
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating the effect of solvent vapours on crystallinity, phase, and optical, morphological and structural properties of organolead halide perovskite films†
Sagar A. Morea,
Rajendra G. Halora,
Raees Shaikha,
Gauri G. Bisena,
Hemant S. Tarkasa,
Swapnil R. Taka,
Bharat R. Badeb,
Sandesh R. Jadkar b,
Jaydeep V. Salia and
Sanjay S. Ghosh*a
b,
Jaydeep V. Salia and
Sanjay S. Ghosh*a
aOptoelectronics/Organic Photovoltaics Laboratory, Department of Physics, Kavayitri Bahinabai Chaudhari North Maharashtra University, Jalgaon-425001, Maharashtra, India. E-mail: ssghosh@nmu.ac.in
bDepartment of Physics, Savitribai Phule Pune University, Pune-411007, Maharashtra, India
First published on 2nd November 2020
Abstract
A comprehensive study regarding the effect of different solvent vapours on organolead halide perovskite properties is lacking. In the present work, the impact of exposing CH3NH3PbI3 films to the vapours of commonly available solvents has been studied. The interaction with perovskite has been correlated to solvent properties like dielectric constant, molecular dipole moment, Gutmann donor number and boiling point. Changes in the crystallinity, phase, optical absorption, morphologies at both nanometer and micrometer scale, functional groups and structures were studied using X-ray diffraction, UV-visible absorption, FE-SEM, FTIR and Raman spectroscopies. Among the aprotic solvents DMSO and DMF vapours deteriorate the crystallinity, phase, and optical, morphological and structural properties of the perovskite films in a very short time, but due to the difference in solvent property values acetone affects the perovskite properties differently. Polar protic 2-propanol and water vapours moderately affect the perovskite properties. However 2-propanol can solvate the organic cation CH3NH3+ more efficiently as compared to water and a considerable difference was found in the film properties especially the morphology at the nanoscale. Nonpolar chlorobenzene vapour minutely affects the perovskite morphology but toluene was found to enhance perovskite crystallinity. Solvent properties can be effectively used to interpret the coordination ability of a solvent. The present study can be immensely useful in understanding the effects of different solvent vapours and also their use for post-deposition processing (like solvent vapour annealing) to improve their properties.
Introduction
Organolead halide perovskites with the general formula ABX3 (where A is an organic cation, B a divalent metal ion, and X a halide) have potential applications in many fields such as photovoltaics,1–3 field-effect transistors (FETs),4,5 light-emitting devices (LEDs),6,7 lasers,8 and photodetectors.9,10 This class of materials has several advantages like solution processability, low-temperature processing requirements, flexibility in terms of composition, and it can be deposited on flexible substrates.11–13 Organolead halide perovskites combine the properties of inorganic materials (such as high carrier mobility and a wide band gap range from ∼1.1 to ∼4 eV) and those of organic materials (such as structural diversity, high-efficiency luminescence, and plastic mechanical properties).14–16 Improved device performances can be achieved by obtaining enhanced optoelectronic properties of the perovskite materials through various methods and by understanding their properties in a better way. The only aspect of organic–inorganic perovskites which hinders the journey to the commercial market is their instability.17 Both intrinsic and extrinsic factors may be responsible for the instability. Prominent among the intrinsic factors are thermal, photochemical, and degradation due to ion migration. Various groups have studied the effect of using different solvents dimethyl sulfoxide (DMSO), dimethylformamide (DMF), γ-butyrolactone, dimethylacetamide, (N-methyl-2-pyrrolidone), and their mixtures for perovskite film formation.18,19 It is reported that the perovskite films are formed through complex intermediates from the precursor solution of DMF, DMSO, and other solvents. Radicchi et al. have investigated the chemistry of typical precursor solutions employed for lead halide perovskite synthesis by a combined experimental and computational approach.20 Many groups have studied the effect of humidity and oxygen on the perovskites.20–22 Hao Xiong et al. have studied solvent vapour annealing of oriented PbI2 films for improved crystallization of perovskite films in the air.23 Jun Luo et al. studied the mechanism and effect of γ-butyrolactone solvent vapour post-annealing on the perovskite of a mesoporous solar cell structure.24 However, a complete study regarding the impact of solvent vapours of different classes on the organolead halide perovskite properties is not present in literature.Different types of solvents are commonly available in the laboratory, and while synthesizing and fabricating the devices, solvent vapours can be available in the surroundings. Therefore, it is important to know the effect of these commonly available solvents of different classes on the organolead halide properties. Also understanding the use of solvent vapours for post-deposition processing (like solvent vapour annealing) to improve perovskite properties. To the best of our knowledge, there is no comprehensive report where the effect of vapours of commonly available solvents from different classes viz. polar protic, polar aprotic and non-polar which can have varied effects and also to different levels on perovskite properties has been studied. In the present work, we have studied the impact of exposing vapours of various solvents like dimethylformamide (DMF), dimethylsulfoxide (DMSO), acetone, water, 2-propanol, chlorobenzene and toluene on the perovskite material for relatively short time up to 30 minutes. We have studied the effect of solvents on absorption, crystallinity, phase, morphology both at the nanometre and micrometre scale and the structure of perovskite. Our results show that vapours of polar protic solvents like DMF and DMSO affect the optical properties, morphology, and crystalline structure of perovskite drastically in a short time. Acetone and the polar protic water and 2-propanol affect some aspects of the perovskites. Vapours of non-polar solvents like chlorobenzene and toluene have a minimal effect on the perovskite properties. Toluene vapour was found to improve the perovskite crystallinity without much affecting the nanoscale morphology. We also propose the use of mixed solvents to obtain tailor-made properties of solvents for obtaining desired properties by vapour exposure.
Materials and methods
All the chemicals and solvents were purchased from Sigma Aldrich. CH3NH3I (MAI) was synthesized in the laboratory, and the synthetic details are provided in the ESI.†CH3NH3PbI3 film deposition
CH3NH3PbI3 film was deposited using a single step spin coating method. A mixture of precursors MAI (3 M) and PbCl2 (1 M) was prepared in DMF. The solutions were stirred at 40 °C for 2 hours, and after that, spin coated on a glass substrate at 3000 rpm for 1 minute. The spun films were then heated at 100 °C for 45 minutes.Pristine PbI2 film deposition
PbI2 film was deposited using spin coating. A PbI2 solution (461 mg mL−1) was prepared in DMF, and this solution was stirred at 70 °C for 2 h. Then, this solution was spin-coated on pre-cleaned glass substrates at 3000 rpm for 1 minute.Solvent vapour exposure
The glass substrates coated with perovskite films were placed in Petri-dish on thicker platforms made by staking of 5 glass slides. 10 mL of required solvents were then poured in each Petri-dish (90 mm diameter). Precautions were taken so that the solvent does not touch the film directly. The Petri-dish was then covered with another glass plate. A schematic diagram showing the film formation and vapour exposure procedure is given in the Fig. 1.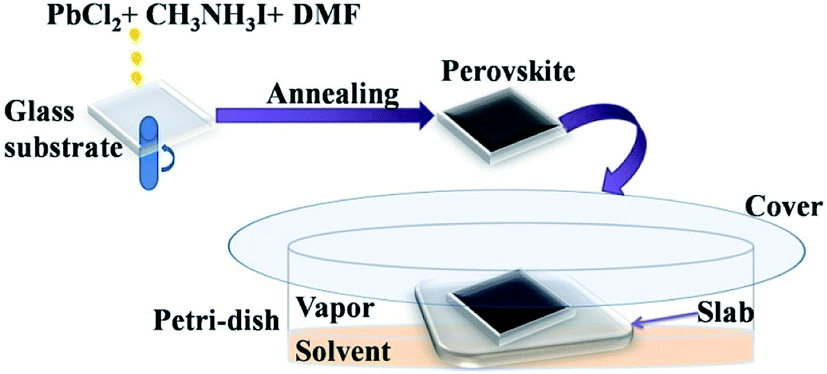 | ||
| Fig. 1 Schematic diagram showing the perovskite film formation and exposing to the solvent vapours. | ||
Characterizations
UV-visible absorption spectrophotometer (UV 1600, Shimadzu) was used to measure the absorption spectra of the samples. Field Emission Scanning Electron Microscope (FE-SEM Hitachi-4800) was used to study the surface morphological changes of the samples by applying an accelerating voltage of 20 kV. D8 advance Bruker X-ray diffractometer with incident source wavelength λ = 1.54 Å in coupling mode was used for the identification of material phase and study the effect on crystalline properties. Fourier transformed infrared (FTIR) spectrometer (PerkinElmer) was used to examine the molecular nature of the sample constituents. Raman spectra were recorded by using (Renishaw inVia microscope Raman, resolution 1 cm−1, excitation source 632.8 nm line of He–Ne laser). Weight of the amount of solvent settled on the exposed film was measured using Shimadzu balance AUW220D with readability 0.01 mg.Results
The following equations govern the reaction between PbCl2 and CH3NH3I in the solution and thereafter during the perovskite phase formation due to thermal annealing after spin coating.
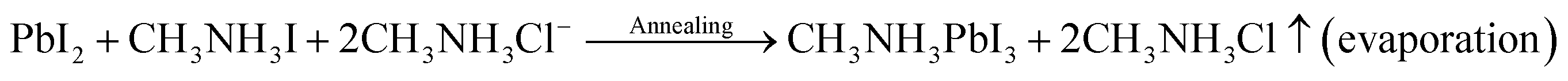
In the precursor complex, PbI2 is formed due to an ion exchange between PbCl2 and CH3NH3I. After thermal annealing, it is converted into the CH3NH3PbI3 phase. There are numerous reports which show that CH3NH3Cl evaporates during thermal annealing process.25 Several studies have suggested that the presence of CH3NH3Cl slow down the film formation process during thermal annealing which therefore results in more crystalline and stable perovskite.26 Due to this reason PbCl2 was preferred in place of PbI2 for perovskite film formation in the present study.
X-ray diffraction study
X-ray diffraction (XRD) pattern of the annealed perovskite film and of those after exposing to different solvent vapours for 30 minutes were recorded (Fig. 2). It shows that, good phase of CH3NH3PbI3 is formed after thermal annealing the spin coated film. Standard diffraction peaks at 14.2°, 28.5° and 43.3° values of 2θ are attributed to (1 1 0), (2 2 0), and (3 0 0) planes respectively.27,28 No signatures of other phases containing chlorine were detected. On exposing the films to different solvent vapour many changes are introduced in the XRD pattern. Few details are given in Table 1. On exposing to DMSO vapour the standard peaks corresponding to perovskite were completely missing. However, many low intensity peaks were observed, prominent among them were at 6.6°, 7.3° and 9.3° values of 2θ. These peaks were attributed to intercalation of DMSO or a mixture of DMSO and the organic part into PbI2 phase. To confirm whether this is due to the intercalation of only DMSO or DMSO plus organic part into the PbI2 phase, we exposed pristine PbI2 film with DMSO vapour. In addition to standard PbI2 peak at 12.7° a new peak at 10.16° value of 2θ was observed (Fig. 2b). Therefore it is expected that the other peaks at 6.6°, 7.3° and 9.3° values of 2θ in the DMSO vapour exposed perovskite film are due to the intercalation of DMSO and organic component of the perovskite into the PbI2 lattice. This is in accordance with previously reported literature.29 This shows that PbI2 and organic component are formed due to the exposure of DMSO vapour. The quality of this PbI2 is however different from that of pure PbI2 film. This is in accordance with the absorption studies shown in the next sub-section. The perovskite film exposed to DMF vapour shows less intense standard perovskite peaks. Additionally peaks at 6.6°, 8.13° and 9.5° values of 2θ were observed. The PbI2 film shows additional peaks at 8.9° and 9.5° after exposing with DMF vapour. Therefore the peaks at 6.6° and 8.13° are attributed to the intercalation of DMF and organic part into PbI2. For acetone exposed perovskite film, additional peaks were observed at 6.6°, 7.3°, 9.3° and 10.6°. Acetone vapour exposed PbI2 film does not show any peak at lower 2θ values. Thus all the peaks at lower 2θ values in the perovskite film are due to the intercalation of acetone and organic part into PbI2. Above results indicate that the polar aprotic solvent vapour exposure leads to the formation of intercalation state or complex between solvent, PbI2, and the organic part. | ||
| Fig. 2 X-ray diffraction pattern of (a) perovskite film before and after exposure to different solvent vapours, (b) PbI2 films before and after exposure to different solvent vapours. | ||
| Solvent vapour | 2θ (degree) values of additional peaks observed upon solvent vapour exposure |
|---|---|
| DMSO | 6.6, 7.3, 9.3 and 11.9 |
| DMF | 6.6, 8.1 and 9.6 |
| Acetone | 6.6, 7.3, 9.3 and 10.6 |
| Water | 12.7 |
| 2-Propanol | 6.6, 7.3, 9.3 |
| Chlorobenzene | No additional peaks |
| Toluene | No additional peaks |
Water vapour exposure for 30 minutes on perovskite film shows a new peak at 12.7°. This peak is attributed to PbI2 (0 0 1) phase.25 This shows that upon exposing to water vapour the beginning of PbI2 formation takes place in 30 minutes time. The standard perovskite peaks were also seen which indicates that complete conversion of perovskite into PbI2 does not take place in this time scale. The perovskite film exposed to 2-propanol vapour showed very low intensity peaks in the lower 2θ values. In analogy with studies on DMF and DMSO vapours these are also attributed to the intercalation of solvent and organic component into the PbI2 phase. Upon exposure with chlorobenzene, no prominent changes in the XRD pattern were observed. However by exposing with non-polar toluene solvent the intensity of main peak improves. This shows enhancement in grain size of perovskite upon exposing with toluene vapour. The crystallite size ‘d’ was calculated using the following Scherer formula:
d = 0.9λ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ, cosθ, |
For the as prepared film crystallite size calculated was 28.8 nm while after exposing to toluene it improved to 32.1 nm.
UV-visible absorption spectroscopy
Fig. 3a shows the UV-visible absorption spectra of the annealed perovskite and PbI2 film. Optical bandgap was calculated by using the following relation:Where h is the planks constant, c is the speed of light in vaccum and λ is the absorption edge.

 | ||
| Fig. 3 UV-visible absorption spectra of (a) annealed perovskite film and PbI2 film (b) perovskite films after exposure to different solvent vapours (c) PbI2 films after exposure to different solvent vapours. | ||
Tabulated values of absorption edge and corresponding calculated optical bandgap are shown in Table 2. It shows the absorption edge at around ∼788 nm for CH3NH3PbI3, while that for PbI2 is at ∼523 nm corresponding to optical bandgap of 1.57 eV and 2.37 eV respectively. The absorption spectrum matches with those given in the literature.30 Fig. 3b shows the absorption spectra of CH3NH3PbI3 films after exposing to different solvent vapours for 30 minutes. The absorption edge remains same for films exposed to 2-propanol, toluene, and chlorobenzene vapours. It decreases slightly to 780 nm when exposed to water vapour. This indicates an increase in optical bandgap due to the partial conversion to PbI2, which is indicated in XRD result also. Minimum changes in films exposed to toluene and chlorobenzene vapours were observed. This shows that there are nearly no changes in the absorption properties of the films upon exposing to these vapours. For the films exposed to DMF, DMSO, and acetone vapours, the absorption spectra change remarkably. DMF vapour exposed film shows blue shifted absorption spectra compared to that of exposed PbI2 film exposed to the same vapour. Fig. 3c shows the absorption spectra of PbI2 film exposed to different vapours. This signifies that by exposing to DMF vapour, perovskite breaks down partially into PbI2 and the organic components giving rise to blue shift. This is again confirmed by the change in colour of perovskite film from black to faint yellow and thereafter transparent after exposure to DMF vapour. The colour of PbI2 film exposed to DMF vapour also turns transparent from initially yellow colour. Film exposed to DMSO vapour also shows blue shifted absorption spectrum compared to that of exposed PbI2 film. Also the colour of film becomes faint yellow initially and then transparent. It can be therefore said that by exposing to DMSO vapour also perovskite breaks down to PbI2 and organic components. The perovskite film which is exposed to acetone vapour shows absorption property completely different from the pure perovskite film. A closer look at the spectrum reveals that it is similar to pristine PbI2 film with small peaks having reduced absorption at 515 nm, 455 nm and 415 nm. This shows that by exposing the perovskite film to acetone vapour also PbI2 and organic components are formed. The decrease in absorption is assigned to the partial dissolution of formed PbI2 in acetone. No changes were observed in the pristine PbI2 absorption spectrum after exposing to acetone. This shows that PbI2 becomes soluble in acetone due to the presence of organic component in perovskite. This is due to strong interaction of I− with PbI2.
| Solvent vapour | Absorption edge (nm)/calculated bandgap (eV) | |
|---|---|---|
| Perovskite | PbI2 | |
| Unexposed | 788/1.57 | 523/2.37 |
| DMSO | 444//2.79 | Broad edge 488/2.54 |
| DMF | 429/2.89 | 471/2.63 |
| Acetone | Clear edge not seen | 523/2.37 |
| Water | 780/1.59 | 523/2.37 |
| 2-Propanol | 788/1.57 | 523/2.37 |
| Chlorobenzene | 788/1.57 | 523/2.37 |
| Toluene | 788/1.57 | 523/2.37 |
The absorption coefficient reduces (2.4 × 104 cm−1 for perovskite to 1.4 × 104 cm−1 for 2-propanol and 0.2 × 104 cm−1 for water vapour exposed samples around the absorption edge) for perovskite film exposed to water vapour and to a lesser extent in film exposed to 2-propanol vapour. This reduction in absorption coefficient by exposing to water vapour may be due to the conversion of perovskite to hydrated forms. It has been reported previously that perovskite can form (CH3NH3)4PbI6·2H2O due to water vapour exposure.31 The changes in absorption spectra of the film exposed to 2-propanol vapour also shows that the perovskite properties changes.
Raman spectroscopy
The above discussion was further supported by Raman spectroscopic studies. The excitation wavelength used in the present study is 632.8 nm which is near resonance for Raman measurement of lead halide perovskite. We found assortment in literature in assigning the Raman bands for CH3NH3PbI3 material. The Raman spectrum for pure PbI2 (2H-polytype) is shown in Fig. 4. The most dominating peak corresponding to PbI2 is at 113 cm−1. For the films unexposed to any vapour, Raman bands obtained at 71, 110 cm−1 were attributed to the Pb–I cage in CH3NH3PbI3 (Fig. 4). The band at 138 cm−1 is attributed to the libration motion of methyl ammonium (MA) cation. The bands at 260 and 350 cm−1 can be assigned to the MA cation torsional mode of CH3NH3PbI3. Raman bands obtained in the present study, their assignment and comparison with literature is given in Table 3. | ||
| Fig. 4 Raman spectra of the PbI2 and perovskite films before and after exposing to different solvent vapours. | ||
| Sr. no. | Observed peak position for CH3NH3PbI3 in cm−1 | Peak positions given in literature31–34 for CH3NH3PbI3 in cm−1 | Assignment |
|---|---|---|---|
| 1 | 71 | 69, 71 | Pb–I stretching |
| 2 | 110 | 110 | Pb–I stretching |
| 3 | 138 | 138 | Libration of organic cation |
| 4 | 260 | 250 | MA torsion mode |
| 5 | 350 | 348 | MA torsion mode |
The Raman spectrum for each perovskite thin film after exposing to different vapours for 30 minutes are also shown in the Fig. 4. Film exposed to DMF vapour shows only two bands at 71 and 110 cm−1. In addition the band at 110 cm−1 was with much reduced intensity than that in the unexposed film. The bands at higher wavenumbers were completely absent after exposing the film to DMF vapour. If the light source intensity and the wavelength of the source are kept same the Raman peak intensity is mainly affected by concentration of the sample or number of molecules and the scattering properties of the sample.35,36 Absence of the peaks at 138, 260 and 350 cm−1 which are corresponding to the modes of organic part of the perovskite, indicates that the concentration of number of scattering molecules and their scattering property is reduced. This indicates towards the dissolution of organic part of CH3NH3PbI3 due to the solubility in DMF. The presence of the peak at 110 cm−1 indicates to the conversion to the PbI2 phase. The reduced intensity of this peak points to the dissolution of the PbI2 phase also in the solvent. A small shift of 3 cm−1 of this peak towards lower wave-number compared to 113 cm−1 of the pure PbI2 phase indicates the increase in bond length and hence the weakening of the bond. The quality of PbI2 is not same to the pure PbI2 film due to the intercalation of organic matter and solvent. This is in agreement with the XRD results shown. These results show the ability of DMF to dissolve both the organic and inorganic counterparts of CH3NH3PbI3. Similar results were obtained by exposing the films to DMSO vapour. The band at 110 cm−1 was broader than the DMF vapour exposed film implying a highly disordered structure of PbI2 formed.
In case of acetone vapour exposure, all the bands for the perovskite were observed, but with slightly different intensities. Small broadening of the peaks corresponding to Pb–I cage at 110 cm−1, and that of peak corresponding to MA libration mode at 138 cm−1 suggests the partial and selective weakening of the bonds corresponding to these modes. In case of 2-propanol vapour exposure, we observed very small changes in the band intensity. Exposing the film to chlorobenzene vapour, no changes in the Raman spectra were observed. Toluene vapour exposed film shows peak shift from 138 cm−1 to 142 cm−1. This shift towards higher wavenumber indicates shortening of the bond length. Bond length is inversely proportional to the bond strength.37 This shift therefore indicates strengthening of the bond corresponding to the libration mode of the MA cation. A lot of research has been done on the degradation of perovskite by water/humidity.38,39 We found that after 30 minutes of exposure perovskite shows a very small decrease in intensity corresponding to Pb–I cage and broadening of CH3NH3PbI3 cage bands.
Scanning electron microscopy
Fig. 5 shows the scanning electron microscope images of perovskite film and that by exposing to different solvent vapours for 30 minutes each. Drastic changes were found in morphology after exposure to DMSO and DMF. Broom like structures are seen in the films in the micrometre scale. Widths of the structures being much smaller in the DMSO vapour exposure film. Similar trend was observed at the nanometre scale images. Acetone vapour exposed film also shows drastic changes in the film morphology. The effect of water vapour shows changes in the shape of the islands found in the film. At the nanometre scale features disappeared in the film which were visible in the as deposited film. 2-Propanol vapour shows changes in the size of islands and voids were found in each individual island increasing the porosity of the film. Minimum effect was found by exposing the films to chlorobenzene and toluene both at the nanoscale as well as micrometre scale. However, as per the results of previous sections it is found that toluene exposure improves the crystallite size. This may be due to very small solubility which is just sufficient to minutely dissolve the small boundaries and thereby fusing them. | ||
| Fig. 5 Scanning electron microscope images in both the nanoscale and microscopic scale of the unexposed and solvent vapour exposed perovskite films. | ||
Fourier transform infrared spectroscopy
FTIR spectra in the range between 4000 cm−1 to 400 cm−1 were recorded which belongs to the absorption of organic materials.40 Therefore the effect of solvent vapour on the organic part of the perovskite can be explained by using FTIR spectroscopy. Fig. 6 shows the FTIR spectra of CH3NH3PbI3 before and after exposure to various solvent vapours. The main vibrational lines in the spectra can be assigned to fundamental modes of the CH3NH3 cation and anharmonic combinations thereof. Three characteristics are commonly examined in the FTIR spectra: peak position, integrated peak intensity, and peak width. The peak positions provide a fingerprint that can be used to identify chemical groups. The integrated intensity is proportional to the concentration of absorbing bonds.40 The peak width is a function of the homogeneity of the chemical bonding. Table 4 shows the characteristic vibrational bands of CH3NH3PbI3 obtained in the present study and that of reported in the literature. All NH3 related peaks are stronger than the CH3 vibrations, mainly due to the positive charge located on the ammonium group. After exposing the films to toluene and chlorobenzene vapour no changes in the vibrational band structures like shift in peak position, integrated peak intensity and peak width were observed. This shows that the vibrational modes, their concentration and the homogeneity remain intact by exposing with these vapours. Since the peak width remains same it implies that defects and bond strains are not introduced in the functional groups. Upon exposing with water vapour no change in the FTIR spectrum was observed. However there are reports where changes in the FTIR spectrum have been reported.42 These are however for long time exposure to water vapour. It can therefore be concluded that water vapour exposure for this time scale is not sufficient to bring major changes in the methyl ammonium part of the material.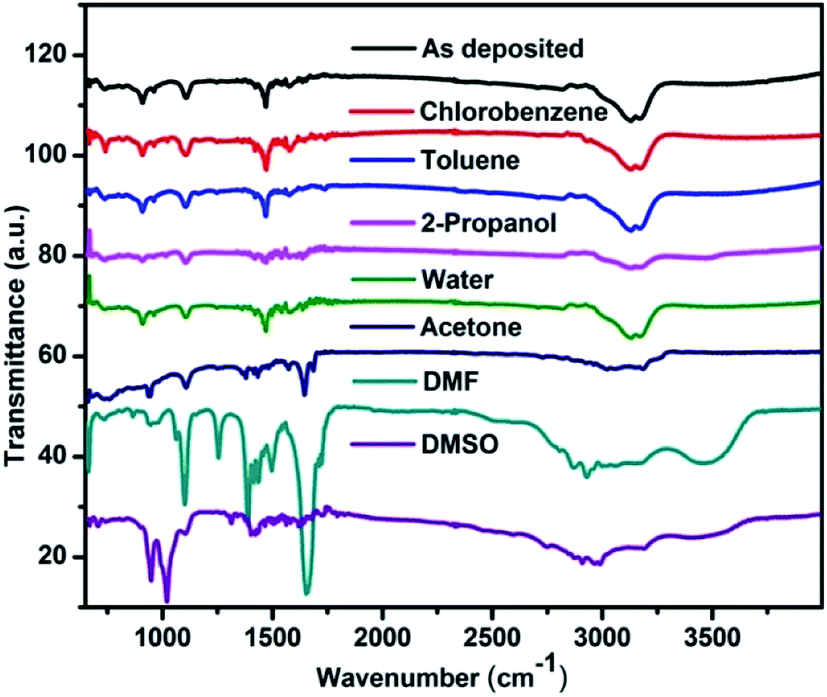 | ||
| Fig. 6 FTIR spectra of the perovskite films before and after exposing to different solvent vapours. | ||
| Vibrational bands observed | Vibrational bands reported40,41 | Assignment for CH3NH3PbI3 |
|---|---|---|
| 908 | 911 | CH3NH3+ rocking |
| 960 | 960 | C–N stretching |
| 1421 | 1425 | AsymCH3 bend |
| 1469 | 1469 | SymNH3+ bend |
| 1577 | 1577 | AsymNH3+ bend |
| 3000 | 3005 | C–H stretch |
| 3100 | 3105 | N–H stretch (sym) |
| 3130 | 3132 | SymNH3+ stretch |
| 3170 | 3179 | AsymNH3+ stretch |
Upon exposing with 2-propanol and acetone vapour the symmetric and asymmetric NH3 stretch integrated peak intensity decreases. This indicates that the concentration of these absorbing bonds reduces upon these solvent vapour exposures. The intensity corresponding to CH3–NH3+ rocking, symmetric and asymmetric NH3+ bend and C–N stretching vibrational modes also reduces which indicates that the concentration of these groups also reduces upon solvent vapour exposure. This is an indication of the dissolution of organic counterpart of the material. Upon acetone vapour exposure a peak at 1645 cm−1 appears, the intensity of which increases with exposure time. Acetone having the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) functional group shows an intense peak at 1715 cm−1 wavenumber.40 The shift in this peak is therefore due to change in chemical environment corresponding to this functional group due to the interaction with perovskite.
O) functional group shows an intense peak at 1715 cm−1 wavenumber.40 The shift in this peak is therefore due to change in chemical environment corresponding to this functional group due to the interaction with perovskite.
When the perovskite samples were exposed to DMF vapour, we see changes in the vibrational band within very short period of time (ESI†). The symmetric and asymmetric CH3 and NH3 stretch peaks becomes broad. This suggests that the homogeneity of these chemical bonds is disturbed due to defects and bond strain. Small shift in bond strengths cause small shifts in peak positions. The net result is broadening of the absorption band. After 30 minutes exposure the peaks disappears. In the finger print region many new peaks appears in the spectrum corresponding to DMF. Similar results were observed for the DMSO vapour exposed film also.
Discussion
CH3NH3PbI3 is an ionic crystal in which the organic part CH3NH3+ is the cation with +1 oxidation state, the metal ion Pb2+ has +2 oxidation state while the halogen I is the anion with −1 oxidation state.43 Crystals of this perovskite are held together by ionic interactions between the organic and inorganic portions as well as hydrogen-bonding interactions between the hydrogens in NH3+ head and the iodine atoms. The hydrogen bonding interactions between the organic cation and halide ions of the perovskite lattice provide structural stability. The effect of particular solvent vapour on the perovskite properties can be explained by considering the solvent properties like dielectric constant, molecular dipole moment, Gutmann donor number, boiling point etc. Few properties of the solvents used in the present study have been given in Table 5 below. Aprotic polar solvents lack O–H or N–H bonds and so do not have hydrogen bonding.44 However, due to the presence of carbonyl or sulfoxide groups they can act as hydrogen bond acceptors. The Pb2+ and CH3NH3+ cations in CH3NH3PbI3 will have the ability to readily bond with the partially negative oxygen, nitrogen or sulphur atoms in aprotic solvents. This bonding enables dissociation of the PbI6 octahedral frame that forms the skeleton of the organic–inorganic perovskite material, thereby resulting in high solubility of the perovskite material in polar aprotic solvents. Therefore, the aprotic solvents have the ability to form PbI2-solvent compounds.| Solvent | Dielectric constant | Dipole moment (D) | Gutmann donor number44 (kcal mol−1) | Boiling point (°C) |
|---|---|---|---|---|
| DMSO | 47.2 | 3.96 | 29.8 | 189 |
| DMF | 36.7 | 3.82 | 26.6 | 153 |
| Acetone | 20.7 | 2.88 | 17 | 56 |
| 2-Propanol | 10.9 | 1.58 | 21.1 | 82.5 |
| Water | 80.1 | 1.85 | 18 | 100 |
| Chlorobenzene | 5.62 | 1.69 | 3.3 | 132 |
| Toluene | 2.38 | 0.37 | 0.1 | 110.6 |
Inspite of the fact that DMSO, DMF and acetone are polar aprotic solvents there is a difference in the effects their solvent vapours have on the perovskite film properties. In general, higher solvent polarity enables better solubility of ionic compounds. DMSO has the largest value of dipole moment and dielectric constant followed by DMF and then acetone. It is therefore perovskite films exposed to DMSO vapour shows no XRD peaks corresponding to CH3NH3PbI3. The combined study of UV-visible absorption, XRD and Raman spectroscopy suggested the formation of solvent and organic component intercalated PbI2 in the DMSO vapour exposed films. Also the bands corresponding to organic component were completely missing in FTIR and Raman spectra. The effect of acetone vapour was minimal due to its low polarity among the polar aprotic solvents. In addition DMSO has the higher value of Gutmann donor number compared to DMF and therefore coordinating ability of DMSO is greater. The Gutmann donor number is a quantitative measure of Lewis basicity and gives the ability of a solvent to solvate cations and Lewis acids.45,47 It is defined as the negative enthalpy value for the 1:1 adduct formation between a Lewis base and the standard Lewis acid antimony pentachloride, in dilute solution in the non-coordinating solvent 1,2-dichloroethane with a zero donor number. The donor number of acetone is much smaller than that of DMSO and DMF. This is an indication of the reduced ability of acetone to solvate the Pb2+ and CH3NH3+ cation as compared to the other two polar aprotic solvents. Our experiments have shown that acetone is not a good solvent for PbI2. However, when CH3NH3I is added, PbI2 becomes readily soluble in acetone. It suggests that as CH3NH3PbI3 comes in contact of acetone vapours, it dissolves the organic part and results in intercalation of acetone and organic component in PbI2. Several new XRD peaks in the perovskite film after exposing to acetone vapour confirms this. FTIR study confirms the fact that acetone easily affects the organic part in CH3NH3PbI3. The above discussion indicates that DMSO has the strongest coordination capability with the Pb2+ and CH3NH3+ cations in CH3NH3PbI3. This coordination ability can be as a monodentate ligand by donating a pair of electrons to the cations through atom of the S–O bond and also by hydrogen bonding.48 Therefore DMF seems to be a weaker field ligand compared to DMSO but stronger than acetone. For the aprotic solvents therefore, the solvent coordination ability follows the same trend as its Gutmann donor number.
In addition to the solubility factor, acetone have comparatively low boiling point compared to DMF and DMSO. It is therefore, once deposited, DMF and DMSO does not evaporate easily from the perovskite surface. This results in more effective dissolution of perovskite and therefore reduction in standard perovskite peak intensity. This effect is most prominent in the case of DMSO which has the highest boiling point of 189 °C as compared to 153 °C for DMF. To confirm this, the exact amount of solvent settled on the exposed film was measured by weighing the coated film before and after exposing with the specific solvent vapours. Results are given in Table 6. It clearly shows that solvents with high boiling point stick more readily to the film after settling on the surface. In the process of dissolution, solute separates into ions or molecules, and molecules of solvent surround each ion or molecule. More the number of molecules, more the dissolution expected.
| Solvent/molecular weight | Weight of solvent after exposing to solvent vapours (mg)/number of moles of solvent |
|---|---|
| DMSO/78.13 | 0.90/1.15 × 10−5 |
| DMF/73.09 | 0.66/9.03 × 10−6 |
| Acetone/58.08 | 0.07/1.20 × 10−6 |
| Water/18.01 | 0.63/3.50 × 10−5 |
| 2-Propanol/60.10 | 0.15/2.47 × 10−6 |
| Chlorobenzene/112.56 | 0.85/7.55 × 10−6 |
| Toluene/92.14 | 0.17/1.84 × 10−6 |
2-Propanol and water used in the present study are polar protic solvents that possess O–H bonds, and so they can participate in hydrogen bonding. These solvents can also serve as acids (sources of protons) and weak nucleophiles (forming bonds with strong electrophiles). Due to the H bonding present in these solvents, anion I− can be solvated. The solubility of PbI2 is very low in these solvents. This is confirmed by the fact that no changes were observed in the absorption and XRD pattern of PbI2 film exposed to these solvent vapours. However, considerable changes were observed in the CH3NH3PbI3 films due to these solvent vapours. Even though both these solvents are polar protic, the degradation mechanism upon exposure to their solvent vapour seems to be different. Negligible or no changes were observed in the FTIR spectra of film exposed to water vapour for 30 min. This shows no changes take place in the methylammonium ion. Raman spectroscopy results show a slight decrease in peak intensity corresponding to Pb–I stretching and libration mode of organic cation, which indicates the decrease in the number of moieties corresponding to these modes. It also shows a decrease in CH3NH3+–I− hydrogen bond and Pb2+–I− bonds. Water is a polar solvent with high dielectric constant and strong hydrogen bonding. Water vapour can, therefore, weaken the above hydrogen bonds in CH3NH3PbI3, leading to the formation of a small amount of PbI2 (As shown in the XRD result) by liberating some CH3NH3+ and I−. X-ray diffraction pattern of 2-propanol vapour exposed perovskite films show new very low-intensity peaks in lower 2θ values. We attribute this to the intercalation of the organic component of perovskite into the PbI2 lattice. This effect is similar but small in magnitude as compared to that of aprotic solvents. This effect was not observed for perovskite film exposed to water vapours. This is due to the ability of 2-propanol to solvate the organic component (as shown in the FTIR spectra). 2-Propanol has a lower polarity compared to water but a higher Gutmann donor number. This is an indication of higher coordination ability of organic cation by 2-propanol as compared to water, inspite of more number of water vapour molecules settled on the film as shown in Table 6.
Both chlorobenzene and toluene have lower values of dipole moment and very low values of Gutmann donor numbers; toluene has lower numbers than chlorobenzene. This shows a very weak ability of toluene to affect the ionic perovskite molecule. Crystallinity changes indicate that toluene has at least some ability to dissolve CH3NH3PbI3. This weak ability is helpful by weakly dissolving the perovskite and thereby healing the interface between grain boundaries at the nanoscale and enhancing the crystallinity. The increased XRD peak intensity indicates this in the film exposed to toluene vapour. It is observed that the coordination ability of different solvents with the cations of CH3NH3PbI3 follows the same trend as the Gutmann donor number. Thus donor number is an important parameter and can be used to interpret the coordination ability of a solvent, with perovskite materials.
Conclusions
We have therefore produced a comprehensive study of the effect of various solvent vapours of different classes on the perovskite properties. CH3NH3PbI3 is ionic and can interact with different solvents, the magnitude of which depends on the solvent properties. We showed that aprotic solvents like DMSO and DMF considerably deteriorates the perovskite properties in a very short time due to their high molecular polarity, Gutmann donor number and high boiling point. Acetone also being an aprotic solvent deteriorates the perovskite properties but moderately, due to its limited ability to solvate the cation and interaction with the polar solute. Water and 2-propanol are both polar protic solvents but interacts differently with perovskite. Due to the higher Gutmann donor number of 2-propanol, it can solvate the organic cation CH3NH3+ more efficiently as compared to water. Having very low values of molecular dipole moment and Gutmann donor numbers, chlorobenzene and toluene are not able to affect perovskite much. The extremely weak ability of toluene vapour to interact with perovskite was helpful in fusing the neighbouring grains and heals the perovskite defects at the nanometre scale to improve the crystallinity. The effect of different solvent has been correlated to the solvent properties like dielectric constant, molecular dipole moment, Gutmann donor number, and boiling point. We show that donor number is an important parameter and can be used to interpret the coordination ability of a solvent with perovskite materials. The present study can be useful in understanding the negative effect of different solvent vapours and also the use of solvent vapours for post-deposition processing (like solvent vapour annealing) to improve their properties. We propose the use of mixed solvents to obtain tailor-made properties of solvents to modify the perovskite properties by vapour exposure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the DST project (DST/TMD/SERI/S38), New Delhi, India is acknowledged. SAM is thankful to UGC for RGNF. SSG is thankful to Dr Sidhanath Bhosale from CSIR-Indian Institute of Chemical Technology, Hyderabad, Dr Shinto Verghese from IACS, Kolkata and Prof. D. Venkataraman, University of Massachusetts Amherst for useful discussions.Notes and references
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS.
- E. D. Juarez, D. G. Quijano, D. F. G. Gutierrez, D. I. G. Gutierrez, J. I. Rodriguez and E. Sanchez, J. Alloys Compd., 2020, 817, 153076 CrossRef.
- J. Kang and J. H. Cho, Phys. Chem. Chem. Phys., 2020, 22, 13347 RSC.
- S. P. Senanayak, B. Yang, T. H. Thomas, N. Giesbrecht, W. Huang, E. Gann, B. Nair, K. Goedel, S. Guha, X. Moya, C. R. McNeill, P. Docampo, A. Sadhanala, R. H. Friend and H. Sirringhaus, Sci. Adv., 2017, 3, 1–11 Search PubMed.
- W. Yu, F. Li, L. Yu, M. R. Niazi, Y. Zou, D. Corzo, A. Basu, C. Ma, S. Dey, M. L. Tietze, U. Buttner, X. Wang, Z. Wang, M. N. Hedhili, C. Guo, T. Wu and A. Amassian, Nat. Commun., 2018, 9, 1–10 CrossRef.
- Z. Zhenfu, J. Liang, W. Zhihai, C. Jiong, Z. Miaomiao and H. Yafei, J. Mater. Sci., 2018, 53, 15430–15441 CrossRef CAS.
- A. Vassilakopoulou, D. Papadatos, I. Zakouras and I. Koutselas, J. Alloys Compd., 2017, 692, 589–598 CrossRef CAS.
- M. M. Stylianakis, T. Maksudov, A. Panagiotopoulos, G. Kakavelakis and K. Petridis, Materials, 2019, 16, 1–28 Search PubMed.
- H. Wang and D. H. Kim, Chem. Soc. Rev., 2017, 46, 5204–5236 RSC.
- X. Zhao, L. Huang, Y. Wang, X. Zhu, L. Li, G. Li and W. Sun, RSC Adv., 2020, 10, 32976–32983 RSC.
- B. Liu, S. Wang, Z. Ma, J. Ma, R. Ma and C. Wang, Appl. Surf. Sci., 2019, 467–468, 708–714 CrossRef CAS.
- Z. Yi, N. H. Ladi, X. Shai, H. Li, Y. Shen and M. Wang, Nanoscale Adv., 2019, 1, 1276 RSC.
- I. K. Popoola, M. A. Gondal and T. F. Qahtan, Renewable Sustainable Energy Rev., 2018, 82, 3127–3151 CrossRef CAS.
- Q. Ou, X. Bao, Y. Zhang, H. Shao, G. Xing and X. Li, Nano Materials Science, 2019, 1, 268–287 CrossRef.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS.
- J. Feng, APL Mater., 2014, 2, 081801 CrossRef.
- R. K. Battula, G. Veerappan, P. Bhyrappa, C. Sudakar and E. Ramasamy, RSC Adv., 2020, 10, 30767–30775 RSC.
- P. H. Huang, Y. H. Wang, J. C. Ke and C. J. Huang, Energies, 2017, 10, 1–8 CAS.
- Y. Li, L. Zhi, G. Ge, Z. Zhao, X. Cao, F. Chen, X. Cui, F. Lin, L. Ci, J. Sun, D. Zhuang and J. Wei, Cryst. Growth Des., 2019, 19, 959–965 CrossRef CAS.
- E. Radicchi, E. Mosconi, F. Elisei, F. Nunzi and F. De Angelis, ACS Appl. Energy Mater., 2019, 2, 3400–3409 CrossRef CAS.
- M. Ralaiarisoa, I. Salzmann, F. S. Zu and N. Koch, Adv. Electron. Mater., 2018, 4, 1–8 Search PubMed.
- Y. Huang, L. Zhao, J. Li, F. Lu and S. Wang, Nanoscale Adv., 2020, 2, 332 RSC.
- H. Xiong, G. DeLuca, Y. Rui, Y. Li, E. Reichmanis, Q. Zhang and H. Wang, Sol. Energy Mater. Sol. Cells, 2017, 166, 167–175 CrossRef CAS.
- J. Luo, R. Z. Qiu, Z. S. Yang, Y. X. Wang and Q. F. Zhang, RSC Adv., 2018, 8, 724–731 RSC.
- J. Chae, Q. Dong, J. Huang and A. Centrone, Nano Lett., 2015, 15, 8114–8121 CrossRef CAS.
- S. Wang, L. K. Ono, M. R. Leyden, Y. Kato, S. R. Raga, M. V. Lee and Y. Qi, J. Mater. Chem. A, 2015, 3, 14631–14641 RSC.
- A. K. Chauhan, P. Kumar, S. R. Pal, S. K. Srivastava and S. Muthiah, J. Mater. Sci., 2017, 52, 10886–10897 CrossRef CAS.
- Y. Zong, Y. Zhou, Y. Zhang, Z. Li, L. Zhang, M. G. Ju, M. Chen, S. Pang, X. C. Zeng and N. P. Padture, Chem, 2018, 4, 1404–1415 CAS.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. Il Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS.
- S. M. Jain, B. Philippe, E. M. J. Johansson, B. W. Park, H. Rensmo, T. Edvinsson and G. Boschloo, J. Mater. Chem. A, 2016, 4, 2630–2642 RSC.
- J. A. Christians, P. A. Miranda Herrera and P. V. Kamat, J. Am. Chem. Soc., 2015, 137, 1530–1538 CrossRef CAS.
- B. W. Park, S. M. Jain, X. Zhang, A. Hagfeldt, G. Boschloo and T. Edvinsson, ACS Nano, 2015, 9, 2088–2101 CrossRef CAS.
- R. Gottesman, L. Gouda, B. S. Kalanoor, E. Haltzi, S. Tirosh, E. Rosh-Hodesh, Y. Tischler, A. Zaban, C. Quarti, E. Mosconi and F. De Angelis, J. Phys. Chem. Lett., 2015, 6, 2332–2338 CrossRef CAS.
- M. Ledinský, P. Löper, B. Niesen, J. Holovský, S. J. Moon, J. H. Yum, S. De Wolf, A. Fejfar and C. Ballif, J. Phys. Chem. Lett., 2015, 6, 401–406 CrossRef.
- J. F. Ready, Industrial applications of lasers, Elsevier, 2nd edn, 1997 Search PubMed.
- R. R. Jones, D. C. Hooper, L. Zhang, D. Wolverson and V. K. Valev, Nanoscale Res. Lett., 2019, 14, 231 CrossRef.
- R. Segovia, G. Qu, M. Peng, X. Sun, H. Shi and B. Gao, Nanoscale Res. Lett., 2018, 13, 7 CrossRef.
- U. G. Jong, C. J. Yu, G. C. Ri, A. P. McMahon, N. M. Harrison, P. R. F. Barnes and A. Walsh, J. Mater. Chem. A, 2018, 6, 1067–1074 RSC.
- M. Salado, L. Contreras-Bernal, L. Caliò, A. Todinova, C. López-Santos, S. Ahmad, A. Borras, J. Idígoras and J. A. Anta, J. Mater. Chem. A, 2017, 5, 10917–10927 RSC.
- A. W. Fountain III, Infrared Spectral Interpretation: A Systematic Approach by B. Smith, Appl. Spectrosc., 1999, 53(6), 208A CrossRef.
- T. Glaser, C. Müller, M. Sendner, C. Krekeler, O. E. Semonin, T. D. Hull, O. Yaffe, J. S. Owen, W. Kowalsky, A. Pucci and R. Lovrinčić, J. Phys. Chem. Lett., 2015, 6, 2913–2918 CrossRef CAS.
- J. Idígoras, A. Todinova, J. R. Sánchez-Valencia, A. Barranco, A. Borrás and J. A. Anta, Phys. Chem. Chem. Phys., 2016, 18, 13583–13590 RSC.
- G. Li, J. Y.-L. Ho, M. Wong and H. S. Kwok, J. Phys. Chem. C, 2015, 119, 26883–26888 CrossRef CAS.
- A. J. Parker, Q. Rev., Chem. Soc., 1962, 16, 163–187 RSC.
- F. Cataldo, Eur. Chem. Bull., 2015, 4, 92–97 CAS.
- D. R. Joshi and N. Adhikari, J. Pharm. Res. Int., 2019, 28, 1–18 Search PubMed.
- Y. Deng, C. H. V. Brackle, X. Dai, J. Zhao, B. Chen and J. Huang, Sci. Adv., 2019, 5, eaax7537, DOI:10.1126/sciadv.aax7537.
- W. L. Reynolds, in Progress in Inorganic Chemistry, Dimethyl Sulfoxide in Inorganic Chemistry, ed. S. Lippard, John Wiley & Sons, Inc., New York, 1970, pp. 1–99 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra07926j |
| This journal is © The Royal Society of Chemistry 2020 |