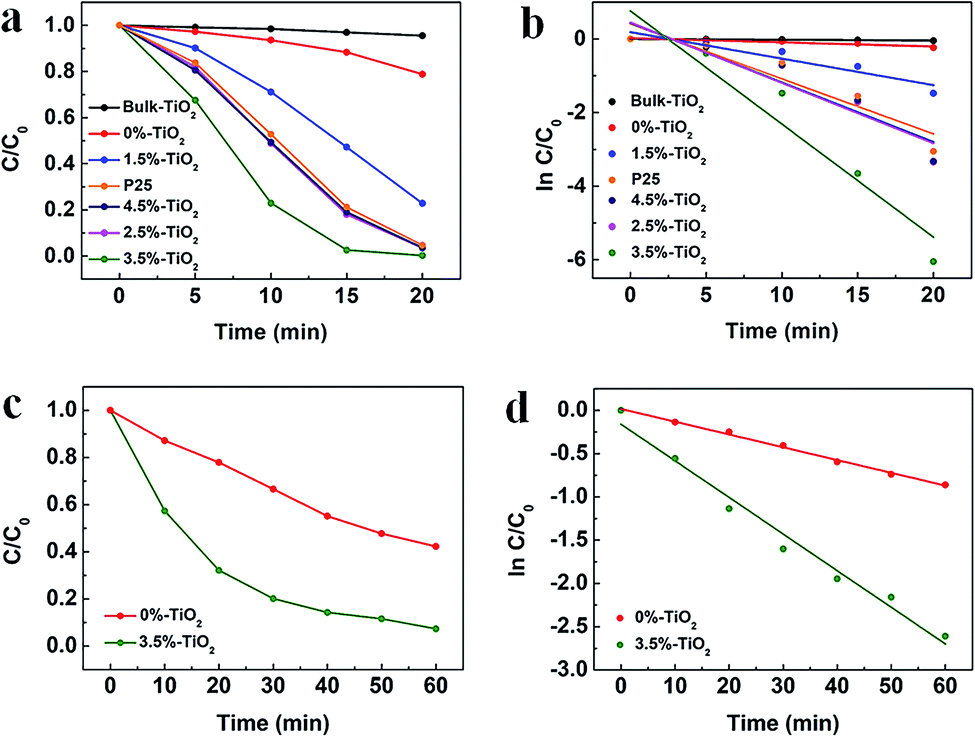
To explore the role of Vo and Fe doping in photocatalytic reactions, we evaluated the photocatalytic performance of X%-TiO2 nanosheets, Bulk-TiO2 and Degussa P25 for dye/antibiotics decomposition under UV-vis light irradiation (Fig. 4). Compared with Bulk-TiO2, 0%-TiO2 nanosheets with rich Vo showed a 5-fold increase in the photocatalytic degradation rate of Rhodamine B (RhB) (Fig. 4a), proving the key role of Vo in accelerating the photocatalytic process. With the introduction of iron ions, the photocatalytic performance of X%-TiO2 nanosheets was further improved, with the RhB degradation rate following the order: 3.5%-TiO2 > 2.5%-TiO2 > 4.5%-TiO2 > P25 > 1.5%-TiO2 > 0%-TiO2 > Bulk-TiO2. The optimized 3.5%-TiO2 realized the almost complete degradation of RhB in only 20 minutes with the removal efficiency of 99.53%, and the corresponding degradation rate as high as 0.3073 min−1 (the plot slope) that were 134 times higher than Bulk-TiO2, twice higher than benchmark P25 (Fig. 4b), and was superior to most of reported photocatalysts (Table S2†). Moreover, the strategy of defect-enhanced photocatalytic activity can be further applied to antibiotics disintegration. The 3.5%-TiO2 nearly decomposed tetracycline hydrochloride (TC-HCl) in 60 minutes photocatalytic reaction (removal efficiency = 92.65%, degradation rate = 0.0423 min−1) (Fig. 4c and d). To examine the photocatalytic stability of 3.5%-TiO2 nanosheets, the recycling experiments for the degradation of RhB and TC-HCl solution were performed. As shown in Fig. S5,† the 3.5%-TiO2 photocatalyst showed no obvious decrease in RhB and TC-HCl photodegradation activity over successive four photocatalytic cycles, indicating the outstanding photocatalytic stability of 3.5%-TiO2. Considering that specific surface area may also contribute to the photocatalytic performance, Brunauer–Emmett–Telle (BET) specific surface area of each sample was measured and used to normalize the photocatalytic performance. As shown in Fig. S6a and b,† the specific surface area of all the X%-TiO2 nanosheets were larger than that of Bulk-TiO2, in which 3.5%-TiO2 nanosheets possessed the highest specific surface area (198.85 m2 g−1) that was approximately 2.8 times higher than Bulk-TiO2 (71.40 m2 g−1), but it was not sufficient to account for the 134-fold increase in photocatalytic degradation rate. Consequently, it would be safe to conclude that specific surface area of each photocatalyst was not the decisive factor dictating the photocatalytic performance, whilst Vo and Fe doping made great contribution to superior photocatalytic performance.
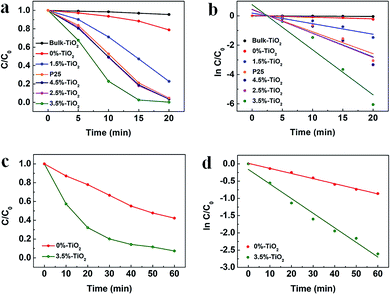 |
| | Fig. 4 (a) Time profiles of RhB degradation and (b) the corresponding plot of ln[C/C0] vs. time for X%-TiO2 nanosheets, Bulk-TiO2 and P25. The solid lines indicate the linear fittings to ln[C/C0] = −kt + α. (c) Time profiles of TC-HCl degradation and (d) the corresponding plot of ln[C/C0] vs. time for 3.5%-TiO2 nanosheets and Bulk-TiO2. (C and C0 present the initial concentration before irradiation and the residual concentration of RhB and TC-HCl). | |
To probe the role of Vo and Fe doping in photocatalytic process, we examined three principal properties: light absorption, charge separation/transfer and surface reaction. The light absorption capacity of photocatalysts determines the generation (amount) of charge carriers. As evidenced by UV-vis diffuse reflectance spectra (UV-DRS) (Fig. S7a and b†), ultrathin 0%-TiO2 nanosheets with plentiful Vo displayed stronger light absorption than Bulk-TiO2, and Fe doping can further enhance visible-light absorption of ultrathin TiO2 nanosheets, which could be ascribed to the interaction between Fe3+ d electrons and the CB or VB of TiO2.39–41 In brief, Vo and Fe doping were verified to improve light absorption properties of anatase, enabling efficient generation of photo-excited charge carriers.
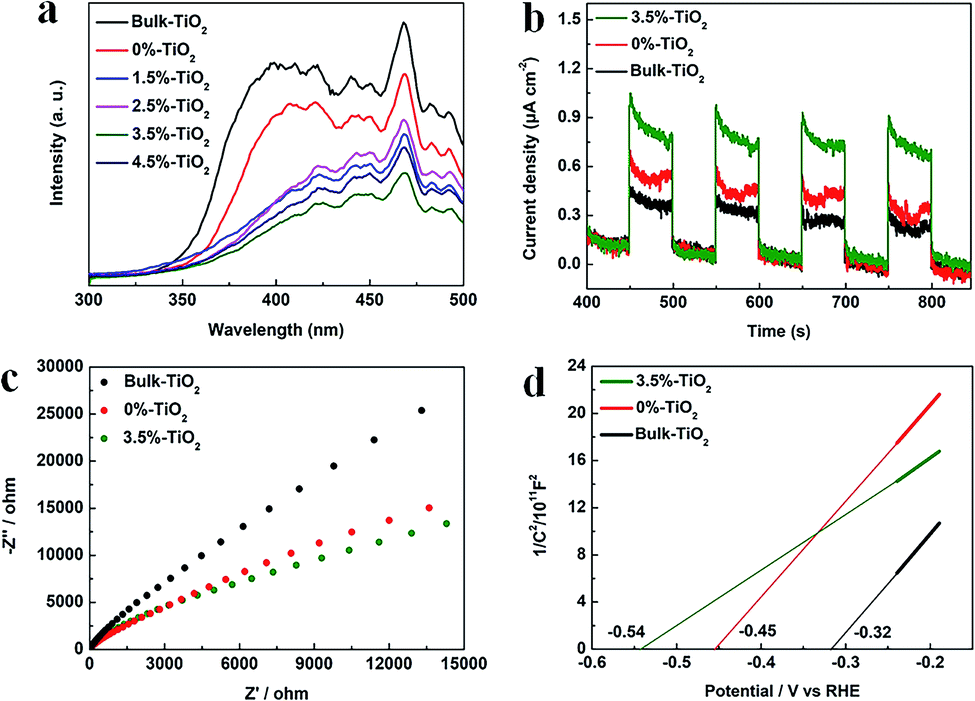
The next step, the separation and transfer of photo-excited charge carriers, is of great significance to improve the availability of surface electrons and holes, thereby dominating overall photocatalytic reaction rate. The photoluminescence (PL) data for X%-TiO2 nanosheets and Bulk-TiO2 were initially collected to investigate the role of Vo and Fe doping in this regard (Fig. 5a). The emission bands of all the samples were in the wavelength of 350–500 nm, which could be explained as the luminescence from localized surface states due to electron–hole recombination.42 Compared with Bulk-TiO2, all the X%-TiO2 nanosheets showed the depressed emission bands and the intensity of PL signals of X%-TiO2 weakened with increasing Fe doping (the weakest PL peak appeared in the 3.5%-TiO2). It turned out that Vo and Fe doping could effectively inhibit the photo-excited electron–hole recombination. To evaluate charge separation/transfer in anatase TiO2, photocurrent responses and electrochemical impedance spectrum (EIS) experiments were performed (Fig. 5b and c), with the result that both 3.5%-TiO2 and 0%-TiO2 exhibited higher photocurrent density and smaller semicircle of Nyquist plots than that seen for Bulk-TiO2 under almost identical testing conditions, corroborating the imperative role of Vo and Fe doping in facilitating separation and migration of photo-excited electrons and holes. The energy band structure of photocatalysts was also a non-negligible factor affecting photocatalytic performance. Subsequently, Mott–Schottky (MS) analyses were implemented to obtain the flat band potentials for Bulk TiO2, 0%-TiO2 and 3.5%-TiO2. The flat band potential of 3.5%-TiO2 was calculated to be −0.54 V vs. the reversible hydrogen electrode (RHE), which was more negative than that determined for 0%-TiO2 (−0.45 V vs. RHE) and Bulk-TiO2 (−0.32 V vs. RHE) (Fig. 5d). It pointed to that Vo and Fe doping could up-shift Fermi level and CB minimum of anatase TiO2, thus imparting TiO2 with high reduction potential of photo-excited electrons that were beneficial to activate O2 molecules for efficient generation of ˙O2− radicals.
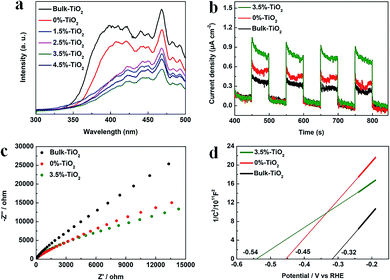 |
| | Fig. 5 (a) PL spectra of X%-TiO2 nanosheets and Bulk-TiO2. (b) Photocurrent responses of 3.5%-TiO2, 0%-TiO2 and Bulk-TiO2 under UV-vis illumination (c) EIS Nyquist plots and (d) Mott–Schottky plots for 3.5%-TiO2, 0%-TiO2 and Bulk-TiO2. | |
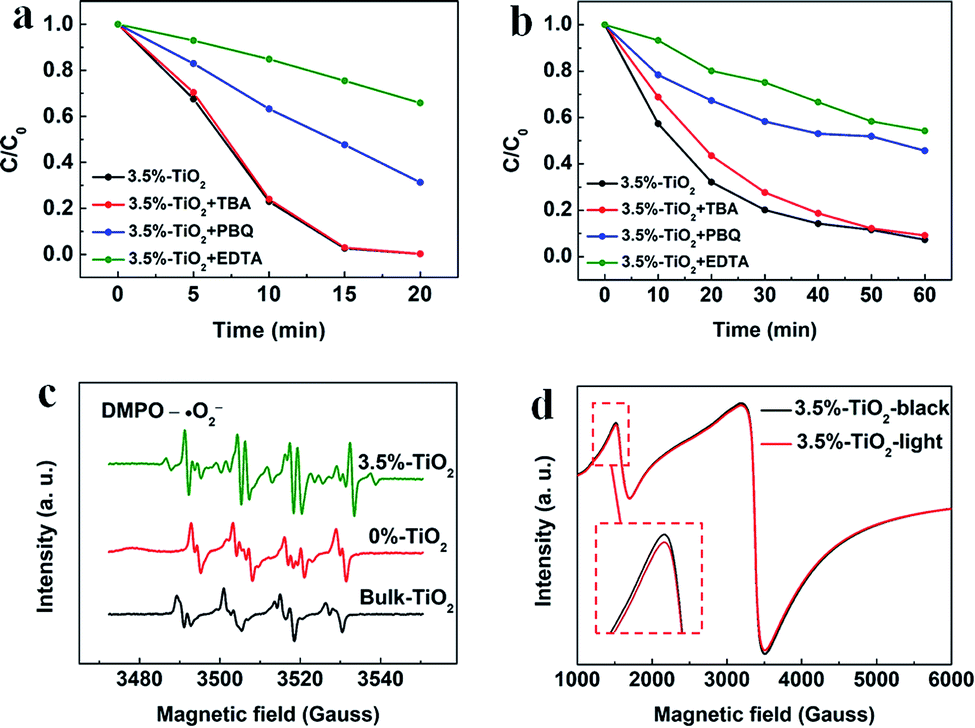
Considering potential mechanism of photocatalytic reaction, main oxidative species in the 3.5%-TiO2 photocatalytic system were detected through trapping experiments. It was found that the photocatalytic performance of 3.5%-TiO2 nanosheets varied with the addition of specific scavengers. In Fig. 6a and b, the photocatalytic performance was apparently suppressed in the presence of PBQ (˙O2− capture) or EDTA (h+ capture), highlighting the pivotal role of ˙O2− and h+ in accelerating the degradation of RhB and TC-HCl, while ˙OH may not directly participate in degradation reaction, due to trivial change in degrading rate when using TBA (˙OH scavenger). Band alignment for 3.5%-TiO2 indicated that both ˙O2− and the photo-generated holes of 3.5%-TiO2 possess sufficient oxidation ability to oxidize RhB and TC-HCl (Fig. S8†), further corroborating the results of trapping experiments. On the basis of discussion above, we assumed that upon UV-vis irradiation, 3.5%-TiO2 could produce more active ˙O2− and h+ than that generated from 0%-TiO2 and Bulk-TiO2, which was further verified by EPR technique. As displayed in Fig. 6c, all the tested photocatalysts exhibited six characteristic peaks of DMPO-˙O2− adducts with the EPR intensity in the order of 3.5%-TiO2 > 0%-TiO2 > Bulk-TiO2, it hinted at the presence of ˙O2− in present system and the production of ˙O2− promoted by Vo and Fe doping. Combining the experimental results (Fig. 5) with corresponding discussion, Vo and Fe doping were proven to be efficient in promoting separation and transfer of photo-generated electron–hole pairs, warranting more surface holes generation. Therefore, it is reasonable to conclude that Vo and Fe doping present in 3.5%-TiO2 were responsible for boosting the production of ˙O2− and h+ that were the decisive oxidative species for degrading RhB and TC-HCl. To further reveal how Vo and Fe doping promote AOS production, we carried out EPR tests of 3.5%-TiO2 with/without light irradiation (Fig. 6d). The EPR signals at g = 4.27 and g = 1.99 were attenuated under UV-vis illumination, which could be inferred to the conversion of Fe3+ into Fe2+ during the photocatalytic process.36 We then proposed possible photocatalytic mechanisms: (1) upon UV-vis irradiation, photo-generated electrons in 3.5%-TiO2 were excited from VB to CB and holes remained in the VB; (2) Fe3+ and Vo could act as electron acceptor (the conversion of Fe3+ to Fe2+) to inhibit electron–hole recombination due to the presence of defect energy levels between VB and CB;31 (3) O2 molecules were activated by photo-generated electrons from electron-rich Fe2+ and/or Vo, then converted into ˙O2−;43,44 and H2O2 might be oxidized to ˙O2− by photogenerated h+ (Fig. S9†);45 (4) the photo-generated holes and ˙O2− were responsible for degrading RhB and TC-HCl;46 (5) partial Fe2+ turned into Fe3+ by H2O2 through Fenton-like process or by O2 though partial oxidation, finalizing a reaction cycle.47
| |
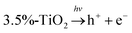 | (1) |
| | |
O2 + Fe2+ → Fe3+ + ˙O2−
| (3-2) |
| | |
H2O2 + h+ → ˙O2− + 2H+
| (3-3) |
| | |
h+ + ˙O2− + RhB/TC-HCl → degradation products
| (4) |
| |
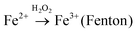 | (5) |
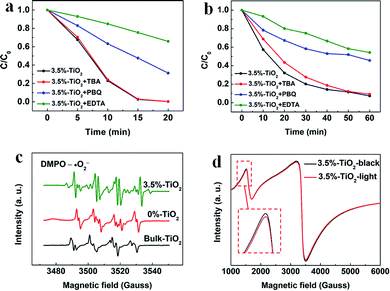 |
| | Fig. 6 The trapping experiments for photocatalytic degradation of (a) RhB and (b) TC-HCl by 3.5%-TiO2 nanosheets. (c) EPR spectra recorded for 3.5%-TiO2, 0%-TiO2 and Bulk-TiO2 in the presence of 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) without adding H2O2. (d) EPR spectra of 3.5%-TiO2 nanosheets with/without light irradiation. | |
3. Conclusions
In summary, ultrathin X%-TiO2 nanosheets with rich Vo and tunable Fe doping were prepared by a facile hydrothermal method for efficient photocatalytic degradation. Compared with Bulk-TiO2, X%-TiO2 nanosheets showed much higher photocatalytic activity in RhB degradation (degradation rate = 0.3073 min−1) and TC-HCl degradation (degradation rate of 0.0423 min−1), which could be attributed to the key role of Vo and Fe doping in promoting AOS production. The in-depth investigation and characterizations revealed that Vo and Fe doping could not only furnish anatase TiO2 with enhanced light absorption and boosted separation and migration of photo-generated electron–hole pairs, but also expedite O2 activation by abundant Vo and reactive Fe2+ with high-reduction-potential electrons for ˙O2− production. In addition, H2O2 may also be oxidized to ˙O2− by photogenerated h+. As a consequence, ˙O2− and h+ obtained on the surface of 3.5%-TiO2 were highly efficient in accelerating photocatalytic degradation of RhB and TC-HCl, as supported by radicals-trapping experiments and in situ EPR tests. This research provides an in-depth understanding of defect-promoted AOS generation and a practical design strategy of photocatalysts to optimize this process for broad photooxidation application.
Conflicts of interest
There are no conflicts to declare.
Funding statement
This work was supported by Zenghe Li as a funder, corresponding author at Beijing Key Laboratory of Environmentally Harmful Chemical Analysis, College of Chemistry, Beijing University of Chemical Technology, Beijing, 100029, PR China.
Notes and references
- C. A. D'Amato, R. Giovannetti, M. Zannotti, E. Rommozzi, S. Ferraro, C. Seghetti, M. Minicucci, R. Gunnella and A. Di Cicco, Appl. Surf. Sci., 2018, 441, 575–587 CrossRef.
- H. Wang, Y. Wu, M. Feng, W. Tu, T. Xiao, T. Xiong, H. Ang, X. Yuan and J. W. Chew, Water Res., 2018, 144, 215–225 CrossRef CAS.
- N. Oturan, J. Wu, H. Zhang, V. K. Sharma and M. A. Oturan, Appl. Catal., B, 2013, 140–141, 92–97 CrossRef CAS.
- F. Deng, L. Zhao, X. Luo, S. Luo and D. D. Dionysiou, Chem. Eng. J., 2018, 333, 423–433 CrossRef CAS.
- U. Iriarte-Velasco, J. I. Álvarez-Uriarte, N. Chimeno-Alanís and J. R. González-Velasco, Ind. Eng. Chem. Res., 2008, 47, 7868–7876 CrossRef CAS.
- M. Ahmad, M. Yousaf, A. Nasir, I. A. Bhatti, A. Mahmood, X. Fang, X. Jian, K. Kalantar-Zadeh and N. Mahmood, Environ. Sci. Technol., 2019, 53, 2161–2170 CrossRef CAS.
- A. Savateev, I. Ghosh, B. König and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 15936–15947 CrossRef CAS.
- X. Sun, X. Luo, X. Zhang, J. Xie, S. Jin, H. Wang, X. Zheng, X. Wu and Y. Xie, J. Am. Chem. Soc., 2019, 141, 3797–3801 CrossRef CAS.
- H. Yang, H. Liu, Z. Hu, J. Liang, H. Pang and B. Yi, Chem. Eng. J., 2014, 245, 24–33 CrossRef CAS.
- M. Aslam, M. T. Qamar, M. T. Soomro, I. M. I. Ismail, N. Salah, T. Almeelbi, M. A. Gondal and A. Hameed, Appl. Catal., B, 2016, 180, 391–402 CrossRef CAS.
- E. Gao and W. Wang, Nanoscale, 2013, 5, 11248–11256 RSC.
- X. Pan, M.-Q. Yang, X. Fu, N. Zhang and Y.-J. Xu, Nanoscale, 2013, 5, 3601–3614 RSC.
- A. Ahmed, M. Naseem Siddique, U. Alam, T. Ali and P. Tripathi, Appl. Surf. Sci., 2019, 463, 976–985 CrossRef CAS.
- H. Xu, Y. Hu, D. Huang, Y. Lin, W. Zhao, Y. Huang, S. Zhang and Y. Tong, ACS Sustainable Chem. Eng., 2019, 7, 5784–5791 CrossRef CAS.
- B. Tryba, A. W. Morawski, M. Inagaki and M. Toyoda, Appl. Catal., B, 2006, 63, 215–221 CrossRef CAS.
- A. A. Isari, A. Payan, M. Fattahi, S. Jorfi and B. Kakavandi, Appl. Surf. Sci., 2018, 462, 549–564 CrossRef CAS.
- J.-J. Li, M. Zhang, B. Weng, X. Chen, J. Chen and H.-P. Jia, Appl. Surf. Sci., 2020, 507, 145133 CrossRef CAS.
- Y. Zhao, Y. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2019, 31, 1806482 CrossRef.
- T. Liu and H. Zhang, RSC Adv., 2013, 3, 16255–16258 RSC.
- X. Zhu, H. Xu, Y. Yao, H. Liu, J. Wang, Y. Pu, W. Feng and S. Chen, RSC Adv., 2019, 9, 40003–40012 RSC.
- M. Pera-Titus, V. García-Molina, M. A. Baños, J. Giménez and S. Esplugas, Appl. Catal., B, 2004, 47, 219–256 CrossRef CAS.
- J. Araña, O. González Díaz, M. Miranda Saracho, J. M. Doña Rodríguez, J. A. Herrera Melián and J. Pérez Peña, Appl. Catal., B, 2001, 32, 49–61 CrossRef.
- C.-C. Chen, S.-H. Hu and Y.-P. Fu, J. Alloys Compd., 2015, 632, 326–334 CrossRef CAS.
- W. Wen, J.-m. Wu, Y.-z. Jiang, S.-l. Yu, J.-q. Bai, M.-h. Cao and J. Cui, Sci. Rep., 2015, 5, 11804 CrossRef.
- C. Chen, L. Kuai, Y. Chen, Q. Wang, E. Kan and B. Geng, RSC Adv., 2015, 5, 98254–98259 RSC.
- X. Zhang, D. Li, J. Wan and X. Yu, RSC Adv., 2016, 6, 17906–17912 RSC.
- G. Cheng, X. Liu, X. Song, X. Chen, W. Dai, R. Yuan and X. Fu, Appl. Catal., B, 2020, 277, 119196 CrossRef CAS.
- J. Liu, D. Qian, H. Feng, J. Li, J. Jiang, S. Peng and Y. Liu, J. Mater. Chem. A, 2014, 2, 11372–11381 RSC.
- N. S. McIntyre and D. G. Zetaruk, Anal. Chem., 1977, 49, 1521–1529 CrossRef CAS.
- J. Xie, R. Jin, A. Li, Y. Bi, Q. Ruan, Y. Deng, Y. Zhang, S. Yao, G. Sankar, D. Ma and J. Tang, Nat. Catal., 2018, 1, 889–896 CrossRef CAS.
- H. Khan and I. K. Swati, Ind. Eng. Chem. Res., 2016, 55, 6619–6633 CrossRef CAS.
- L. Sun, R. Li, W. Zhan, Y. Yuan, X. Wang, X. Han and Y. Zhao, Nat. Commun., 2019, 10, 2270 CrossRef.
- L. Wang, M. Ghoussoub, H. Wang, Y. Shao, W. Sun, A. A. Tountas, T. E. Wood, H. Li, J. Y. Y. Loh, Y. Dong, M. Xia, Y. Li, S. Wang, J. Jia, C. Qiu, C. Qian, N. P. Kherani, L. He, X. Zhang and G. A. Ozin, Joule, 2018, 2, 1369–1381 CrossRef CAS.
- T. Wu, X. Zhu, Z. Xing, S. Mou, C. Li, Y. Qiao, Q. Liu, Y. Luo, X. Shi, Y. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 18449–18453 CrossRef CAS.
- A. Molea, V. Popescu, N. A. Rowson, I. Cojocaru, A. Dinescu, A. Dehelean and M. Lazăr, Ind. Eng. Chem. Res., 2015, 54, 7346–7351 CrossRef CAS.
- M. Graetzel and R. F. Howe, J. Phys. Chem. C, 1990, 94, 2566–2572 CrossRef CAS.
- K. Nagaveni, M. S. Hegde and G. Madras, J. Phys. Chem. B, 2004, 108, 20204–20212 CrossRef CAS.
- Q. Jin, M. Fujishima and H. Tada, J. Phys. Chem. C, 2011, 115, 6478–6483 CrossRef CAS.
- X. Yang, C. Cao, L. Erickson, K. Hohn, R. Maghirang and K. Klabunde, Appl. Catal., B, 2009, 91, 657–662 CrossRef CAS.
- Y. Cong, J. Zhang, F. Chen, M. Anpo and D. He, J. Phys. Chem. C, 2007, 111, 10618–10623 CrossRef CAS.
- M. I. Litter and J. A. Navío, J. Photochem. Photobiol., A, 1996, 98, 171–181 CrossRef CAS.
- X. Huang, L. Wang, J. Zhou and N. Gao, Water Res., 2014, 57, 1–7 CrossRef CAS.
- Y. Shi, X. Wang, X. Liu, C. Ling, W. Shen and L. Zhang, Appl. Catal., B, 2020, 277, 119229 CrossRef CAS.
- L. Zhang, Q. Liu, Y. Chai, F. Yang, M. Cao and W.-L. Dai, J. Phys. Chem. C, 2018, 122, 12900–12912 CrossRef CAS.
- T. Hirakawa and Y. Nosaka, Langmuir, 2002, 18, 3247–3254 CrossRef CAS.
- X. Zhang, L. Li, Y. Zeng, F. Liu, J. Yuan, X. Li, Y. Yu, X. Zhu, Z. Xiong, H. Yu and Y. Xie, ACS Appl. Nano Mater., 2019, 2, 7255–7265 CrossRef CAS.
- J. Wang, Z. Liu† and R. Cai, Environ. Sci. Technol., 2008, 42, 5759–5764 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08116g |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Shiqi Xua and
Zenghe Li
c,
Shiqi Xua and
Zenghe Li