
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComprehensive DFT investigation of transition-metal-based new quaternary Heusler alloys CoNbMnZ (Z = Ge, Sn): compatible for spin-dependent and thermoelectric applications†
Ab Quyoom Seh and
Dinesh C. Gupta*
and
Dinesh C. Gupta*
Condensed Matter Theory Group, School of Studies in Physics, Jiwaji University, Gwalior-474011, India. E-mail: sosfizix@gmail.com
First published on 9th December 2020
Abstract
The hunt for high spin polarization and efficient thermoelectric materials has endured for decades. In this paper, we have explored the structural, mechanical stability, magneto-electronic, and thermoelectric properties of two new quaternary Heusler alloys, CoNbMnZ (Z = Ge, Sn), using first-principles simulation methods. The alloys are stable, showing a Y1-type phase and ferromagnetic nature. Based on a generalized gradient approximation method, the alloys exhibit metallic nature; upon employing a modified version of the Becke–Johnson potential, both alloys demonstrate half-metallic nature, with gaps of 0.43 and 0.45 eV, which is a precursor for high spin polarization in these alloys. The alloys also follow the necessary Slater–Pauling rule condition MT = ZT − 24 for half-metallicity and they have a total magnetic moment of 1 μB. Elastic parameters convey the mechanical stabilities of these alloys, with Debye temperatures of 518 K and 445 K. These materials act as anisotropic media with respect to longitudinal and transverse sound velocities. Possible energy efficiency and thermoelectric applications were scrutinized via computing Seebeck coefficients, electrical and electronic lattice thermal conductivities, and, lastly, power factors. The highest S values for Ge- and Sn-based alloys are 60.43 and 68.2 μV K−1, respectively, and the highest power factors are 32 and 35 μW K−2 cm−1, respectively, suggesting potential efficient applications in thermoelectric power generation.
Introduction
In recent decades, Heusler alloys have captured a lot of attention due to their possible utilization in various technologies, like spintronics,1–4 giant magnetoresistance devices,5 shape memory alloys,6 and thermoelectric devices.7,8 This is because these alloys are mostly ferromagnetic with high Curie temperatures.9,10 Heusler alloys also exhibit unique behavior at the Fermi level, with half-metallicity, giving rise to 100% spin polarization.1,2,11 The high spin polarized currents make them suitable for enhancing the performances and properties of conventional semiconductor devices. These materials can be incorporated into spin filters, and spin-injection and magneto-optical devices.12,13 The investigation of such materials has gained the interest of researchers, and a large number of theoretical and experimental studies have been carried out to find new materials with fascinating properties. Besides the many variants of Heusler alloys, recently, new materials called quaternary Heusler alloys are being investigated thoroughly. To date, a large number of such alloys has been investigated both theoretically and experimentally.1,2,13–16 Among these alloys, Co-based examples are of particular interest due to their high Curie temperatures and high spin polarization at the Fermi energy (EF), making them prospective candidates for use in spin-dependent devices.17 These materials can also find applications as energy-conserving materials.18,19According to statistical results, a significant percentage (more than 60%) of energy is lost as waste heat throughout the world. The depletion of energy resources and degradation of the environment have forced the scientific world to find alternative efficient and eco-friendly energy technologies. The disastrous consequences emerging from the combustion of fossil fuels have caused severe problems to human health and energy reservoirs due to environmental pollution and energy wastage. Hence, the search for energy-efficient and eco-friendly materials is currently a vital challenge globally.20 One of the fundamental solutions to such problems is the concept of thermoelectricity, which enables the direct conversion of heat energy into electrical energy. Thermoelectric devices do not require any environmentally harmful fluids like chlorofluorocarbons or fullerenes, nor do they release harmful gases into environment; for this reason, these are called green-energy materials.21 Heusler compounds have been found to be compatible for converting dissipated heat into electric energy.22 These compounds are providing new sustainable energy technology approaches to overcome energy crises.
As mentioned above, a huge number of quaternary Heusler alloys have been investigated that exhibit fascinating properties. Their high spin polarization, compatible lattice structures, and high thermoelectric efficiency with conventional half-metallic ferromagnetism have inspired us to design new quaternary Heusler half-metallic CoNbMnZ (Z = Ge, Sn) alloys. Since no experimental or theoretical information is available about these alloys, we performed detailed analysis, which is vital for understanding the structural, mechanical stability, strength, and natural properties of these material for experimental synthesis and technological applications. The authenticity of the results for these alloys can be confirmed via comparing the obtained results with other Heusler alloy families, especially with the nearest-neighbor family CoX′NbGa (X = Cr, Mn, Fe).23 We will also explore how the results are dependent on electron correlations in the narrow d-bands of transition-metal atoms.
Methodology
Calculations of total energies and electronic properties were done using DFT. The aforementioned alloys were analyzed by utilizing Wein2k simulation code based on the full-potential linearized augmented plane wave method (FP-LAPW). The solutions to Kohn–Sham equations are obtained through an iterative method. Since the exchange–correlation potentials are not exactly known, we have employed two different methods, the Perdew–Burke–Ernzerhof generalized gradient approximation (GGA-PBE) and integrated modified Becke–Johnson GGA (GGA + mBJ) methods.24,25 Highly correlated electron systems, including transition metal (TM) complexes that possess d- and f-states, are not effectively estimated via the GGA method. The modified Becke–Johnson method is used to overcome erroneous estimated values when using the GGA method. To restrict the number of plane-waves, a cut-off parameter of RMTKMAX = 6 has been employed, where the former represents the largest reciprocal lattice vector and the latter is the smallest muffin tin sphere radius. For Brillouin zone (BZ) integration, a tetrahedral method following Monkhorst–Pack conventions with a dense k-mesh of 3000 points has been used. The RMT values of Co, Nb, Mn, and Ge are chosen in such a way as to prevent charge leakage to the interstitial region and, thus, total energy convergence is also ensured. The energy and charge convergence criteria are set to 10−7 e nm−1 and 10−4 e, respectively, so as to ensure that the self-consistent calculations are accurate and fully converged.In order to calculate the transport properties of the compounds, BoltzTraP2 code26 using constant relaxation time approximations for the charge carriers is employed. This approximation is based on the fact that these parameters do not vary readily with energy on the kBT scale.27 The electrical conductivity (σ), electronic thermal conductivity (κe), and Seebeck coefficient (S) values can be determined through the following relationships:28

where υαK depicts the α component of the group velocity with the wave vector k.

To determine the transport coefficients, a large k-point mesh of 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 k points was used in order to obtain more precise results.
000 k points was used in order to obtain more precise results.
Structural
The crystal framework associated with the XX′YZ quaternary Heusler alloys consists of four interpenetrating fcc lattices with the space group F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m. The prototype structure of these alloys is LiMgPdSn-type, which is also called a Y-type structure. The structural description of an XX′YZ alloy is such that the atom X with the most valance electrons occupies the 4c site of the lattice. The sp atom Z will occupy the 4a position, and X′ and Y are placed at the 4b and 4d positions, respectively. Two types of interactions, viz ionic and covalent, occur in a Heusler-type structure. This is because the 4a and 4c sites of the unit cell form a ZnS-type lattice, and the bonding between the atoms has covalent nature. The sites 4a and 4b form an octahedron with NaCl-type structure, and the interactions among the atoms have strong ionic character.29 Thus, overall, quaternary Heusler alloys depict polar covalent bonding character.
3m. The prototype structure of these alloys is LiMgPdSn-type, which is also called a Y-type structure. The structural description of an XX′YZ alloy is such that the atom X with the most valance electrons occupies the 4c site of the lattice. The sp atom Z will occupy the 4a position, and X′ and Y are placed at the 4b and 4d positions, respectively. Two types of interactions, viz ionic and covalent, occur in a Heusler-type structure. This is because the 4a and 4c sites of the unit cell form a ZnS-type lattice, and the bonding between the atoms has covalent nature. The sites 4a and 4b form an octahedron with NaCl-type structure, and the interactions among the atoms have strong ionic character.29 Thus, overall, quaternary Heusler alloys depict polar covalent bonding character.
Depending upon the occupancy of atoms in the unit cell, there are three inequivalent possible structural types. The occupancy of Wyckoff positions in the Y1 phase is X = (0.75, 0.75, 0.75), X′ = (0.50, 0.50, 0.50), Y = (0.25, 0.25, 0.25), and Z = (0, 0, 0). For the Y2 and Y3 phases, the atoms are situated at X = (0.25, 0.25, 0.25), Y = (0.50, 0.50, 0.50), X′ = (0.75, 0.75, 0.75), and Z = (0, 0, 0), and X = (0.50, 0.50, 0.50), Y = (0.75, 0.75, 0.75), X′ = (0.25, 0.25, 0.25), and Z = (0, 0, 0), respectively.30 These structural configurations are shown in Fig. S1(a–c) in the ESI.†
The physical properties (structural, electronic, magnetic, and transport) are greatly affected if there is any structural disorder or distortion; for this reason, we perform structural optimization in all possible phases. Structural optimization has been carried out to calculate the total energy and lattice constants, and to find the stable or ground state of the material via varying the unit-cell volume. Energy vs. volume curves are plotted in Fig. 1(a and b). From the plots it is evident that Y1 has the least energy and, hence, is the most stable phase, and it is also the most stable phase for other similar CoX′NbGa (X = Cr, Mn, Fe) alloys.23 The large energies in the Y2 and Y3 phases are attributed to the changing positions of atoms. In CoNbMnZ (Z = Ge, Sn) alloys, since Co is the atom with the maximum number of valence electrons, it will occupy the 4c (1/4, 1/4, 1/4) site. The p-block element Z has four outermost electrons and is the most electronegative atom in the present alloy, so it will occupy the 4a (0, 0, 0) site. Similarly, the third and fourth atoms (Mn, Nb) have seven and five valence electrons, respectively, so they will take up the 4b (1/2, 1/2, 1/2) and 4d (3/4, 3/4, 3/4) sites of the unit cell. The resultant ordered structure is called a Y1-type structure. The various equilibrium lattice parameters (a), bulk moduli (B), pressure derivative of bulk modulus ( ), and total energies in two magnetic states are presented in Table 1. The obtained lattice constants in the most stable phase for CoNbMnGe and CoNbMnSn are 0.614 and 0.625, respectively, which are comparable to the lattice constant of CoNbMnGa (0.596 nm).
), and total energies in two magnetic states are presented in Table 1. The obtained lattice constants in the most stable phase for CoNbMnGe and CoNbMnSn are 0.614 and 0.625, respectively, which are comparable to the lattice constant of CoNbMnGa (0.596 nm).
 | ||
| Fig. 1 Energy optimization curves of CoNbMnZ ((a) Ge, (b) Sn) in different phases. | ||
 ), ground state energy (E0, eV), and relaxed lattice constant (a, nm) of CoNbMnGe and CoNbMnSn alloys with different structure types
), ground state energy (E0, eV), and relaxed lattice constant (a, nm) of CoNbMnGe and CoNbMnSn alloys with different structure types
| Phase | V | B |
|
E0 | a | |
|---|---|---|---|---|---|---|
| CoNbMnGe | Y1 | 0.05257 | 225.46 | 4.24 | −230430.39230 | 0.614 |
| Y1-NM | 0.05282 | 216.63 | 4.51 | −230430.37695 | 0.590 | |
| Y2 | 0.05246 | 197.38 | 5.95 | −230430.34900 | 0.614 | |
| Y3 | 0.05633 | 149.50 | 5.04 | −230429.70450 | 0.621 | |
| CoNbMnSn | Y1 | 0.5923 | 145.33 | 5.24 | −341406.75163 | 0.6257 |
| Y1-NM | 0.05865 | 197.33 | 4.55 | −341406.72558 | 0.6142 | |
| Y2 | 0.05633 | 149.76 | 4.79 | −341406.084796 | 0.6216 | |
| Y3 | 0.06415 | 129.75 | 4.50 | −341405.806459 | 0.6301 |

Electronic properties
The novel properties of a material directly stem from the crystal structure and the electronic band profile. The electronic properties can be analyzed using GGA and mBJ methods, and by incorporating a Hubbard term with GGA. We have scrutinized the spin-polarized band structures of CoNbMnZ (Z = Ge, Sn) alloys along high-symmetry Brillouin zone (BZ) directions via GGA and mBJ techniques. Fig. 2(a and b) depicts the band structures of both alloys obtained via GGA; it is evident from the plots that many energy levels make metallic intersections at the Fermi level in the spin-up channel, hence creating metallic character. However, in spin-down orientation, the Fermi level is deserted, except at the X-point of the BZ for CoNbMnGe and the L-point of the BZ for CoNbMnSn. Upon zooming-in closely to the band structure, it is found that the energy band crosses the Fermi level, hence confirming the metallic character in the spin-down channel as well. Since the GGA method underestimates the band gap, we have incorporated the mBJ method into GGA in order to obtain the precise band structure. The mBJ band structure calculations are very effective compared to GGA. The elucidation of band structure using mBJ is presented in Fig. 3(a and b). From the band plots it is evident that the energy states are crossing the Fermi level in the majority spin channel, indicating metallic character, whereas in the spin-down state, neither the valence nor conduction band touches the Fermi level, giving rise to half-metallic nature with indirect band gaps of 0.43 and 0.45 eV at the Γ and X points. | ||
| Fig. 2 Representations of the band structures of CoNbMnZ (Z = (a) Ge, (b) Sn) using the GGA approximation. | ||
 | ||
| Fig. 3 The band structures of CoNbMnZ (Z = (a) Ge, (b) Sn) obtained via the mBJ method. | ||
The electronic properties of CoNbMnZ (Z = Ge, Sn) alloys can be further explored via plotting partial density of states (pDOS). pDOS gives information about the energy states occupied in a particular energy interval. In order to find the contributions of atomic energy levels to the band structure, we have plotted the pDOS of CoNbMnZ (Z = Ge, Sn) using the GGA approximation, as shown in Fig. S5(a and b) [shown in the ESI].† From both plots it is obvious that many energy states make metallic intersections with the Fermi level in the spin-up channel. Upon observing the pDOS in the minority spin channel, it can be seen that some energy levels cross the Fermi level and, hence, the alloys exhibit metallic nature in the spin-down channel as well. Since GGA usually underestimates the band gap, we employed mBJ, and the pDOS plots obtained using the mBJ method are shown in Fig. 4(a and b) for both alloys in the different spin channels. From the figures it is obvious that the alloys are metals in the majority spin channel, whereas in the minority channel none of the energy levels exists at the Fermi level; hence, the alloys show perfect half-metallic character. The band structures and pDOS of CoX′NbGa (X = Cr, Mn, Fe) alloys also show half-metallic nature.
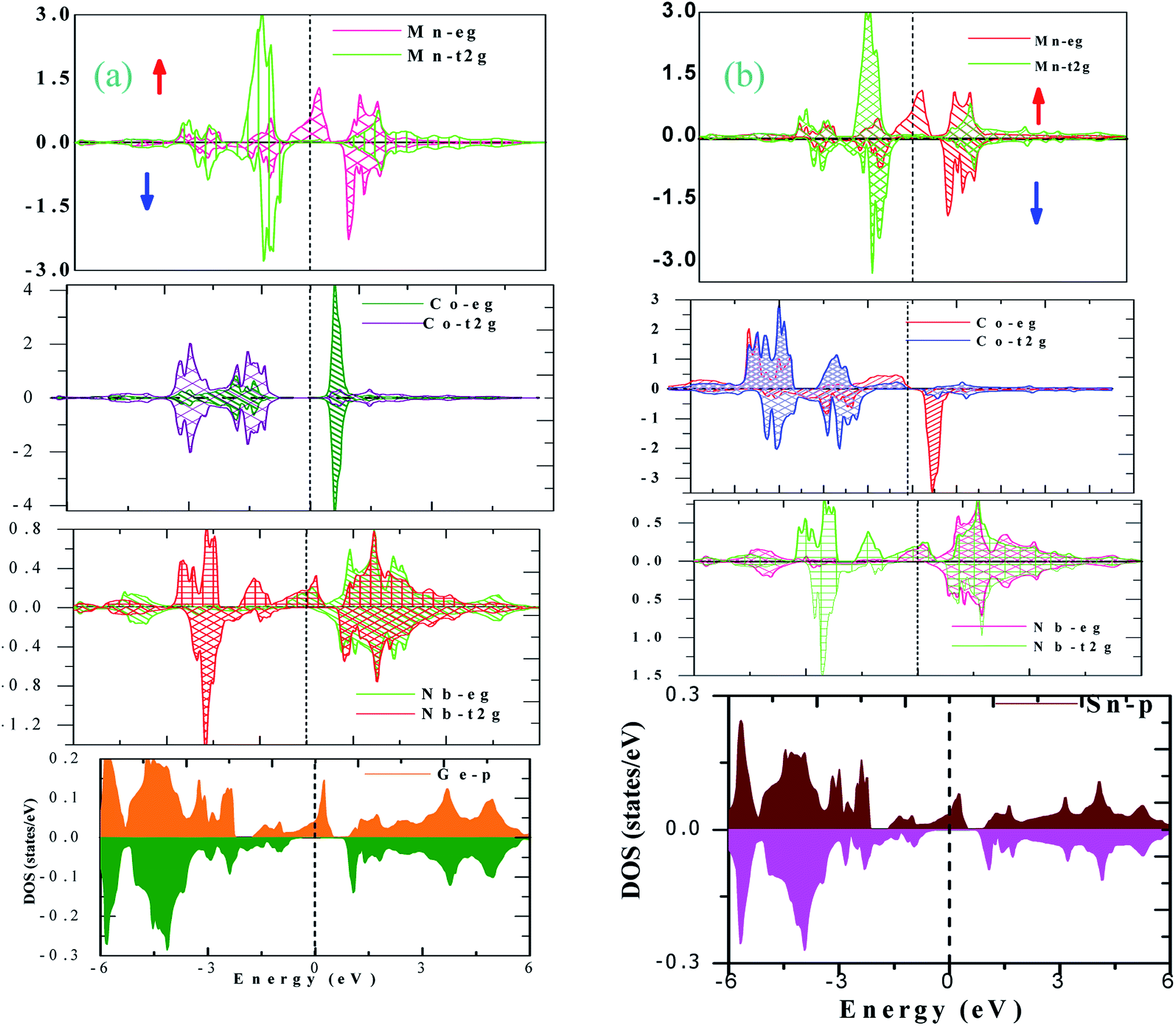 | ||
| Fig. 4 The atom-resolved pDOS plots of both alloys ((a) Z = Ge; (b) Z = Sn) obtained using the mBJ method. | ||
Origin of the half-metallic gap
The origin of the half-metallic nature of CoNbMnZ (Z= Ge, Sn) can be explored via presenting the possible hybridization among the constituent atoms schematically. Transition metal atom d-states in the presence of a crystal field are decomposed into doubly degenerate eg (dx2–y2, dz2) and triply degenerate t2g (dxy, dyz, dzx) states. The appearance of a band gap in the minority spin state and the half-metallicity of the Heusler compounds originates from the d–d hybridization of transition metal atoms. In the present alloys, Z is the most electronegative element and Nb has an exceptional electronic configuration (4d45s1). Therefore, the s-state electrons, two from Mn and one from both Nb and Co, will be accumulated by Z to complete its octet and achieve a minimum energy state. Thus, Z makes a negligible contribution to the DOS in the neighborhood of the Fermi level. Considering the d–d hybridization of the Mn and Nb atoms, according to Jahn–Teller theory, the degeneracy of the d-orbitals is lifted due to their mutual interaction and the d states of each transition metal atom hybridize according to their symmetry; this creates five bonding (2eg, 3t2g) and five anti-bonding (2eu, 3t1u) orbitals.31 Mn and Nb have five and four d-electrons, respectively. The possible orbital hybridization between the Mn and Nb d-states is profiled in Fig. 5(a). From the figure, it is evident that the MnNb-3t2g state is completely filled, hence the Mn and Nb bonding 3t2g state lies wholly in the valence band and the corresponding anti-bonding state lies in the conduction band. The 2eg state of MnNb has only three electrons, while Co has 8 electrons, 6 of which are in the 3t2g state and 2 are in the 2eg state, which may be due to s-Rydberg destabilization of a metal complex;32 therefore, the 2eg state of MnNb will couple with the Co-2eg state of Co, and this is depicted in Fig. 5(b). From the figure, it is obvious that the 2eg state is completely paired; one electron remains unpaired and this unpaired electron occupies the 3t1u state. The resultant hybridization is shown in Fig. 5(c); due to the intermixed character of the energy bands, both the Co and Mn-eg states lie in the valence band in the respective spin channels and their anti-bonding states lie in the conduction band. The unpaired electron in the 3t1u state is responsible for the metallic nature of the alloy in the spin-up channel, 100% spin polarization, and the magnetic moment of 1 μB according to the Slater–Pauling rule. | ||
| Fig. 5 (a) The hybridization of Mn–Nb. (b) The resultant hybridization of MnNb with Co. (c) The overall hybridization of the alloys. | ||
Electron density plots
Confirmation of the possible d–d hybridization of the materials under study is obtained via charge density plots and contour graphs, as portrayed in Fig. 6(a and b). Electron density plots play a vital role in the interpretation of the chemical and physical properties of a material based on electron clouds, hence denoting the nature of bond formation between different atoms. To obtain a clear perception of all atoms, we have chosen the 〈110〉 plane along the diagonal direction of the C1b structure.33 It is clear from the plots that the shape of the charge density cloud of Nb is dumbbell-type along the Z-atom direction, indicating an ionic interaction. This may be due to the more electronegative character of Z compared with Nb. From these plots, it is also evident that the Co charge density is spherically symmetric, while the Mn density is nearly spherical, demonstrating that Co and Mn interact covalently, as is evident from the orbital hybridization diagram. | ||
| Fig. 6 Electron density plots of both alloys ((a) Z = Ge; (b) Z = Sn) along the 〈111〉 plane direction. | ||
Magnetic properties
The magnetic nature of a material can be described based on its total magnetic moment. Table 2 presents the absolute and individual magnetic moments of CoNbMnZ (Z = Ge, Sn) alloys using GGA and mBJ methods. The alloys follow the Slater–Pauling rule MT = ZT − 24, with a total magnetic moment of 1 μB, which is an integral value, and, hence, the alloys are perfect half-metallic materials. μint describes the interstitial magnetic moment. From the table it is obvious that μint has almost zero value, and we can say that there is almost zero charge leakage to the interstitial regions of the alloys. The prominent contribution to the total magnetic moment comes from Co atoms. The contributions from the Z and Nb atoms to the magnetic moment are negligible in comparison to the Co and Mn atoms. The magnetic moments calculated via GGA and mBJ are almost same. Since the number of valence electrons is 25, there are 12 occupied states in both channels, while the extra electron is responsible for the magnetic moment and spin-polarization in the alloys. This can be further confirmed by examining the CoX′NbGa (X = Cr, Mn, Fe)23 family of alloys, where the authors gave detailed descriptions of the magnetic moments of CoCrNbGa and CoFeNbGa and the zero magnetic moment of CoMnNbGa.23| Magnetic moment (μB) | CoNbMnGe | CoNbMnSn | ||
|---|---|---|---|---|
| GGA | mBJ | GGA | m-BJ | |
| μint | −0.01 | −0.04 | 0.03 | −0.06 |
| μCo | 0.44 | 0.46 | 0.44 | 0.45 |
| μZ | 0.02 | 0.07 | −0.05 | −0.03 |
| μNb | 0.19 | 0.14 | 0.24 | 0.25 |
| μMn | 0.34 | 0.37 | 0.39 | 0.39 |
| μTot | 0.98 | 1.0 | 0.99 | 1.0 |
Cohesive energy
Theoretically, the stability of a material can be determined based on the cohesive energy (Ecoh), mechanical stability and formation energy. The stability of a material can be correlated with its cohesive energy, which is defined as the energy required to split the material into its individual constituents, hence indicating the bond strength of a compound.34 The cohesive energy, therefore, is a measure of the inter-molecular energy of a substance. The Ecoh values of the alloys under investigation can be calculated using the formula below, which involves the sum of the individual atomic energies minus the total energy of the compound:where ECo, ENb, EMn, and EZ are the energies of isolated atoms, and ETot is the bulk energy of the considered material. The higher the cohesive energy value, the more stable the structure will be. The computed value of ECoh for CoNbMnGe is 5.43 eV per atom and it is 5.17 eV per atom for CoNbMnSn, affirming that the materials are stable.
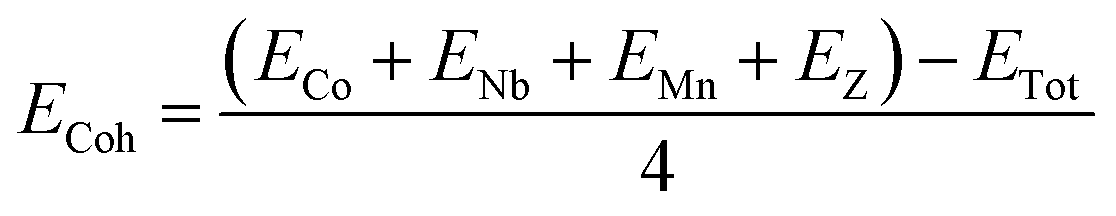
Mechanical properties
The mechanical properties of a material define its ability to regain and recover its original configuration after the cessation of stress. Elastic constants (Cij) are indispensable for exploring the mechanical properties of materials. These properties determine the favorable conditions for initiating and retaining the quality of the crystal structure. The mechanical constants are linked to some thermodynamic parameters, like specific heat, the Debye temperature, and the Gruneisen parameter. Elastic properties fundamentally determine the mechanical strength of a material, for example, the fracture toughness, sound velocity, load deflection, thermoelastic stress, and, indirectly, the melting temperature, which are basic requirements for determining technologically favorable materials. Most importantly, these parameters provide valuable information regarding the nature of the forces exerted on atoms and the bonding and stiffness of the material. For cubic crystals, only three independent elastic tensor components are needed, they are C11, C12, and C44. The first represents unidirectional squeezing along the principal crystallographic directions. C11 undergoes a large change because the volume change is very sensitive to temperature and pressure variations. The constants C12 and C44 determine resistance to shear deformation and are linked to elasticity of shape, which is a shear constant. Transverse strain or shear causes deformation without a change in volume. Thus, C12 and C44 are expected to be less sensitive to temperature and pressure compared with C11.Once these elastic constants are obtained, the elastic behavior can be studied via calculating other elastic parameters, shear (G) and bulk modulus (B), which are calculated through the Voigt–Reuss–Hill approximation.35,36
Some other important elastic parameters, such as Poisson's ratio (ν), anisotropic factor (A), and Young's modulus (E), can be calculated using the following relationships:37

The magnitude of the resistance of a material when its volume is changed is defined through B, while the limit of the occurrence of plastic deformation upon shear stress is measured through the shear modulus. The values obtained for ν, B, and G from the alloys are given in Table 3. The values of bulk and shear modulus are positive, which is a true indication of the structural stability of the materials. The stiffness of a material depends on Y. The higher the value of Y, the stiffer a material will be. Pugh proposed the index of ductility ratio (B/G), which is also called Pugh's ratio, which characterizes the plastic behavior of a compound. The critical value of B/G is 1.75. This is one of the most important and fundamental ratios that determines whether a material will show a brittle or ductile nature. If the ratio is greater than 1.75, the material shows ductile behavior, otherwise it shows a brittle nature.38 The calculated value of B/G for CoNbMnGe is 2.24 and it is 2.14 for CoNbMnSn, indicating that the materials possess ductile natures. This can also be confirmed from Cauchy's pressure: C′ = C12 − C44. A positive C′ value for a material corresponds to ductility, while a negative value of C′ indicates a brittle nature. For CoNbMnZ (Z = Ge, Sn) the values of C′ are positive, hence showing ductile natures.
| Alloy | C11 | C12 | C44 | Bv | BR | B | GV | GR | G | Y | ν | CP | B/G |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CoNbMnGe | 306.05 | 162.46 | 138.37 | 210.32 | 210.32 | 210.32 | 111.74 | 75.88 | 93.81 | 245.01 | 0.30 | 24.09 | 1.93 |
| CoNbMnSn | 251.62 | 103.41 | 83.36 | 152.81 | 152.81 | 152.81 | 79.65 | 62.72 | 71.18 | 184.84 | 0.29 | 20.05 | 1.12 |
The Poisson coefficient is a dimensionless parameter that provides good insight into the behavior of a material and the characteristics of the bonding forces existing within the material. The limiting values of this coefficient are 0.25 and 0.5, describing the lower and upper limits for central forces to exist within the constituents of an alloy. The value of ν is 0.1 for covalent compounds. For ionic and metallic bonding, the values are 0.25 and 0.33, respectively. We know that both ionic and covalent interactions exist in the C1b unit cell of a quaternary Heusler alloy. The computed Poisson ratio value for the CoNbMnGe alloy is 0.30 and it is 0.29 for CoNbMnSn, suggesting that the metallic bonding nature is dominant. Furthermore, ν can also be used to determine whether a compound has a brittle or ductile nature, because the value of this ratio depends on the nature of bonding and is different for different materials (it varies in different materials). If the Poisson ratio is less than 0.25, then the material will exhibit a brittle nature. The values of ν for the alloys under investigation is greater than 0.25 indicating these materials are ductile in nature.
The Zener anisotropy factor plays an important role when examining the extent of isotropy of a material. It is denoted by A and is defined in terms of lattice constants. For a perfect isotropic material, the value of A = 1 (unity). If the value of A deviates from unity (either greater or smaller), then the material possesses anisotropy. When the mechanical properties of a material alter along different directions then it is said to be an anisotropic material. A high value of elastic anisotropy in a material is associated with the chance of micro-cracks arising in a compound. Since the alloys exhibit high anisotropy, the velocities of transverse and longitudinal waves will be different along different directions. These waves propagate along three different modes: one longitudinal and two transverse modes. The calculated values of these modes along different directions are presented in Tables 4 and 5.
| Parameter | vt | vl | vm | θD | A |
|---|---|---|---|---|---|
| CoNbMnAs | 3886 | 6403 | 4479 | 518 | 1.93 |
| CoNbMnSb | 2780 | 5170 | 3660 | 445 | 1.12 |
| Propagation direction | Mode of vibration | Ceff | CoNbMnGe | CoNbMnSn |
|---|---|---|---|---|
| [100] | vl | C11 | 6110 | 5220 |
| vt1 | C44 | 4112 | 3010 | |
| vt2 | C44 | 4112 | 3010 | |
| [110] | vl | (C11 + C12 + 2C44)/2 | 6749 | 5310 |
| vt1 | C44 | 4112 | 3010 | |
| vt2 | (C11 − C12)/2 | 3000 | 2830 | |
| [111] | vl | (C11 + 2C12 + 4C44)/3 | 6947 | 5344 |
| vt1 | (C11 − C12 + C44)/3 | 3889 | 2890 | |
| vt2 | (C11 − C12 + C44)/3 | 3889 | 2890 |
Thermoelectric properties
The thermoelectric properties were elucidated using semi-classical Boltzmann transport theory. Thermoelectric materials provide an alternate approach for power generation and refrigeration. These materials can also act as heat pumps or solid-state refrigerators that do not use any moving parts. The performance of a thermoelectric material is governed by a dimensionless figure of merit, which is defined as where the terms have their usual meanings.39 Thermoelectric transport measurements, coupled with microstructure studies and theoretical modeling, show that the ZT improvement is the result of low lattice thermal conductivity due to increased phonon scattering by grain boundaries and structural defects.
where the terms have their usual meanings.39 Thermoelectric transport measurements, coupled with microstructure studies and theoretical modeling, show that the ZT improvement is the result of low lattice thermal conductivity due to increased phonon scattering by grain boundaries and structural defects.
Electrical conductivity
The variations of electrical conductivity σ are shown in Fig. 7(a and b). It is evident from the plots that σ decreases with temperature in the majority spin channel, which infers the metallic nature of the materials. Since the electrical conductivity of a metal is given by the equation σ = neμ, σ depends on the concentration and mobility of carriers. As the value of ‘e’ is constant and ‘n’ is almost independent of temperature in metals, the decreasing trend can be attributed to the mobility behavior in the material. The values of σ at 50 K are 8.0 × 105 W Ω−1 m−1 and 7.73 × 105 W Ω−1 m−1, which decrease with temperature and reach minimum values of 7.56 × 105 W Ω−1 m−1 and 7.17 × 105 W Ω−1 m−1 at 900 K in the spin-up channels of the alloys. As the alloys are semiconducting in the spin-down channel and the conductivity of a semiconductor is given by the equation σ = ni|e|(μe + μh), with a rise in temperature, the concentration of carriers ‘ni’ increases due to the transition of electrons from the valence to the conduction band. The magnitude of μe + μh will decrease with an increase in temperature, but this decrease will not overcome the increasing trend of ni; hence, the conductivity increases. The values of σ in the spin-down channels increase, and maximum values of 0.07 × 105 W Ω−1 m−1 and 0.42 × 105 W Ω−1 m−1 are attained at 900 K, hence signifying the semiconducting nature of the alloys. | ||
| Fig. 7 The variation of electrical conductivity with temperature for CoNbMnZ (Z = (a) Ge, (b) Sn) alloys. | ||
Thermal conductivity
One of the conditions that a promising thermoelectric material must meet is that the thermal conductivity should be as low as possible. The lower the value of κe, the more thermo-efficient the material will be. The variations of electronic thermal conductivity with temperature are plotted in Fig. 8(a and b). From the plots it is obvious that κe shows an increasing trend in both spin channels. It can be seen from the graphs that the variation of κe in the majority spin state is greater compared to the spin-down channel. This can be attributed to interactions between the carriers and the anisotropy of lattice forces. The values of k at room temperature in the spin-up state are 0.0197 W m−1 K−1 and 0.093 W m−1 K−1 and these increase linearly to maximum values of 3.37 W m−1 K−1 and 1.59 W m−1 K−1 at 900 K. The k-values in the minority spin state at room temperature are 0.010 W m−1 K−1 and 0.002 W m−1 K−1; a nonlinear trend is seen, and peak values of 0.755 W m−1 K−1 and 0.3833 W m−1 K−1 are obtained at 900 K. The small increase of κe is due to the semiconducting nature of the alloys in the spin-down state. | ||
| Fig. 8 The change in electronic thermal conductivity of CoNbMnZ (Z = (a) Ge, (b) Sn) alloys with temperature. | ||
Seebeck coefficient
The variations of the Seebeck coefficient in the spin-up channels for CoNbMnZ (Z = Ge, Sn) are depicted in Fig. 9. The values of S in the entire temperature range are positive in both spin channels, signifying the presence of holes as majority carriers. It is clear from the plots that S increases with an increase in temperature, from 7.68 and 8.93 at 50 K to 62.45 and 70.55 at 900 K in the majority spin channels of both alloys. The variations of S in the minority spin channels are profiled in Fig. 10. As the materials are semiconducting in the spin-down channels, they have minimum carrier concentrations at lower temperatures. The Seebeck coefficient has an inverse relation with carrier concentration (n), as shown in the equation:
 | ||
| Fig. 9 Seebeck coefficients in the spin-up channel. | ||
 | ||
| Fig. 10 Seebeck coefficients in the spin-down channel. | ||
Hence, maximum values are seen at very low temperatures. As the temperature is increased, the electrons in the valence band obtain sufficient energy to be excited into the conduction band, creating electron–hole pairs; due to this, the carrier concentration increases gradually. Hence, this results in a decrease of S in the minority spin channels. From the figure, it is evident that the values of S in the spin-down channels at 50 K are 471.25 and 767.50; these values decrease readily with temperature and attain minimum values of 120.72 and 53.20 at 900 K for the alloys. The total S value is determined using the two current model. The total thermopower is the average value of Seebeck coefficients from both spin configurations, weighted based on their corresponding conductivities. The variations of the total S values are plotted in Fig. 11; it is evident that the value of S increases from 50–900 K and achieves maximum values of 65.57 and 69.62. The thermoelectric parameters follow the same trend as shown by CoX′NbGa (X = Cr, Mn, Fe) alloys.
 | ||
| Fig. 11 The change in the total Seebeck coefficient with T (K). | ||
The power factor (PF) of a material is determined by a weighted average of the Seebeck coefficient and the total electrical conductivity, i.e., PF![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S2σ. It describes the efficiency of a material, and a high value of PF implies higher efficiency. The variations of the PF are depicted in Fig. 12 for both alloys. It is evident from the plots that the PF increases with temperature and reaches maximum values of 32 and 35.
S2σ. It describes the efficiency of a material, and a high value of PF implies higher efficiency. The variations of the PF are depicted in Fig. 12 for both alloys. It is evident from the plots that the PF increases with temperature and reaches maximum values of 32 and 35.
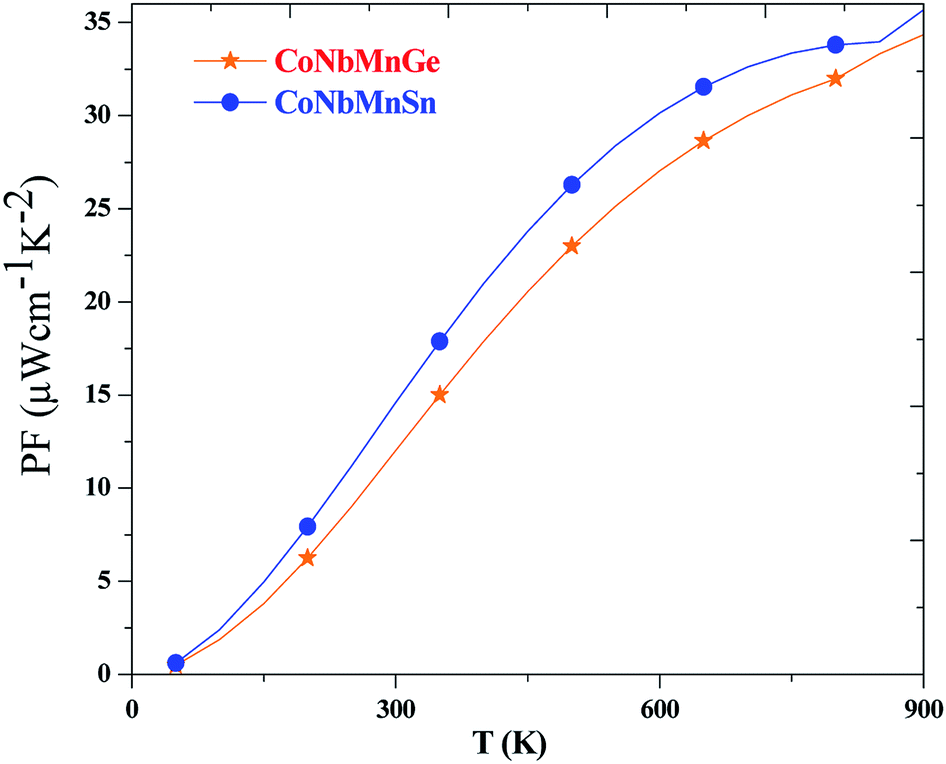 | ||
| Fig. 12 The change in the power factor of CoNbMnZ (Z = Ge, Sn) with temperature. | ||
To further confirm the relevance, we compare the pDOS, band structures, and thermoelectric parameters and study whether they follow the same trend. The band structure and DOS plots shown in Fig. 1–4 convey CoNbMnZ (Z = Ge, Sn) to be half metallic with metallic character in the majority spin channel. Upon analyzing the electric conductivity and Seebeck coefficients, we observe that electric conductivity decreases with temperature, while the Seebeck coefficient increases, in the spin-up channel of CoNbMnZ (Z = Ge, Sn) (see Fig. 6 and 8). Since these behaviors of σ and S are a peculiar characteristic of metals, this confirms that CoNbMnZ (Z = Ge, Sn) behaves as a metal in the majority spin channel. However, in the spin-down channel of CoNbMnZ (Z = Ge, Sn), the reverse effect is observed, i.e., S decreases while σ increases, suggesting the semiconducting nature in this spin orientation. Thus, in a nutshell, it can be concluded that both the band structures and thermoelectric properties suggest the half-metallic character of CoNbMnZ (Z = Ge, Sn). The resemblance in results between the electronic and transport properties authenticates our investigation.
Conclusions
We have presented detailed information about the structural, electronic band structure, magnetic, elastic, and thermoelectric properties of CoNbMnZ (Z = Ge, Sn) alloys. Band structure analysis through GGA indicates that both alloys are metallic. However, upon the inclusion of mBJ, perfect half-metallic nature is observed, with indirect band gaps of 0.43 and 0.45 eV at the Γ- and X-points of the Brillouin zone. The states at the Fermi level are 100% spin polarized. The total magnetic moment per formula unit is 1 μB, following the Slater–Pauling rule MT = ZT − 24. Charge density analysis reveals polar-covalent-type bonding. The mechanical stability is elucidated via calculating the elastic constants. Pugh's ratio, Poisson's ratio, and Cauchy's pressure values show the ductile nature of the alloys. The anisotropic factor value is more than 1, indicating that the properties are directionally dependent. Furthermore, the transport properties of CoNbMnZ (Z = Ge, Sn) reveal some interesting results. The value of S shows an increasing trend over the whole temperature range, attaining maximum values of 60.43 and 68.20 for the alloys, and the positive value of S predicts that holes are the majority carriers. The maximum power factor values are 32 and 35 at 900 K, also suggesting possible thermoelectric applications for these alloys.Conflicts of interest
The authors hereby declare that we have no conflict of interest.References
- T. M. Bhat and D. C. Gupta, J. Magn. Magn. Mater., 2017, 435, 173 CrossRef CAS.
- S. Yousuf and D. C. Gupta, J. Phys. Chem. Solids, 2017, 108, 109 CrossRef CAS.
- S. Yousuf and D. C. Gupta, J. Alloys Compd., 2018, 735, 1245 CrossRef CAS.
- S. A. Khandy and D. C. Gupta, Semicond. Sci. Technol., 2017, 32, 125019 CrossRef.
- Y. Sutou, Y. Imano, N. Koeda, T. Omori, R. Kainuma, K. Ishidab and K. Oikawa, Appl. Phys. Lett., 2004, 85, 4358 CrossRef CAS.
- Z. Wen, T. Kubota, T. Yamamoto and K. Takanashi, Sci. Rep., 2015, 5, 18387 CrossRef CAS.
- S. Yousuf and D. C. Gupta, Phys. B, 2018, 534, 5 CrossRef CAS.
- S. Yousuf and D. C. Gupta, Indian J. Phys., 2017, 91, 33 CrossRef CAS.
- L. Bainsla, M. M. Raja, A. K. Nigam and K. G. Suresh, J. Alloys Compd., 2015, 651, 631 CrossRef CAS.
- A. Kundu, S. Ghosh, R. Banerjee, S. Ghosh and B. Sanyal, Sci. Rep., 2017, 7, 1803 CrossRef.
- R. A. de Groot, F. M. Mueller, P. G. van Engen and K. H. J. Buschow, Phys. Rev. Lett., 1983, 50, 2024 CrossRef CAS.
- X. Y. Dong, C. Adelmann, J. Q. Xie and C. J. Palmstrøma, Appl. Phys. Lett., 2005, 86, 102107 CrossRef.
- V. Alijani, S. Ouardi, G. H. Fecher, J. Winterlik, S. S. Naghavi, X. Kozina, G. Stryganyuk, C. Felser, E. Ikenaga, Y. Yamashita, S. Ueda and K. Kobayashi, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 224416 CrossRef.
- L. Bainsla, A. I. Mallick, M. M. Raja, A. A. Coelho, A. K. Nigam, D. D. Johnson, A. Alam and K. G. Suresh, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 045201 CrossRef.
- L. Bainsla and K. G. Suresh, Appl. Phys. Rev., 2016, 3, 031101 Search PubMed.
- S. Singh and D. C. Gupta, J. Alloys Compd., 2019, 806, 1292 CrossRef CAS.
- T. M. Bhat and D. C. Gupta, J. Electron. Mater., 2016, 45, 6012 CrossRef CAS.
- F. Casper, T. Graf, S. Chadov, B. Balke and C. Felser, Semicond. Sci. Technol., 2012, 27, 063001 CrossRef.
- T. Zhu, C. Fu, H. Xie, Y. Liu and X. Zhao, Adv. Energy Mater., 2015, 5, 1500588 CrossRef.
- T. M. Tritt, H. Böttner and L. Chen, MRS Bull., 2008, 33, 366 CrossRef CAS.
- P. Ying, X. Liu, C. Fu, X. Yue, H. Xie, X. Zhao, W. Zhang and T. Zhu, Chem. Mater., 2015, 27, 909 CrossRef CAS.
- H. Zhu, J. Mao, Y. Li, J. Sun, Y. Wang, Q. Zhu, G. Li, Q. Song, J. Zhou, Y. Fu, R. He, T. Tong, Z. Liu, W. Ren, L. You, Z. Wang, J. Luo, A. Sotnikov, J. Bao, K. Nielsch, G. Chen, D. J. Singh and Z. Ren, Nat. Commun., 2019, 10, 270 CrossRef.
- N. A. Koshi and R. John, Eur. Phys. J. B, 2019, 92, 86 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3868 CrossRef.
- F. Tran and P. Blah, Phys. Rev., 2009, 102, 4 Search PubMed.
- G. K. H. Madsen and D. J. Singh, Comput. Phys. Commun., 2006, 175, 67 CrossRef CAS.
- B. Ryu and M. W. Oh, J. Korean Ceram. Soc., 2016, 53, 273 CrossRef CAS.
- S. Yousuf and D. C. Gupta, J. Alloys Compd., 2018, 738, 501 CrossRef CAS.
- T. Graf, C. Felser and S. S. P. Parkin, Prog. Solid State Chem., 2011, 39, 1 CrossRef CAS.
- T. M. Bhat and D. C. Gupta, J. Electron. Mater., 2018, 47, 2042 CrossRef CAS.
- Z. H. Liu, Y. J. Zhang, E. K. Liu, G. D. Liu, X. Q. Ma and G. H. Wu, J. Phys. D: Appl. Phys., 2015, 48, 1 Search PubMed.
- W. H. E. Schwarz, J. Chem. Educ., 2010, 87, 444 CrossRef CAS.
- T. M. Bhat and D. C. Gupta, Mater. Res. Express, 2017, 4, 1 Search PubMed.
- S. A. Khandy and D. C. Gupta, J. Phys. Chem. Solids, 2019, 135, 109079 CrossRef CAS.
- A. Reuss, Z. Angew. Math. Mech., 1929, 9, 49 CrossRef CAS.
- R. Hill, J. Mech. Phys. Solids, 1963, 11, 375 Search PubMed.
- X. K. Liu, W. Zhou, Z. Zheng and S. M. Peng, J. Alloys Compd., 2014, 615, 975 CrossRef CAS.
- I. H. Bhat, T. M. Bhat and D. C. Gupta, J. Phys. Chem. Solids, 2018, 119, 251 CrossRef CAS.
- S. A. Mir and D. C. Gupta, J. Alloys Compd., 2020, 854, 1–10 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra08007a |
| This journal is © The Royal Society of Chemistry 2020 |