
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePycnidiophorones A–D, four new cytochalasans from the wetland derived fungus Pycnidiophora dispersa†
Chen Zhaoa,
Gaoran Liub,
Xingzhong Liub,
Lan Zhanga,
Lin Li*a and
Ling Liu *b
*b
aDepartment of Pharmacy, Xuanwu Hospital of Capital Medical University, National Clinical Research Center for Geriatric Diseases, Beijing Engineering Research Center for Nervous System Drugs, Beijing Institute for Brain Disorders, Key Laboratory for Neurodegenerative Diseases of Ministry of Education, Beijing 100053, China. E-mail: linlixw@126.com
bState Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, People's Republic of China. E-mail: liul@im.ac.cn
First published on 5th November 2020
Abstract
Pycnidiophorones A–D (1–4), four new cytochalasans with a rare 5/6/6/5/6 pentacyclic skeleton incorporating the unique 12-oxatricyclo[6.3.1.02,7]dodecane core, and six known depsidones (5–10) were isolated from cultures of the wetland-soil-derived fungus Pycnidiophora dispersa. Their chemical structures were unambiguously determined using NMR spectroscopic data. The absolute configurations of 1 and 3 were assigned by electronic circular dichroism (ECD) calculations. Compounds 1–10 showed moderate cytotoxicity against a panel of five human tumor cell lines.
Introduction
Fungi are important sources of bioactive natural products, which play a significant role in research and development of new drugs or lead compounds.1 Fungal species from unique ecological niches are more likely to produce structurally unique and biologically active secondary metabolites, presumably due to their highly adapted metabolic systems that evolved during the natural selection process.2–6 On the basis of this consideration and the documented success in finding new bioactive natural products from special types of fungi, we initiated chemical studies of the fungal species isolated from wetland environments, which have the special dual characteristics of water and land. As an indispensable part of wetland ecosystems, microorganisms including fungi play an irreplaceable role in energy flow and material transformation. Moreover, secondary metabolites from wetland-derived fungi showed a wide range of bioactivities including antibacterial, antifungal, cytotoxic, and α-glucosidase inhibitory activities.7–14 Our prior investigations of the wetland-derived fungus Paraconiothyrium sporulosum grown in liquid-substrate fermentation cultures led to isolation of a series of bioactive metabolites.15 As part of our program to discover structurally unique and biologically active secondary metabolites from wetland-derived fungi, a strain of Pycnidiophora dispersa, isolated from the Baiyangdian Lake, Hebei Province, People's Republic of China, was subjected to scale-up fermentation on rice. Fractionation of the EtOAc extract prepared from the cultures afforded four new 5/6/6/5/6 pentacyclic cytochalasans, pycnidiophorones A–D (1–4), and six known depsidones (5–10; Fig. 1). Details of the isolation, structure elucidation, and cytotoxicity of these compounds are reported herein.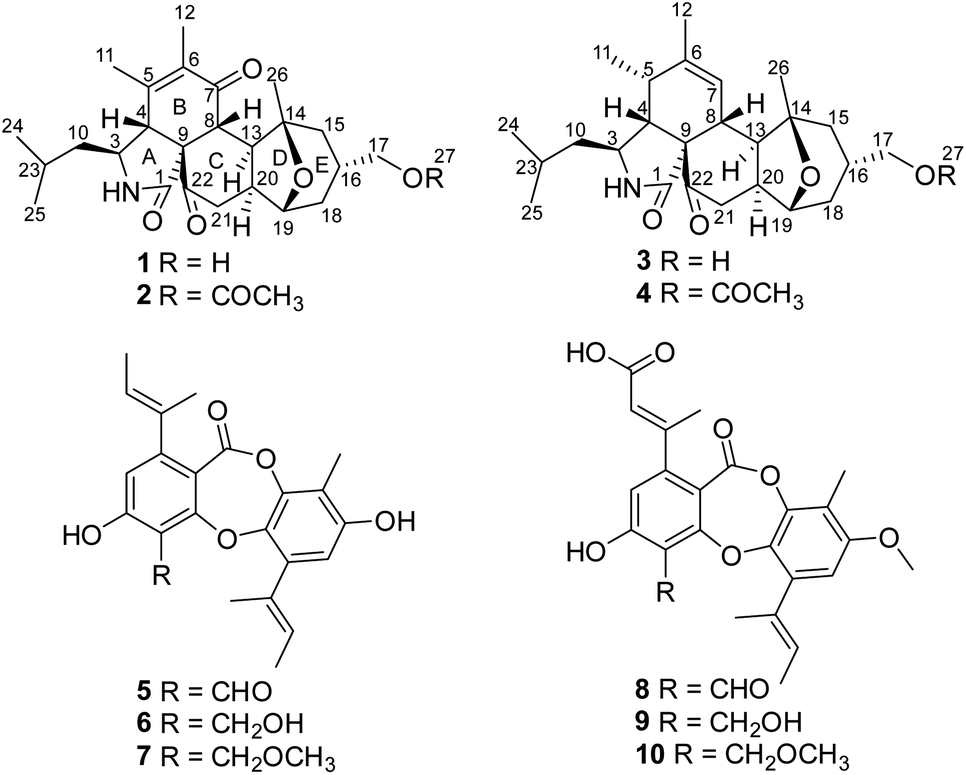 | ||
| Fig. 1 Structures of compounds 1–10. | ||
Results and discussion
The molecular formula of pycnidiophorone A (1) was determined to be C25H35NO5 (9 degrees of unsaturation) on the basis of HRESIMS. Analysis of its nuclear magnetic resonance (NMR) data (Table 1) revealed the presence of one exchangeable proton (–NH–, δH 5.85), five methyl groups, five methylene units (one oxygenated), eight methines (one oxygenated and one nitrogenated), two sp3 quaternary carbons with one oxygenated, two olefinic carbons, one amide carbon (δC 170.8), one α,β-unsaturated ketone carbon (δC 198.7) and one ketone carbon (δC 208.1). These data accounted for all 1H and 13C resonances except for one exchangeable proton, suggesting that 1 was a pentacyclic compound. The 1H–1H COSY NMR data (Fig. 2) of 1 gave the C-4–C-3–C-10–C-23–C-24–C-25, C-8–C-13–C-20–C-21 and C-15–C-16–C-17–C-18–C-19–C-20 three proton spin systems, and the remaining connection was established by HMBC correlations (Fig. 2). HMBC correlations from H-3 to C-1, H-4 to C-1 and C-3, and from 2-NH to C-4 and C-9 completed α-ketopyrrolidine unit (ring A) with isobutyl attached at C-3. The correlations from H-4 to C-5 and C-6, H-8 to C-1, C-7 and C-9, H3-11 to C-4, C-5 and C-6, and from H3-12 to C-5, C-6 and C-7 permitted the completion of cyclohexenone unit (ring B) with two methyls located at C-5 and C-6, respectively, establishing the isoindole-1,6(2H)-dione substructure. Other HMBC cross-peaks from H-4, H-8 and H-20 to the ketone carbon C-22 (δC 208.1), and from H2-21 to C-9 and C-22 implied that C-22 was located between C-9 and C-21 to form cyclohexanone subunit (ring C) fused with the isoindole-1,6(2H)-dione at C-8 and C-9. Further analysis of the HMBC correlations from H-8 to C-14, H-13 and H3-26 to C-14 and C-15, and from H2-15 to C-13 and C-26 connected both C-13 and C-26 to C-14, completing the cycloheptane moiety fused the cyclohexanone at C-13 and C-20. Considering the chemical shift values for C-14 (δC 81.0) and C-19 (δC 78.5) and the unsaturation requirement for 1, the two carbons were attached to the same oxygen to form 8-oxabicyclo[3.2.1]octane moiety (rings D and E) by default, which was further confirmed by an key HMBC correlation from H-19 to C-14. The exchangeable proton was located at C-17 by default, which supported by the chemical shift value for C-17 (δC 67.2). Thus the planar structure of 1 was established.| Pos. | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δCa, mult. | δHb (J in Hz) | δCc, mult. | δHd (J in Hz) | δCe, mult. | δHf (J in Hz) | δCc, mult. | δHd (J in Hz) | |
| a Recorded at 125 MHz in CDCl3.b Recorded at 500 MHz in CDCl3.c Recorded at 150 MHz in CDCl3.d Recorded at 600 MHz in CDCl3.e Recorded at 100 MHz in CDCl3.f Recorded at 400 MHz in CDCl3. | ||||||||
| 1 | 170.8, qC | 170.6, qC | 173.6, qC | 173.7, qC | ||||
| 2-NH | 5.85, br, d | 5.88, br, d | 5.80, s | 6.28, s | ||||
| 3 | 56.6, CH | 3.26, d (8.9) | 56.5, CH | 3.25, m | 51.9, CH | 3.09, m | 51.9, CH | 3.06, m |
| 4 | 50.9, CH | 3.13, br, s | 50.7, CH | 3.13, br, s | 51.7, CH | 2.66, t (4.3) | 51.8, CH | 2.62, t (4.4) |
| 5 | 146.8, qC | 146.8, qC | 35.0, CH | 2.36, m | 35.1, CH | 2.33, m | ||
| 6 | 132.4, qC | 132.3, qC | 139.7, qC | 139.9, qC | ||||
| 7 | 198.7, qC | 198.2, qC | 127.3, CH | 5.49, s | 127.2, CH | 5.44, br, s | ||
| 8 | 45.8, CH | 2.93, d (11.3) | 45.8, CH | 2.89, d (11.2) | 36.8, CH | 2.42, d (12.2) | 36.8, CH | 2.36, br, d (12.0) |
| 9 | 57.7, qC | 57.8, qC | 63.9, qC | 64.2, qC | ||||
| 10a | 46.4, CH2 | 2.09, m | 46.4, CH2 | 2.07, m | 47.4, CH2 | 1.70, m | 47.5, CH2 | 1.66, m |
| 10b | 1.61, m | 1.57, m | 1.32, m | 1.24, m | ||||
| 11 | 18.6, CH3 | 1.90, s | 18.6, CH3 | 1.89, s | 13.6, CH3 | 1.16, d (7.2) | 13.7, CH3 | 1.12, d (7.2) |
| 12 | 12.4, CH3 | 1.80, s | 12.4, CH3 | 1.78, s | 20.2, CH3 | 1.79, s | 20.3, CH3 | 1.76, br, s |
| 13 | 39.7, CH | 3.43, m | 40.1, CH | 3.46, m | 45.0, CH | 2.89, dd (11.9, 9.0) | 45.4, CH | 2.90, dd (12.0, 8.7) |
| 14 | 81.0, qC | 80.9, qC | 81.0, qC | 80.9, qC | ||||
| 15a | 39.9, CH2 | 1.98, m | 40.4, CH2 | 1.91, m | 40.2, CH2 | 1.91, dd (14.1, 8.2) | 40.8, CH2 | 1.90, dd (14.2, 8.4) |
| 15b | 1.88, m | 1.76, d (14.1) | 1.61, dd (14.2, 1.6) | |||||
| 16 | 31.8, CH | 2.11, m | 28.5, CH | 2.22, m | 31.8, CH | 2.13, m | 28.7, CH | 2.19, m |
| 17a | 67.2, CH2 | 3.85, m | 68.4, CH2 | 4.36, m | 66.8, CH2 | 3.82, m | 68.3, CH2 | 4.24, m |
| 17b | 4.20. m | |||||||
| 18a | 29.7, CH2 | 2.08, m | 29.9, CH2 | 2.07, m | 29.8, CH2 | 2.09, dd (13.9, 8.4) | 30.1, CH2 | 2.06, dt (14.2, 5.3) |
| 18b | 1.53, m | 1.46, m | 1.56, d (13.9) | 1.49, d (14.2) | ||||
| 19 | 78.5, CH | 3.82, m | 77.9, CH | 3.81, d (4.7) | 79.0, CH | 3.84, d (3.3) | 78.7, CH | 3.81, d (5.3) |
| 20 | 41.1, CH | 2.90, m | 41.4, CH | 2.96, m | 41.7, CH | 3.01, ddd (12.7, 6.9, 6.9) | 41.9, CH | 3.02, ddd (12.2, 8.7, 5.1) |
| 21a | 43.0, CH2 | 2.61, dd (17.0, 5.0) | 42.8, CH2 | 2.61, dd (17.1, 4.6) | 43.2, CH2 | 2.56, m | 43.2, CH2 | 2.52, m |
| 21b | 2.51, dd (17.0, 13.0) | 2.48, dd (17.1, 12.9) | ||||||
| 22 | 208.1, qC | 207.9, qC | 210.8, qC | 210.7, qC | ||||
| 23 | 25.2, CH | 1.60, m | 25.2, CH | 1.58, m | 25.4, CH | 1.55, m | 25.3, CH | 1.53, m |
| 24 | 23.4, CH3 | 0.99, d (6.0) | 23.4, CH3 | 0.97, d (5.5) | 23.7, CH3 | 0.97, d (6.5) | 23.9, CH3 | 0.91, d (6.5) |
| 25 | 21.8, CH3 | 0.96, d (5.9) | 21.8, CH3 | 0.94, d (5.9) | 21.3, CH3 | 0.93, d (6.5) | 21.3, CH3 | 0.88, d (6.5) |
| 26 | 22.5, CH3 | 1.05, s | 22.5, CH3 | 1.04, s | 23.9, CH3 | 1.23, s | 24.0, CH3 | 1.19, s |
| 27 | 171.0, qC | 170.9, qC | ||||||
| 28 | 21.0, CH3 | 2.04, s | 21.2, CH3 | 2.03, s | ||||
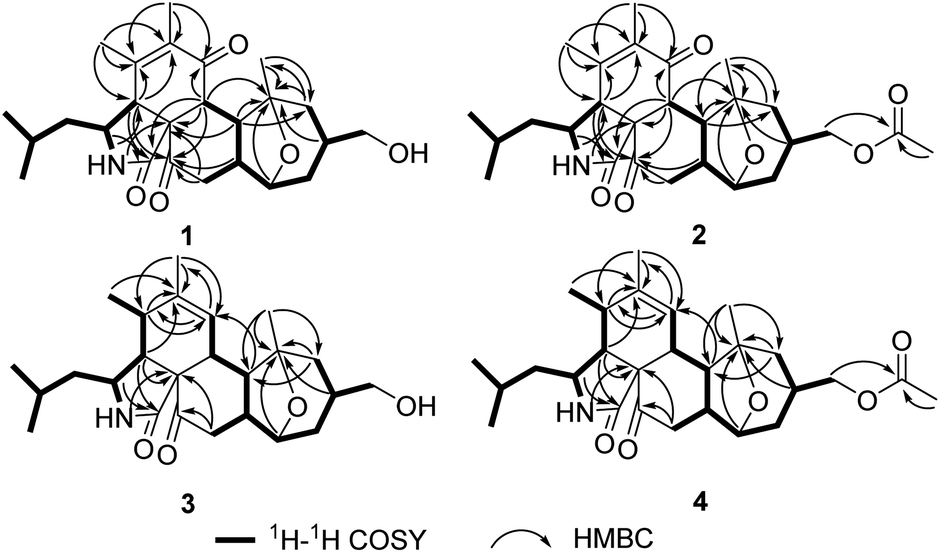 | ||
| Fig. 2 Key 1H–1H and HMBC correlations of 1–4. | ||
The relative configuration was determined by analysis of the 1H–1H coupling constants and NOESY correlations (Fig. 3). The large trans-diaxial-type J values of 11.3 Hz for H-8/H-13 revealed their axial orientations. NOESY correlations of H-8 with H-4 and H3-26, and of H-4 with H2-10 determined the relative configurations of the isoindole-1,6(2H)-dione moiety which are the same as their counterparts in the trichoderone B.16 The other NOESY correlations of H-13 with H2-17, H-19 and H-20, and of H2-17 with H-20 indicated that the fused and bridged rings were oriented in a trans fashion about the central tetrahydropyran ring, thus establishing the relative configuration of 1.
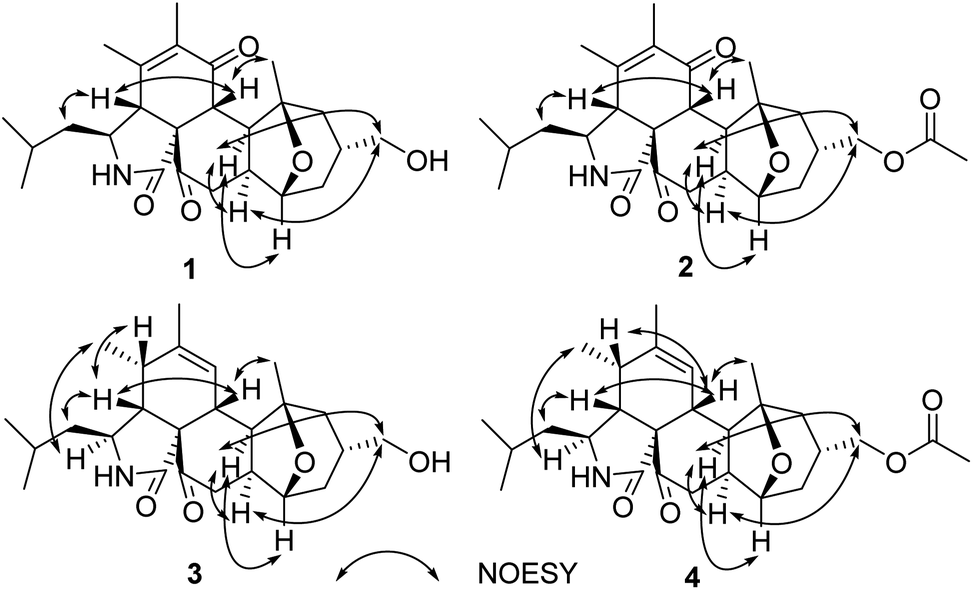 | ||
| Fig. 3 Key NOESY correlations of 1–4. | ||
The absolute configuration of 1 was assigned by comparison of the experimental and simulated ECD spectra generated by the time dependent density functional theory (TDDFT) for two enantiomers (3S,4R,5Z,8R,9R,13S,14S,16R,19R,20S)-1 (1a) and (3R,4S,5Z,8S,9S,13R,14R,16S,19S,20R)-1 (1b). The MMFF94 conformational search and DFT re-optimization at the B3LYP/6-311G(d,p) level yielded one significant conformer for each configuration (Fig. S25†). The overall calculated ECD spectra of 1a and 1b were then generated by Gaussian broadening (Fig. 4). The experimental ECD spectrum of 1 was nearly identical to the calculated ECD spectrum for 1a, suggesting the 3S,4R,5Z,8R,9R,13S,14S,16R,19R,20S absolute configuration for 1.
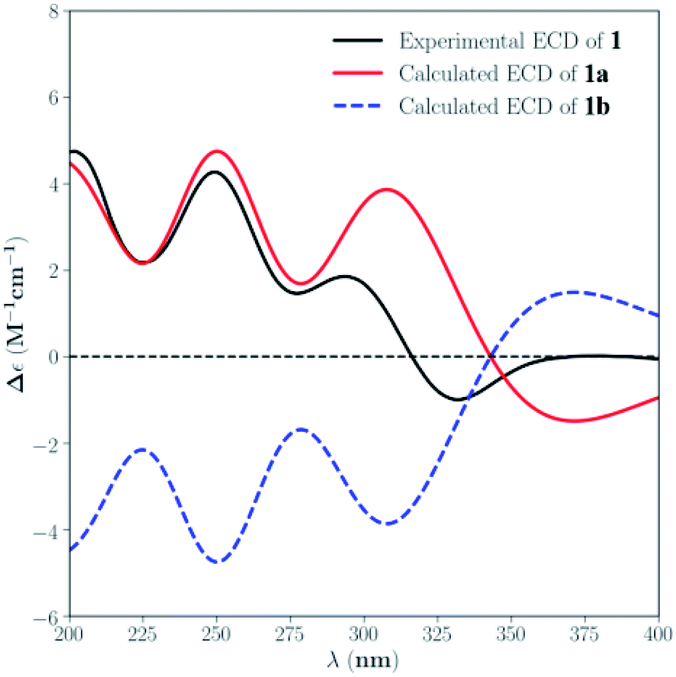 | ||
| Fig. 4 Experimental ECD spectrum of 1 in MeOH and the calculated ECD spectra of 1a and 1b. | ||
The molecular formula of pycnidiophorone B (2) was determined to be C27H37NO6 (10 degrees of unsaturation) based on HRESIMS and the NMR data (Table 1), which is 42 mass units higher than 1. Analysis of the 1H and 13C NMR data for 2 revealed the presence of structural features similar to those found in 1, except that the oxygenated methylene protons signals (H2-17) at 3.85 ppm was significantly downfield (δH 4.36 and 4.20). In addition, the NMR resonances corresponding to an acetyl group (δC/δH 21.0/2.04; 171.0) were observed, indicating that the C-17 oxygen of 2 is acylated, which was supported by HMBC correlations (Fig. 2) from H2-17 to the carboxylic carbon (δC 171.0) of the acetyl group. Therefore, 2 was determined as the C-17 monoacetate of 1. The relative configuration of 2 was deduced as shown by analysis of the 1H–1H coupling constants and NOESY correlations (Fig. 3) and by analogy to 1. The ECD spectrum of 2 was nearly identical to that of 1 (Fig. S31 and S32†), indicating that the absolute configuration of 2 was the same as that of 1.
The molecular formula of pycnidiophorone B (3) was determined to be C25H37NO4 (8 degrees of unsaturation) based on HRESIMS and NMR data (Table 1), which was 14 mass units less than that of 1. Analysis of its NMR data revealed the presence of the same partial structure (rings A, C–E) as that found in 1, except that those corresponding to the cyclohexenone ring (ring B) in 1 were different in 3. Specifically, the resonances for the conjugated ketone subunit (C-5–C-7) of ring B in 1 were replaced by those for a methine unit (δH/δC 2.36/35.0, C-5) and one C-6/C-7 olefin (δH/δC 5.49/127.3; 139.7) in the spectra of 3, indicating that the double bond at C-5/C-6 was transferred to C-6/C-7 to form the cyclohexene ring. Such observation was also confirmed by relevant 1H–1H COSY and HMBC correlations (Fig. 2) from H-5 to C-6 and C-7, H-7 to C-5 and C-12, H3-12 to C-5, C-6 and C-7, and from H-13 to C-7. On the basis of these data, the gross structure of 3 was established as shown.
By comparison of their 1H–1H coupling constants (Table 1) and NOESY data (Fig. 3), compound 3 was deduced to have the same relative configuration as 1 except for the newly formed chiral center C-5. The relative configuration of C-5 was further assigned as shown by NOESY correlations (Fig. 3) of H-3 with H3-11, H-4 with H-5 and H2-10, and of H-5 with H-8.
The absolute configuration for 3 was also deduced by comparison of the experimental and calculated ECD spectra for the enantiomers (3R,4S,5R,6Z,8S,9R,13S,14R,16S,19S,20R)-3 (3a) and (3S,4R,5S,6Z,8R,9S,13R,14S,16R,19R,20S)-3 (3b). The MMFF94 conformational search followed by B3LYP/6-311G(d,p) DFT reoptimization afforded the lowest energy conformers, which were further filtered based on the Boltzmann-population rule, resulting in one significant conformer for each configuration (Fig. S26†). The experimental ECD spectrum of 3 matched well to the calculated curve of 3b (Fig. 5), suggesting the 3S,4R,5S,6Z,8R,9S,13R,14S,16R,19R,20S absolute configuration.
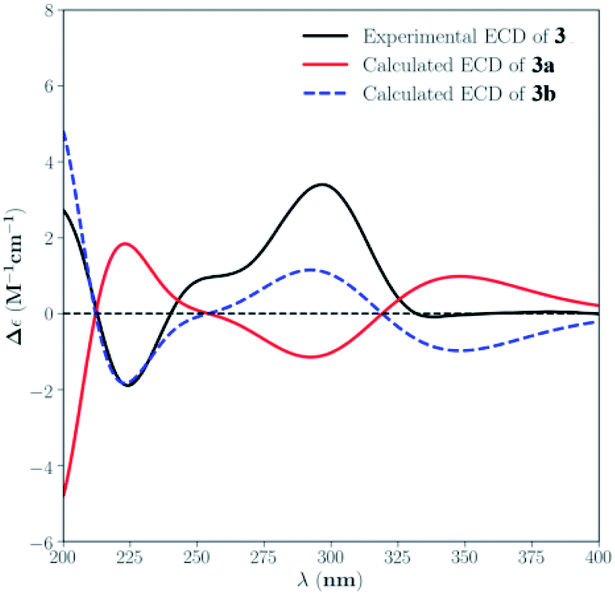 | ||
| Fig. 5 Experimental ECD spectrum of 3 in MeOH and the calculated ECD spectra of 3a and 3b. | ||
Pycnidiophorone D (4) was assigned a molecular formula of C27H39NO5 (9 degrees of unsaturation) by HRESIMS, which is 42 mass units higher than 3. Interpretation of NMR data (Table 1) for 4 revealed the presence of structural features similar to those found in 3, except that the oxymethylene signals (H2-17) was downfield (δH 4.24 in 4; 3.82 in 3) and resonances corresponding to an acetyl group (δC/δH 21.2/2.03; 170.9) were observed in 4. HMBC correlations (Fig. 2) from H2-17 to the carboxylic carbon C-27 (δC 170.9) indicate that the C-17 oxygen of 4 is acylated. Therefore, 4 was determined as the C-17 monoacetate of 3. The relative and absolute configurations of 4 were deduced to be the same as those of 3 by comparison of its NOESY correlations (Fig. 3) and ECD spectrum with those of 3 (Fig. S33 and S34†).
The known depsidones 5–10, with a typical 6/7/6 tricyclic core bearing a central seven-membered lactone, were identified as 4-formyl-3,8-dihydroxy-9-methyl-1,6-bis(1-methyl-1-propenyl)-11H-dibenzo[b,e][1,4]dioxepin-11-one (5),17 3,8-dihydroxy-4-(hydroxymethyl)-9-methyl-1,6-bis(1-methyl-1-propenyl)-11H-dibenzo[b,e][1,4]dioxepin-11-one (6),17 3,8-dihydroxy-4-(methoxymethyl)-9-methyl-1,6-bis(1-methyl-1-propenyl)-11H-dibenzo[b,e][1,4]dioxepin-11-one (7),17 auranticin B (8),18 auranticin A (9),18 pilobolusone C (10),19 respectively, by comparison of their NMR and MS data with those reported.
Compounds 1–10 were tested for cytotoxicity against a panel of five human tumor cell lines, HeLa, PC-3, A549, HepG-2 and HL-60 (Table 2). Compound 1 showed cytotoxicity to A549, HepG-2 and HL-60 cells with IC50 values of 7.6, 7.0, and 8.8 μM, respectively. Compound 5 showed cytotoxicity to A549 cells, with an IC50 value of 11.4 μM.
| Compound | IC50a (μM) | ||||
|---|---|---|---|---|---|
| HeLa | PC-3 | A549 | HepG-2 | HL-60 | |
| a IC50 values were averaged from at least three independent experiments.b No activity was detected at 100 μM.c Positive control. | |||||
| 1 | 99.0 ± 0.8 | 46.2 ± 0.7 | 7.6 ± 1.2 | 7.0 ± 2.6 | 8.8 ± 1.7 |
| 2 | 95.1 ± 3.4 | 91.4 ± 2.2 | NAb | 41.2 ± 0.4 | 57.0 ± 0.9 |
| 3 | 75.9 ± 1.9 | NAb | 27.4 ± 1.2 | NAb | 26.0 ± 0.6 |
| 4 | 71.0 ± 3.4 | NAb | 31.5 ± 0.4 | NAb | NAb |
| 5 | 45.3 ± 0.4 | 22.2 ± 3.2 | 11.4 ± 0.3 | 15.3 ± 4.0 | 22.3 ± 1.4 |
| 6 | 36.1 ± 1.5 | NAb | 35.8 ± 2.2 | NAb | NAb |
| 7 | 53.4 ± 3.7 | 58.1 ± 4.1 | 59.4 ± 2.6 | 24.6 ± 0.9 | 57.6 ± 0.4 |
| 8 | 42.2 ± 2.0 | NAb | 33.7 ± 0.8 | 42.2 ± 1.4 | 22.8 ± 3.1 |
| 9 | NAb | 21.8 ± 2.5 | 13.0 ± 3.0 | 61.2 ± 1.1 | 21.9 ± 2.0 |
| 10 | NAb | 23.4 ± 0.6 | 66.9 ± 1.4 | 86.8 ± 2.3 | NAb |
| Cisplatinc | 11.7 ± 0.2 | 5.6 ± 0.6 | 11.8 ± 0.5 | 9.3 ± 0.4 | 15.7 ± 1.0 |
Experimental
General experimental procedures
An analytical automatic polarimeter (Rudolph Research) was used to record optical rotations of the isolated compounds, and a Shimadzu Biospec-1601 spectrophotometer was used to measure the ultraviolet (UV) spectra. A Nicolet Magna-IR 750 spectrophotometer was used to record the infrared (IR) spectra. 1H and 13C NMR data were acquired with Inova-400, Inova-500 and Inova-600 spectrometers using solvent signals (CDCl3; δH 7.26/δC 77.7) as references. The HMBC and HMQC experiments were optimized for 8.0 and 145.0 Hz, respectively. ESIMS and HRESIMS data were obtained on an Agilent Accurate-Mass-Q-TOF LC/MS G6550 instrument equipped with an ESI source. CD spectra were recorded on a JASCO J-815 spectropolarimeter. All HPLC analysis and separation were performed using an Agilent 1260 instrument (Agilent, USA) equipped with a variable-wavelength UV detector.Fungal material
The strain P. dispersa was isolated from the soil samples that were collected at Baiyangdian Lake, Hebei Province, P. R. China, in December, 2010. The fungus was identified by morphological observation, and sequence (Genbank accession no. JX076943.1) analyses of the ITS region of the rDNA. The identified P. dispersa strain was cultured on Potato Dextrose Agar (PDA) at room temperature for 10 days, and the resulting agar plugs were cut into small pieces (0.5 × 0.5 × 0.5 cm3) under aseptic conditions. Thirty pieces were inoculated into six 250 mL Erlenmeyer flasks, each containing 50 mL media (0.4% glucose, 1% malt extract, and 0.4% yeast extract; pH 6.5), which were then incubated at room temperature on an orbital shaker at 200 rpm for 5 days to prepare the seed culture. Spore inoculum was prepared by suspension in sterile, distilled H2O to give a final spore/cell suspension of 1 × 106 mL−1. Fermentation was carried out in 48 Fernbach flasks (500 mL), each containing 80 g of rice. Distilled H2O (120 mL) was added to each flask, and the contents were soaked overnight before autoclaving at 15 psi for 30 min. After cooling to room temperature, each flask was inoculated with 5.0 mL of the spore inoculum and incubated at 25 °C for 20 or 30 days.Extraction and isolation
The fermented culture (incubated for 20 days) was extracted repeatedly with ethyl acetate (EtOAc; 4 × 4.8 L), and the organic solvent was evaporated to dryness under vacuum to afford the crude extract (4.2 g). Subsequently, the crude extract was fractionated by vacuum liquid chromatography (VLC) on silica gel with gradient elution of petroleum ether (PE)–EtOAc–MeOH. The fractions eluted with 91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 (PE–EtOAc)–100:2 (EtOAc–MeOH) were combined (1.9 g) and separated by ODS C18 column chromatography (H2O–MeOH). The subfraction (215.0 mg) eluted with 50:50 (H2O–MeOH) was purified by reversed phase (RP) HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 55% CH3CN in H2O for 35 min; 2 mL min−1) to afford 10 (23.0 mg, tR 21.0 min) and 7 (4.5 mg, tR 27.0 min). The subfraction (178.0 mg) eluted with 65:70 (H2O–MeOH) was purified by RP HPLC (79% MeOH in H2O for 60 min; 2 mL min−1) to afford 8 (2.0 mg, tR 49.6 min). The subfraction (214.0 mg) eluted with 25:75 (H2O–MeOH) was purified by RP HPLC (82% MeOH in H2O for 45 min; 2 mL min−1) to afford 9 (6.0 mg, tR 39.0 min).
:9 (PE–EtOAc)–100:2 (EtOAc–MeOH) were combined (1.9 g) and separated by ODS C18 column chromatography (H2O–MeOH). The subfraction (215.0 mg) eluted with 50:50 (H2O–MeOH) was purified by reversed phase (RP) HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 × 250 mm; 55% CH3CN in H2O for 35 min; 2 mL min−1) to afford 10 (23.0 mg, tR 21.0 min) and 7 (4.5 mg, tR 27.0 min). The subfraction (178.0 mg) eluted with 65:70 (H2O–MeOH) was purified by RP HPLC (79% MeOH in H2O for 60 min; 2 mL min−1) to afford 8 (2.0 mg, tR 49.6 min). The subfraction (214.0 mg) eluted with 25:75 (H2O–MeOH) was purified by RP HPLC (82% MeOH in H2O for 45 min; 2 mL min−1) to afford 9 (6.0 mg, tR 39.0 min).
The fermentation material (incubated for 30 days) was extracted repeatedly with EtOAc (4 × 4.8 L), and the organic solvent was evaporated to dryness under vacuum to afford 4.5 g crude extract. Similarly, the crude extract was fractionated by silica gel VLC using PE–EtOAc–MeOH gradient elution. The fractions eluted with 88:12 (PE–EtOAc)–85:15 (EtOAc–MeOH) were combined (2.8 g) and separated by silica gel column chromatography (CC) using petroleum ether (PE)–EtOAc gradient elution. The subfraction of CC (98.0 mg) eluted with 80:20 (PE–EtOAc) was purified by RP HPLC (55% CH3CN in H2O for 60 min; 2 mL min−1) to afford 5 (1.6 mg, tR 56.0 min). The subfractions of CC (160.2 mg) eluted with 65:35 (PE–EtOAc)–56:44 (PE–EtOAc) were further combined and separated by Sephadex LH-20 CC (1:1 MeOH–CH2Cl2), and purified by RP HPLC (65% CH3CN in H2O for 30 min; 2 mL min−1) to afford 2 (4.2 mg, tR 12.8 min) and 4 (2.0 mg, tR 19.5 min). Meanwhile, 6 (1.4 mg, tR 35.0 min) was obtained by RP HPLC (47% CH3CN in H2O for 45 min; 2 mL min−1). The subfractions of CC (362.5 mg) eluted with 10:90 (PE–EtOAc)–100:2 (EtOAc–MeOH) were further combined and separated by Sephadex LH-20 CC (MeOH), and purified by RP HPLC (70% MeOH in H2O for 30 min; 2 mL min−1) to afford 1 (2.3 mg, tR 11.8 min) and 3 (1.9 mg, tR 22.5 min).
ε) 212 (2.79), 250 (1.79) nm; IR (neat) νmax 3368 (br), 2928, 1694, 1449, 1379, 1214, 1033 cm−1; CD (c 1.2 × 10−3 M, MeOH) λmax (Δε) 250 (+4.27), 293 (+1.85), 332 (−0.99) nm; 1H and 13C NMR data see Table 1; HMBC data (CDCl3, 500 MHz) 2-NH → C-4, 9; H-3 → C-1, 5; H-4 → C-1, 3, 5, 6, 10, 22; H-8 → C-1, 7, 9, 14, 22; H-10a → C-3, 25; H-10b → C-3, 25; H3-11 → C-4, 5, 6; H3-12 → C-5, 6, 7, 11; H-13 → C-9, 14, 15; H-15a → C-13, 14, 16, 17, 18; H-15b → C-13, 14, 16, 17, 26; H-16 → C-15, 17, 19; H2-17 → C-15, 18; H-18a → C-15, 16, 17, 19; H-18b → C-15, 16, 17, 19; H-19 → C-14, 15, 16, 20, 21; H-20 → C-8, 13, 15, 22; H-21a → C-9, 20, 22; H-21b → C-13, 19, 22; H-23 → C-25; H3-24 → C-10, 23, 25; H3-25 → C-10, 23, 24; H3-26 → C-14, 15; NOESY correlations (CDCl3, 500 MHz) H-4 ↔ H2-10, H-8; H-8 ↔ H3-26; H-13 ↔ H2-17, H-19, H-20; H2-17 ↔ H-20; HRESIMS m/z 430.2582 [M + H]+ (calcd for C25H36NO5, 430.2589).ε) 205 (1.11), 250 (0.34) nm; IR (neat) νmax 2936, 2866, 1734, 1718, 1437, 1369, 1246, 1054, 1032 cm−1; CD (c 5.3 × 10−4 M, MeOH) λmax (Δε) 249 (+6.71), 293 (+2.62), 331 (−1.54) nm; 1H and 13C NMR data see Table 1; HMBC data (CDCl3, 600 MHz) H-3 → C-1, 4; H-4 → C-1, 3, 5, 6, 10, 13, 22; H-8 → C-1, 7, 9, 14, 20, 22; H-10a → C-3, 23, 24, 25; H-10b → C-3, 23, 24, 25; H3-11 → C-4, 5, 6, 12; H3-12 → C-5, 6, 11; H-13 → C-7, 8, 14, 15, 21; H-15a → C-13, 14, 16, 17, 18; H-15b → C-26; H-16 → C-27; H2-17 → C-15, 18, 27; H-18a → C-15, 17, 19, 20; H-18b → C-15, 17, 19, 20; H-19 → C-14, 15, 21; H-20 → C-8, 15, 22; H-21a → C-9, 13, 19, 20, 22; H-21b → C-9, 13, 19, 20, 22; H-23 → C-3, 10; H3-24 → C-10, 23, 25; H3-25 → C-10, 23, 24; H3-26 → C-13, 14, 15; H3-28 → C-27; NOESY correlations (CDCl3, 600 MHz) H-4 ↔ H2-10, H-8; H-8 ↔ H3-26; H-13 ↔ H2-17, H-19, H-20; H2-17 ↔ H-20; HRESIMS m/z 472.2692 [M + H]+ (calcd for C27H38NO6, 472.2699).ε) 215 (3.06) nm; IR (neat) νmax 3368, 2929, 1710, 1683, 1442, 1382, 1223, 1032 cm−1; CD (c 9.6 × 10−4 M, MeOH) λmax (Δε) 224 (−1.90), 296 (+3.40) nm; 1H and 13C NMR data see Table 1; HMBC data (CDCl3, 400 MHz) 2-NH → C-3, 9; H-3 → C-1, 5, 23; H-4 → C-1, 3, 5, 6, 9, 10; H-5 → C-3, 6, 7; H-7 →C-5, 12; H-10a → C-4, 24, 25; H-10b → C-4, 24, 25; H3-11 → C-4, 5, 6; H3-12 → C-5, 6, 7; H-13 → C-7, 8, 20; H-15a → C-13, 14, 16, 17, 26; H-15b → C-14, 16, 17; H-16 → C-14, 15, 17, 18, 19; H2-17 → C-15, 16, 18; H-18a → C-16, 17, 19, 20; H-18b → C-16, 17, 20; H-19 → C-14, 16, 21; H-20 → C-13, 14, 18, 21; H2-21 → C-9, 13, 19, 20, 22; H3-24 → C-10, 23, 25; H3-25 → C-10, 24; H3-26 → C-13, 14, 15, 16; NOESY correlations (CDCl3, 400 MHz) H-3 ↔ H3-11; H-4 ↔ H-5, H-8, H2-10; H-8 ↔ H3-26; H-13 ↔ H-19, H-20; H2-17 ↔ H-13, H-20; HRESIMS m/z 416.2791 [M + H]+ (calcd for C25H38NO4, 416.2799).ε) 214 (2.79); IR (neat) νmax 3337 (br), 2955, 1712, 1686, 1444, 1369, 1225, 1033 cm−1; CD (c 2.2 × 10−3 M, MeOH) λmax (Δε) 224 (−2.49), 297 (+3.71) nm; 1H and 13C NMR data see Table 1; HMBC data (CDCl3, 600 MHz) 2-NH → C-3, 9; H-3 → C-1, 5, 23; H-4 → C-1, 3, 5, 6, 9, 10; H-5 → C-3, 6, 7; H-7 →C-5, 12; H-10a → C-4, 24, 25; H-10b → C-4, 24, 25; H3-11 → C-4, 5, 6; H3-12 → C-5, 6, 7; H-13 → C-7, 8, 20; H-15a → C-13, 14, 16, 17, 26; H-15b → C-16, 17; H-16 → C-14, 15, 17, 18, 19; H-17a → C-15, 18, 27; H2-17 → C-15, 16, 27; H-18a → C-16, 17, 19, 20; H-18b → C-16, 17, 20; H-19 → C-14, 16, 21; H-20 → C-13, 14, 18, 21; H2-21 → C-9, 13, 19, 20, 22; H3-24 → C-10, 23, 25; H3-25 → C-10, 23, 24; H3-26 → C-13, 14, 15, 16; H3-28 → C-27; NOESY correlations (CDCl3, 600 MHz) H-3 ↔ H3-11; H-4 ↔ H-5, H-8, H2-10; H-5 ↔ H-8; H-8 ↔ H3-26; H-13 ↔ H-19, H-20; H2-17 ↔ H-13, H-20; H-19 ↔ H-20; HRESIMS m/z 458.2905 [M + H]+ (calcd for C27H40NO5, 458.2905).Computational details
Conformational analyses for 1 and 3 were performed via the Molecular Operating Environment (MOE) version 2009.10 (Chemical Computing Group, Canada) software package with LowModeMD at the MMFF94 force field. The MMFF94 conformers were further optimized using DFT at the B3LYP/6-311G(d,p) basis set level in MeOH with the IEFPCM model. The stationary points have been checked as the true minima of the potential energy surface by verifying that they do not exhibit vibrational imaginary frequencies. The 25 lowest electronic transitions were calculated, and the rotational strengths of each electronic excitation were given using both dipole length and velocity representations. ECD spectra were simulated in SpecDis20 using a Gaussian function with half-bandwidths of 0.30 eV. The overall ECD spectra were then generated according to Boltzmann weighting of each conformer. The systematic errors in the prediction of wavelength and excited-state energies are compensated by employing UV correlation. All quantum computations were performed using the Gaussian 09 package.21
Cytotoxicity bioassays
Inhibitory effects on cell proliferation were determined using the Cell Counting Kit-8 (Dojindo China Co., Ltd.) assay.22 Briefly, the cell lines were seeded in 96-well plates at a density of 1.0 × 104 cells per well and incubated to allow adhesion. Cells were obtained from the Department of Clinical Pharmacology in Chinese PLA General Hospital. HeLa, A549, HepG2 and HL-60 cells were cultured in DMEM/high glucose (Hyclone) supplemented with 10% FBS (Hyclone) at 37 °C with 5% CO2, while PC-3 cells were cultured in Ham's F12K (Bio-Channel) supplemented with 10% FBS. Subsequently, the test compounds were added to the cells at concentrations of 0.1–100 μM and incubated for 24 h. At the end of the period, 10 μL of CCK-8 reagent was added and allowed to incubate at 37 °C for 4 h. Colorimetric absorbance was measured using an Enspire 2300 multilabel reader (PerkinElmer, Waltham, MA, USA) at 450 nm to obtain the optical density values. Three duplicate wells were used for each concentration, and all the tests were repeated three times. The IC50 values of the test compounds were calculated using a nonlinear regression model (GraphPad Software, San Diego, CA, USA) and presented as the mean ± standard deviation.Conclusions
In conclusion, pycnidiophorones A–D (1–4), four new cytochalasans possessing a rare 6/5/6/6/5 pentacyclic skeleton, which was incorporated in a 12-oxatricyclo[6.3.1.02,7]dodecane core, and six known depsidones have been isolated from the crude extract of P. dispersa. Their structures were elucidated based on NMR spectroscopic data and ECD calculations. Pycnidiophorone A (1) is structurally related to the known fungal metabolite trichoderone B,16 but differs by having one hydroxymethyl group at C-16 instead of the hydroxy group at C-18. While pycnidiophorone C (3) differs from the known aspergilin PZ23 by having one hydroxymethyl group at C-16 instead of the hydroxy group at C-18. Compounds 2 and 4 are the acetylation products of 1 and 3 at C-17, respectively. Until now, only nine cytochalasan alkaloids with 5/6/6/5/6 pentacyclic system (aspergilin PZ, trichoderones A and B, trichalasin H, flavichalasines C–E, spicochalasin A and xylastriasan A) have been reported previously.16,23–27 Compounds 1–10 showed moderate cytotoxicity against a panel of five human tumor cell lines. To our knowledge, this is the first report that cytochalasans and depsidones were isolated and identified from the genus Pycnidiophora. The discovery of these new bioactive compounds enriches the structural diversity of cytochalasans and would attract much attention for further investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Key R&D Program of China (2018YFC0311002), the National Natural Science Foundation of China (32022002, 21772228 and 21977113), the Beijing Hospitals Authority Ascent Plan (DFL20190803), and Capital Science and Technology Leading Talent Training Project (Z191100006119017).Notes and references
- A. Schueffler and T. Anke, Nat. Prod. Rep., 2014, 31, 1425–1448 RSC.
- L. Liu, Y. Li, S. Liu, Z. Zheng, X. Chen, L. Guo and Y. Che, Org. Lett., 2009, 11, 2836–2839 CrossRef CAS.
- M. Liu, Y. Yan, L. Shen, Z. Hu and Y. Zhang, Chin. J. Nat. Med., 2019, 17, 935–944 Search PubMed.
- X. Luo, Y. Lin, Y. Lu, X. Zhou and Y. Liu, Chin. J. Nat. Med., 2019, 17, 149–154 Search PubMed.
- L. Liu, Y. Li, L. Li, Y. Cao, L. Guo, G. Liu and Y. Che, J. Org. Chem., 2013, 78, 2992–3000 CrossRef CAS.
- J. Ren, F. Zhang, X. Liu, L. Li, G. Liu, X. Liu and Y. Che, Org. Lett., 2012, 14, 6226–6229 CrossRef CAS.
- L. Zhang, B. Feng, Y. Sun, H. Wu, S. Li, B. Liu, F. Liu, W. Zhang, G. Chen, J. Bai, H. Hua, H. Wang and Y. Pei, Tetrahedron Lett., 2016, 57, 645–649 CrossRef CAS.
- R. Liu, Q. Gu, W. Zhu, C. Cui, G. Fan, Y. Fang, T. Zhu and H. Liu, J. Nat. Prod., 2006, 69, 871–875 CrossRef CAS.
- T. Sun, R. Kuang, G. Chen, S. Qin, C. Wang, D. Hu, B. Wu, X. Liu, X. Yao and H. Gao, Molecules, 2016, 21, 1184–1195 CrossRef.
- T. Sun, J. Zou, G. Chen, D. Hu, B. Wu, X. Liu, X. Yao and H. Gao, Acta Pharm. Sin. B, 2017, 7, 167–172 CrossRef.
- J. He, D. Qin, H. Gao, R. Kuang, Y. Yu, X. Liu and X. Yao, Molecules, 2014, 19, 20880–20887 CrossRef.
- J. Tian, P. Li, X. Li, P. Sun, H. Gao, X. Liu, P. Huang, J. Tang and X. Yao, Bioorg. Med. Chem. Lett., 2016, 26, 1391–1396 CrossRef CAS.
- J. He, Z. Mu, H. Gao, G. Chen, Q. Zhao, D. Hu, J. Sun, X. Li, Y. Li, X. Liu and X. Yao, Tetrahedron, 2014, 70, 4425–4430 CrossRef CAS.
- J. He, H. Liang, H. Gao, R. Kuang, G. Chen, D. Hu, C. Wang, X. Liu, Y. Li and X. Yao, J. Asian Nat. Prod. Res., 2014, 16, 1029–1034 CrossRef CAS.
- C. Zhao, P. Fu, Y. Zhang, X. Liu, F. Ren and Y. Che, Molecules, 2018, 23, 1263–1271 CrossRef.
- G. Ding, H. Wang, L. Li, A. Chen, L. Chen, L. Chen, H. Chen, H. Zhang, X. Liu and Z. Zou, Eur. J. Org. Chem., 2012, 2516–2519 CrossRef CAS.
- P. Pittayakhajonwut, A. Dramae, S. Madla, N. Lartpornmatulee, N. Boonyuen and M. Tanticharoen, J. Nat. Prod., 2006, 69, 1361–1363 CrossRef CAS.
- G. Poch and J. Gloer, J. Nat. Prod., 1991, 54, 213–217 CrossRef CAS.
- Q. Rajachan, S. Kanokmedhakul, K. Kanokmedhakul and K. Soytong, Planta Med., 2014, 80, 1635–1640 CrossRef.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Y. Zhang, Z. Zhang, B. Wang, L. Liu and Y. Che, Bioorg. Med. Chem. Lett., 2016, 26, 1885–1888 CrossRef CAS.
- Y. Zhang, T. Wang, H. Hua and B. Peng, J. Antibiot., 2002, 55, 693–695 CrossRef CAS.
- L. Chen, Y. Liu, B. Song, H. Zhang, G. Ding, X. Liu, Y. Gu and Z. Zou, Fitoterapia, 2014, 96, 115–122 CrossRef CAS.
- G. Wei, D. Tan, C. Chen, Q. Tong, X. Li, J. Huang, J. Liu, Y. Xue, J. Wang, Z. Luo, H. Zhu and Y. Zhang, Sci. Rep., 2017, 7, 42434–42445 CrossRef CAS.
- Z. Lin, T. Zhu, H. Wei, G. Zhang, H. Wang and Q. Gu, Eur. J. Org. Chem., 2009, 3045–3051 CrossRef CAS.
- C. Lei, Z. Yang, Y. Zeng, Y. Zhou, Y. Huang, X. He, G. Li and X. Yuan, Nat. Prod. Res., 2017, 32, 7–13 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: UV, IR, HRESIMS, NMR spectra of compounds 1–4; ECD calculations of compounds 1–4. See DOI: 10.1039/d0ra08072a |
| This journal is © The Royal Society of Chemistry 2020 |