
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclohexamer [-(D-Phe-azaPhe-Ala)2-]: good candidate to formulate supramolecular organogels†
Mohamed I. A. Ibrahim*ab,
Guillaume Pickaerta,
Loïc Stefan a,
Brigitte Jamart-Grégoirea,
Jacques Bodiguela and
Marie-Christine Averlant-Petit*a
a,
Brigitte Jamart-Grégoirea,
Jacques Bodiguela and
Marie-Christine Averlant-Petit*a
aLaboratoire de Chimie-Physique Macromoléculaire (LCPM), UMR 7375, CNRS, Université de Lorraine, Nancy, France. E-mail: marie.averlant@univ-lorraine.fr
bLaboratory of Marine Chemistry, Marine Environment Division, National Institute of Oceanography and Fisheries, NIOF, Egypt. E-mail: ibrahimmohamed2030@gmail.com
First published on 7th December 2020
Abstract
Molecular self-assembly is a fascinating process which has become an area of great interest in supramolecular chemistry, as it leads in certain cases to molecular gels. Organogels formulated from low molecular weight compounds (LMWOGs) have attracted much interest in the past decades due to their applications as new soft materials. Herein, we report on the ability of the cyclic pseudopeptide cyclo-[-(D-Phe-azaPhe-Ala)2-] (2) to self-assemble in some aromatic solvents and to form organogels driven by non-covalent forces, mainly hydrogen bonding and π-stacking interactions. Comprehensive FTIR and NMR studies emphasized that this cyclic aza-peptide adopts a β-turn conformation at low concentration in toluene, while an equilibrium between the monomeric states (intramolecular forces) and the supramolecular structures (intra- and intermolecular forces) is established at high concentration (gel state). Rheological investigations of the organogels highlight the dependence of their stiffness (up to ∼4 kPa) and sol/gel transition temperatures (up to 100 °C) as a function of the solvent and concentration of gelator used. The formulation of fibrous structures confirmed the phenomenon of self-assembly. Finally, we found that cyclo-[-(D-Phe-azaPhe-Ala)2-] is an effective organogelator for application in phase selective gelation (PSG) of organic solvents from aqueous/organic mixtures with recovery percents up to 96%.
Introduction
Recently, gels have experienced a great growth in interest from the scientific community as they have been involved in various applications due to their promising physical, mechanical and morphological properties.1–4 Over more than 150 years, many attempts have evolved to define and describe a “gel”, started by Graham,5 and then Dorothy Jordon Lloyd,6 Hermans,7 and Ferry.8 Recently, Weiss and Terech have reported general criteria to classify a substance as a gel, namely a continuous microscopic structure with macroscopic dimensions and a substance showing solid-like properties in its rheological behavior.9 Gels can be classified in several ways based on their origin (natural or synthetic), constitution (polymeric or molecular), the type of cross-linking that creates the 3D network (chemical or physical), and the medium of gelation (hydrogels, organogels, xerogels, or aerogels).10Firstly, gels were obtained from polymers11 and in the last two decades authors have reported molecular gels obtained from low molecular weight gelators (LMWGs). Molecular gels, which are achieved through self-assembly process to yield highly ordered 3D hierarchical structures, have attracted much interest in the field of supramolecular chemistry due to their promising applications as new soft materials.12,13 In contrast to the polymeric gel, LMWGs monomers must first self-assemble before entanglement and eventually gel the solvent.14 The behaviour of physical gels are controlled by weak non-covalent interactions such as hydrogen bonding, π-stacking, electrostatic interactions, metal coordination, ion–ion interactions, dipole–dipole interactions, van der Waals interactions, etc.9,13,15–22 These weak forces control the ability of a gel to readily interconvert between the gel–sol states under the influence of external stimuli such as pH, ionic strength of the medium, temperature, ultrasound, magnetic field, light, etc.2,23,24
Low molecular weight organogelators (LMWOGs) have been derived from various systems including hydrocarbons, fatty acids, saccharides, steroids, amides, amino acids, ureas, aromatic molecules, metal complexes and dendrimers.1–3,25–28 Recently, LMWGs from peptides, pseudopeptides or their cyclic analogs have revealed greater advantages as they: (i) can easily be synthesized, (ii) can multi-responsive, (iii) may be controlled over their physical and chemical properties, and (iv) exert higher mechanical and thermal stabilities.12,29,30
Serendipitously, our group reported their first low molecular weight organogelator in 2010 after cooling a refluxed solution of an amino acid derivative in toluene.31 Subsequently, Moussodia et al. developed a series of pseudopeptides of the cyclo 1:1-[α/α-Nα-Bn-hydrazino]-mers family able to self-assemble into supramolecular ordered structures through a network of intermolecular hydrogen bonds which immobilize organic solvent molecules to form organogels.32
In a previous study33 we overviewed in details the synthesis and structural studies of homo- and heterochiral cyclo hexamer 2:1-[α/aza]-(L- or D-Phe-azaPhe-Ala)2 in solid and solution states. Afterwards, both molecules were tested for their ability to form gels in various solvents. Interestingly, only heterochiral analogue cyclo 2:1-[D-Phe-azaPhe-Ala]2 (2) could self-assemble in some solvents, precisely aromatic solvents to form thermoreversible organogels in which the gels exhibit the phenomenon of gel-to-sol phase transition reversibly by the influence of temperature.
In the current work, the characterizations of the LMWOG (2) have been established by studying its molecular and supramolecular structures, morphology, chemical and physical behaviors using several sophisticated techniques including NMR, FTIR, SEM and TEM in order to emphasize the existence of certain conformations (e.g. β-turn, α-helix, β-sheet, etc.) and to obtain a virtual image for the 3D hierarchical structures of the gel system. Furthermore, rheological measurements have been carried out to assess the mechanical and thermal stabilities of the organogels formulated in different organic solvents.
Experimental
Synthesis
The general stepwise strategy used to synthesize homo- and heterochiral cyclo 2:1-[α/aza]-(L- or D-Phe-azaPhe-Ala)2 hexamers (1) and (2), respectively (Fig. 1) with all the characteristic spectroscopic, X-ray diffraction and HRMS features were described and reported in the previous studies.33,34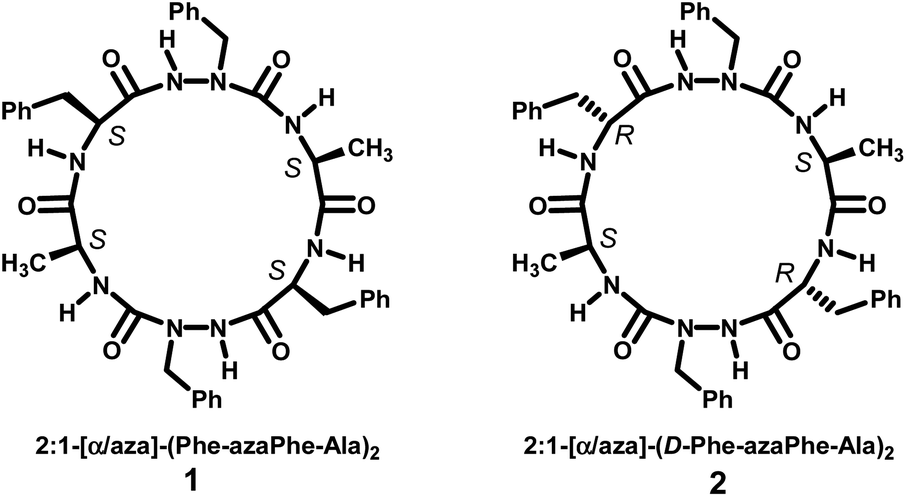 | ||
| Fig. 1 Molecular structures of homo- and heterochiral cyclo 2:1-[α/aza]-hexamers. | ||
Organogels formation, gelation tests and minimum gelation concentrations (MGCs) measurements
The gelation ability of both cyclic pseudopeptides (1) and (2) were tested in various organic solvents (e.g. n-hexane, CCl4, toluene, etc.) by “stable-to-inversion of test tube” method.35 Gelation experiments were performed by weighing 10.0 mg of the compound in a transparent glass tube in which the solvent was subsequently added dropwise to reach final weight of 1.0 g mixture, and a final concentration of 1.0 wt%. Then, the suspension was refluxed to form clear solution which was cooled down to room temperature. The results of gelation tests were recorded for all tested solvents.The minimum gelation concentration (MGC)
It refers to the lowest concentration that leads to gel formation. The MGCs were determined in all the gelled solvents (benzene, chlorobenzene, toluene and p-xylene) using the “stable-to-inversion of test tube” method. It involves the continuous dilution for a gel sample until a concentration below which a gravitational flow of the gel occurs in the inverted glass vial: this concentration is defined as the minimum gelation concentration (MGC).35Gel-to-sol transition temperature (Tg) measurement
The gel-to-sol transition temperature (Tg) at the MGC for each solvent was determined by the “falling ball method’’ through placing the gel-containing glass vial in a thermostated oil bath and slowly raising the temperature of the bath at a rate of 2 °C min−1. The Tg was defined by the temperature at which the gel melted and the ball breaks through the gel.36–38 The value of the Tg at the MGC was the average of a triplicate measurements. Additionally, in this study, the Tg in the different gels were measured using several physico-chemical techniques36 including NMR, FTIR and rheological measurements.NMR spectroscopic studies
All NMR spectra (1H, ROESY) were recorded using a Bruker AvanceII NMR spectrophotometer (300 and 600 MHz) in benzene-d6, chlorobenzene-d5, toluene-d8, and p-xylene-d10 as solvents. The chemical shifts are reported in ppm (δ) relative to tetramethylsilane (TMS) used as an internal standard (δ = 0 ppm). Additionally, concentration and temperature dependent NMR experiments for the organogels from the different gelled solvents were performed.FTIR spectroscopic studies
The FTIR spectra were recorded with Bruker Tensor 27 and referenced to the residual solvent resonances. The device is equipped with thermostated controlled heating unit by which temperature-dependent FT-IR experiments were performed. In addition, attenuated total reflectance (ATR-FTIR) measurements were operated on solids, xerogels (air-drying) and aerogel (CO2-drying), in which each solid was loaded over a trough plate comprising of a germanium single crystal. All the spectra were acquired in the 4000–400 cm−1 range with a resolution of 4.0 cm−1 over 128 scans, taken into consideration the background subtraction from each spectrum to correct for atmospheric interferences.Rheological measurements
The rheological experiments were carried out on Advanced Rheometer-AR2000 (TA instruments) operating in oscillatory mode with a 20 mm parallel plate geometry with serrated surfaces to prevent sliding due to the liquid film expelled by certain dilute samples37 (diameter gap was adjusted to 1000 μm). In order to determine the linear viscoelastic region (LVR) at which the oscillatory experiments were operated, each gel sample was subjected to an oscillatory stress sweep experiments (OSS); (G′ and G′′ were measured as a function of oscillatory stress39,40 (0.6–6366 Pa) at a constant angular frequency of 0.628 rad s−1). Then a dynamic oscillatory time sweep (OTS)41 was performed for 15 minutes with an angular frequency of 0.628 rad s−1, and applied stress of 1.5 Pa which has been chosen within the LVR from the OSS. After that, each sample was subjected to an oscillatory frequency sweep experiment (OFS)41 over a range of frequencies (0.1–62.83 rad s−1) and applied stress of 1.5 Pa obtained from OSS step at 25 °C. Finally, in order to obtain the sol-to-gel transition temperature (thermal properties of the gel), G′ and G′′ for each gel sample were studied over a range of temperatures sufficient for melting the gel to solution through oscillatory temperature sweep experiment, from which Tg can be determined at the cross point between the G′ and G′′ curves.14Morphological study
Removal of toluene solvent from the organogel of (2) using a supercritical CO2 drying process led to the formation of the corresponding aerogel,42 while xerogels from (2) were obtained by slow evaporation of the trapped solvent molecules (benzene, toluene, chlorobenzene or p-xylene) from gel samples in air.38 To get a visual insight into the aggregation and morphology of the formed gels from (2) at a nanoscale, high-resolution scanning electron microscopy (HRSEM) (Environmental Quanta FEG 600-FEI, high vacuum, acceleration voltage of 15 kV, large field detector), and transmission electronic microscope (TEM) (Philips CM200 microscope, 200 kV, equipped with an EDXS spectrometer) were employed. While the dried samples (aerogel and xerogels) for SEM analysis were coated with gold (15 Å) during 2 min by physical vapor deposition,38 TEM images for the aerogel sample from toluene were recorded without pre-treatment.Phase selective gelation strategy (PSG), potential application
Mixture of the organic solvents of interest (benzene, toluene, chlorobenzene and p-xylene; 1.2 mL each) was poured into salty water (NaCl 3.5 w/v%, 10 mL) which was stained with CuSO4 for better visualization of the two-phases.29,43 The entire organic layer from the biphasic mixture could be efficiently gelled by adding a highly concentrated solution (12.5 w/v%) of gelator (2) from ethanol as a co-solvent at room temperature (25 mg of compound (2) in 200 μL ethanol). By taking the advantages of the thermo-reversibility of the physical organogels from (2) (maintained by weak non-covalent interactions): the organic phase was separated from aqueous phase by simple filtration and then the gel melts upon heating, and the organic phase is subsequently distilled off in a round bottom flask.43,44Results and discussion
Gelation behaviors
It has been reported that the gelation phenomenon is the equilibrium state between solubility and phase separation.28 It is achieved when the intermolecular forces (repulsion and attractive) in the gel system are balanced.45 Tests of gelation propensity for each diastereoisomers (1) and (2) in various organic solvents were done, which reflected that only compound (2) exerted gelation properties. Results of gelation tests are reported in Table 1. Compound (2) was found to be soluble in alcohols (e.g. methanol, ethanol, pentan-3-ol), dimethyl sulfoxide (DMSO), N,N-dimethyl formamide (DMF) and no gelation was observed even after several hours in a refrigerator. The insolubility of the cyclopseudopeptide (2) in non-polar solvents like n-hexane or carbon tetrachloride (CCl4), suggests unbalanced forces (hydrophobic interactions ≪ hydrophilic interactions) essential to trigger the gelation process. Finally, transparent gels were observed with compound (2) in some aromatic solvents including benzene, chlorobenzene, toluene and p-xylene (Table 1 and Fig. S1†). However, by increasing the concentrations the gels become less transparent which can be attributed either to the aggregation phenomena, or to a change of the size and/or the shape of objects formed by the gelator molecules (2).| Solvent | MGC (wt%) | Tge (±2 °C) | Tgf (°C) | Tgg (°C) | Tgh (°C) |
|---|---|---|---|---|---|
| a 0.5 wt% = 5.95 mmol L−1 (toluene); 6.01 mmol L−1 (benzene); 5.91 mmol L−1 (p-xylene); and 7.62 mmol L−1 (chlorobenzene).b G = gel.c I = insoluble.d S = soluble.e Gelation temperature at MGC determined by “falling ball” method.f Gelation temperatures at (c = 0.5 wt%) determined by NMR.g Gelation temperatures at (c = 0.5 wt%) determined by FTIR.h Gelation temperatures at (c = 0.5 wt%) determined by rheology. | |||||
| Benzene | Gb 0.41 | 38 | 65–70 | 55–60 | 47.8 |
| Toluene | G 0.30 | 45 | 70–75 | 70–75 | 67.4 |
| p-Xylene | G 0.34 | 40 | 70–75 | 75–80 | 87.2 |
| Chlorobenzene | G 0.38 | 39 | 70–75 | 55–60 | 52.9 |
The minimum gelation concentrations (MGCs), which refer to the minimum concentrations that lead to gelation,35 are summarized in Table 1. The MGCs of (2) vary from 0.30 to 0.41 wt% in the different gelled solvents. The highest gelation efficiency for (2) was observed in toluene (MGC = 0.30 wt%) for which a single molecule of (2) can gel about ∼2640 toluene molecules. The Tg values of organogels from (2) at their MGCs were determined by the “falling ball” method36,38,46 and reported in Table 1 with temperature range = 38.3–45.3 °C, depending of the solvent. All gels maintain their stabilities over a period of months at room temperature. Thus, the gelator (2) obeys the criteria of a stable gel as reported by Abdallah and Weiss.22
Formation of LMW organogels is an evidence of the self-assembly of the gelator molecules to form interconnected three-dimensional network which entrap solvent molecules, leading to gel formation that stabilized via non-covalent interactions.27,31,47–49
To achieve a comprehensive understanding of the structural behavior of compound (2) both in its gel and soluble forms (i.e., in its self-assembled and monomeric forms), we performed a series of NMR and FTIR investigations at different temperatures (above and below the Tg) and different concentrations (above and below the MGC). As discussed herein before, gelator (2) demonstrates the best gelation efficiency for toluene, one of the most common solvents used practically compared to the other gelled solvents.50 Moreover, the physico-chemical properties (e.g., boiling temperature) of toluene are adequate and moderate for most of the analyses and has been consequently selected as the solvent for all our experiments.
NMR spectroscopic studies at low concentration
1H NMR spectrum of compound (2) was recorded in toluene-d8 at low concentration (1.0 mmol L−1, 293 K) to avoid the intermolecular interactions.The spectrum of the molecule (2) in toluene-d8 shows well resolved signals with at least two conformers, suggesting that the molecule behaves as previously observed in CDCl3 (equilibrium between more than one conformers at low concentration).33 The duplicated signals reveal the presence of an equilibrium between two conformers (2A) and (2B) (Fig. S2†). Moreover, the chemical shifts of the NH protons of Ala residues are around ∼6.3 ppm, supposing the involvement of these NHs in hydrogen bonds. This is confirmed by following the concentration effect using NMR and FTIR studies (discussed below). In toluene-d8, it is also observed that the protons of methylene groups of the benzyl moieties are magnetically non-equivalent. All these data suggest that β-turn conformations, previously been observed in CDCl3,33 might occur in toluene. In order to confirm the existence of β-turn, 2D NMR (ROESY) in toluene-d8 (1.0 mmol L−1, 293 K) was operated for compound (2), (Fig. S3†). In case of conformer (2A), the NH (D-Phe) signal is superimposed and overlapped with the aromatic protons of toluene and it is difficult to determine all the dipolar coupling constants associated with this proton. Conformer (2A) reveals moderate correlation between CαH (D-Phe) and NH (azaPhe) and strong correlation between NH (azaPhe) and NH (Ala).
Similarly, the protons of the CαH (D-Phe), NH (azaPhe), and NH (Ala) of conformer (2B) are well separated from conformer (2A) showing strong correlation between CαH (D-Phe) and NH (azaPhe) in addition to moderate correlation between NH (azaPhe) and NH (Ala).
These correlations are characteristic of the presence of βII-turn conformation (Fig. S3†).51,52 In a previous work and from the structural studies of acyclic series,34 it was noticed that changing the absolute configuration of the Phe residue to (R) instead of (S), has led to a change in the conformation from βII to βII′, so both conformers (2A) and (2B) adopt βII′-turn conformation (Fig. 2) which supports the 3D supramolecular organization. Based on X-ray results and NMR studies in chloroform33 or in toluene (this study), authors could suggest that molecule (2) adopts majorly two βII′-turn conformations in solid and solution states.
 | ||
| Fig. 2 ROE correlations of β-turn conformations in the two conformers (2A) and (2B) from ROESY experiment in toluene-d8 (1.0 mmol L−1, 293 K). | ||
Concentration-dependent 1H NMR
It has been reported that the NMR signals of the gelator molecules completely disappear in the gel state and the signals are only detected in the liquid state in case of “dry gel”. Indeed, the fibers formed by the gelators in the gel can be considered as crystals and, consequently, the molecular motion of the gelator molecules within the fibers becomes very limited and the solvent molecules are excluded from the fiber.53Concentration-dependent NMR study of (2) was performed at 25 °C in toluene-d8 from 0.1 mmol L−1 (solution state) to 6.0 mmol L−1 (gel state) (Fig. 3a). The spectra show that the signals of the gelator molecules are still visible even in the gel state, which suggests that the gelator molecules are in equilibrium between their free and self-assembly (i.e. in fibers) states. This behavior is consistent with the principle of “wet gel” as described by Sakurai et al. in which the solvent molecules are incorporated into the gel fibers.31,54–56
 | ||
| Fig. 3 (a) Concentration-dependent 1H NMR (600 MHz, 300 K) spectra show chemical shift (δ ppm) of NH protons for compound (2) in toluene-d8 (c = 0.1 to 6.0 mmol L−1), and (b) temperature-dependent 1H NMR (300 MHz) of gelator (2) in toluene-d8 (c = 0.5 wt% = 5.95 mmol L−1). | ||
One can notice that the signal of the NH proton (D-Phe) of conformer (2B) is deshielded from 5.76 ppm to 6.15 ppm and the signal becomes broad when the concentration exceeds 3.0 mmol L−1, due to the sol to gel transition (Fig. 3a). These observations confirm that the gel state of (2) corresponds to molecules assembled by intermolecular hydrogen bonds.37,57–62 Unfortunately, the signal of the NH (D-Phe) of the conformer (2A) is overlapped with the toluene signals and it therefore not possible to predict its contribution in the organogel formation. In addition, no chemical shift variations for the signals of the NH or the aromatic protons of the (azaPhe) moieties have been observed for both conformers which mainly stabilize the 3D supramolecular structure in case of CDCl3 through π-stacking in the previous study.33
Temperature-dependent 1H NMR
Studying the property of thermal reversibility of a gel provides more detailed information about the physical changes and the mechanism of the supramolecular formation.36 Temperature of gelation (Tg) is the most often reported parameter which can be determined by multiple methods, including the “falling ball” experiment, differential scanning calorimetry (DSC), rheological measurement, as well as spectroscopic techniques such as nuclear magnetic resonance (NMR), UV-vis, circular dichroism (CD) and Fourier transform infra-red (FTIR).36,63 The transition temperatures determined by spectroscopic techniques are often different from those determined from rheology, since the former detect changes in primary rather than tertiary organizations.36,37 Accordingly, studying the thermal behaviors of the gels from (2) in all of the gelled solvents (toluene, benzene, chlorobenzene and p-xylene) were investigated using temperature-dependent NMR spectroscopy from gel state to solution state (Fig. S4a–c†). The 1H NMR spectra for gelator (2) in the different gelled solvents reflected similar behaviors upon raising temperature, and the gels show close Tg at the same concentration (Table 1).In the case of toluene-d8, temperature-dependent 1H NMR spectra of (2) at (c = 0.5 wt%, 5.95 mmol L−1) were recorded in the temperature range from 27 °C to 100 °C in order to follow the evolution of the proton signals in transition from gel to solution state. Although the signals of the molecule in the gel state are less visible in the 1H NMR spectrum (Fig. 3b), the peaks are still detected. This result suggests that some of the gelling molecules still keep good mobility in the fibers.31,47,49,53,55,56 When increasing the temperature, gradual improvement of the spectral resolution and sharpness of signals are observed just after 65 °C which reflects the liquid behavior of the gelling molecules (Fig. 3b). In addition, the NH (azaPhe) protons' signals of conformers (2A) and (2B) are shifting linearly upfield with the increase of the temperature. This shielding reflects the impact of broken hydrogen bonds with temperature and then destruction of the supramolecular structure. So, we could suppose that the NH (azaPhe) protons are directly involved in the supramolecular construction. The protons of NH (D-Phe) were less influenced by the temperature increase from 27 °C to 100 °C, which means that these NH protons are not involved in intermolecular hydrogen bonds. Interestingly, coalescence of CH2 (azaPhe) protons for conformers (2A) and (2B) is observed when the temperature rises over 65 °C and they become chemically equivalent in the solution state (Fig. 3b) which can be interpreted as the fast equilibrium between the two conformers. Finally, NH (Ala) protons chemical shift is not impacted by the variation of temperature, strongly suggesting that NH (Ala) is involved in intramolecular hydrogen bonds as in the crystalline state.33
Thus, NMR studies of (2) at low concentrations (solution) in toluene suggested that the two conformers are structured in a βII′-turn conformation (Fig. 2), while at high concentrations (gel), the gelator molecules are organized into supramolecular structures stabilized by intermolecular hydrogen bonds (Fig. 3a and b). However, NMR is not a method of choice to evaluate the contribution of π-stacking interactions in the self-assembly process. To study such phenomenon, FTIR experiments were subsequently carried out.
FTIR spectroscopic studies
In order to identify the role of the driving forces particularly hydrogen bonds in the formation of the gels, concentration-dependent and temperature-dependent FTIR measurements were performed on gel samples from (2) in the different gelled solvents. In the next sections, we will present in details the behaviors of gels formed with toluene-d8.Concentration-dependent FTIR analysis
The experiments were carried out on gelator (2) using the concentration range (0.8 mmol L−1 to 3.0 mmol L−1) in toluene-d8 at 25 °C (Fig. 4). The spectra showed two characteristic domains corresponding to the NH and CO stretching regions. At low concentration (0.8 mmol L−1), the NH region reveals the presence of free NH band around 3408 cm−1 belongs to the two free NH from D-Phe and azaPhe. In addition, a broad band around 3340 cm−1, corresponding to the bound NH protons of Ala, confirms the presence of intramolecular hydrogen bonds at the monomeric state, as previously observed in chloroform (Fig. 4a).34 Interestingly, the presence of an additional band (not observed in chloroform) around 3390 cm−1 might be related to the solvation effect by toluene molecules inducing NH–π interactions.64,65 | ||
| Fig. 4 (a, b) Concentration-dependent FTIR of (2) in toluene-d8 (c = 0.8 to 3.0 mmol L−1), and (c, d) temperature-dependent FTIR spectra from 25 °C to 90 °C of toluene-d8 gel from (2) at (c = 0.5 wt%; 5.95 mmol L−1); (c) NH stretching region, and (d) CO stretching region. | ||
Regarding the CO region at 0.8 mmol L−1, four bands were observed and assigned according to the FT-IR wavenumbers of the linear precursor (Fig. 4b).33,34 The CO of the Ala residues are involved in intramolecular hydrogen bonds at ν = 1655 cm−1, while the other CO groups are in free states in toluene solution, as noticed from their higher wavenumbers: 1721 cm−1, 1704 cm−1, and 1680 cm−1 corresponding to the free CO of D-Phe, Ala, and azaPhe residues, respectively (Fig. 4b). Moreover, a small band around 1629 cm−1 characteristic of a β-turn conformation in small peptides has been observed.66 The spectrum of (2) in toluene-d8 is similar to spectra obtained in CDCl3 in previous work, suggesting that the conformation of (2) in toluene-d8 at low concentration might be close to conformations observed in CDCl3.33,34
An increase of the concentration from 0.8 mmol L−1 to 3.0 mmol L−1 leads to an increase of the intensity of the band at 3340 cm−1 (bound NH) and the rising of one new NH band at 3288 cm−1 (corresponding to more bound NH) (Fig. 4a). These observations are consistent with the NMR studies which reveal the deshielding of the NH protons of D-Phe in conformer (2B) when increasing concentration. According to these results, we can suppose that this NH proton is involved in intermolecular hydrogen bonds. Additionally, the band at 3408 cm−1, assigned to unbound NH protons (free state) of (D-Phe) and (azaPhe), increases with increasing the concentration (Fig. 4a). The existence of the signals at high concentration confirms the NMR hypothesis that our gelator molecules are still mobile within the fibers (“wet gel”, see above). In contrast, the band at 3390 cm−1 (bound NH) has no significant changes when increasing the concentration, suggesting the confinement of the toluene molecules when increasing the concentration (gel state) since NH–π interactions have no much variations.
When the concentration of (2) increases from 0.8 mmol L−1 to 3.0 mmol L−1, the intensity of all the CO bands increases, which indicates that most of the CO groups are involved in intra- and intermolecular hydrogen bonds (Fig. 4b). Unfortunately, we have not observed any decrease in the intensity of the free CO bands from which we cannot predict which CO group is bound intermolecularly with the NH (D-Phe). Furthermore, at 3.0 mmol L−1 the intensity of the band at 1629 cm−1 increases, which may be assigned to the self-assembly of the gelator molecules via intermolecular hydrogen bonds at gel state (Fig. 4b). Consequently, concentration-dependent FTIR studies suggested the presence of equilibrium between the monomeric state and the supramolecular structure at the gel state.
Interestingly, comparison of the ATR-FTIR spectra of the aerogel, xerogel and the crystal of (2) demonstrate similar IR signatures for the three states (Fig. S5a and b†). Accordingly, we could suggest that molecules (2) self-assemble in a close manner in gel and in the solid states and they have almost the same molecular packing.28,31,53,67,68
Temperature-dependent FTIR studies
The thermal behaviors of gelator (2) in the different gelled solvents were studied following the wavenumber evolution of amide CO and NH groups during raising temperature high enough to break the intermolecular interactions and recover the solution state.IR spectra were recorded for gelator (2) in gelled solvents (benzene, chlorobenzene, toluene and p-xylene) at 0.5 wt%. The gel was heated until it turned into solution state (Fig. S6a–c†). This method was also used to determine the sol-to-gel transition temperature which corresponds to the collapsing of the physical interactions.36 The Tg values of the different gels at 0.5 wt% are summarized in Table 1.
Heating the toluene-d8 gel from 25 °C to 90 °C leads to a gradual collapse of all three NH bands at 3286, 3325 and 3410 cm−1 in addition to the band at 3202 cm−1 that may be related to other intermolecular forces formed in the gel state, in favor of one band located at 3345 cm−1 (Fig. 4c). This observation could be interpreted as the consequence disruption of the intermolecular hydrogen bonds, NH–π interactions and other intermolecular forces responsible for the gel formation.32 The band at 3345 cm−1 (bound NH) is close to the ones obtained in isotropic chloroform or toluene solutions, and is characteristic for an intramolecular hydrogen bound NH, which was assigned to Ala moieties.33
The NH and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bands give rise to same conclusions. The gel state showed four CO bands at 1629, 1655, 1683, and 1709 cm−1 at 25 °C (Fig. 4d). Heating the gel from 25 °C to 90 °C resulted in almost disappearance of the band at 1629 cm−1 and the decrease of the intensity of the CO wavenumber at 1655 cm−1 confirming the involvement of these CO groups in intra- and intermolecular hydrogen bonds. Moreover, the intensities of the bands at 1683, and 1709 cm−1 increase with raising the temperature with the appearance of a new band at 1725 cm−1 which has a close value from what was previously observed in isotropic solutions of chloroform and toluene (free state).33 Since the spectra recorded at high temperatures are very similar to the one obtained for (2) in isotropic solutions of chloroform or toluene, we could propose that the bands located at 1629 cm−1 and 1655 cm−1 result from intermolecular hydrogen bonds in the gel state. It is important to note that by crossing the Tg around 72.5 °C, the band at 1656 cm−1 still exists even at 90 °C: this band must correspond to the CO (Ala) involved in intramolecular hydrogen bonds in solution state.33
O stretching bands give rise to same conclusions. The gel state showed four CO bands at 1629, 1655, 1683, and 1709 cm−1 at 25 °C (Fig. 4d). Heating the gel from 25 °C to 90 °C resulted in almost disappearance of the band at 1629 cm−1 and the decrease of the intensity of the CO wavenumber at 1655 cm−1 confirming the involvement of these CO groups in intra- and intermolecular hydrogen bonds. Moreover, the intensities of the bands at 1683, and 1709 cm−1 increase with raising the temperature with the appearance of a new band at 1725 cm−1 which has a close value from what was previously observed in isotropic solutions of chloroform and toluene (free state).33 Since the spectra recorded at high temperatures are very similar to the one obtained for (2) in isotropic solutions of chloroform or toluene, we could propose that the bands located at 1629 cm−1 and 1655 cm−1 result from intermolecular hydrogen bonds in the gel state. It is important to note that by crossing the Tg around 72.5 °C, the band at 1656 cm−1 still exists even at 90 °C: this band must correspond to the CO (Ala) involved in intramolecular hydrogen bonds in solution state.33
The Tg at 0.5 wt% from toluene-d8 was determined by studying the change in the intensities of the NH free band at 3410 cm−1 and the bound CO band at 1628 cm−1 in which they showed total disappearance when heating (Fig. 4c and d). As depicted in Fig. S7,† the intensities of these two bands decrease sharply when heating from 25 °C to 65 °C. Then, the curves show weak change in their intensities accompanied by semi-steady state behavior, corresponding to the collapsing of most of the physical intermolecular interactions in the gel. The sol-to-gel transition can be considered at the inflection point of the two curves, i.e. Tg = 72.5 °C for 0.5 wt% from toluene-d8.
Temperature-dependent FTIR experiments suggest that all formed gels from the different solvents are supported mainly by intermolecular hydrogen bonds stabilizing the gel fiber organization. In addition, gelator (2) shows similar behaviors in the different gelled solvents (Fig. S6a–c†), suggesting that the mechanism of gelation is very similar from these solvents.
In addition to the structural studies using NMR and FTIR techniques, further insight into studying the mechanical properties and the morphology for the different gels from (2) have gained our interests.
Rheological measurements
Rheological experiments provide information about the mechanical strength and rigidity of the gels through measuring two main parameters: storage (elastic) modulus (G′) corresponding to the ability of the deformed material to store energy, and the loss (viscous) modulus (G′′) which reflects the flowing behavior of the gels upon applying external stress.29 Generally, true physical gels reveal G′ > G′′ in gel state, and G′′ > G′ in the solution state.69–72 The thermal property of gels can also be evaluated through an oscillatory temperature sweep experiment in terms of the transition temperature (Tg).29In these experiments, organogels from (2) were prepared in different organic solvents (benzene, chlorobenzene, toluene and p-xylene) at different concentrations. All the oscillatory experiments for all the gels were performed within the linear viscoelastic region (LVR) which has been investigated through two main experiments, namely oscillatory stress sweep (OSS) and oscillatory time sweep (OTS) experiments (Fig. S8 and S9†). In the OSS experiments, all the studied organogels show higher G′ values than G′′ at initial. By a gradual increase of the applied stress, the values of both G′ and G′′ remain nearly unaltered and get deviated from linearity beyond a certain stress. The stress at which G′′ becomes higher than G′ is referred to the yield stress.39,40 All the gels at 0.5 wt% showed close yield stresses at the crossover point in the range (38–48 Pa) (Fig. S8†). In the OTS experiments, the recovery of the gel represented by G′ from the deformed state has been studied at as a function of time (15 minutes), and the data (Fig. S9†) show that all the gels are mostly recovered within the time span of 100 seconds.
The oscillatory frequency sweep experiments (OFS) carried out for gel samples at 0.5 wt%, show that the both G′ and G′′ are regulated as a function of angular frequency (ω) at a constant applied stress.73,74 For all the organogels, G′ is higher than G′′ by at least two order of magnitude and the two curves do not cross over the entire ω range = 0.1–62.83 rad s−1 (Fig. 5). The storage moduli of all the organogels G′ was in the order p-xylene > chlorobenzene > benzene > toluene, and show moderate mechanical strengths (104 > G′ > 102 Pa) in comparison with other previously reported supramolecular gels.44,75–80
 | ||
| Fig. 5 Oscillatory frequency sweep experiments (OFS) for organogels of (2); (c = 0.5 wt%, σ = 1.5 Pa, T = 25 °C). | ||
Further elucidation of the thermal property of our gels from (2) in the different solvents was investigated by measuring the Tg through the oscillatory temperature sweep experiments for each gel sample at different concentrations (Fig. S10a–d†). Transition temperatures (Tg) corresponding to certain concentrations could be obtained at the cross point between the G′ and G′′ curves when plotting against temperature.14 The Tg for the gel at 0.5 wt% from toluene reveals Tg ∼ 67 °C (Fig. S11†). All the Tgs at 0.5 wt% from the different gelled solvents are summarized in Table 1.
We also examined the sol-to-gel transition temperature as a function of concentration by operating oscillatory temperature sweep experiment of each concentration for each solvent individually (Fig. S10a–d†). All the gels have the same trend in which their Tgs increase with increasing concentration (Fig. S12†), in agreement with earlier reports.38,45,46 Regarding the case study of the gel in toluene, the OFS experiment for 0.5 wt% gel from (2) was performed at the Tg = 65 °C. The result plotted in Fig. S13† reveals that both G′ and G′′ increase linearly keeping G′ > G′′ over the entire range of the applied angular frequency supposing that the gel from toluene acquires the behaviors of the true physical organogel and not the viscoelastic fluid.69–71
Rheological studies showed that all the organogels from (2) have nearly similar thermal and mechanical properties. Consequently, gelator (2) self-assembles by the same way in the different gelled solvents leading to the formation of nearly resemble supramolecular 3D arrangement which are almost maintained by the same intermolecular interactions that keep gel stability.
Morphological study
The gelation process of (2) reflects a great self-assembly ability of this aza-family to form fibrous network through the combination of non-covalent interactions, Fig. 6. | ||
| Fig. 6 (a, b) SEM images, and (c) TEM image of aerogel obtained from organogel of (2) in toluene. | ||
SEM images of the aerogel of toluene from (2) exhibit an interconnected network of thin fibers-like structure with a thickness of a few tens of nanometers (Fig. 6a and b). The morphology of the aerogel (2) from toluene was also confirmed by TEM images (Fig. 6c) which clearly show the formation of self-assembled interconnected fibrillar networks. The thickness of the fibril ranges from 40–50 nm with variable lengths of about several micrometers, are consistent with the results from SEM. In the other hand, the SEM images of the xerogels of (2) from p-xylene, benzene, chlorobenzene and toluene could not show a clear fiber structure from which we let us easily suppose that the xerogels could not conserve the self-assembly of compound (2) (Fig. S15†).
Phase selective gelation (PSG), potential application
In the recent decades, water pollution has become a serious problem to mankind where the aquatic systems have suffered from several hazardous, mainly the organic contaminants that enter through several oil spill accidents and human activities.38,43,81,82 Separation of the oil phase, especially the aromatic components which are hardly decomposed naturally,78,79 from water is essential because of their toxicity which brings a huge damage to aquatic ecosystems.43 In this context, bioremediation is a commonly available process which has used to clean up an oil spill.83 Chemical treatment has also been suggested for oil recovery by using several chemical materials such as dispersants,84 sorbents,85–88 and solidifiers.89Unfortunately, some of these materials are toxic to aquatic life and/or not biodegradable naturally, and in certain cases, the recovery of the oil is really difficult.44 Phase selective gelation (PSG) of organic phase from organic/water mixtures is being considered as a promising tool in water purification.43,44 The ideal PSG treatment must fulfill some requirements: (a) the gelator must exhibit efficient and selective gelation ability at room temperature, (b) the gelator have to be easily synthesized from cheap starting materials, (c) the gel must have high mechanical stability to facilitate the separation of oil and gelator from a biphasic mixture, (d) the oil have to be easily recovered from the gel phase materials, and (e) the gelator should be recyclable for reuse.44,90
One of the major drawback of some PSG described in the literature is the heating-cooling protocol necessary to induce gelation, which makes it inoperable for the removal of oil spills due to the high flammability of most oil components.43
To overcome this problem, many methods have been reported to trigger effective phase selective gelation process (PSG) at room temperature by: (a) changing the pH of the medium,91 (b) mechanical shaking,79,92 or sonication,93 (c) addition of slightly pre-warmed concentrated solutions of the suitable gelator to an oil/aqueous mixture,43 and recently phase selective gelation at room temperature can be achieved by (d) addition of pre-dissolved gelator in a co-solvent and it seems to be a practical procedure in real situation for water purification.44,81,94–96
In this regard, Bhattacharya and Ghosh in 2001 reported the first phase selective gelation of oil by an alanine-based amphiphilic gelator in the presence of water.97 Then, several other groups have reported very interesting LMWGs with PSG ability.29,38,80,82,92,98 John and co-workers recently demonstrated model solidifiers for oil spills by using an ethanolic solution of sugar-based gelators in the oil/water mixture at room temperature.90 In addition, sugar-based gelators have also been used by Prathap and Sureshan for phase selective gelation by addition of pre-warmed solution of gelator into the biphasic oil/water mixture.95
In order to evaluate the potential application of PSG triggered by (2) for natural polluted water with organic contaminants, comparative experiments using biphasic systems containing pure water, and synthetic salty water (NaCl 3.5 w/v%) have been done. Thus, gelator (2) was firstly solubilized in ethanol and subsequently added to the organic/aqueous mixture (Fig. S16†) to trigger the gelation process. The results show that gelator (2) has good gelation ability at low concentration (∼0.5 wt%), even in the presence of salts. Moreover, gelator (2) able to gel the organic phase (aromatic solvents) efficiently with good recovered percentages after a simple distillation process (77–96%) (Table S1†). Interestingly, gelator (2) was found to be an appropriate molecule for phase selective gelation of several organic solvents, including benzene, toluene, chlorobenzene and p-xylene, from an organic/aqueous mixture within 2 minutes due to its: (i) excellent organogelation ability at room temperature, (ii) water insolubility, and (iii) high mechanical rigidity in the gelled organic solvents.
Conclusions
We have reported in this work the investigation of the conformation of molecule (2) in toluene which behaves similarly as in chloroform, i.e., self-assembling via the formation of a β-turn at low concentration. At high concentrations or gel state, the organogelator (2) containing peptide bonds, aza moieties, and aromatic amino acids establishes non-covalent interactions to self-assemble, mainly hydrogen bonds and NH–π interactions. These interactions play a vital role in the stability of the self-assembled 3D network fibrous structure that entraps solvent molecules forming the gel at the macroscopic level. Furthermore, the organogels of (2) from the different gelled solvents showed similar thermal and mechanical behaviors as observed by rheological measurements. Finally, the gelator show high gelation efficiency of organic solvents from organic/aqueous mixture. This study emphasized the self-assembly phenomenon by azapeptides that may lead to the formation of soft materials (organo-/hydro-/oleogels) and this will be our interests in the forthcoming studies to be involved in promising applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge Dr Halima Alem-Marchand for SEM and TEM experiments; Institut Jean Lamour, UMR 7198 CNRS-Université de Lorraine. Thanks to Mr Olivier Fabre for performing all the NMR experiments, Laboratoire de Chimie Physique Macromoléculaire (UMR 7375 CNRS, Université de Lorraine). Thanks to the NMR facilities in the Faculté des Sciences et Technologies, Université de Lorraine for the 600 MHz NMR experiments. M. C. A.-P. and L. S. thank the Centre National de la Recherche Scientifique (CNRS) for funding.References
- S. Banerjee, R. K. Das and U. Maitra, J. Mater. Chem., 2009, 19, 6649–6687 RSC
.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef CAS
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS
- T. Graham, Philos. Trans. R. Soc. London, 1861, 151, 183–224 CrossRef
- D. J. Lloyd, Colloid Chemistry, ed. J. Alexander, The Chemical Catalogue Company, New York, USA, 1926, vol. 1, p. 767 Search PubMed
- P. H. Hermans, Colloid Science, ed. H. R. Kruyt, Elsevier, Amsterdam, 1949, vol. II, ch. XII, p. 484 Search PubMed
- J. D. Ferry, Viscoelastic Properties of Polymers, Wiley, New York, 1980, p. 391 Search PubMed
- R. G. Weiss and P. Terech, Molecular Gels: Materials with Self-Assembled Fibrillar Networks, Springer, 2006 Search PubMed
- Gels handbook, ed. Y. Osada, K. Kajiwara, T. Fushimi, O. Hirasa, Y. Hirokawa, T. Matsunaga, T. Snimomura, L. Wang and H. Ishida, Academic Press, San Diego, 2001 Search PubMed
- A. H. Clark, Gels and Gelling, Physical Chemistry of Foods, Marcel Dekker, New York, 1996, pp. 263−305 Search PubMed
- S. M. M. Reddy, G. Shanmugam, N. Duraipandy, M. S. Kiran and A. B. Mandal, Soft Matter, 2015, 11, 8126–8140 RSC
- C.-W. Liu, M. Su, X.-L. Li, T. Xue, N. Liu, J. Yin, Y.-Y. Zhu and Z.-Q. Wu, Soft Matter, 2015, 11, 5727–5737 RSC
- M. A. Rogers and J. H. J. Kim, Food Res. Int., 2011, 44, 1447–1451 CrossRef CAS
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC
- M. D. Loos, B. L. Feringa and J. H. V. Esch, Eur. J. Org. Chem., 2005, 3615–3631 CrossRef
- B.-K. An, D.-S. Lee, J.-S. Lee, Y.-S. Park, H.-S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232–10233 CrossRef CAS
- C. Wang, D. Zhang and D. Zhu, J. Am. Chem. Soc., 2005, 127, 16372–16373 CrossRef CAS
- S. Tanaka, M. Shirakawa, K. Kaneko, M. Takeuchi and S. Shinkai, Langmuir, 2005, 21, 2163–2172 CrossRef CAS
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef CAS
- T. Tu, W. Fang, X. Bao, X. Li and K. H. Dotz, Angew. Chem., Int. Ed., 2011, 50, 6601–6605 CrossRef CAS
- D. J. Abdallah and R. G. Weiss, Langmuir, 2000, 16, 352–355 CrossRef CAS
- P. Jana, S. Maity, S. K. Maity, P. K. Ghorai and D. Haldar, Soft Matter, 2012, 8, 5621–5628 RSC
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS
- D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237–1247 CrossRef CAS
- F. Fages, Top. Curr. Chem., Springer, 2005, vol. 256 Search PubMed
- T. Ishi-i and S. Shinkai, Top. Curr. Chem., 2005, 258, 119–160 CrossRef CAS
- M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS
- T. Kar, S. Mukherjee and P. K. Das, New J. Chem., 2014, 38, 1158–1167 RSC
- A. B. Seabra and N. Duran, Peptides, 2013, 39, 47–54 CrossRef CAS
- F. Allix, P. Curcio, N. P. Quoc, G. Pickaert and B. Jamart-Gregoire, Langmuir, 2010, 26, 16818–16827 CrossRef CAS
- R. O. Moussodia, S. Acherar, E. Romero, C. Didierjean and B. Jamart-Gregoire, J. Org. Chem., 2015, 80, 3022–3029 CrossRef CAS
- M. I. A. Ibrahim, Z. Zhou, C. Deng, C. Didierjean, R. Vanderesse, J. Bodiguel, M.-C. Averlant-Petit and B. Jamart-Grégoire, Eur. J. Org. Chem., 2017, 4703–4712 CrossRef CAS
- Z. Zhou, C. Deng, C. Abbas, C. Didierjean, M. C. Averlant-Petit, J. Bodiguel, R. Vanderesse and B. Jamart-Gregoire, Eur. J. Org. Chem., 2014, 7643–7650 CrossRef CAS
- Y. Y. Chen, H. Wang, D. W. Zhang, J. L. Hou and Z. T. Li, Chem. Commun., 2015, 51, 12088–12091 RSC
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS
- P. Terech, C. Rossat and F. Volino, J. Colloid Interface Sci., 2000, 227, 363–370 CrossRef CAS
- G. L. Feng, H. H. Chen, J. H. Cai, J. W. Wen and X. B. Liu, Soft Matter, 2014, 12, 403–410 CrossRef CAS
- F. M. Menger and A. V. Peresypkin, J. Am. Chem. Soc., 2003, 125, 5340–5345 CrossRef CAS
- Z. Yang, G. Liang and B. Xu, Chem. Commun., 2006, 738–740 RSC
- S. Sathaye, A. Mbi, C. Sonmez, Y. C. Chen, D. L. Blair, J. P. Schneider and D. J. Pochan, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 34–68 CAS
- F. Placin, J.-P. Desvergne and F. Cansell, J. Mater. Chem., 2000, 10, 2147–2149 RSC
- J. Bachl, S. Oehm, J. Mayr, C. Cativiela, J. J. Marrero-Tellado and D. D. Diaz, Int. J. Mol. Sci., 2015, 16, 11766–11784 CrossRef CAS
- S. Basak, J. Nanda and A. Banerjee, J. Mater. Chem., 2012, 22, 11658–11664 RSC
- N. Zweep, A. Hopkinson, A. Meetsma, W. R. Browne, B. L. Feringa and J. H. van Esch, Langmuir, 2009, 25, 8802–8809 CrossRef CAS
- P. Terech, D. Pasquier, V. Bordas and C. Rossat, Langmuir, 2000, 16, 4485–4494 CrossRef CAS
- K. Hanabusa, M. Yamada, M. Kimura and H. Shirai, Angew. Chem., Int. Ed., 1996, 35, 1949–1951 CrossRef CAS
- R. E. Melendez, A. J. Carr, B. R. Linton and A. D. Hamilton, Struct. Bonding, 2000, 96, 31–61 CrossRef CAS
- J. H. van Esch, Langmuir, 2009, 25, 8392–8394 CrossRef CAS
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson Prentice Hall, New York, 3rd edn, 2008 Search PubMed
- H. J. Lee, H. M. Park and K. B. Lee, Biophys. Chem., 2007, 125, 117–126 CrossRef CAS
- M. Thormann and H. J. Hofmann, J. Mol. Struct.: THEOCHEM, 1999, 469, 63–76 CrossRef CAS
- G. Cassin, C. de Costa, J. P. M. van Duynhoven and W. G. M. Agterof, Langmuir, 1998, 14, 5757–5763 CrossRef CAS
- C. Geiger, M. Stanescu, L. Chen and D. G. Whitten, Langmuir, 1999, 15, 2241–2245 CrossRef CAS
- K. Sakurai, Y. Jeong, K. Koumoto, A. Friggeri, O. Gronwald, S. Sakurai, S. Okamoto, K. Inoue and S. Shinkai, Langmuir, 2003, 19, 8211–8217 CrossRef CAS
- R. Wang, C. Geiger, L. Chen, B. Swanson and D. G. Whitten, J. Am. Chem. Soc., 2000, 122, 2399–2400 CrossRef CAS
- D. C. Duncan and D. G. Whitten, Langmuir, 2000, 16, 6445–6452 CrossRef CAS
- B. Huang, A. R. Hirst, D. K. Smith, V. Castelletto and I. W. Hamley, J. Am. Chem. Soc., 2005, 127, 7130–7139 CrossRef CAS
- V. Kral, S. Pataridis, V. Setnicka, K. Zaruba, M. Urbanova and K. Volka, Tetrahedron, 2005, 61, 5499–5506 CrossRef CAS
- J. Makarevic, M. Jokic, Z. Raza, Z. Stefanic, B. Kojic-Prodic and M. Zinic, Chem.–Eur. J., 2003, 9, 5567–5580 CrossRef CAS
- F. S. Schoonbeek, J. V. van Esch, R. Hulst, R. M. Kellogg and B. L. Feringa, Chem.–Eur. J., 2000, 6, 2633–2643 CrossRef CAS
- K. M. Sureshan, K. Yamaguchi, Y. Sei and Y. Watanabe, Eur. J. Org. Chem., 2004, 4703–4709 CrossRef CAS
- X. W. Du, J. Zhou, J. F. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS
- S. Dautrey, J. Bodiguel, A. Arrault and B. Jamart-Grégoire, Tetrahedron, 2012, 68, 4362–4367 CrossRef CAS
- W. Y. Sohn, V. Brenner, E. Gloaguen and M. Mons, Phys. Chem. Chem. Phys., 2016, 18, 29969–29978 RSC
- E. Vass, M. Kurz, R. K. Konat and M. Hollósi, Spectrochim. Acta, Part A, 1998, 54, 773–786 CrossRef
- D. J. Abdallah, S. A. Sirchio and R. G. Weiss, Langmuir, 2000, 16, 7558–7561 CrossRef CAS
- M. George and R. G. Weiss, J. Am. Chem. Soc., 2001, 123, 10393–10394 CrossRef CAS
- A. M. Smith, R. M. L. Turner, A. Saianiand and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS
- B. Adhikari, J. Nanda and A. Banerjee, Soft Matter, 2011, 7, 8913–8922 RSC
- J. Nanda and A. Banerjee, Soft Matter, 2012, 8, 3380–3386 RSC
- A. J. Kleinsmann and B. J. Nachtsheim, Chem. Commun., 2013, 49, 7818–7820 RSC
- S. K. Mandal, T. Kar, D. Das and P. K. Das, Chem. Commun., 2012, 48, 1814–1816 RSC
- S. Bhattacharya, A. Srivastava and A. Pal, Angew. Chem., Int. Ed., 2006, 45, 2934–2937 CrossRef CAS
- S. Mukherjee and B. Mukhopadhyay, RSC Adv., 2012, 2, 2270–2273 RSC
- D. R. Trivedi and P. Dastidar, Chem. Mater., 2006, 18, 1470–1478 CrossRef CAS
- T. Zhang, Y. Wu, L. Gao, Z. Song, L. Zhao, Y. Zhang and J. Tao, Soft Matter, 2013, 9, 638–641 RSC
- M. J. Huertas, E. Duque, L. Molina, R. Rossello-Mora, G. Mosqueda, P. Godoy, B. Christensen, S. Molin and J. L. Ramos, Environ. Sci. Technol., 2000, 34, 3395–3400 CrossRef CAS
- J. Peng, K. Liu, X. Liu, H. Xia, J. Liu and Y. Fang, New J. Chem., 2008, 32, 2218–2224 RSC
- S. Debnath, A. Shome, S. Dutta and P. K. Das, Chem.–Eur. J., 2008, 14, 6870–6881 CrossRef CAS
- M. Konda, I. Maity, D. B. Rasale and A. K. Das, ChemPlusChem., 2014, 79, 1482–1488 CrossRef CAS
- S. L. Yu, X. Q. Dou, D. H. Qu and C. L. Feng, J. Mol. Liq., 2014, 190, 94–98 CrossRef CAS
- R. J. Swannell, K. Lee and M. Mcdonagh, Microbiol. Rev., 1996, 60, 342–365 CrossRef CAS
- R. J. Fiocco and A. Lewis, Pure Appl. Chem., 1999, 71, 27–42 CAS
- H. M. Choi and R. M. Cloud, Environ. Sci. Technol., 1992, 26, 772–776 CrossRef CAS
- F. Wang, S. Yu, M. Xue, J. Ou and W. Li, New J. Chem., 2014, 38, 4388–4393 RSC
- H. Zhu, S. Qiu, W. Jiang, D. Wu and C. Zhang, Environ. Sci. Technol., 2011, 45, 4527–4531 CrossRef CAS
- S. Kizil, K. Karadag, G. O. Aydin and H. B. Sonmez, J. Environ. Manage., 2015, 149, 57–64 CrossRef CAS
- É. Pelletier and R. Siron, Environ. Toxicol. Chem., 1999, 18, 813–818 CrossRef
- S. R. Jadhav, P. K. Vemula, R. Kumar, S. R. Raghavan and G. John, Angew. Chem., Int. Ed., 2010, 49, 7695–7698 CrossRef CAS
- T. Kar, S. Debnath, D. Das, A. Shome and P. K. Das, Langmuir, 2009, 25, 8639–8648 CrossRef CAS
- M. Xue, D. Gao, K. Liu, J. Peng and Y. Fang, Tetrahedron, 2009, 65, 3369–3377 CrossRef CAS
- M. Suzuki, T. Sato, H. Shirai and K. Hanabusa, New J. Chem., 2006, 30, 1184–1191 RSC
- S. Mukherjee, C. Shang, X. Chen, X. Chang, K. Liu, C. Yua and Y. Fang, Chem. Commun., 2014, 50, 13940–13943 RSC
- A. Prathap and K. M. Sureshan, Chem. Commun., 2012, 48, 5250–5252 RSC
- L. Yan, G. Li, Z. Ye, F. Tian and S. Zhang, Chem. Commun., 2014, 50, 14839–14842 RSC
- S. Bhattacharya and Y. Krishnan-Ghosh, Chem. Commun., 2001, 185–186 RSC
- R. D. Trivedi, A. Ballabh and P. Dastidar, Chem. Mater., 2003, 15, 3971–3973 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07775e |
| This journal is © The Royal Society of Chemistry 2020 |